

Abstract
Pathogenic mutations in MLH1, MSH2, PMS2, and MSH6 compromise DNA mismatch repair mechanisms and in the heterozygous state result in Lynch syndrome, which is typified by a predisposition to endometrial, ovarian, colorectal, gastric, breast, hematologic, and soft tissue cancers. Rarely, germline pathogenic aberrations in these genes are associated with the development of primary central nervous system tumors. We present a report of an adult female with no prior cancer history who presented with a multicentric, infiltrative supratentorial glioma involving both the left anterior temporal horn and left precentral gyrus. Surgical treatment and neuropathological/molecular evaluation of these lesions revealed discordant isocitrate dehydrogenase (IDH) status and histologic grade at these spatially distinct disease sites. A frameshift alteration within the MLH1 gene (p.R217fs*12, c.648delT) was identified in both lesions and subsequently identified in germline testing of a blood sample, consistent with Lynch syndrome. Despite distinct histopathologic features and divergent IDH status of the patient's tumors, the molecular findings suggest that both sites of intracranial neoplasia may have developed as a consequence of underlying monoallelic germline mismatch repair deficiency. This case illustrates the importance of characterizing the genetic profile of multicentric gliomas and highlights the oncogenic potential of germline mismatch repair gene pathogenic alterations within central nervous system gliomas.
Keywords: neoplasm of the central nervous system
CASE PRESENTATION
A 36-yr-old female with a history of cortical venous sinus thrombosis presented to our institution complaining of worsening headaches and seizures. Brain imaging obtained on admission revealed noncontiguous, T2/FLAIR hyperintense, non–contrast enhancing lesions involving the left temporal pole and left precentral gyrus. These findings raised concern for a multicentric low-grade infiltrating glioma. Though surgery was recommended, the patient opted to not pursue surgery at the time and was discharged with a plan for further follow-up. She was readmitted 1 month later for recurrent seizures, with radiographic evidence of disease progression and clinical concern for elevated intracranial pressure and local mass effect. She underwent an emergent, left-sided decompressive frontotemporal craniectomy and resection of her left anterior temporal pole (Fig. 1A). This was followed 2 wk later by a redo frontotemporal craniotomy for resection of the frontal tumor involving the precentral gyrus (Fig. 1B).
Figure 1.

(A) Preoperative (left) and postoperative (right) FLAIR sequence showing T2 hyperintensity in the anterior medial temporal lesion (arrow) before and after resection. (B) Demonstrates a similar appearing lesion involving the left frontal precentral gyrus. (C) Hematoxylin and eosin (H&E) and immunohistochemical staining of temporal and frontal tumor samples. Neither sample displayed microvascular proliferation or necrosis. Note the absence of IDH1 R132H staining in the frontal tumor sample and the ATRX loss and positive p53 stain in tumor tissue from both tumor sites. (D) Venn diagram depicting shared and distinct mutations of the somatic (tumoral) and germline (nontumoral) tissue samples. Scale bar: 50 microns, applies to all panels (C).
On histopathology, the patient's intracranial tumors were found to be of different World Health Organization (WHO) grade and isocitrate dehydrogenase (IDH) status, with histological evidence for shared pathogenic alterations in ATRX and TP53 (Fig. 1C). The temporal lobe lesion was mildly hypercellular with minimal cytologic atypia, with atypical cells showing immunoreactivity for IDH1 mutant protein (p.R132H) accompanied by strong labeling with p53 immunohistochemistry and loss of nuclear ATRX consistent with TP53 and ATRX mutations, respectively. No necrosis, microvascular proliferation, or mitotic activity was identified. Based on these findings, an integrated diagnosis of astrocytoma, IDH-mutant, central nervous system (CNS) WHO grade 2 was given for the temporal lesion. Examination of the left frontal lesion showed a hypercellular astrocytic glioma with increased mitotic activity without microvascular proliferation or necrosis. Immunohistochemistry was negative for the IDH1 p.R132H mutant protein and other IDH1 and IDH2 mutations were excluded by next-generation sequencing (NGS). Similar to the temporal lesion, loss of ATRX immunoreactivity and p53 overexpression was observed, consistent with gene mutations. Overall, the left frontal lesion best aligned to diffuse high-grade astrocytoma, IDH-wild-type, not elsewhere classified (NEC), at least CNS WHO grade 3. The “NEC” qualifier reflected that molecular information was available, but the tumor did not align to a specific WHO entity. Although the frontal lobe tumor was proven to be IDH-wild-type, the absence of necessary histological or genetic features required for the diagnosis of glioblastoma, IDH-wild-type precluded that designation. Specifically, there was no microvascular proliferation, necrosis, or evidence for EGFR amplification, whole-chromosome 7 gain and 10 loss (+7/−10), or TERT promoter region mutation.
Next-generation DNA sequencing of the tumor specimens confirmed these histopathology findings and revealed a shared pathogenic MLH1 frameshift alteration in both lesions (p.R217fs*12; Fig. 1D). Tumor-only sequencing could not definitively establish somatic versus germline alterations. However, it was noted that the MLH1 alteration had a higher variant allele frequency (VAF 0.68) compared to the candidate tumor-only driving alterations including alterations of IDH1 (VAF 0.09) and ATRX (VAF 0.14), suggesting that the MLH1 alteration could be germline and, therefore, present in both the neoplastic and background nonneoplastic cells. In light of the MLH1 mutation noted in both resection samples, further family history was obtained and was notable for early colorectal cancer deaths in the patient's maternal grandfather and uncle at ages 38 and 41, respectively. This prompted medical genetics consult for evaluation of Lynch syndrome. Subsequent sequencing from a blood sample detected the same pathogenic, heterozygous p.R217Afs*12 (c.648delT) MLH1 gene mutation, confirming a diagnosis of Lynch syndrome and aligning with the patient's family history of multigenerational, early colorectal cancer deaths. Lynch syndrome related phenotypic features of the patient and her affected family members are presented on Table 1.
Table 1.
Lynch syndrome phenotypic features
| Lynch syndrome clinical features | Proband | Grandfather (maternal) | Uncle (maternal) | Comments |
|---|---|---|---|---|
| Endometrial carcinoma | No | No | No | — |
| Colorectal neoplasm | No | Yes | Yes | Only feature noted in immediate family members |
| Gastric carcinoma | No | Unknown | Unknown | — |
| Ovarian carcinoma | No | N/A | N/A | — |
| Extracolonic neoplasmsa | No | Unknown | Unknown | — |
| Glioma | Yes | Unknown | Unknown | Sole presenting feature of patient in question |
| Breast malignancies | No | Unknown | Unknown | — |
| Sarcoma | No | Unknown | Unknown | — |
| Leukemia | No | Unknown | Unknown | — |
List of clinical features based on the OMIM clinical synopsis (# 120435; Lynch Syndrome 1).
aIncluding small bowel, biliary tract, urothelial, and bladder.
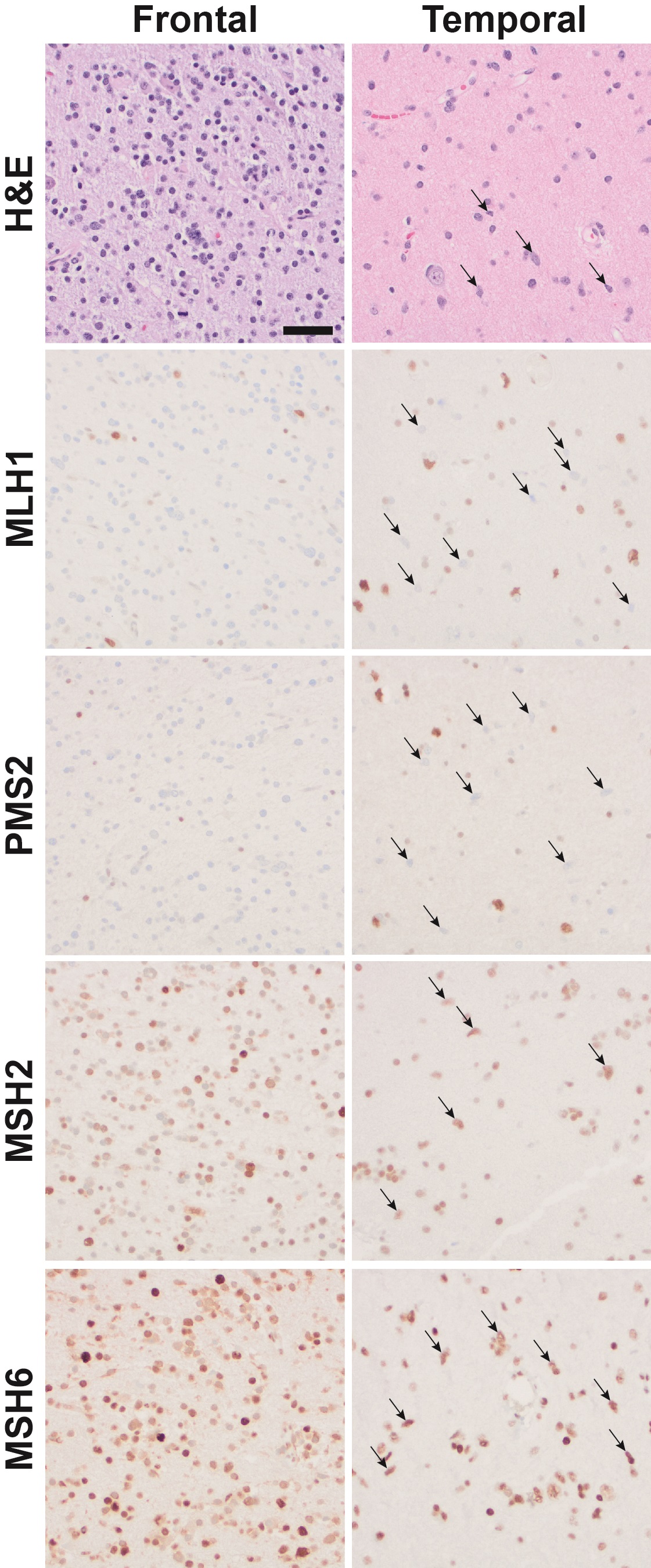
Genetic features of the tumors and germline are depicted in Figure 1D, with the complete detected variants listed in Supplemental Table 1; MGMT promoter methylation testing was negative for hypermethylation in both lesions. Consistent with the mismatch repair–deficient (MMRD) state, both tumors showed increased tumor mutation burden at 24.5 and 31.1 mutations per megabase for the temporal and frontal tumors, respectively. Immunohistochemical stains for mismatch repair proteins Msh2, Msh6, Mlh1, and Pms2 showed loss of tumor cell reactivity for Mlh1 and Pms2 in both tumors, a pattern compatible with MLH1 mutation and concomitant instability and loss of immunoreactivity for Pms2 (Supplemental Fig. 1). High microsatellite instability (MSI) status was detected in the frontal lobe tumor with 18 of 226 MSI sites altered (8.0%; cutoff > 5% required for MSI-High). MSI was not increased in the temporal lobe tumor; however, the sensitivity of this analysis would be limited by low tumor cell fraction in that sampling and by bulk tumor analysis (Touat et al. 2020). Loss of immunoreactivity for the Mlh1 protein in the frontal lobe tumor was associated with detection of a second pathogenic MLH1 frameshift alteration (p.D450fs*41, c.1348delG). In the temporal lobe tumor, loss of Mlh1 protein immunoreactivity was consistent with a second pathogenic MLH1 alteration; however, no mutations were detected by sequencing, possibly as a limitation of the low tumor cell content or non-sequence-related inactivating alterations such as MLH1 promoter hypermethylation.
Postoperatively, the patient completed 6 weeks of adjuvant fractionated radiotherapy with concurrent temozolomide, which was complicated by severe anemia. The patient was counseled on solid tumor screening recommendations for Lynch syndrome. She also elected to pursue an additional course of temozolomide considering interval imaging findings of nodular extensions along the margins of the frontal resection cavity.
TECHNICAL ANALYSIS
NGS Panels
The GeneTrails Comprehensive Solid Tumor Panel (CSTP) is a customizable NGS test that can identify clinically relevant genetic alterations. The CSTP is comprised of DNA and RNA amplicon-based libraries capable of providing tumor mutation burden estimates and detecting single-nucleotide polymorphisms, insertions/deletions, mutations, copy-number alterations, fusion events, and MSI status based on analysis of 226 short tandem repeats. The panel covers 225 cancer-associated genes (199 whole-exon, 26 hotspot) with a sequencing footprint of 0.61 megabases, and fusion coverage of 21 target genes by RNA-based sequencing (full gene list available at https://knightdxlabs.ohsu.edu/home/test-details?id=GeneTrails+Comprehensive+Solid+Tumor+Panel). Sequence variants are identified using FreeBayes and MuTect2 algorithms in a custom analysis pipeline. The average read depths in this analysis were 1938 (temporal) and 1585 (frontal). For germline testing, we used the CancerNext-Expanded assay. This is a NGS panel that allows for concurrent analysis of 77 genes associated with solid neoplasms across multiple organ systems. Genes are analyzed via next-generation or Sanger sequencing of coding domains as well as flanking 5′ and 3′ all introns and untranslated regions. MGMT methylation status was ascertained through methylation specific polymerase chain reaction (PCR) for regions of the MGMT promoter gene region.
Variant Interpretation
The c.648delT mutation in the coding exon 8 of the MLH1 gene results from a single-nucleotide deletion at position 648, causing a translational reading frame shift that generates an alternative stop codon downstream to the alteration (p.R217Afs*12, Chr 3:37053559 c.648delT, GRCh37) (Table 2). This alteration is predicted to result in pathogenic loss of function mutation by premature protein truncation or nonsense-mediated mRNA degradation. Heterozygous pathogenic alterations in the MLH1 gene are responsible for Lynch syndrome/hereditary nonpolyposis colorectal cancer (HNPCC), an autosomal dominant cancer predisposition syndrome. This patient was also found to be a heterozygous carrier of a pathogenic alteration arising from a G to A substitution after exon 48 of the ATM gene which is associated with ataxia telangiectasia (Chr1 1:108199966 c.7307 + 1G > A, GRCh37) (Table 2). The clinical significance of this alteration in the heterozygous state is less well-established but has been linked to increased cancer risk (Thompson et al. 2005).
Table 2.
Germline Lynch syndrome confirmatory workup: CancerNext-Expanded panel sequencing results from nontumoral tissue testing
| Gene/genomic location | Chromosome | Transcript | HGVS DNA ref (if genic) | HGVS protein ref | Variant type | Predicted effect | Allele frequency | Target coverage |
|---|---|---|---|---|---|---|---|---|
| MLH1 | 3 | NM_000249.3 | c.648delT | p.R217Afs*12 | Deletion | Frameshift | Not reported | Not reported |
| ATM | 11 | NM_000051.3 | c.7307 + 1G > A | N/A | Substitution | Splice site | Not reported | Not reported |
SUMMARY
Multicentric gliomas are defined as concurrent, noncontiguous primary CNS tumors in separate cerebral lobes or hemispheres and represent ∼2% of all gliomas (Mishra et al. 1990; Salvati et al. 2003). Although their exact pathogenesis is poorly understood, it has been hypothesized that they arise as spatially distinct sites of disease from a common progenitor population with early predisposing genetic aberrations (Leung et al. 2000; Wan et al. 2014; Hayes et al. 2017). Subsequent acquired somatic mutations further drive tumorigenesis, resulting in multicentric tumors that can have distinct histopathologic and molecular characteristics. In line with these postulates, the somatic mutations private to this patient's frontal and temporal tumors in the context of a germline MLH1 mutation suggests that the natural history of her lesions diverged after accumulating a shared set of genetic aberrations, including the ATRX and TP53 mutations, in the setting of mismatch repair deficiency.
The features of primary MMRD brain tumors have been described in previous reports (Dodgshun et al. 2020; Suwala et al. 2021; Kim et al. 2022). In one series, Kim et al. (2022) found that these gliomas to have higher tumor mutational burden compared to MMR intact controls, similar to both of our patient's tumors. In a large series of 32 patients with IDH-mutant MMRD gliomas, Suwala et al. (2021) found that MMRD tumors have a high frequency of ATRX and TP53 mutations, which is consistent with the genotypic profile of our patient's IDH-mutant temporal lobe lesion. Dodgshun et al. (2020) showed that replication repair–deficient high-grade gliomas have significantly fewer differentially methylated positions at CpG islands compared to gliomas with intact replication repair. Although our study does not address a genome-wide methylation pattern in this patient's tumors, we note that both tumor sites tested negative for MGMT promoter hypermethylation, which is very rare in IDH-mutant gliomas.
Multicentric gliomas can potentially reveal clinically unexpected tumor predisposition syndromes. In a study of four adult patients with spatially distinct low-grade gliomas, Hayes et al. (2017) identified a patient who was found to have germline TP53 mutation indicative of Li–Fraumeni syndrome. This series also included a patient with different IDH1 mutations in spatially distinct sites of disease (p.R132H and p.R132C). However, the occurrence of discordant IDH status (i.e., IDH-mutant and IDH-wild-type) in multicentric glioma appears to be very rare and reported mostly in childhood and adolescence in the setting of constitutional biallelic mismatch repair deficiency. The series of Dodgshun et al. (2020) includes one such case, identified in the setting of biallelic pathogenic MSH6 variants leading to Msh6 protein deficiency, and Galuppini et al. (2018) reported a similar case involving an adolescent female with biallelic MSH6 alterations. In a report of an adult with IDH-discordant multicentric glioma from Lombardi et al. (2018), mismatch repair proteins were found to be intact by immunohistochemistry. The case that we report here could, therefore, represent the first report of multicentric glioma with discordant IDH status occurring in an adult with Lynch syndrome. The presence of a few shared genetic alterations at both tumor sites suggests a shared precursor lesion to both tumors. There are rare reports of recurrent/progressive IDH-mutant low-grade gliomas in vivo that have lost the mutant IDH1 allele because of chromosomal deletions or amplifications of the wild-type IDH1 allele (Mazor et al. 2017). Such a mechanism could be considered in our case. However, the context of concurrent, noncontiguous tumors is different from the post-treatment recurrent/progressive gliomas that are shown to have IDH1 loss, and we do not observe copy-number loss of the IDH1 locus in our patient's frontal lobe tumor by NGS copy-number data. Still, we cannot exclude the possibility of copy-neutral loss of heterozygosity, which is not assessed on our panel. Nonetheless, it is of clinical interest that a tumor syndrome was not suspected in our patient until tumor NGS results raised this possibility. Detection of such syndromes have significant ramifications for diagnosis and treatment planning. Thus, patients with multicentric primary brain neoplasms should be strongly considered for evaluation for underlying germline syndromes.
MMRD gliomas have a poor clinical outcome, likely due in part to their distinct genetic features and resistance to the DNA alkylating agent temozolomide, which requires intact DNA mismatch repair for its genotoxic effect (Roos et al. 2007). Suwala et al. (2021) reported a median survival of 15 mo in their cohort of primary IDH-mutant MMRD gliomas—dramatically shorter than other forms of IDH-mutant brain tumors. Ionizing radiation (IR) exposure during glioma treatment and surveillance imaging studies, specifically computerized tomography (CT), may drive further mutagenesis in this patient population as well. Some studies have suggested that IR may contribute to DNA mismatch repair defects in colorectal carcinoma, but the mechanistic links between IR and MMR is uncertain (Rigter et al. 2018; Sun et al. 2023). Considering the poor response to standard of care adjuvant therapy, there is a need for further preclinical and clinical research to identify treatment regimens for gliomas in patients with Lynch syndrome. Despite great interest in immune-checkpoint inhibitors as a treatment for MMRD gliomas, clinical outcomes have been disappointing outside of some successes in pediatric constitutional biallelic MMR deficiency (Bouffet et al. 2016; Hodges et al. 2017; Henderson et al. 2022). In a retrospective cohort, patients treated with checkpoint inhibitors experienced substantially worse overall survival compared to those treated with systemic agents (Touat et al. 2020). Future trials are needed to better assess the efficacy of targeted oncotherapies and explore whether alternative or combinatorial treatment regimens may confer superior survival benefit.
In summary, we report here a case of histopathologically distinct, multicentric frontal and temporal astrocytomas with divergent IDH status arising in the setting of clinically unsuspected Lynch syndrome. This case demonstrates that deep molecular profiling and histological correlation between disease sites in multicentric gliomas can inform the diagnosis and potentially reveal clinically significant genetic findings.
ADDITIONAL INFORMATION
Data Deposition and Access
The ATM variant is listed in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession number VCV001066672.6. The patient did not consent to public deposition of raw sequencing data.
Ethics Statement
This was a case report that does not constitute human subject's research nor require Institutional Review Board (IRB) review according to the policy by the Oregon Health & Science University IRB. Informed consent was obtained by the patient for collection and reporting of clinical data. This study was conducted in accordance with the Declaration of Helsinki.
Acknowledgments
We thank the patient for allowing us to collect and synthesize her clinical data, making this report possible.
Author Contributions
H.T., C.N., M.D.W., and S.G.B. wrote the manuscript. H.T., C.N., C.S., A.M.R., P.A., R.F.B., and M.D.W. collected and analyzed the data. H.T., C.S., and M.D.W. generated figures and tables. M.D.W. oversaw the study. All authors critically reviewed the manuscript prior to submission.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Tejus A. Bale
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Bouffet E, Larouche V, Campbell BB, Merico D, De Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V. 2016. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34: 2206–2211. 10.1200/JCO.2016.66.6552 [DOI] [PubMed] [Google Scholar]
- Dodgshun AJ, Fukuoka K, Edwards M, Bianchi VJ, Das A, Sexton-Oates A, Larouche V, Vanan MI, Lindhorst S, Yalon M. 2020. Germline-driven replication repair-deficient high-grade gliomas exhibit unique hypomethylation patterns. Acta Neuropathol 140: 765–776. 10.1007/s00401-020-02209-8 [DOI] [PubMed] [Google Scholar]
- Galuppini F, Opocher E, Tabori U, Mammi I, Edwards M, Campbell B, Kelly J, Viel A, Quaia M, Rivieri F. 2018. Concomitant IDH wild-type glioblastoma and IDH1-mutant anaplastic astrocytoma in a patient with constitutional mismatch repair deficiency syndrome. Neuropathol Appl Neurobiol 44: 233–239. 10.1111/nan.12450 [DOI] [PubMed] [Google Scholar]
- Hayes J, Yu Y, Jalbert LE, Mazor T, Jones LE, Wood MD, Walsh KM, Bengtsson H, Hong C, Oberndorfer S, et al. 2017. Genomic analysis of the origins and evolution of multicentric diffuse lower-grade gliomas. Neuro Oncol 20: 632–641. 10.1093/neuonc/nox205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson JJ, Das A, Morgenstern DA, Sudhaman S, Bianchi V, Chung J, Negm L, Edwards M, Kram DE, Osborn M, et al. 2022. Immune checkpoint inhibition as single therapy for synchronous cancers exhibiting hypermutation: an IRRDC study. JCO Precis Oncol 6: e2100286. 10.1200/PO.21.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, De Groot J, Li S, Overwijk WW. 2017. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 19: 1047–1057. 10.1093/neuonc/nox026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lim KY, Park JW, Kang J, Won JK, Lee K, Shim Y, Park C-K, Kim S-K, Choi S-H. 2022. Sporadic and Lynch syndrome-associated mismatch repair-deficient brain tumors. Lab Invest 102: 160–171. 10.1038/s41374-021-00694-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SY, Yuen ST, Chan TL, Chan ASY, Ho JWC, Kwan K, Fan YW, Hung KN, Chung LP, Wyllie AH. 2000. Chromosomal instability and p53 inactivation are required for genesis of glioblastoma but not for colorectal cancer in patients with germline mismatch repair gene mutation. Oncogene 19: 4079–4083. 10.1038/sj.onc.1203740 [DOI] [PubMed] [Google Scholar]
- Lombardi G, Della Puppa A, Gardiman MP, Rossi S, Candiotto C, Zanatta L, Bertorelle R, De Rossi A, Fassan M, Zagonel V. 2018. Discordance of IDH mutational status between lesions in an adult patient with multifocal glioma. Neuro Oncol 20: 1142–1143. 10.1093/neuonc/noy080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, Smirnov IV, Marshall R, Souza CF, Shen Y, et al. 2017. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci 114: 10743–10748. 10.1073/pnas.1708914114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra H, Haran R, Singh J, Joseph T. 1990. Multicentric gliomas: two case reports and a review of the literature. Br J Neurosurg 4: 535–539. 10.3109/02688699008993805 [DOI] [PubMed] [Google Scholar]
- Rigter LS, Snaebjornsson P, Rosenberg EH, Atmodimedjo PN, Aleman BM, Ten Hoeve J, Geurts-Giele WR, van Ravesteyn TW, Hoeksel J, Meijer GA, et al. 2018. Double somatic mutations in mismatch repair genes are frequent in colorectal cancer after Hodgkin's lymphoma treatment. Gut 67: 447–455. 10.1136/gutjnl-2016-312608 [DOI] [PubMed] [Google Scholar]
- Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, Kaina B. 2007. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 26: 186–197. 10.1038/sj.onc.1209785 [DOI] [PubMed] [Google Scholar]
- Salvati M, Caroli E, Orlando ER, Frati A, Artizzu S, Ferrante L. 2003. Multicentric glioma: our experience in 25 patients and critical review of the literature. Neurosurg Rev 26: 275–279. 10.1007/s10143-003-0276-7 [DOI] [PubMed] [Google Scholar]
- Sun M, Moquet J, Ellender M, Bouffler S, Badie C, Baldwin-Cleland R, Monahan K, Latchford A, Lloyd D, Clark S, et al. 2023. Potential risks associated with the use of ionizing radiation for imaging and treatment of colorectal cancer in Lynch syndrome patients. Fam Cancer 22: 61–70. 10.1007/s10689-022-00299-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwala AK, Stichel D, Schrimpf D, Kloor M, Wefers AK, Reinhardt A, Maas SL, Kratz CP, Schweizer L, Hasselblatt M. 2021. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 141: 85–100. 10.1007/s00401-020-02243-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. 2005. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst 97: 813–822. 10.1093/jnci/dji141 [DOI] [PubMed] [Google Scholar]
- Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, Cortes-Ciriano I, Birzu C, Geduldig JE, Pelton K. 2020. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580: 517–523. 10.1038/s41586-020-2209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan KR, King NK, Low SY, Sitoh Y-Y, Lee HY, Wong CF, Ng WH. 2014. Synchronous multicentric glioblastoma with PNET and O subtypes: possible pathogenesis. Surg Neurol Int 5: 31. 10.4103/2152-7806.128182 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
{kind=link}
Data Availability Statement
The ATM variant is listed in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession number VCV001066672.6. The patient did not consent to public deposition of raw sequencing data.