

ABSTRACT
During macroautophagy/autophagy, precursor cisterna known as phagophores expand and sequester portions of the cytoplasm and/or organelles, and subsequently close resulting in double-membrane transport vesicles called autophagosomes. Autophagosomes fuse with lysosomes/vacuoles to allow the degradation and recycling of their cargoes. We previously showed that sequential binding of yeast Atg2 and Atg18 to Atg9, the only conserved transmembrane protein in autophagy, at the extremities of the phagophore mediates the establishment of membrane contact sites between the phagophore and the endoplasmic reticulum. As the Atg2-Atg18 complex transfers lipids between adjacent membranes in vitro, it has been postulated that this activity and the scramblase activity of the trimers formed by Atg9 are required for the phagophore expansion. Here, we present evidence that Atg9 indeed promotes Atg2-Atg18 complex-mediated lipid transfer in vitro, although this is not the only requirement for its function in vivo. In particular, we show that Atg9 function is dramatically compromised by a F627A mutation within the conserved interface between the transmembrane domains of the Atg9 monomers. Although Atg9F627A self-interacts and binds to the Atg2-Atg18 complex, the F627A mutation blocks the phagophore expansion and thus autophagy progression. This phenotype is conserved because the corresponding human ATG9A mutant severely impairs autophagy as well. Importantly, Atg9F627A has identical scramblase activity in vitro like Atg9, and as with the wild-type protein enhances Atg2-Atg18-mediated lipid transfer. Collectively, our data reveal that interactions of Atg9 trimers via their transmembrane segments play a key role in phagophore expansion beyond Atg9ʹs role as a lipid scramblase.Abbreviations: BafA1: bafilomycin A1; Cvt: cytoplasm-to-vacuole targeting; Cryo-EM: cryo-electron microscopy; ER: endoplasmic reticulum; GFP: green fluorescent protein; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MCS: membrane contact site; NBD-PE: N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine; PAS: phagophore assembly site; PE: phosphatidylethanolamine; prApe1: precursor Ape1; PtdIns3P: phosphatidylinositol-3-phosphate; SLB: supported lipid bilayer; SUV: small unilamellar vesicle; TMD: transmembrane domain; WT: wild type
KEYWORDS: Autophagosome, lipid transfer, membrane contact site, phagophore, scramblase
Introduction
Macroautophagy, hereafter autophagy, is a catabolic pathway conserved among eukaryotes that delivers intracellular material to lysosomes or vacuoles for degradation. Autophagy is a key regulator of cellular homeostasis as it mediates the clearance of cytoplasmic material and damaged or superfluous organelles, and thus ensures cell survival during stress conditions, such as nutrient starvation. Autophagy is initiated by the assembly of the phagophore assembly site (PAS), which results in the de novo formation of the phagophore. This cup-shaped cistern expands from an initial disc and sequesters the cytoplasmic cargo and upon fusion of its extremities, forms a double-membrane vesicle termed autophagosome. Autophagosomes then fuse with the vacuoles in yeast and plants, or with endosomes prior to fusion with the lysosomes in higher eukaryotes [1–4].
Autophagosomes biogenesis is coordinately catalyzed by a conserved set of proteins known as the Atg (autophagy related) proteins, which hierarchically assemble and get activated upon induction of autophagy [1,5]. A key activator of autophagosome formation is the Atg1/ULK kinase complex. Under normal growth conditions, this complex is phosphorylated by the nutrient-sensitive target of rapamycin complex 1 (TORC1), and thus kept inactive. However, lack of nutrients inactivates TORC1 and promotes the dephosphorylation of the Atg1/ULK kinase complex [1]. This results in Atg1/ULK kinase complex nucleation through phase separation and acts as the initial scaffold structure for the PAS formation [6–8].
The membrane source for the de novo autophagosome formation has been a subject of intense investigation [9]. Mostly based on studies carried out in the yeast Saccharomyces cerevisiae, it has been proposed that both COPII-coated vesicles [10,11] and Atg9-containing vesicles [12,13] are recruited to the PAS, probably providing the primary membrane material necessary to nucleate the initial phagophore [14]. Atg9-containing vesicles are derived from the Golgi and thought to be cytoplasmic membrane reservoirs that shuttle between this location and the PAS [13,15]. Atg9 is the only conserved integral membrane protein within the highly conserved core Atg machinery [16]. Atg1 kinase complex-mediated phosphorylation of Atg9 is required for its efficient interaction with Atg2 and Atg18 [17]. The Atg9, Atg2 and Atg18 proteins localize to the extremities of the expanding phagophore [5,18] and mediate essential membrane contact sites (MCSs) between the phagophore and the endoplasmic reticulum (ER) in yeast [19] or the mitochondria-associated membrane (MAM) in mammalian cells [20]. Indeed, the Atg2-Atg18 complex can tether liposomes in vitro [21,22]. The peculiar distribution of Atg9 on the phagophore might rely on the biophysical properties of this transmembrane protein and is required for the sequential recruitment of Atg2 and Atg18 at this location [19]. Recent findings have pointed out that the Atg2-Atg18 complex is a key component of the phagophore expansion [21,22]. That is, yeast Atg2 and mammalian ATG2A catalyze the transfer of phospholipids between model membranes, which is enhanced by the presence of Atg18 or its homolog WDR45/WIPI4 [23–25]. Surprisingly, a minimal N-terminal fragment of ATG2A can transfer lipids in vitro, and rescues autophagy in ATG2A ATG2B double-knockout cells [24], suggesting that the activity of Atg2 as a lipid transfer protein is the major contributor during autophagosome formation. The current model is that the complex formed by Atg9, Atg2, and Atg18 establishes MCSs between the phagophore and the ER to mediate the lipid supply required for the phagophore expansion into an autophagosome.
In this context, the role of Atg9 had remained enigmatic until recently, when cryo-electron microscopy (cryo-EM) structures of yeast Atg9 and human ATG9A were published [26–28]. It turns out that Atg9 and ATG9A have 4 transmembrane domains (TMDs) and form homotrimers that have lipid scramblase activity. It has been postulated that scramblase activity of Atg9 proteins equilibrates the lipids delivered by Atg2-Atg18 among the two leaflets of the lipid bilayer of the phagophore, thus promoting its expansion [26,27,29].
We have previously shown that the integrity of the interaction between Atg9, Atg2 and Atg18 is required for autophagy progression but the presence of Atg9 and Atg2 in the phagophore is not enough to complete this process, even when phagophore-ER MCSs are still present unless those concentrate to the extremities of this intermediate structure [19]. According to the cryo-EM structures, Atg9 proteins are trimers [26–28]. This arrangement creates a network of hydrophilic cavities within each Atg9 protomer that may allow the passage of the phospholipids between the leaflets of the membrane [26,27,29]. Importantly, the TMDs are also involved in the interactions between the Atg9 protomers. In particular, the TMDs contacting the Atg9 protomers are rich in phenylalanines, which are suitable to mediate protein-protein interactions and to coordinate protein conformational changes [28]. In agreement, all the cryo-EM studies reported more than one conformational state for this homotrimer complex [26,28,29].
In this study, we present evidence that the transmembrane region of Atg9 plays a critical role during the initial phase of phagophore expansion. We identified a single point mutation in the third TMD of Atg9 that completely abolishes its function in vivo without compromising the trimer assembly, recruitment to the PAS and interaction with the Atg2-Atg18 complex in vivo. Moreover, Atg9 lipid scramblase activity and its enhancement of the Atg2-Atg18 complex-mediated lipid transfer in vitro are not affected by this mutation. These observations suggest that the role of Atg9 at the phagophore-ER MCSs goes beyond the Atg9 scramblase activity.
Results
A screen for Atg9 function at the PAS
We previously showed that Atg9 has a key role in positioning the lipid transfer Atg2-Atg18 complex at the MCSs between the ER and the edges of the growing phagophore [19]. Intriguingly, the presence of both Atg9 and Atg2 at the phagophore is not sufficient to promote autophagy if the interaction between these two proteins is impaired, suggesting that the integrity of this complex at the phagophore-ER MCSs is essential [19]. As the scramblase activity of Atg9 coupled to the lipid transfer mediated by Atg2 could constitute a central machinery for membrane expansion, we set out to identify mutants in Atg9 that would specifically impair phagophore expansion. We therefore performed multiple structure-based sequence alignments to identify conserved amino acids that could affect the activity of Atg9 proteins. We performed this lack-of-function screening before the structures of Atg9 were published, though we hypothesized a lipid scramblase activity. For our search, we took the “credit card” mechanism into consideration to explain the scrambling of lipids and used the structure of the Nectria hematococca TMEM16 scramblase as a template [30,31]. We put particular emphasis on residues that could flank a hydrophilic groove and favor the passage of lipids, commonly located at the border of the TMD regions. We also focused on highly conserved positions located in the predicted TMDs, as these are the most conserved regions among the Atg9 sequences, and on charged residues flanking the transmembrane regions, as they could serve as lipid interaction sites (Figures 1B, S1 and S2). We then mutated candidate residues to alanines to abolish electrostatic interactions between charged residues and phospholipids or protein-protein interactions between aromatic residues. Additionally, we mutated D509, E510 and R673 to tryptophan, as these are equivalent to positions that were proposed to be required for the scrambling activity of human ATG9A [28].
Figure 1.
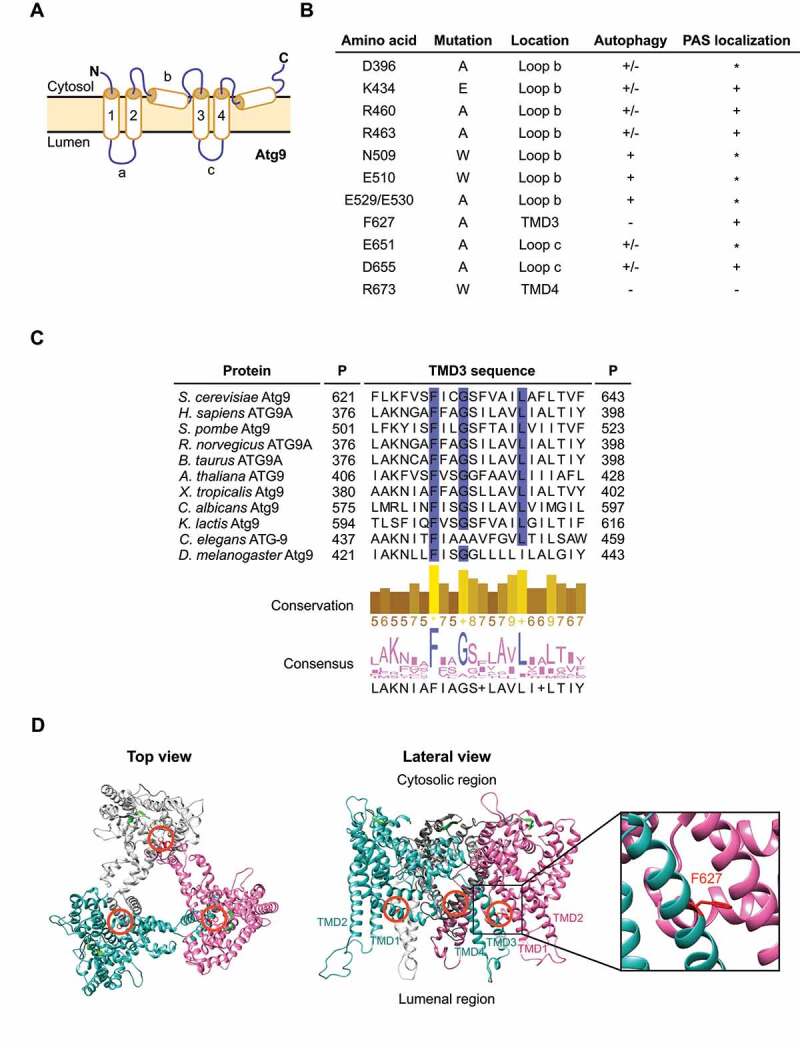
Screen for identification of Atg9 mutants. (A) Schematic representation of the S. cerevisiae Atg9 topology. As recently described, Atg9 presents 4 transmembrane domains (TMDs) (1 to 4) connected by 3 loops (a to c), two luminal and one cytosolic. The N and C termini possess one membrane helix which enter and exit through the cytosolic side of the membrane. (B) Overview of the Atg9 mutations generated in this study. Their position in the amino acid sequence and location within Atg9, i.e., the TMD or loop, are indicated based to the schematic representation in panel A. The effect of mutations on the autophagic flux and Atg9 localization to the PAS are detailed. The asterisk (*) highlights those mutants that although not easily detected at the PAS, did not display an autophagy defect. (C) Alignment of the amino acid sequences in the region that includes the TMD3 of Atg9 from different organisms. P, position of the first and last showed residues. The conservation and the consensus for each position are indicated and visualized as histograms. The conservation of physical-chemical properties is scored from 0 to 11. The highest score, i.e. 11, are highlighted with an asterisk while scores 10, which indicate conservation of properties but not conservation in the residue identity, are marked with a plus. Highly conserved residues are also highlighted in blue. (D) Homotrimeric Atg9 structure from S. cerevisiae was modelled using the structures of SpAtg9 as the template. The top view from the cytoplasmic side (left panel) and the lateral view from the side of the membrane (right panel) are shown. Colored chains (white, blue and magenta) correspond to each Atg9 monomer. Orange circles indicate the location of the F627 residue of each monomer, which is also colored in red. The β-sheet colored in green correspond to the 766–770 motif located in the cytosolic C-terminal domain. The lateral view and the inset show the position of F627 in the TMD3, at the interface between the monomers.
We subsequently expressed C-terminally GFP-tagged Atg9 wild-type (WT) and mutants from an integrative plasmid in atg9∆ cells, which also expressed mCherry-tagged Atg8, and stained vacuoles with CMAC. This approach allowed us to score for both Atg9 localization to the PAS, i.e., by assessing colocalization of the GFP signal with the perivacuolar mCherry-Atg8 puncta, and autophagy progression by examining mCherry-Atg8 delivery into the vacuole. Several Atg9 mutants, such as Atg9K434E and Atg9R460A, showed a partial defect in autophagy, while still localizing to the PAS (Figures 1B and S3). Similar to what has been reported for human ATG9A [28], the R673W mutation in Atg9 blocked autophagy, but we could not observe this mutant at the PAS (Figure 1B). Thus, it is very likely that the lack of function of Atg9R673W is due to a trafficking defect rather than an impairment in protein activity. The Atg9F627A mutant emerged as a particularly interesting hit, since it localized to the PAS but completely blocked autophagy (Figures 1B and S3). Sequence alignment shows that F627 is one of the most conserved residues within the third TMD of Atg9 (Figure 1C), which is consistent with our finding that this residue is essential for autophagy.
To understand the potential role of F627 in the Atg9 function, we then used the available Atg9 structures [26–28] to model S. cerevisiae Atg9 and analyze the position of these residues in the Atg9 trimeric structure. We found that F627 is at an interface between adjacent Atg9 monomers in the homotrimer complex (Figure 1D). F627 is not part of the identified hydrophilic grooves and cavities involved in lipid scrambling [26], indicating that subtle mutations at the TMD interface between Atg9 protomers can have a dramatic effect on Atg9 function.
Atg9F627A completely blocks both selective and bulk autophagy
The result of the screen suggested an impairment in the autophagic flux in cells expressing Atg9F627A. To determine the severity of the defect, we first analyzed the vacuolar transport of the precursor Ape1 (prApe1) protease, a cargo of the biosynthetic selective type of autophagy known as cytoplasm-to-vacuole targeting (Cvt) pathway, which becomes a cargo of autophagosomes during bulk autophagy [32]. Vacuolar delivery of prApe1 can be assessed by western blot since this form is processed into an active mature form (Ape1) upon delivery into the vacuole [33]. The prApe1 was efficiently processed in WT cells and this was completely blocked in atg9∆ cells upon nitrogen starvation (Figure 2A), as expected [34]. The observed defect in the Cvt pathway of the atg9∆ cells was rescued when complemented with WT Atg9, but not with Atg9F627A.
Figure 2.
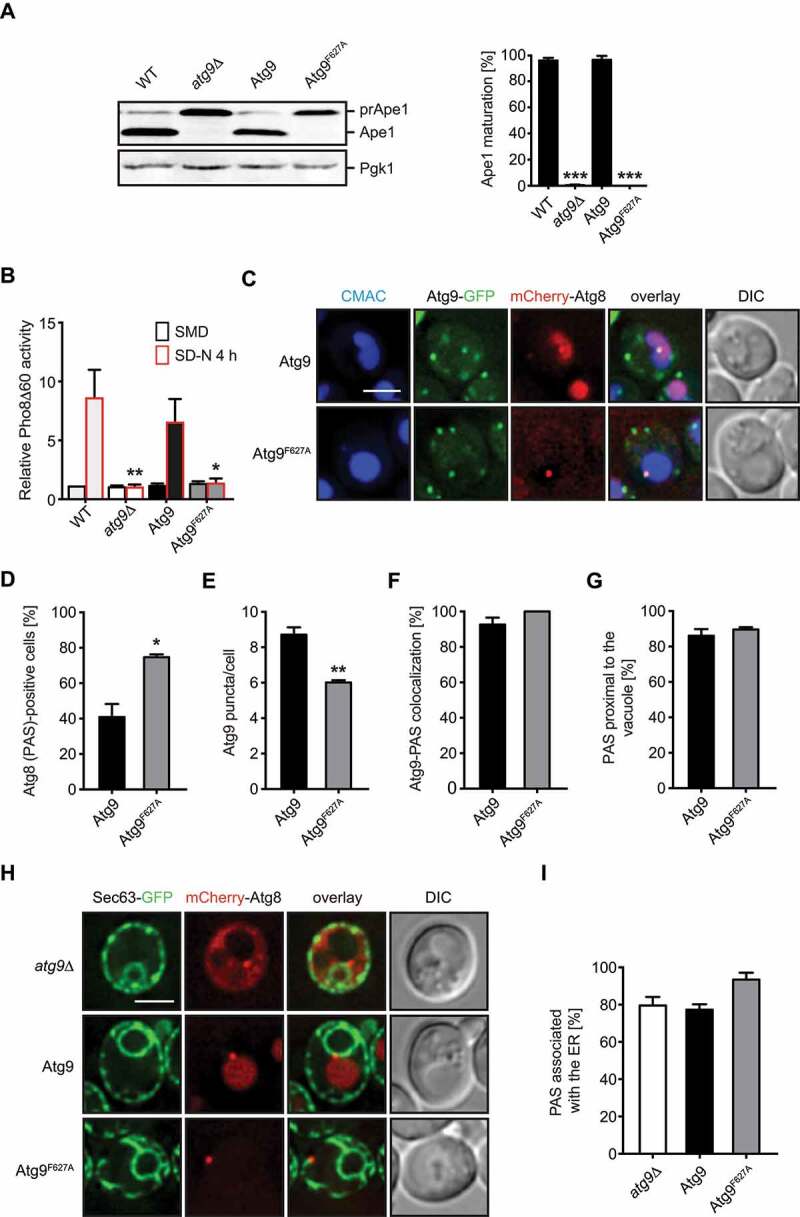
The Cvt pathway and bulk autophagy are blocked in cells expressing Atg9F627A, although its recruitment to the PAS and the PAS positioning are not affected. (A) WT (SEY6210), atg9∆ (RGY751) and atg9∆ cells carrying an integrative plasmid expressing either Atg9-GFP (RGY871, Atg9) or Atg9F627A-GFP (RGY874, Atg9F627A) were grown to a log phase in YPD medium before being nitrogen starved for 4 h in SD-N medium. Proteins were precipitated with 10% trichloroacetic acid and analyzed by western blot using anti-Ape1 and anti-Pgk1 antibodies. Pgk1 served as the loading control. The percentage of prApe1 maturation was quantified and asterisks indicate significant differences with the WT cells. (B) WT (YTS159), atg9∆ (FRY244) and atg9∆ cells expressing either tagged Atg9-GFP (RGY883, Atg9) or AtgF627A-GFP (RGY872, Atg9F627A) were grown to a log phase in YPD or nitrogen starved in SD-N medium for 4 h before measuring Pho8∆60 activity in cell lysates. Pho8∆60 activity was expressed relative to the WT control in YPD medium. Asterisks indicate significant differences with then nitrogen-starved atg9∆ strain carrying WT Atg9. (C) Subcellular distribution of Atg9. The atg9∆ strain expressing mCherry-Atg8, a PAS marker protein, and either Atg9-GFP (RGY745) or Atg9F627A-GFP (RGY884), was grown to an early log phase in YPD medium, nitrogen-starved in SD-N medium for 1 h and labelled with CMAC, a dye specifically staining the vacuole, before being imaged. DIC, differential interference contrast. Scale bar: 2 μm. (D) Quantification of the percentage of cells in panel C with mCherry-Atg8-positive PAS. Asterisks highlight significant differences with cells expressing WT Atg9. (E) Quantification of Atg9-GFP puncta per cell in the experiment shown in panel C. Asterisks indicate significant differences with cells carrying WT Atg9. (F) Statistical evaluation of the percentage with mCherry-Atg8-positive PAS also positive for Atg9-GFP in the experiment depicted in panel A. (G) Quantification of the percentage of mCherry-Atg8-positive PAS in close proximity of the vacuole in panel C. (H) The atg9∆ strain co-expressing endogenous Sec63-GFP, an ER marker protein, and mCherry-Atg8, and carrying an empty vector (RGY760, atg9∆) or an integration plasmid expressing Atg9-13xMYC (RGY762, Atg9) or Atg9F627A-13xMYC (RGY876, Atg9F627A), were imaged as in Figure 2C. DIC, differential interference contrast. Scale bar: 2 μm. (I) Statistical evaluation of the percentage of mCherry-Atg8-positive PAS associated with the ER in the experiment shown in panel A.
As a second assay, we monitored the targeting of a cytosolic form of Pho8 lacking the N-terminal transmembrane domain, i.e., Pho8∆60, to the vacuolar lumen upon autophagy induction. Upon processing in the vacuole, the phosphatase activity of Pho8∆60 can be measured, providing a quantification of the autophagic flux [33]. Thus, Pho8∆60 activity is enhanced upon nitrogen starvation in WT cells but remains unchanged in cells lacking ATG9 (Figure 2B). Similar to the observation with prApe1 processing, the autophagy impairment of the atg9∆ mutant was complemented by expression of WT Atg9 but not of Atg9F627A (Figure 2B). These data show that Atg9F627A is completely deficient in autophagy progression.
Atg9F627A localizes to the PAS
To analyze the function of F627 in more detail, we expressed Atg9F627A-GFP under the control of its endogenous promoter in cells carrying mCherry-Atg8 and then analyzed cells after nitrogen starvation to induce autophagy (Figure 2C). Atg9F627A behaved almost like the WT counterpart, as neither the colocalization of Atg9 with the PAS nor the proximity of the PAS to the CMAC-stained vacuole were altered (Figures 2F and G). However, we observed more PAS-positive structures (Figure 2D) and lower Atg9 punctate structures (Figure 2E), which is indicative of a defect in autophagosome formation and autophagy progression [35].
Efficient lipid transfer to the phagophore requires the PAS being localized adjacently to the ER. We therefore asked whether the subcellular distribution of the PAS was altered in the presence of Atg9F627A mutant by using cells that express Sec63-GFP, an ER marker protein, and mCherry-Atg8. As shown in Figures 2H and I, ATG9 deletion or the expression of either Atg9 or Atg9F627A did not affect the PAS association with the ER. Collectively, we concluded that the Atg9F627A mutant is efficiently transported to the PAS, which is correctly formed in proximity of both the ER and the vacuole.
The F627A mutation inhibits phagophore expansion
Next, we asked whether the phagophore expansion is influenced by the F627A mutation in Atg9. To visualize phagophore formation under a fluorescence microscope, we took advantage of the giant prApe1 assay, in which prApe1 overexpression leads to the formation of large phagophores that can be resolved by fluorescence microscopy [5]. As shown in Figure 3A, the extremities of the mCherry-Atg8-labelled phagophores were associated with the ER, visualized with the Sec63-GFP chimera, in the strain carrying WT Atg9 cells as expected [5,18]. In atg9∆ or Atg9F627A-expressing cells, in contrast, phagophore elongation did not occur. This phenotype is quite distinct from cells expressing the Atg2[PM1] mutant, which does not bind Atg9. Here, the phagophore-ER MCSs are mis-localized but the phagophore is still partially expanded [19]. To confirm this result, we also used the W303 yeast background because phagophores are visible without prApe1 overexpression [18]. We observed that complementation of the atg9∆ knockout with WT Atg9 generates bigger and brighter autophagosomal intermediates than in atg9∆ cells, whereas the expression of Atg9F627A resulted in membranous intermediates with similar sizes than when deleting ATG9 (Figures 3B and C), but with more intensity (Figures 3B and D). In agreement with above and the previous report [19], the strain carrying Atg2[PM1] generated small phagophores compared to cells expressing WT Atg9, but bigger than those present in atg9∆ or Atg9F627A cells (Figures 3B and C). In contrast to the atg9∆ mutant, which has a defect in both the phagophore nucleation and the downstream recruitment of the ATG machinery [36], cells expressing Atg9F627A, but also Atg2[PM1], had higher levels of mCherry-Atg8 conjugated to the phagophore (Figures 3B and D), a protein that requires multiple Atg proteins for its recruitment to the PAS and is important for phagophore expansion [37,38]. This result suggests that the F627 mutation does not affect the early events during autophagosome biogenesis that involve Atg9, i.e., PAS formation and phagophore nucleation, although this mutant has a dramatic impact on the phagophore elongation.
Figure 3.

The Atg9F627A mutant blocks phagophore expansion. (A) The atg9∆ strain carrying Sec63-GFP and mCherry-Atg8, was transformed with both the pDP105 plasmid, which expresses giant Ape1 upon addition of cupper to the medium, and an empty vector (RGY760, atg9∆) or an integration plasmid expressing Atg9-13xMYC (RGY762, Atg9) or Atg9F627A-13xMYC (RGY876, Atg9F627A). The atg2∆ mutant carrying an integrative plasmid expressing Atg2[PM1]-TAP (RGY698, Atg2[PM1]) was used as control. For the giant Ape1 oligomers formation, cells were grown as described in the Materials and Methods section. Scale bars: 2 μm (main images) and 0.5 μm (insets). (B) The atg9∆ knockout in the W303 background, expressing mCherry-V5-Atg8 (RGY898, atg9∆), was transformed with an empty vector or integrative plasmids expressing GFP-tagged Atg9 (RGY900, Atg9) or Atg9F627A (RGY901, Atg9F627A), and imaged as in Figure 2C. The atg2∆ knockout in the W303 background expressing mCherry-V5-Atg8 and carrying an integrative plasmid expressing TAP-tagged version of Atg2[PM1] (RSGY042, Atg2[PM1]) was used as control. DIC, differential interference contrast. Scale bar, 2 μm. (C and D) Quantification of the average size in nm2. (C) and intensity of the fluorescent signal in a.u. (D) of the mCherry-V5-Atg8 puncta in the experiment shown in panel B. Asterisks indicate significant differences with nitrogen-starved atg9∆ strain expressing the empty vector.
The Atg9F627A mutant has reduced self-interaction but no scramblase activity defects
To determine a possible reason for this striking block in autophagy, we next analyzed Atg9 self-interaction, which very likely drives its trimerization at the phagophore [27,28,39]. Therefore, we co-expressed from the endogenous ATG9 promoter WT and mutant Atg9-GFP and Atg9-13xMYC and examined Atg9-self-interaction by GFP-immunoprecipitation. We used three different detergents for the cell lysis, two non-ionic detergents, Triton X-100 and Tween-20, and the zwitterionic detergent CHAPS, to reveal possible alterations in the binding affinity. WT Atg9-GFP but not WT Atg9-mScarlet, efficiently co-isolated Atg9-13xMYC in the presence of all the tested detergents (Figure 4A), demonstrating the specificity of our assay. A similar interaction was detected between Atg9F627A proteins when the immunoprecipitation was carried in the presence of either Tween-20 or CHAPS (Figure 4). However, when we used the non-ionic detergent Triton X-100, we observed an almost 90% reduction in the co-isolation efficiency of the Atg9F627A variants (Figures 4A and B). Altogether, these data suggest that the stability of the Atg9 trimers is subtly affected by the F627A mutation.
Figure 4.

The F627A mutation does not have a major effect on Atg9 oligomerization and scramblase activity. (A-D) The atg9∆ strain expressing Atg9-GFP and Atg9-13xMYC (RGY881), Atg9F627A-GFP and Atg9F627A-13xMYC (RGY882) or Atg9-13xMYC and Atg9-mScarlet (RGY895) were grown in YPD to a log phase and nitrogen starved in SD-N for 1 h. Cell lysates generated in a buffer containing 1% Triton X-100 (A), 0.5% Tween 20 (B) or 1% CHAPS (C) were subsequently immunoprecipitated using GFP-trap beads (IP:GFP). Immunoprecipitates were analyzed by western blot using antibodies against GFP and MYC. (D) Quantification of the experiments shown in panels A, B and C. The ratio between Atg9-13xMYC:Atg9-GFP was determined by quantifying the intensity of the corresponding bands and subsequently expressing it relative to the cells carrying Atg9-GFP. Asterisks highlight significant differences with cells expressing WT Atg9-GFP. (C and D) Stoichiometric analysis of Atg9 oligomers by single-molecule fluorescence microscopy. The oligomeric state of Atg9 in membranes was determined by TIRF imaging of a SLBs containing Atg9-GFP or Atg9F627A-GFP (see Figure S4A-D). The occurrence percentage of Atg9-GFP (C) and Atg9F627A-GFP (D) oligomer species calculated as the averaged values from 3 different experiments ±SD. Data are provided after correction for partial labelling. (E) Schematic representation of the fluorescence-based lipid scramblase assay. NBD-PE is incorporated into protein-free or Atg9-containing liposomes. The fluorescence of NBD in the outer leaflet of the lipid bilayer can be quenched using the non-permeable reducing agent dithionite. As a result, the fluorescence of protein-free liposomes is reduced to 50% of the initial intensity. A scramblase activity leads to a quenching of fluorescence greater than 50%, as it exposes the phospholipids from the inner to the outer leaflet. (F) Scramblase activity was analyzed by measuring the fluorescence signal of NBD over time, in liposomes containing Atg9, Atg9F627A (protein: phospholipid ratio = 1:1500) or protein-free, before and after the addition of 30 mM dithionite. The fluorescence intensity (FI) was recorded every 5 s over a total period of 800 s. The plot shows the FI relative to the average initial intensity recorded before the addition of dithionite, followed by the addition of 0.1% Triton X-100 (TX-100). The data correspond to the mean with the standard error (±SE) of measurements from three independent experiments. The data were analyzed using a Kruskal-Wallis test followed by Dunn’s Multiple Comparison test with a significance level = 0.01. Asterisks indicate significant differences between the control (protein-free) and protein containing conditions (Atg9 and Atg9F627A). There are no significant differences between Atg9 and Atg9F627A conditions. The fact that the fluorescence decay is extremely fast and stabilizes after a few seconds, suggests that the remaining intensity is probably due to the presence of protein-free vesicles.
Recently, several studies have revealed that Atg9 is a lipid scramblase, which uniformly distributes lipids between the two lipid bilayers of the phagophore membrane and thus promotes its expansion [26,27,29]. The flipping of lipids could occur through a central pore within the core of the Atg9 trimer [26–28], which is proposed as the catalytic site of the Atg9. If this is indeed the case, then the integrity of the trimer is essential to support Atg9 scrambling activity. As the stability of Atg9 is affected when the F627 is mutated, we decided to analyze the stoichiometry of the Atg9F627A to determine whether its trimerization is impaired. To this end, we took advantage of a biophysical approach that counts the number subunits in biomolecular complexes by analyzing the fluorescence of single molecules on supported lipid bilayers (SLBs). The approach is based on stepwise photobleaching of the samples followed by a classification that correlates the oligomeric specie with the number of photobleaching steps that they exhibit. We have previously shown that the trimeric structure of Atg9-GFP is conserved after the extraction and reconstitution of the protein into model vesicles, which can be accurately determined by using the automated software Stoichiometry Analysis Software (SAS) to perform the analysis of the fluorescence microscopy images [40]. Thus, we purified Atg9-GFP and Atg9F627A-GFP, reconstituted them into small unilamellar vesicles (SUVs), and generated SLB before imaging. We analyzed the single-molecule fluorescence of the individual oligomers by super-resolution microscopy, before and after stepwise photobleaching. We observed that the stoichiometry of the Atg9F627A mutant was the same of Atg9 (Figure 4C, 4D and S4A-D), indicating that mutation of F627 does not prevent the trimer formation nor massively destabilizes and disrupts the protein structure.
Even though the F627 residue is not located near the potential catalytic site, and the mutant remained as a trimer, we could not exclude that F627 could somehow be involved in the Atg9-mediated lipid scrambling. We therefore tested the lipid scramblase activity of the Atg9F627A mutant using an in vitro assay based on the sensitivity of the nitrobenzoxadiazole (NBD) fluorophore to reducing environmental conditions [31,41]. For this, the fluorescent lipid NBD-PE was reconstituted into liposomes lacking or containing purified Atg9 proteins (Figure 4E). Upon reconstitution of proteoliposomes, NBD-PE is present in the inner and outer leaflet, and an eventual scramblase activity would lead to continuous back and forth lipid transfer between both leaflets. Once the baseline is achieved, the membrane-impermeable reducing agent dithionite is added and the fluorescence is determined over time. If no lipid scrambling takes place, only the NBD-PE exposed to the solution is quenched. Conversely, a lipid scramblase activity leads to a reduction of the NBD-PE signal over time.
When we recorded the fluorescence of our liposomes, the protein-free liposomes showed a reduction above 50% after the addition of the reducing agent, indicating that only the NBD in the outer leaflet was quenched in the absence of a scramblase, as expected (Figure 4F). In contrast, liposomes containing either WT Atg9 or Atg9F627A showed a further reduction in the fluorescence of the NBD, indicating that the NBD-PE was scrambled between both leaflets and that both proteins had comparable lipid scramblase activity. The reduction of the fluorescence intensity depended on the amount of protein used for the vesicle preparation, further underlying the specificity of the reaction (Figure S4E). To ensure that the increased reduction of the NBD by dithionite is due to the enzymatic activity of Atg9 and not an increased permeability of the liposomes, we performed a fluorescence protection assay to test the accessibility of the dithionite to soluble 2-NBD-glucose (2-NBDG), a fluorescent analog of glucose that can be trapped in the interior of liposomes. We measured the fluorescence of 2-NBDG over time and observed that most of the signal remained constant after the addition of dithionite. We observed a rapid and small reduction in fluorescence after the addition of dithionite, which was due to the quenching of some remaining 2-NBDG in the liposome solution after preparation. Importantly, the fluorescence of trapped 2-NBDG was quenched down to background levels after solubilization by Triton X-100 addition, indicating that the dithionite does not have access to the interior of the liposomes during the measurement and cannot reduce the NBD unless the liposomes are lysed (Figure S4F). Together, our data reveal that the severe autophagy defect caused by the F627A mutation is not due to an impairment of Atg9 self-interaction or Atg9 lipid scramblase activity.
Atg2-Atg18 are recruited to the PAS, but their binding to Atg9 is reduced in the F627A mutant
Atg9 recruits and forms a complex with Atg2 and Atg18 during phagophore elongation [19]. To determine whether Atg9F627A is still able to recruit the Atg2-Atg18 complex to the PAS, we studied whether GFP-tagged Atg2 or Atg18 in cells expressing Atg9 or Atg9F627A colocalized with mCherry-Atg8. This fluorescence microscopy analysis showed that the F627A does not compromise the recruitment and assembly of the Atg2-Atg18 complex at the PAS (Figures 5A-D). We also explored the colocalization between mCherry-Atg8 and Atg1-GFP, Atg14-GFP or Atg16-GFP in cells expressing either Atg9 or Atg9F627A, to determine whether the F627A mutation affects the recruitment to the PAS of the Atg1 kinase complex, the autophagy-specific phosphatidylinositol 3-kinase complex and the Atg12 conjugation system, respectively [1]. As shown in Figure S6, the F627A mutation did not alter the recruitment of the key modules of the Atg core machinery. In fact, Atg1 and Atg16 more significantly colocalized with mCherry-Atg8 in the strain expressing Atg9F627A, underscoring that the F627A mutation impairs the autophagic flux.
Figure 5.

Atg2 and Atg18 are recruited to the PAS and Atg9-Atg2-Atg18 complex is formed in cells expressing Atg9F627A. (A) The atg9∆ strain carrying endogenous Atg2-GFP, mCherry-Atg8 and either an empty vector (RGY742, atg9∆) or an integrative plasmid expressing Atg9-13xMYC (RGY748, Atg9) or Atg9F627A-13xMYC (RGY875, Atg9F627A), was imaged as in Figure 2C. DIC, differential interference contrast. Scale bar: 2 μm. (B) Percentage of the mCherry-Atg8-positive PAS that are also positive for Atg2-GFP in the experiment shown in panel A. Asterisks highlight significant differences with the atg9∆ mutant transformed with the plasmid expressing WT Atg9. (C) The atg9∆ mutant expressing endogenous Atg18-GFP, mCherry-Atg8 and an empty vector (RGY877, atg9∆) or an integrative plasmid expressing Atg9-13xMYC (RGY878, Atg9) or Atg9F627A-13xMYC (RGY879, Atg9F627A), was examined as in Figure 2C. DIC, differential interference contrast. Scale bar: 2 μm. (D) Quantification of the percentage of mCherry-Atg8-positive PAS that are also positive for Atg18-GFP in the experiment depicted in panel H. Asterisks indicate significant differences with atg9∆ cells transformed with the plasmid expressing WT Atg9. (E and F) The atg9∆ strain expressing Atg2-TAP, Atg18-13xMYC and Atg9-GFP (RGY885), or Atg9F627A-GFP (RGY886), or WT cells containing Atg2-TAP and Atg18-13xMYC (RSGY012) were grown in YPD to a log phase and nitrogen starved in SD-N for 1 h. Cell lysates in a buffer containing either 1% Triton X-100 (E) or 1% CHAPS (F) were subsequently immunoprecipitated using GFP-trap beads (IP:GFP). Immunoprecipitates were analyzed by western blot using antibodies against GFP, TAP and MYC. (G and H) Quantifications of the experiments shown in panels E and F, respectively. Ratios between Atg2-TAP:Atg9-GFP and Atg18-13xMYC:Atg9-GFP were determined by quantifying the intensity of the corresponding bands and subsequently expressing the values relative to the cells carrying Atg9-GFP. Asterisks highlight significant differences with the cells expressing WT Atg9-GFP.
We have previously reported that the localization of Atg9, Atg2 and Atg18 to the PAS is not sufficient to promote the phagophore expansion, but their assembly into a complex formation is essential [19]. Thus, we studied whether the F627A mutation could affect the strength of the interaction between Atg9 and the Atg2-Atg18 complex. For that, we performed co-immunoprecipitation experiments of Atg9-GFP or Atg9F627A-GFP in cells also expressing Atg2-TAP and Atg18-13xMYC, using two different detergents, i.e., Triton X-100 and CHAPS, to evaluate the strength of the Atg9-Atg2-Atg18 interaction. Even though Atg9F627A-GFP was more efficiently purified than wild-type Atg9-GFP (Figures 5E and F), the corresponding amount of co-isolated Atg2 and Atg18 was apparently reduced in cells carrying Atg9F627A-GFP irrespectively of the used detergent (Figures 5G and H). Since the purification of Atg9F627A was increased, making it difficult to distinguish whether the reduction in co-isolation of Atg2 and Atg18 is due to reduced complex formation or because more Atg9 is solubilized and purified, we also performed a reverse immunoprecipitation. Here, we pulled down Atg2-GFP and examined co-isolated Atg9-TAP or Atg9F627A-TAP (Figures S5A and S5B). Using this approach, the Atg2-Atg9 complex could be detected when cells were lysed with a buffer containing CHAPS but not with the one that included Triton X-100. Nonetheless, this reverse immunoprecipitation in the presence of CHAPS clearly showed that Atg2 interacts to the same extent with Atg9 and Atg9F627A (Figures S5A and S5B). Finally, we also repeated the Pho8∆60 assay at 25°C since low temperatures could stabilize trimer formation of the mutant Atg9F627A if altered by the F627A change, leading to a rescue of the autophagic flux defect of the Atg9F627A-expressing cells. However, autophagy in the Atg9F627A-expressing strain was blocked to the same extent than in both the atg9∆ knockout and the same cells grown at 30°C (Figure S5C). We concluded that the Atg9F627A mutation does not abolish the Atg9 self-interaction and the Atg9-Atg2-Atg18 complex formation, although the stabilization of the Atg9 trimer and integrity of this complex are lessened when the F627 residue is mutated.
Atg9 supports lipid transfer independently of its TMD interactions
Atg2 has been identified as a tether for the phagophore-ER MCSs [19], in which it probably transfers lipids from the ER to the expanding phagophore [21,23–25]. The membrane tethering properties of Atg2 involve the binding on one side of highly curved membranes via an N-terminal amphipathic helix, and on the other side of phosphatidylinositol-3-phosphate (PtdIns3P)-containing membranes together with Atg18 [21,23–25]. To test whether Atg9 contributes to the tethering process, we analyzed the Atg2-Atg18 complex-mediated tethering in the absence or presence of Atg9. For this, we used an in vitro fluorescence-based assay that determines the attachment of the fluorescent light vesicles to non-labeled heavy liposomes. Based on their different density, these liposomes can be easily separated by centrifugation [42]. Purified Atg2-Atg18 complex was added to a mixture of SUVs containing NBD-PE as the donor liposomes (D) and heavy liposomes carrying PtdIns3P as the acceptor liposomes (A). Interaction of SUVs with heavy liposomes was determined by measuring the relative fluorescence in the pellet fraction after centrifugation, which contains heavy liposomes and the tethered SUVs (Figure 6A). Tethering was only observed when the Atg2-Atg18 complex was added to the assay mixture and abolished when proteinase K was included, indicating that the observed signal is protein-mediated, and it is a consequence of tethering instead of membrane fusion (Figure 6B). Interestingly, the addition of Atg9 or Atg9F627A to either the acceptor (Atg9[A] or Atg9F627A[A]) or the donor liposomes (Atg9[D] or Atg9F627A[D]), or in both, led to almost a doubling of the tethering in comparison to samples with Atg2-Atg18 complex only. These data suggest that Atg9 participates in Atg2-Atg18 complex-mediated membrane tethering and agrees with our results from the Atg2-Atg9 pull-down experiments. That is, the F627 mutation does not affect the interaction of Atg9 with its binding partners Atg2 and Atg18, and consequently does not compromise the contribution of Atg9 to membrane tethering.
Figure 6.

Atg9 contribution to tethering and lipid transfer activities of the Atg2-Atg18 complex. (A) Schematic representation of the liposome-based tethering assay. SUVs containing the fluorescently labeled lipid NBD-PE constitute the donor vesicles (D). Heavy liposomes reconstituted in buffer containing 5% sucrose are not labeled and constitute the acceptor vesicles (A). Tethering was measured by detecting the NBD fluorescence intensity that is present in the fraction of heavy liposomes after centrifugation. (B) For the tethering assay, SUVs and heavy liposomes were incubated in the presence of the Atg2-Atg18 complex or buffer as control. Atg9 or Atg9F627A were incorporated in donor vesicles (D), acceptor vesicles (A) or both. After incubation, half of the sample was incubated with 1 mg/ml proteinase K (ProtK). Heavy liposomes and the tethered donor vesicles were separated from non-attached SUVs by centrifugation and the fluorescence intensity of NBD was measured in the pellet fraction. The plot shows the fluorescence intensity (FI) of NBD normalized to the fluorescence intensity in the control condition without Atg9 or Atg9F672A (protein free vesicles) and incubated in the presence of the Atg2-Atg18 complex. The data corresponds to the mean ±SE of measurements from four independent experiments (C) Schematic representation of the FRET-based in vitro lipid transfer assay. The NBD fluorescence is quenched by rhodamine in donor vesicles. After lipid transfer to non-labeled acceptor vesicles, the fluorophores are diluted and the signal of NBD can be detected. (D) For the lipid transfer assay, donor vesicles and acceptor vesicles containing Atg9, Atg9F627A or protein-free, were mixed and incubated with 50 nM of the purified Atg2-Atg18 complex (+Atg2-Atg18) or buffer (-Atg2-Atg18) as control. The NBD fluorescence intensity was measured every 1 min over a period 2 h. The plot shows the fluorescence intensity (FI) of NBD at each point relative to the initial fluorescence signal (FI0). The data corresponds to the mean ±SE of measurements from three independent experiments. The data were analyzed using a Kruskal-Wallis test followed by a Dunn’s Multiple Comparison test with a significance level = 0.01. The asterisks indicate that the means of curves corresponding to Atg9 or Atg9F627A vary significantly in respect to the control condition (+Atg2-Atg18). The difference between Atg9 and Atg9F627A, however, is not significant.
We next analyzed whether Atg9 also promotes Atg2-Atg18 complex-mediated lipid transfer. For this, we used liposomes containing both NBD-PE and rhodamine-PE as donor liposomes and Atg9-containing liposomes without fluorophores as acceptor liposomes (Figure 6C). Due to the Förster/fluorescence resonance energy transfer (FRET) between the fluorophores, the basal fluorescence intensity of NBD in donor liposomes was low and almost undetectable (Figure 6D, black line). When donor and acceptor vesicles were incubated in the presence of the Atg2-Atg18 complex, lipids were transferred to the non-labeled liposomes and the NBD fluorescence increased, as lipids were diluted in the acceptor vesicles over time (Figure 6D, purple line)[23–25]. When Atg9 was included in the acceptor liposomes, the overall signal of the lipid transfer almost doubled, both in the initial kinetics and the absolute efficiency (Figure 6D, green line). Importantly, no significant differences were observed between Atg9 and Atg9F627A (Figure 6D, green and red lines).
As our observations indicate that Atg2-Atg18 complex-mediated tethering is favored independently of Atg9 localization on donor or acceptor liposomes, we also tested whether the lipid transfer activity of this complex was also affected. Surprisingly, lipid transfer activity also increased when Atg9 was present in donor vesicles or in both, donor and acceptor liposomes (Figure S7). We thus conclude that Atg9 contributes both to tethering and lipid transfer activity of the Atg2-Atg18 complex, the latter possibly being influenced by more tethering between liposomes and/or Atg9 scramblase activity. Additionally, the F627A mutation does not affect the contribution of Atg9 in the Atg2-Atg18 complex-mediated lipid transfer between adjacent membranes.
Mammalian ATG9AF382A blocks autophagy in human cells
To examine whether the function of the F627 is conserved among species, we next examined the corresponding residue, i.e., F382 (Figure 1C), in mammalian ATG9A. We transfected GFP-tagged ATG9A and the ATG9AF382A into ATG9A−/− knockout cells [43]. Both proteins colocalized to the same extent with ATG16L1 (Figures 7A and B), a phagophore marker protein [38], indicating that the F382A mutation does not affect ATG9A trafficking. To measure the autophagic flux, we used the same cells and compared them to those just expressing GFP and fed or starved them in the absence or the presence of bafilomycin A1 (BafA1), a lysosomal inhibitor [38]. We then scored both the number of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-positive dots, which principally represent autophagosomes, by fluorescence microscopy and the conversion of non-lipidated LC3 (LC3-I) into lipidated LC3 (LC3-II) by western blot. Under starvation and in the absence of BafA1, ATG9A−/− cells expressing GFP formed LC3 puncta at a lower frequency than the ones transfected with the plasmid carrying GFP-ATG9AF382A, which exhibited a higher number of LC3 puncta per cell, similar to the ATG9A−/− cells expressing GFP-ATG9A cells (Figures 7C and D). This observation suggests that in contrast to cells lacking ATG9A, the ones expressing ATG9AF382A are able to assemble the ATG machinery like WT cells. Upon addition of BafA1, the number of LC3-positive puncta per cell did not significantly change in ATG9A−/− cells expressing either GFP or GFP-ATG9AF382A, whereas BafA1 addition led to more puncta in ATG9A−/− cells expressing GFP-ATG9A, in both fed and starvation conditions (Figures 7C and D). Consistently, starved cells expressing GFP-ATG9A displayed high levels of LC3-II in the presence of BafA1 (Figures 7E and F), indicating an enhanced autophagic flux [4]. In contrast, LC3-II formation was not enhanced in ATG9A−/− cells expressing either GFP or GFP-ATG9AF382A (Figures 7E and F). Taken together, those data show that a mutation at the conserved interface between ATG9A protomers results in a similar early block in autophagy also in mammalian cells, without affecting the assembly of the autophagy machinery.
Figure 7.

The ATG9AF382A-expressing mammalian cells display an autophagy defect. (A) For the localization of ATG9A, U2OS ATG9A−/− cells were transfected with a plasmid expressing GFP (GFP), GFP-ATG9A (WT) or GFP-ATG9AF382A (F382A) for 24 h before subjecting them to nutrient starvation in EBSS for 2 h. Cells were then fixed and processed for immunofluorescence using anti-ATG16L1 antibodies. Dashed squares indicate the position of the insets while arrows denote colocalizations. Scale bar: 5 μm. (B) Quantification of the experiment shown in panel A. Co-localization between GFP-ATG9A and ATG16L1 was assessed using the Pearson’s correlation coefficient. (C) U2OS ATG9A−/− cells were transfected as in panel A and treated with 200 nM BafA1 for 2 h, or starved in EBSS in the presence or the absence of 200 nM BafA1 for 2 h. Cells were then fixed and processed for immunofluorescence with anti-LC3 antibodies. Representative images of the EBSS+BafA1 are shown. Scale bar: 5 μm. (D) Quantification of the experiment described in panel C. The number of LC3 puncta per cell was analyzed using automatic counting and data are represented as fold increase compared to the WT fed condition. Values are means of three independent experiments ±SD. At least 30 cells were analyzed per conditions. Asterisks and violet bar indicate significant differences. (E) U2OS ATG9A−/− cells were transfected and treated as in panel C, but then lysed and processed for western blot analysis using antibodies against GFP, LC3 and actin. ACTA1/actin served as the loading control. (F) Quantification of the autophagic flux in the experiment described in panel E by determining the LC3-II:LC3-I ratio for each sample. Data are means of three independent experiments ±SD. Asterisks indicate significant differences.
Discussion
In this study, we set out to identify critical residues in Atg9 to better understand the molecular function of this protein at the phagophore. Our data uncover that a conserved single mutation, F627A in yeast Atg9 and F382A in mammalian ATG9A, unexpectedly blocks phagophore expansion. F627 and F382 residues are in the third TMD of Atg9 and localize at the interface between the different Atg9 protomers in the homotrimer (Figures 1A and D). The Atg9F627A mutant efficiently traffics to the phagophore, recruits the lipid transfer protein Atg2 and its binding partner Atg18, but is unable to promote phagophore expansion (Figures 2–5). Our data suggest that the F627A mutation weakens the interaction among Atg9 monomers. It could thus influence its binding to Atg2-Atg18, at least by co-immunoprecipitation analyses, but this does not result in a major destabilization of these interactions in vivo. In fact, the Atg2-Atg18 complex is efficiently recruited to the PAS in Atg9F627A-expressing cells, and Atg9F627A does not display a defect in trafficking and PAS assembly of an Atg9 mutant that is unable to self-interact [39]. These observations are consistent with our results showing that Atg9F627A still trimerize. Importantly, the F627A mutation does neither impair the scramblase activity of Atg9, which very likely depends on its trimeric conformation, nor its contribution to the Atg2-Atg18 complex-mediated lipid transfer, which we show that is strongly promoted by the presence of Atg9 (see below). Given that also the corresponding mutation in ATG9A similarly blocks autophagy in human cells, we conclude that the interfaces between Atg9 molecules have a conserved key role in phagophore formation beyond the identified role as lipid scramblases.
How does Atg9 then function at the phagophore? Upon autophagy induction, it has been shown that Atg9 interacts with components of the Atg machinery, especially components of the Atg1 kinase complex, such as Atg11, Atg13 and Atg17 [7,44,45] and very likely participate in phagophore nucleation. Subsequently, Atg9 concentrates at the extremity of the expanding phagophore, where it orchestrates the sequential recruitment of Atg2 and Atg18, and the establishment of the ER-phagophore MCSs [19]. Our findings also agree with a role of Atg9 as an acceptor of lipids that arrive from the ER to the phagophore via Atg2, and these lipids need to be uniformly distributed between the two leaflets of the phagophore membrane. This lipid flux might be coupled to ER-resident lipid scramblases, such as TMEM41B and VMP1 [46,47]. Our observations support this hypothesis as the Atg2-Atg18-mediated lipid transfer is favored when a scramblase activity is located in both donor and acceptor vesicles, suggesting that the continuous lipid flipping between the leaflets increases the efficiency of lipid transfer between donor and acceptor membranes, which probably occur on their external lipid layer.
Cryo-electron microscopy structural analyses revealed that Atg9 forms symmetric homotrimers that appears to act as a wedge in the membrane, which could explain its accumulation at the highly curved edges of the expanding phagophores [26–28]. The Atg9 protomers interact via their TMDs, and the F627A mutation is exactly there. A previous report identified residues at amino acid positions 766–770, in the C-terminal tail close to the fourth TMD, to be involved in Atg9 self-interaction [39]. This region was not resolved in any of the reported structures and its contribution to Atg9 protomer self-interaction thus remains unclear. We know that Atg9F627A can self-interact and maintain the trimeric stoichiometry, but we cannot exclude that F627 stabilize interactions that determine critical features in the structure of Atg9. Thus, the F627A mutation could result in an aberrant interaction among the subunit that impairs partition to highly curved regions, its clustering or even its capacity to bend the membrane, as was proposed for ATG9A based on simulations experiments [28].
As underlined above, Atg9F627A has scramblase activity as the WT protein (Figure 4), and it efficiently contributes to Atg2-Atg18 complex-mediated lipid transfer (Figure 6). It is possible that either the available assays are not stringent enough to recapitulate the in vivo contribution of Atg9 in lipid transfer and distribution, or that the robust binding between Atg9 proteins via their TMDs has functions beyond the identified role of Atg9 in lipid scrambling. For example, they could be important for the association between Atg9 trimers to form higher oligomers, which could lead to the establishment of a protein patch at the extremities on the phagophore. These protein patches in turn could be important to assemble other components that the phagophore-ER MCSs, like for example lipid synthesizing enzymes [48,49], which ultimately are key for the lipid transfer at phagophore-ER MCSs. The identified F627A residue may thus interfere not only with the stability of the Atg9 trimer, but also with possible higher oligomer formation. It cannot also be excluded that a slight disturbance in the Atg9-Atg9 membrane interface also changes cytoplasmic loops, altering possible binding sites for other proteins. At this stage, we have no evidence that the F627A mutation causes a change in the Atg9 interactome. Future cryo-EM analyses of vesicle-embedded Atg9 may clarify some of these different aspects.
We cannot exclude that the in vitro assays widely used to in vitro examine the scramblase and lipid transfer activity are not sensitive enough to resolve the effect of the F627A mutation on these activities. This would explain why phagophores of cells carrying Atg9F627A remain very small despite the presence of Atg2 and Atg18. Scramblase activity is measured by a single established assay where the distribution of the lipid anchored NBD fluorophore is determined after dithionite addition [31,41,50,51]. As convenient as the assay is, it has only limited kinetic resolution. Moreover, Atg9’s exact function may only be uncovered if the Atg2-Atg18 complex is also present in the scramblase assay to recapitulate the in vivo situation. It is thus possible that the local environment at the ER-phagophore MCSs and the exact positioning of Atg2-Atg18 complex toward Atg9 is critical enough that the Atg9 trimers need to provide an exact interface to allow efficient lipid transfer from Atg2 to Atg9, although in mammalian cells, ATG9A does not appear to associate transiently with phagophores [52]. Such an interface may be bypassed in the liposome environment if sufficient Atg2-Atg18 complex and Atg9 is present. Furthermore, recent data suggest that local production of phospholipids at the phagophore-ER MCSs contributes to growth of the phagophore [48]. Thus, another open question is whether the TMDs at the interface of the Atg9 protomers contribute to lipid transfer in the presence of ongoing lipid synthesis.
The identification of Atg2 as a lipid transfer protein at the ER-phagophore MCSs, and the structural and functional characterization of Atg9 as a lipid scramblase, have been a breakthrough in our understanding of autophagosome biogenesis. As revealed here, however, even slight disturbances at the TMD interface of Atg9 protomers have drastic consequences that extend beyond the identified Atg9 scramblase activity. Our study thus reveals that there is much to be learned about how Atg9 exactly contributes to the growth of the phagophore membrane.
Materials and methods
Strains and media
The S. cerevisiae strains used in this study are listed in Table S1. For gene deletion, the ATG8 and ATG9 coding regions were replaced with the kanamycin-resistance gene flanked by loxP sites (loxP-kanMX-loxP) or hygromycin-resistance gene (hphNT1) using PCR primers containing approximately 60 bases of identity to the regions flanking the targeted open reading frame [53,54]. Gene knockouts were verified by PCR and prApe1 maturation analysis [33].
PCR-based integration of GFP tag at the 3’ end of ATG1, ATG2, ATG14, ATG16, ATG18 and SEC63 was used to generate strains expressing C-terminal fusion proteins under the control of the native promoters. The plasmid templates for integration were pFA6a-GFP(S65T)-HIS3MX6, [53] and pFA6a-GFP(S65T)-NatMX6, [13]. Expression of tagged proteins was analyzed by live-cell imaging and western blot, and prApe1 processing was used to test the functionality of the generated fusion proteins.
Yeast cells were grown in rich medium (YPD; 1% yeast extract, 2% peptone, 2% glucose) or synthetic minimal media (SMD; 0.67% yeast nitrogen base, 2% glucose, and amino acids and vitamins as needed). Starvation experiments were conducted in synthetic media lacking nitrogen (SD-N; 0.17% yeast nitrogen base without amino acids, 2% glucose) or by treating the cells with 400 nM rapamycin (LC Laboratories, R-5000).
Cell lines and cell culture
U2OS (ATCC, HTB-96™) and U2OS ATG9A−/− [43] were cultured in Dulbecco’s modified Eagle medium (DMEM; Life Technologies, 31966) supplemented with 10% fetal bovine serum and penicillin-streptomycin (Thermo Fisher Scientific, 15140163). For starvation experiments, cells were washed twice with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and treated in Earle’s Balanced Salt Solution (EBSS; Life Technologies, 24010) supplemented with 200 nM bafilomycin A1 (Bioaustralis, BIA-B1012). Cells were kept at 37°C under 5% CO2 humidified atmosphere. For DNA transfection, cells were forward transfected using Xfect (Takara Bio, 631318) according to the manufacturer’s instructions.
Plasmids
The pRS404, pRS405, pRS406, pCumCheV5ATG8(406) and pDP105/pRS315-CUP1pr-BFP-APE1 plasmids have been described elsewhere [55–57].
The pATG9-GFP[405] plasmid was generated by successive cloning in the integrative plasmid pRS405 of an ATG9 genomic fragment (including endogenous promoter) with ApaI and PacI and of the GFP gene with PacI and AscI (from pFA6a-GFP[S65T]-TRP1 [53],). pATG9-GFP[405] was used as a template to create the plasmids expressing the mutant versions of Atg9 (indicated in Figure 2C). Mutations were introduced by site-directed mutagenesis using Phusion™ High-Fidelity DNA-Polymerase (Thermo Fisher Scientific, F305S). The correct introduction of the point mutations was verified by DNA sequencing.
To create the plasmids expressing WT and the F627A mutant tagged with 13xMYC (pATG9-13xMYC[405] and pATG9F627A-13xMYC[405]), the sequence coding for the GFP gene was replaced with the one of the 13xMYC tag as PacI-SacI fragment excised from pFA6a-GFP(S65T)-TRP1 [53]. pATG9-13xMYC[404] and pATG9F627A-13xMYC[404] plasmids were generated by replacing the vector backbone of pATG9-13xMYC[405] with the one from the pRS404 vector using ApaI and SacI. To create the plasmids expressing WT and F627A mutant tagged with TAP, the sequence coding for GFP was replaced with the TAP tag as PacI-AscI fragment using pATG2-TAP(405) [19] as a template. pATG9-GFP[406] and pATG9F627A-GFP[406] constructs were obtained by cloning the corresponding sequence as a KpnI-SacI fragment into the pRS406 vector, using pATG4-ATG4-TAP(406) (Reggiori lab collection) as a template. pATG9-mScarlet[405] was generated by replacing the GFP gene with the one of encoding the mScarlet tag as a PacI-AscI fragment using pABH1-ATG13-mScarlet (plasmid from C. Kraft, University of Freiburg, Freiburg, Germany) as the donor template. pATG2[PM1]-TAP[404] was generated by cloning ATG2[PM1] mutant [19] and the gene coding for the TAP tag as ApaI-PacI and PacI-AscI fragments, respectively, into the pRS404 vector. Correct integration of the different constructs was verified by western blotting and/or DNA sequencing.
To generate the plasmids expressing Atg9 and the Atg9F627A tagged with 3x-FLAG tag (pATG9-3xFLAG[416] and pATG9F627A-3xFLAG[416]) or with GFP-3x-FLAG (pATG9-GFP-3xFLAG[416] and pATG9F627A-GFP-3xFLAG[416]), the coding sequences of Atg9 and Atg9F627A were amplified from pATG9-GFP[405] using the primers 5´Atg9 BamHI (CGCGGATCCATGGAGAGAGATGAATAC) and 3´Atg9 ClaI (CAGATCGATTCTTCCGACGTCAGACTTC) or 3´Atg9 SmaI (TTTCCCGGGTCTTCCGACGTCAGACTT). The PCR products were digested with BamHI and ClaI and inserted in the centromeric plasmid 3xFLAG[416] (pRS416 GAL1p-3xFLAG) or were digested with BamHI and SmaI and inserted in the centromeric plasmid mGFP-3xFLAG[416] (pRS416 GAL1p-GFP-3xFLAG), using the same restriction sites.
ATG9A was cloned into the peGFP-C1 vector (Reggiori lab collection) as a KpnI-HindIII fragment generated by PCR from pMXs-puro-RFP-ATG9A (Addgene, 60609; deposited by Noboru Mizushima), to create the peGFP-ATG9A vector. The peGFP-ATG9AF382A plasmid was generated from peGFP-ATG9A by site directed mutagenesis. Correctness of the inserted mutation was verified by DNA sequencing.
In silico analysis of Atg9 sequence for identification of conserved residues
Protein sequences of Atg9 from different model organisms were collected from the Uniprot database (http://www.uniprot.org/) and were aligned using the software MUSCLE [58]. The figure of the alignment was prepared using Jalview (http://www.jalview.org/).
In silico modeling of yeast Atg9 structure
The structure modeling of Atg9 from Saccharomyces cerevisiae was performed using SWISS-MODEL (https://swissmodel.expasy.org/interactive). The cryo-EM structure of Atg9 from Schizosaccharomyces pombe (SpAtg9) was obtained from the UniProt database and was used as template. The analysis of the structure and the molecular graphics were performed using the software UCSF Chimera (https://www.cgl.ucsf.edu/chimera/)[59].
Fluorescence microscopy
To examine bulk autophagy, cells were grown in YPD medium before transferring them in SD-N medium for 1 h. Vacuoles were stained with CellTracker™ Blue 7-amino-4-chloromethylcoumarin dye (CMAC; Thermo Fisher Scientific, C2110) as previously described [60]. Images were acquired with a DeltaVision Elite RT microscope system (GE Healthcare, Applied Precision), equipped with a UPLASPO 100× oil/1.40 NA objective, a pco.edge 5.5 sCMOS camera (PCO) and a seven-color InsightSSI solid-state illumination system (GE Healthcare, Applied Precision). Images were generated by collecting a Z-stack of 22 pictures with focal planes 0.20 μm apart to cover the entire volume of a yeast cell. Image stacks were then deconvolved using the SoftWoRx software (Applied Precision). Photoshop CC and Illustrator CC software (Adobe) were used for figure preparation. A single focal plane is shown in the figures. Percentage of Atg8-positive cells, number of Atg9 puncta per cell, percentage of Atg8-positive structures associated with the ER and to the vacuole, and the degree of colocalization between mCherry-V5-Atg8 and Atg9-GFP, Atg1-GFP, Atg2-GFP, Atg14-GFP, Atg16-GFP or Atg18-GFP was determined by analyzing ≥100 cells from three independent experiments. The mean size and intensity of the fluorescent signals of the mCherryV5-Atg8 puncta in the W303 background were quantified in ≥20 cells from raw (non-deconvolved) images of three independent experiments using ImageJ (National Institutes of Health).
To generate giant prApe1 oligomers, cells carrying the pDP105 plasmid were grown to an exponential phase in SMD containing 250 μM of CuSO4 (reaching 0.6 OD600), before to be treated with 400 nM rapamycin for 1 h to induce autophagy [19].
Mammalian cells were cultured in 24-well plates on coverslips, fixed with 4% paraformaldehyde and washed once with PBS. Cells were subsequently permeabilised with 0.1% Triton X-100 (Sigma-Aldrich, 93443) in PBS for 10 min and blocked with blocking buffer (1% bovine serum albumin [Sigma-Aldrich, 93443], 0.1% Tween 20 [Sigma-Aldrich, 93773], PBS). Primary (rabbit anti-LC3B, Novus Biologicals, NB600-1384; or rabbit anti-ATG16L1, Abgent, AP1817b) and secondary (Alexa Fluor 568-conjugated goat polyclonal anti-rabbit IgG; Thermo Fisher Scientific, A-11011) antibodies were diluted in blocking buffer and sequentially incubated for 1 h. Coverslips were then mounted on slides using ProLong® Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, P-36931). Images were acquired on a Deltavision Elite microscope using PLAPON 60× oil objective and SoftWoRx 6 acquisition and integrated deconvolution software (Applied Precision). Analysis was then performed using the Icy software [61].
Purification of Atg9 and the Atg2-Atg18 complex
Atg9 and Atg9F627A were overexpressed in yeast as 3xFLAG or 3xFLAG-GFP fusion proteins under the control of GAL1 promoter. Atg2 and Atg18 were co-overexpressed in the same yeast strain under the control of the GAL1 promoter to purify them as a complex. Atg18 was expressed as a TAP tag fusion protein with a Tobacco etch virus (TEV) protease cleavage site. Cells were grown at 30°C in the presence of 2% galactose (Carl Roth GmbH, 4987.3) up to an OD600 of 2, then they were harvested and resuspended in lysis buffer (500 mM NaCl, 1.5 mM MgCl2, 50 mM HEPES, pH 7.5, for Atg2-Atg18 complex; 250 mM NaCl, 1.5 mM MgCl2, 50 mM HEPES, pH 7.5, for Atg9), containing the protease inhibitor mix FY (SERVA Serving Scientific, 39104.02) and 1 mM PMSF (Carl Roth GmbH, 63672), in a 1:1 (g pellet:ml buffer) ratio. Resuspended cells were frozen in liquid nitrogen as little drops and lysed in a Freezer Mill 6875D (SPEX samplePrep) in 10 cycles each 2 min at 12 counts per second (cps).
For the purification of Atg9 and Atg9F627A, the lysates were clarified by centrifugation at 3,200 x g for 15 min at 4°C and the cleared supernatants were centrifuged at 100,000 x g for 90 min to pellet the membrane fractions. The membranes were resuspended in lysis buffer containing 2% n-dodecyl β-D-maltoside (DDM; Carl Roth GmbH, CN26.4) and were incubated for 2 h at 4°C with mild agitation. The resuspended membranes were centrifuged at 100,000 x g for 45 min to remove the insoluble material. The supernatants were diluted to reach a final concentration of 0.2% DDM and incubated with anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220), according to the manufacturer specifications. The elution of the proteins was performed in lysis buffer containing 0.1% DDM and an excess of FLAG peptide (0.5 mg/ml).
For the purification of the Atg2-Atg18 complex the lysate was clarified by centrifugation at 3,200 x g for 15 min at 4°C and the cleared supernatant was centrifuged at 100,000 x g for 45 min at 4°C. The supernatant was incubated with IgG-Sepharose 6-Fast Flow beads (Cytiva, 17096901) according to the manufacturer specifications. The complex was eluted by incubation with His-TEV-protease for 60 min at 16°C. His-TEV-protease was purified using Ni-NTA agarose according to the specification of the manufacturer (Macherey-Nagel, 745400.25).
Lipid scramblase assay
The liposomes used for the lipid scramblase assay were prepared as described [41]. Phosphatidylcholine (POPC) and phosphatidylglycerol (POPG) (Avanti Polar lipids, 850457, 840457) were mixed in a 9:1 ratio, dried under vacuum, and resuspended in buffer A (50 mM HEPES-NaOH, pH 7.4, 100 mM NaCl). The lipid suspension was sonicated for 10 min and then subjected to five cycles of freeze-thawing. The liposomes were extruded through a membrane with a 400 nm pore size (ten passages) (Whatman, 10417104), followed by extrusion through a membrane with a 200 nm pore size (five passages) (Whatman, 800281). For the incorporation of the fluorescently-labeled lipid N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (NBD-PE; Invitrogen, N360) and the proteins Atg9 or Atg9F627A, the liposomes were destabilized by incubation with 0.34% DDM for 3 h, followed by the addition of 0.5% NBD-PE and the purified protein. The protein was incorporated in a protein:phospholipid (g:g) ratio (PPR) of 1:1500, 1:3000, or 1:6000. For protein-free controls, the destabilized liposomes were incubated with an equivalent volume of the elution buffer used for the purification of Atg9. The detergent was gradually removed by the addition of BIO-BEADS SM2 adsorbent Media (BIO-RAD, 1523920) as described [41].
For the scramblase assay, the liposome suspensions were diluted 20 times in buffer A (50 mM HEPES-NaOH, pH 7.4, 100 mM NaCl) and stirred in a cuvette at 23°C. The fluorescence of NBD was recorded over time on the Spectrofluorometer FP-6500 (Jasco Inc.) with an excitation wavelength of 470 nm and an emission wavelength of 530 nm. After 200 s, sodium dithionite was added to a final concentration of 30 mM, followed by the addition of 0.1% Triton X-100 after 700 s. The fluorescence was recorded for an additional 100 s. The fluorescence intensity was normalized to the average of the initial intensity before the addition of dithionite (during the first 200 s).
2-NBD glucose protection assay
The liposomes and proteoliposomes were prepared as described for the lipid scrambling assay, except that 1 mM 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG; Invitrogen, N13195) was added during the steps of the destabilization with detergent and incorporation of the protein (PPR = 1:1500), instead of the fluorescently-labeled phospholipid NBD-PE. After preparation, the liposomes were dialyzed against buffer A to remove the excess of 2-NBDG. The dialysis was performed using Slide-A-Lyzer dialysis cassettes 3.5-kDa molecular mass cut off (Thermo Fisher Scientific, 66330). The concentration of phospholipids was calculated by the quantification of phosphates (see below), and an equivalent amount of liposomes from each sample was diluted in buffer A to measure the fluorescence of 2-NBDG. The measurement of fluorescence over time was done as described for the scramblase assay. The fluorescence of 2-NBDG showed a slight decrease due to bleaching that was observed before the addition of dithionite. To correct this reduction, the rate of bleaching was calculated from a linear regression of the fluorescence values measured in the absence of dithionite. The slope obtained from the regression (counts per second) was used as the correction factor. Finally, the fluorescence intensity was normalized to the average of the initial intensity before the addition of dithionite.
Phosphate quantification
The concentration of phospholipids in the liposomes was determined by the quantification of phosphates. For this, 25 µl of the liposome suspensions were incubated with 300 μl of 70% perchloric acid (Merck, 7601903) at 190°C for 1 h. Then, the samples were cooled down to room temperature and 2 ml of water were added, followed by the addition of 400 µl of 1.25% ammonium molybdate (Merck, 1182) and 400 μl of 5% ascorbic acid (Carl Roth GmbH, 35251) both freshly prepared. Samples were vortexed, incubated at 95°C for 5 min, and cooled to room temperature. The absorbance was measured at 797 nm, and the concentration of phosphate was calculated from a NaH2PO4 standard curve performed in parallel.
Tethering and lipid transfer assays
Liposome preparation was performed by mixing lipids in the indicated compositions, dried under vacuum, and resuspended in the indicated buffers to a final concentration of 2 mM. SUVs for lipid transfer assay were composed of 46 mol% dioleoyl-phosphatidylcholine (DOPC), 30 mol% dioleoyl-PE (DOPE), 20 mol% dioleoyl-phosphatidylserine (DOPS; Avanti Polar Lipids, 850375, 850725, 840035), 2 mol% NBP-PE (Invitrogen, N360), 2 mol% Egg Liss Rhodamine PE (Avanti Polar Lipids, 810146). SUVs for tethering assay were composed of 48 mol% DOPC, 30 mol% DOPE, 20 mol% DOPS, 2 mol% NBP-PE. Resuspension of SUVs was done in buffer C (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) and SUVs were sonicated 15 min followed by extrusion up to 50 nm through a Nucleopore Track Filter Membrane (Whatman, 800281, 800309, 800308). Acceptor vesicles contained 45 mol% DOPC, 30 mol% DOPE, 20 mol% DOPS, 1 mol% PtdIns3P, and they were resuspended in buffer D (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5% sucrose [AppliChem, 131621211]). For Atg9 or Atg9F627A incorporation, 1 µmol of the lipids were destabilized by incubation with 35 mM CHAPS (Carl Roth GmbH, 14792) 2 h at room temperature, followed by the addition of 10 μg of protein and incubation for 1 h at 4°C. Then, samples were diluted 10 times with the respective buffer C or D, and then dialyzed overnight using Slide-A-Lyzer dialysis cassettes with 20 MWCO (Thermo Fisher Scientific, 66003) and by adding 1 g of Bio-beads SM2 adsorbent Media per liter of buffer. Reconstituted liposomes were freeze-thawed twice.
For the tethering assay, donor and acceptor vesicles were mixed in a molar ration 1:1 and incubated in the presence of either the purified complex Atg2-Atg18 to a final concentration of 170 nM or buffer as control, for 1 h at 23°C. Subsequently, samples were divided in two aliquots, and 1 mg/ml of proteinase K (ICN Biomedicals Inc., 193504) was added to one of them. Samples were incubated 1 h at 37°C and then centrifuged at 15,000 x g for 10 min. The pellet containing the heavy liposomes and the attached SUVs was washed three times with buffer C. The NBD fluorescence in the pellet fraction of each sample was measured in duplicates and was normalized to the average of the total NBD-fluorescence intensity.
For the lipid transfer assay, donor and acceptor vesicles were mixed in a molar ratio of 1:3, and the purified complex Atg2-Atg18 was added to a final concentration of 50 nM. The NBD fluorescence was recorded immediately after the addition of the complex and for 90 min at 1 min intervals. The fluorescence intensity was normalized to the initial intensity (at time 0). The fluorescence was measured in the SpectraMax M3 Microplate reader (Molecular Devices LLC) with an excitation wavelength of 460 nm and emission wavelength of 535 nm.
Supported lipid bilayer preparation
Liposomes containing Atg9-GFP or Atg9F627A-GFP were prepared by mixing 60 mol% DOPC, 30 mol% DOPE and 10 mol% DOPS. The lipid mix was dried under vacuum, resuspended in buffer A (300 mM KCl, 20 mM HEPES, pH 7.4) and sonicated for 15 min. The generated SUVs were destabilized by incubation with 35 mM CHAPS for 1 h at 23°C and then incubated for 30 min at 4°C with the purified proteins in a protein:phospholipid (g:g) ratio of 1:800, or with buffer for the protein-free control. The detergent was removed by dialysis as described above for the preparation of the liposomes used for the tethering and scrambling assays. Atg9 oligomers were imaged on SLBs formed on freshly cleaned microscopy slides. Glass microscopy slides were treated by immersion into a 3:1 mixture of sulphuric acid (Carl Roth GmbH, X9441) with hydrogen peroxide (30%; Sigma-Aldrich, 95294) for at least 1 h. Slides were rinsed with deionized water and sonicated in water for 5 min. Immediately before usage, slides were dried under a nitrogen stream. Atg9-GFP and Atg9F627A-GFP proteoliposomes mixture was diluted 1:10 or 1:40 in buffer A, incubated on the slides for 10 min, at 37°C, in the presence of 3 mM CaCl2, and washed 15 times with buffer A in order to remove CaCl2 and non-fused vesicles.
Single-molecule fluorescence microscopy and analysis
All experiments were performed on a custom-designed TIRF microscope system for a total of 1500–2000 frames under 30-ms exposure time. Laser excitation from a 400 nm laser (max power 400 mW – 75% lp) was coupled into a single-mode polarization-maintaining fiber to a TIRF module connected to an Olympus IX83 inverted microscope (IX3-ZDC, Olympus). Images were collected with a 100x oil-immersion objective ((UPLAPO100xOHR) and additionally magnified by 1.6x (IX3-CAS, Olympus). The final magnification and pixel size were 160x and 100 nm, respectively. Fluorescence was filtered by a four-line polychroic mirror (zt405/488/561/640rpc, Chroma, 3 mm) and rejection band filter (zet405/488/561/647 TIRF, Chroma). An iXon Ultra EMCCD Camera (Andor Technologies) was used. The images acquired were used for a stoichiometric characterization by brightness analysis using Stoichiometry Analysis Software [40]. The calibration dataset necessary to obtain the intensity value corresponding to the monomeric intensity was obtained by partial bleaching of the sample or by sampling monomeric Halo-tagged GFP on PLL-PEG-OMe:PLL-PEG-HTL 99:1 functionalized chips.
Co-immunoprecipitation experiments
Yeast cell cultures were grown to a log phase in YPD medium and then equivalents of 100 OD600 were transferred into SD-N medium for 1 h, harvested by centrifugation, and resuspended in 1 ml of lysis buffer (20 mM Tris-HCl, pH 8.0, 150 mM KCl, 5 mM MgCl2, containing 1% Triton X-100 (Sigma-Aldrich, 93443), or 0.5% Tween-20 [Sigma-Aldrich, 93773], or 1% CHAPS [Thermo Scientific™, 28300]), supplemented with 1 mM PMSF (Sigma-Aldrich, P7626) and the cOmplete™ Protease Inhibitor Cocktail (Roche, 11697498001). Cells were lysed by vortexing in presence of glass beads (Soda Lime Glass Beads, 0.4–0.6 mm; Thomas Scientific, 5663R5O) for 5 min at 4°C, and lysates were subsequently cleared by centrifugation at 13,000 x g for 5 min at 4°C. The supernatants were incubated with 50 µl of pre-washed GFP-Trap®_A agarose (ChromoTek, gta) on a rotating wheel at 4°C for 2 h. Beads were washed once with 1 ml lysis buffer, once with lysis buffer containing 300 mM KCl, once with lysis buffer containing 500 mM KCl, once again with lysis buffer containing 300 mM KCl, and finally one more time with the lysis buffer [62]. Protein complexes were eluted by resuspension in sample buffer and boiling. Proteins were then separated by SDS-PAGE and detected with specific antibodies after western blot.
Western blotting
Western blot membranes with yeast proteins were probed with monoclonal anti-MYC (Santa Cruz Biotechnology, clone 9E10, sc-40), anti-GFP (Clontech, 632381) and anti-V5 (Invitrogen, R960-25), polyclonal anti-TAP (Invitrogen, CAB1001), anti-Ape1 [13] or anti-Pgk1 antibodies [63]. Secondary antibodies were Alexa Fluor® 680-conjugated goat anti-rabbit (Invitrogen, A-21109) or anti-mouse IgG (Invitrogen, A-21058), and HRP-conjugated goat anti-rabbit (LI-COR Biosciences, 926–80011) or rabbit anti-mouse (Sigma-Aldrich, A9044). WesternSure® PREMIUM Chemiluminiscent Substrate (LI-COR Biosciences, C90829-03) was used to visualized HRP-conjugated secondary antibodies, Detection of proteins and quantification was performed using an Odyssey® Fc Imaging System (LI-COR Biosciences). The percentage of prApe1 maturation was calculated by dividing the densiometric amount of Ape1 by the one of total Ape1 (i.e. prApe1 + Ape1).
Mammalian cells grown in 12-well plates were washed once with PBS and harvested in 50 µl of Laemmli buffer (65.8 mM Tris-HCI, pH 6.8, 2.1% SDS, 26.3% [w:v] glycerol, 0.01% bromophenol blue, 5% β-mercaptoethanol). The lysates were incubated on ice for 20 min, sonicated and finally boiled at 95°C for 5 min. Equal protein amounts were separated by SDS-PAGE, and after standard western blotting, proteins were detected using specific primary antibodies (rabbit anti-LC3B [Novus Biologicals, NB600-1384], mouse anti-actin [EMD Millipore, MAB1501] or rabbit anti-ATG16L1 [Abgent, AP1817b]) followed by incubation with fluorescently-labelled secondary antibodies (Alexa Fluor® 680 conjugated goat anti-rabbit, [Invitrogen, A-21109]; or anti-mouse IgG, [Invitrogen, A-21058]). Signals were captured with an Odyssey Imaging System (LI-COR Biosciences) and signal intensities for each sample (densitometric values) normalized to the actin protein levels. Densitometric values were quantified on western blots at non-saturating exposures using the ImageJ software [64–66].
Pho8∆60 assay
Measurement of bulk autophagy by Pho8∆60 assay was performed at either 25°C or 30°C as previously described [33]. The enzymatic activity was calculated by the linear method, i.e., comparing the slopes of the experimental samples and normalizing the Pho8∆60 activity of WT cells during SMD conditions to 1 and expressed in arbitrary units (a.u.).
Statistical analyses
Data represent the average of three independent biological replicates (excluding Atg9-13xMYC-Atg9-GFP [1% CHAPS] and Atg2-TAP-Atg9-GFP immunoprecipitations depicted in Figures 4D and 5F, respectively), ± standard deviation (SD). The data shown are those of a representative experiment. Data were statistically analyzed with GraphPad Prism 6 (GraphPad Software Inc.), using the paired two-tailed Student’s t-test. All comparisons with a p value less than 0.05 (p < 0.05) were considered statistically significant. p < 0.001, p < 0.01, and p < 0.05 are indicated with triple, double, or single asterisks, respectively. Non-significant results are not indicated in the figures. For the in vitro enzymatic assays, the data was analyzed with the GraphPad Prism 6 (GraphPad Software Inc.). The plots represent the mean and the standard error (±SE) from three independent experiments, except for the scrambling titration curves. Data from lipid transfer and scrambling assays were statistically analyzed by Kruskal-Wallis test, followed by Dunn´s Multiple Comparison test with a significance level = 0.01 (99% confidence intervals).
Supplementary Material
Acknowledgments
The authors thank Claudine Kraft for reagents and Cristina Paulino for discussions. This work was funded by grants from the Deutsche Forschungsgemeinschaft (UN111/13-1) to C.U., the SFB 944 (project P26) to K.C., and ZonMW TOP (91217002), Open Competition ENW-KLEIN (OCENW.KLEIN.118), Marie Skłodowska Curie ETN (765912) and Novo Nordisk Foundation (0066384) to F.R.
Funding Statement
This work was supported by the Horizon 2020 [Marie Skłodowska Curie ETN (765912)]; Novo Nordisk Foundation [0066384]; ZonMW Top [91217002]; Deutsche Forschungsgemeinschaft; [SFB 944, P26] [UN111/13-1]; Open Competition ENW-KLEIN [OCENW.KLEIN.118].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2136340
References
- [1].Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Bio. 2020;21:439–458. [DOI] [PubMed] [Google Scholar]
- [2].Reggiori F, Ungermann C. Autophagosome maturation and fusion. J Mol Biol. 2017;429:486–496. [DOI] [PubMed] [Google Scholar]
- [3].Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol. 2018;218:757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy. 2021;17:1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Suzuki K, Akioka M, Kondo-Kakuta C, et al. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J Cell Sci. 2013;126:2534–2544. [DOI] [PubMed] [Google Scholar]
- [6].Fujioka Y, JMd A, Noshiro D, et al. Phase separation organizes the site of autophagosome formation. Nature. 2020;578:301–305. [DOI] [PubMed] [Google Scholar]
- [7].Suzuki SW, Yamamoto H, Oikawa Y, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci. 2015;112:3350–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamamoto H, Fujioka Y, Suzuki SW, et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell. 2016;38:86–99. [DOI] [PubMed] [Google Scholar]
- [9].Gómez-Sánchez R, Tooze SA, Reggiori F. Membrane supply and remodeling during autophagosome biogenesis. Curr Opin Cell Biol. 2021;71:112–119. [DOI] [PubMed] [Google Scholar]
- [10].Shima T, Kirisako H, Nakatogawa H. COPII vesicles contribute to autophagosomal membranes. J Cell Biol. 2019;218:1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sanchez-Wandelmer J, Ktistakis NT, Reggiori F. ERES: sites for autophagosome biogenesis and maturation? J Cell Sci. 2015;128:185–192. [DOI] [PubMed] [Google Scholar]
- [12].Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198:219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mari M, Griffith J, Rieter E, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190:1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sawa-Makarska J, Baumann V, Coudevylle N, et al. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science. 2020;369:eaaz7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reggiori F, Tucker KA, Stromhaug PE, et al. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90. [DOI] [PubMed] [Google Scholar]
- [16].Ungermann C, Reggiori F. Atg9 proteins, not so different after all. Autophagy. 2018;14:1456–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Papinski D, Schuschnig M, Reiter W, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53:471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Graef M, Friedman JR, Graham C, et al. ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell. 2013;24:2918–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gómez-Sánchez R, Rose J, Guimarães R, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol. 2018;217:2743–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tang Z, Takahashi Y, He H, et al. TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep. 2019;28:1744–1757.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kotani T, Kirisako H, Koizumi M, et al. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc Natl Acad Sci. 2018;115:201806727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chowdhury S, Otomo C, Leitner A, et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc Natl Acad Sci. 2018;58:201811874–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Osawa T, Kotani T, Kawaoka T, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol. 2019;26:281–288. [DOI] [PubMed] [Google Scholar]
- [24].Valverde DP, Yu S, Boggavarapu V, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol [Internet]. 2019;218:1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. elife. 2019;8:e45777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Matoba K, Kotani T, Tsutsumi A, et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol. 2020;27:1185–1193. [DOI] [PubMed] [Google Scholar]
- [27].Maeda S, Yamamoto H, Kinch LN, et al. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat Struct Mol Biol. 2020;27:1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guardia CM, Tan X-F, Lian T, et al. Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep. 2020;31:107837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Orii M, Tsuji T, Ogasawara Y, et al. Transmembrane phospholipid translocation mediated by Atg9 is involved in autophagosome formation. J Cell Biol. 2021;220:e202009194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ballesteros A, Swartz KJ. Lipids surf the groove in scramblases. Proc Natl Acad Sci. 2018;115:201809472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bushell SR, Pike ACW, Falzone ME, et al. The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat Commun. 2019;10:3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Baba M, Osumi M, Scott SV, et al. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J Cell Biol. 1997;139:1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guimaraes RS, Delorme-Axford E, Klionsky DJ, et al. Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae. Methods. 2015;75:141–150. [DOI] [PubMed] [Google Scholar]
- [34].Noda T, Kim J, Huang W-P, et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the cvt and autophagy pathways. J Cell Biol. 2000;148:465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Reggiori F, Shintani T, Nair U, et al. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Suzuki K, Kubota Y, Sekito T, et al. Hierarchy of Atg proteins in pre‐autophagosomal structure organization. Genes Cells. 2007;12:209–218. [DOI] [PubMed] [Google Scholar]
- [37].Xie Z, Nair U, Klionsky DJ. Atg8 CONTROLS PHAGOPHORE expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041. [DOI] [PubMed] [Google Scholar]
- [39].He C, Baba M, Cao Y, et al. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell. 2008;19:5506–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Danial JSH, Quintana Y, Ros U, et al. Systematic assessment of the accuracy of subunit counting in biomolecular complexes using automated single-Molecule brightness analysis. J Phys Chem Lett. 2022;13:822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ploier B, Menon AK. A fluorescence-based assay of phospholipid scramblase activity. J Vis Exp. 2016;(115):54635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lees JA, Messa M, Sun EW, et al. Lipid transport by TMEM24 at ER–plasma membrane contacts regulates pulsatile insulin secretion. Science. 2017;355:eaah6171–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Claude-Taupin A, Jia J, Bhujabal Z, et al. ATG9A protects the plasma membrane from programmed and incidental permeabilization. Nat Cell Biol. 2021;23:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sekito T, Kawamata T, Ichikawa R, et al. Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells. 2009;14:525–538. [DOI] [PubMed] [Google Scholar]
- [45].He C, Song H, Yorimitsu T, et al. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol. 2006;175:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ghanbarpour A, Valverde DP, Melia TJ, et al. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc Natl Acad Sci. 2021;118:e2101562118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li YE, Wang Y, Du X, et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J Cell Biol. 2021;220:e202103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schütter M, Giavalisco P, Brodesser S, et al. Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell. 2020;180:135–149.e14. [DOI] [PubMed] [Google Scholar]
- [49].Nishimura T, Tamura N, Kono N, et al. Autophagosome formation is initiated at phosphatidylinositol synthase?enriched ER subdomains. EMBO J. 2017;36:1719–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Malvezzi M, Andra KK, Pandey K, et al. Out-of-the-groove transport of lipids by TMEM16 and GPCR scramblases. Proc Natl Acad Sci. 2018;115:201806721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Goren MA, Morizumi T, Menon I, et al. Constitutive phospholipid scramblase activity of a G protein-coupled receptor. Nat Commun. 2014;5:5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Orsi A, Razi M, Dooley HC, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Longtine M, McKenzie A, Demarini D, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. [DOI] [PubMed] [Google Scholar]
- [54].Gueldener U, Heinisch J, Koehler GJ, et al. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23–e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rieter E, Vinke F, Bakula D, et al. Atg18 function in autophagy is regulated by specific sites within its β-propeller. J Cell Sci. 2013;126:593–604. [DOI] [PubMed] [Google Scholar]
- [57].Pfaffenwimmer T, Reiter W, Brach T, et al. Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg19. EMBO Rep. 2014;15:862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- [60].Stefan CJ, Blumer KJ. A syntaxin homolog encoded by VAM3 mediates down-regulation of a yeast G protein-coupled receptor*. J Biol Chem. 1999;274:1835–1841. [DOI] [PubMed] [Google Scholar]
- [61].de CF, Dallongeville S, Chenouard N, et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods. 2012;9:690–696. [DOI] [PubMed] [Google Scholar]
- [62].Reggiori F, Wang C-W, Stromhaug PE, et al. Vps51 is part of the yeast Vps fifty-three tethering complex essential for retrograde traffic from the early endosome and Cvt vesicle completion*. J Biol Chem. 2003;278:5009–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sánchez-Wandelmer J, Kriegenburg F, Rohringer S, et al. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat Commun. 2017;8:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Robinson JS, Klionsky DJ, Banta LM, et al. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cheong H, Nair U, Geng J, et al. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.