

Abstract
This protocol describes a bacterial three-hybrid (B3H) assay, an in vivo system that reports on RNA-protein interactions and can be implemented in both forward and reverse genetic experiments. The B3H assay connects the strength of an RNA-protein interaction inside of living Escherichia coli cells to the transcription of a reporter gene (here, lacZ). We present protocols to (1) insert RNA and protein sequences into appropriate vectors for B3H experiments, (2) detect putative RNA-protein interactions with both qualitative and quantitative readouts, and (3) carry out forward-genetic mutagenesis screens. The B3H assay builds on a well-established bacterial two-hybrid (B2H) system for genetic analyses. As a result, protein-protein interactions can be assessed in tandem with RNA interactions with a B2H assay to ensure that protein variants maintain their functionality. The B3H system is a powerful complement to traditional biochemical methods for dissecting RNA-protein interaction mechanisms: RNAs and proteins of interest do not need to be purified, and their interactions can be assessed under native conditions inside of a living bacterial cell. Once cloning has been completed, an assay can be completed in under a week and a screen in one to two weeks.
Keywords: RNA-protein interactions, bacterial genetics, three-hybrid assay, RNA-binding proteins
INTRODUCTION
RNA plays important roles in gene regulation, not only as a precursor to protein as messenger RNA (mRNA), but also as a post-transcriptional regulator of gene expression. Bacterial gene expression can be regulated through interactions of both coding and non-coding RNAs with RNA-binding proteins such as Hfq, CsrA, and ProQ1–6. Because of their ability to control gene expression, RNA-protein interactions are important in many biological contexts and tools to detect and probe these interactions are critical to advancing this field. A variety of methods have been developed to identify and study RNA-protein interactions7–11, and this bacterial three-hybrid (B3H) assay provides a complementary approach which builds on the tools of bacterial genetics.
The B3H assay can be used to confirm putative RNA-protein interactions and to assess how mutations in an RNA or protein affect the strength of an interaction. The system utilizes E. coli reporter cells with the same transcriptional reporter as a well-established bacterial two-hybrid (B2H) assay for protein-protein interactions (Fig. 1)12–14. Reporter cells carry a single-copy reporter on an F’ episome, with a test promoter upstream of a lacZ reporter gene. To conduct a B3H experiment, three plasmids are transformed into reporter cells to express the necessary hybrid components:
Figure 1.
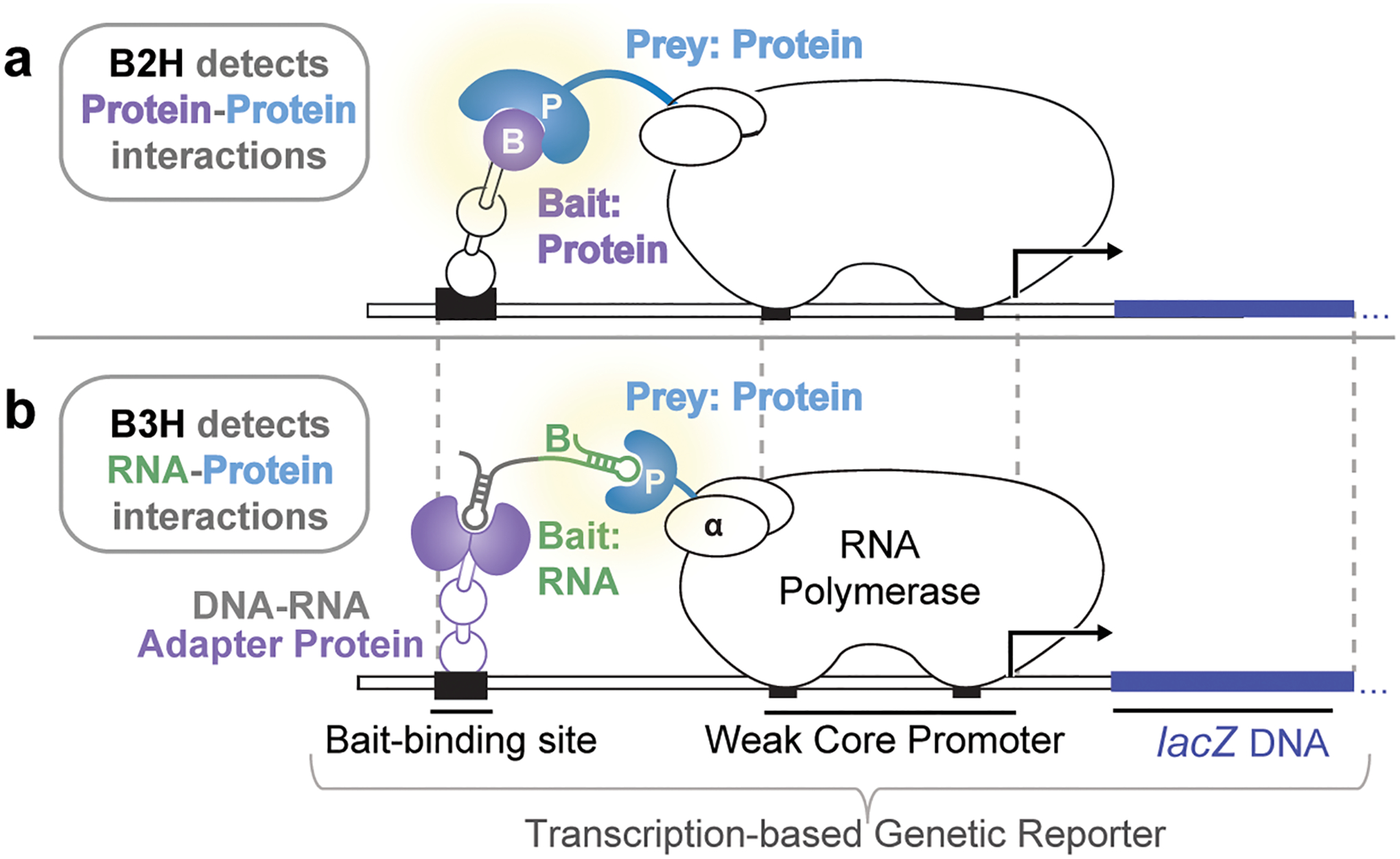
General schematic of bacterial two- and three-hybrid systems. a, Schematic of the B2H system. Protein moiety B(ait) is fused to the bacteriophage λ CI protein (λCI) and protein moiety P(rey) is fused to the N-terminal domain of the α subunit of RNAP (α-NTD). Interaction between Bait and Prey proteins activates transcription from the test promoter directing transcription of a lacZ reporter gene on a single copy F’ episome. b, Schematic of the B3H system. RNA moiety B(ait) is fused to the MS2 RNA hairpin (MS2hp) and the protein moiety P(rey) is fused to the α-NTD as in a. The MS2hp-binding moiety MS2 coat protein (MS2CP) is fused to λCI to create a DNA-RNA adapter protein that tethers the RNA Bait upstream of the test promoter. Interaction between Bait RNA and Prey protein activates transcription of the same reporter system used in the B2H assay shown in (a).
1. pAdapter: encodes the DNA-RNA adapter – a fusion protein consisting of the DNA binding-protein λCI and the RNA-binding coat protein from bacteriophage MS2 (MS2CP; Fig. 2a). In the most recent implementations of the assay, the expression of this DNA-RNA adapter is controlled by a constitutive promoter, but the original promoter was inducible by isopropyl-β-D-thiogalactoside (IPTG)12,15. The DNA-RNA adapter protein (λCI-MS2CPprotein binds to an OL2 “bait-binding site” upstream of the test promoter (Fig. 1b).
Figure 2.
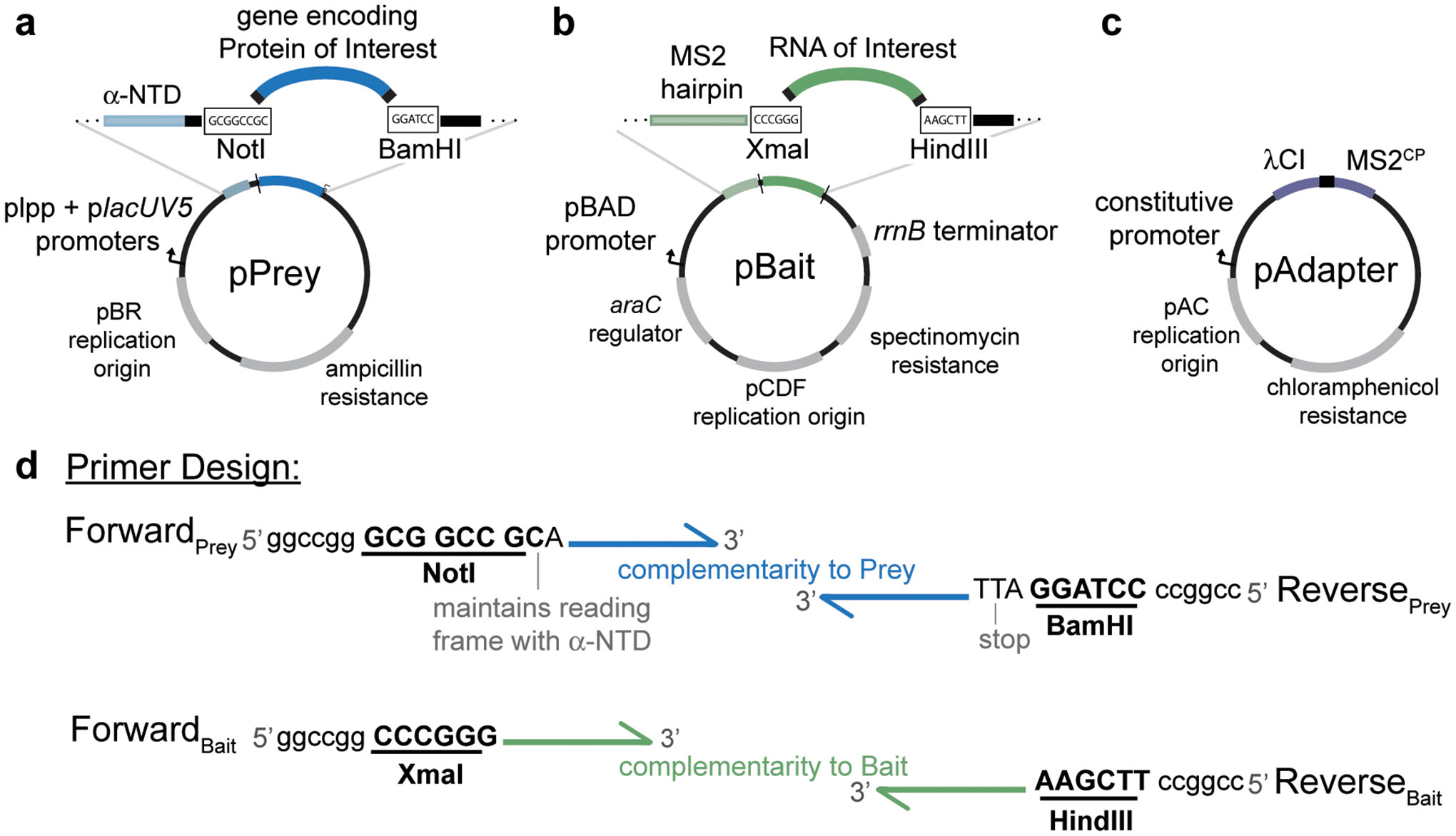
Plasmid maps of vectors used in B3H experiments highlighting key restriction sites used for cloning, promoters driving the expression of hybrid components and antibiotic resistance conferred by each plasmid. a, pPrey encodes the protein of interest tethered to α-NTD. Restriction enzymes NotI and BamHI downstream of α-NTD can be used for cloning different prey proteins into the plasmid. b, pBait encodes the hybrid-RNA containing an MS2hp and the RNA of interest. Restriction enzymes XmaI and HindIII downstream of MS2hp can be used for cloning different RNA baits into the plasmid. c, pAdapter encodes the DNA-RNA adapter fusion protein λCI-MS2CP. See Table 1 for additional plasmid details. d, Schematic representation of design of forward and reverse primers to clone a protein of interest into pPrey (top) and an RNA of interest into pBait (bottom). Restriction sites are bolded and underlined, and example “landing pads” to assist with restriction digestion of PCR products are shown at the 5’ ends of primers; these sequences provide additional dsDNA downstream of the restriction site, but the exact sequence of these is not critical. Note that an extra nucleotide following the NotI site is essential to maintain the reading frame of the α-NTD with the prey protein of interest. If you are cloning a domain of a protein that does not contain its own stop codon, one should be added upstream of the BamHI restriction site of the reverse primer.
2. pBait: encodes a hybrid RNA containing an MS2 hairpin (MS2hp) upstream of the RNA of interest (Fig. 2b). The MS2hp interacts with the adapter protein through the MS2CP moiety, and the RNA of interest is available to interact with the protein of interest (Fig. 1b). Expression of this hybrid RNA is under the control of arabinose.
3. pPrey: encodes the protein of interest tethered to the N-terminal domain of the α subunit (α-NTD) of RNA polymerase (RNAP) (Fig. 2c). Expression of this fusion protein is under the control of IPTG. Although small amounts of IPTG can boost signal, larger amounts can be toxic for cells15.
When these three plasmids are present in reporter cells, the DNA-RNA adapter binds to the hybrid RNA and tethers it upstream of the test promoter. An interaction between the RNA- and protein- of interest stabilizes RNAP at the promoter and stimulates transcription of the reporter gene lacZ (Fig. 1b); activity of the gene product β-galactosidase (β-gal) can then be measured. An increase in β-gal activity over basal levels provides information about the strength of the RNA-protein interaction being studied.
The protocol below details the steps to perform this assay in addition to necessary cloning information to insert the desired RNA ‘bait’ and protein ‘prey’ into the pBait and pPrey vectors. See Table 1 for a list of plasmids used in the assay and for cloning.
Table 1.
Plasmids for use in the B3H assay or for use as vectors for cloning new plasmids with Baits and Preys of interest.
| Plasmid Type | Plasmid Info | Expresses | Description | Name | Details | Ref |
|---|---|---|---|---|---|---|
| pAdapter | pAC; p15A origin; CmR | λCI-MS2CP | Encodes λCI fused via 12aa linker (AAAEFPGIHPGM) to a multimerization-resistant MS2CP; transcription driven by various promoters. | pKB989 | Under control of lacUV5 promoter | 12 |
| pCW17 (addgene #: 174664) |
Under control of a constitutive promoter | 16 | ||||
| p35u4 (addgene #: 174786) |
Under control of a constitutive promoter | 15 | ||||
| λCI-empty | Negative control plasmid lacking MS2CP (encodes λCI alone) | pAC-λCI (addgene #: 53730) |
corresponds to pKB989 | 13 | ||
| pRM16 (addgene #: 174787) |
corresponds to pCW17 | |||||
| pRM17 (addgene #: 174788) |
corresponds to p35u4 | |||||
| pPrey | pBR322 origin; CarbR | α-Hfq | Encodes residues 1–248 of α fused via three alanine residues to prey protein, under the control of tandem lpp and lacUV5 promoters; Cloning sites: NotI + BamHI |
pKB817 (addgene #174661) |
.Prey: full-length wild-type Ec Hfq | 12 |
| α-ProQ | pKB949 | Prey: full-length wild-type Ec ProQ | 16 | |||
| α-empty | Encodes full-length rpoA (α), under the control of tandem lpp and lacUV5 promoters; should not be used for cloning, as it lacks restriction sites. | pBR-α (addgene #53731) |
Negative control plasmid lacking prey protein | 13 | ||
| pBait | pCDF; CloDF13 origin; SpecR; | 1XMS2hp- ChiX |
Encodes a single MS2 hairpin sequence upstream of Bait RNA sequence, under the control of an arabinose inducible promoter; Cloning sites: XmaI + HindIII |
pCH6 (addgene #174662) |
RNA Bait: E. coli chiX |
16 |
| 1XMS2hp- cspE 3’UTR |
pSP10 | RNA Bait: E. coli 3’UTR of cspE |
16 | |||
| 1XMS2hp- SibB |
pSP14 | RNA Bait: E. coli sibB. |
16 | |||
| 1XMS2hp- OxyS |
pCH9 | RNA Bait: E. coli oxyS. |
16 | |||
| 1xMS2hp -empty |
pCH1 (addgene #174663) |
A 14bp multiple cloning site is present between XmaI and HindIII sites. | 16 |
Development of the protocol
This assay was developed from the B2H assay which has been previously used to detect protein-protein interactions13,14. In the B2H assay (Fig. 1a), the bait and prey are two proteins of interest13,14; in the B3H system (Fig. 1b), the prey is a protein and the bait is an RNA of interest12. The prey fusion protein used in the B3H assay remains identical to the B2H prey. This similarity allows the B3H and B2H assays to be easily used together to probe both RNA- and protein- interactions of a given protein.
The B3H assay was first used to detect interactions between the RNA-chaperone protein Hfq and its small RNA (sRNA) binding partners9. It has subsequently been used to detect interactions between the global RNA-binding protein ProQ and sRNAs and 3’UTRs16,17. In order to reliably detect ProQ-RNA interactions, the pBait vector was modified to express bait RNAs behind a single MS2hp to eliminate background interaction between ProQ and the second MS2 hairpin16; such 1xMS2hp constructs are also suitable for Hfq-sRNA interactions. Recent modifications to the system include the development of new pAdapter constructs that increase signal-to-noise15,16, and the development of visual, qualitative readout for reverse genetic analysis17.
When used to detect RNA interactions with both Hfq and ProQ, the B3H assay has been able to identify mutant proteins with lowered abilities to bind RNA12,16. The effects of RNA-sequence modifications on binding to both proteins have also been analyzed17. The ability to test effects of both mutant RNAs and proteins facilitates mechanistic studies of RNA-protein interactions.
Advantages and Limitations
Traditionally, mechanistic studies of RNA-protein interactions have been conducted in vitro. In such approaches, interactions between purified proteins and labeled RNAs can be detected using electrophoretic mobility shift assays, filter binding, or fluorescence spectroscopy7,8,18,19. While in vitro studies have many advantageous applications, the requirement of purifying proteins makes mutagenesis studies with a large number of samples a laborious process. In addition, selection of buffer conditions is somewhat arbitrary and can affect RNA structure and/or RNA-protein interactions.
By detecting RNA-protein interactions in vivo, the tools of molecular genetics become fully accessible. The well-established yeast three-hybrid (Y3H), two-hybrid (Y2H), and B2H assays have demonstrated the benefits of using molecular genetics to dissect molecular interactions9,13,14,20,21. Each of these is an in vivo assay, which avoids the need to purify proteins and RNAs as in traditional biochemical in vitro studies. The B3H is a helpful addition to this genetic toolbox as it allows bacterial interactions to be studied under more native cytoplasmic conditions than the yeast hybrid assays and allows for the use of RNAs with bacterial intrinsic terminators at their 3’ ends which would not be recognized by yeast polymerases. Additionally, bacteria provide fast growth and high transformation efficiency. It is ultimately useful to have tools to search for and dissect RNA-protein interactions in multiple model organisms.
One limitation of the B3H assay – indeed, of any genetic interaction assay – is that the apparent loss of an interaction of a protein or RNA variant could be due to protein misfolding or degradation inside of the cell. For this reason, we recommend using Western or Northern blots to confirm the stable expression of proteins or RNAs, and/or control B2H assays to confirm normal functionality of proteins. There is the additional concern that the hybrid RNA could form an unexpected secondary structure. This issue could cause false negatives if the sequence of the RNA-of-interest is unavailable to bind to the protein.
Another limitation of the B3H assay is the relatively low signal-to-noise ratio for many RNA-protein interactions. We are currently working on ways to boost signal to noise and have recently improved the pAdapter component of this assay by placing it under the control of a constitutive promoter instead of an IPTG inducible promoter15. It appears that our assay requires high-affinity interactions and that lower binding affinity interactions are not as reliably detected. When a panel of Hfq variants with single amino acid substitutions was compared side by side, the B3H assay detected interactions with KD values up to 25–100 nM, depending on the identity of the RNA and the assay readout15.
Applications
In principle, the B3H assay can be used to research a variety of RNA-binding proteins. To date, we have used the B3H assay to understand the interactions between sRNAs/mRNA-3’UTRs and the global RNA-binding proteins ProQ and Hfq12,15–17. The use of this assay in combination with mutagenesis can aid in the determination of mechanistic contributions of specific amino acid residues or domains of a protein as well as certain RNA motifs12,15,16.
Testing the effects of individual mutations is a helpful tool for dissecting RNA-protein interactions, however, using a forward genetic screen can broaden the search for mechanistic information. The B3H assay can be used with plasmid mutagenesis libraries to facilitate the testing of large groups of RNA and protein variants for interactions (Fig. 3). This technique has been used as part of a mechanistic study with the protein ProQ16, and for a proof-of-principle screen with the protein Hfq, which identified many residues in Hfq that have been shown to be involved in RNA binding in vitro12,22,23.
Figure 3.
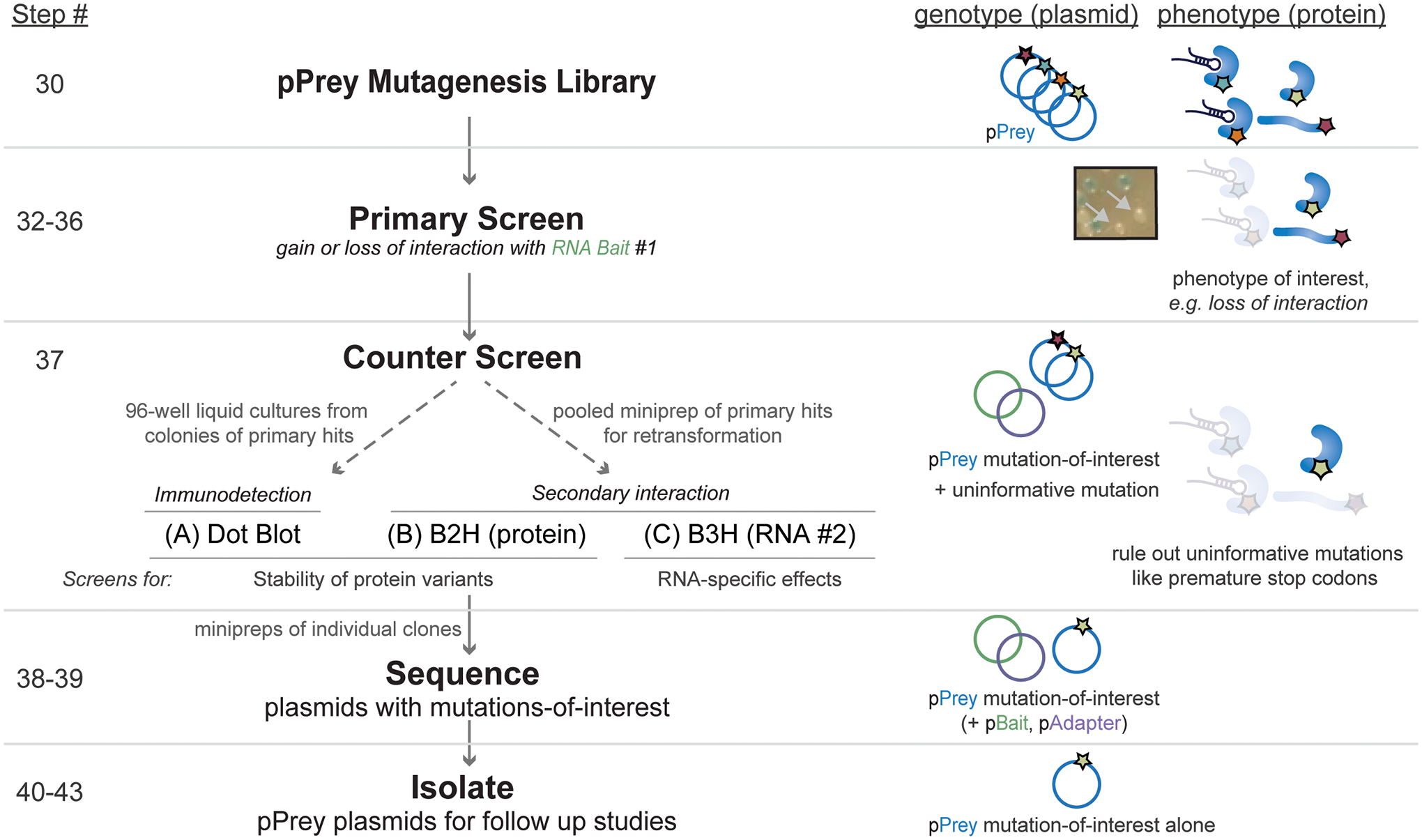
Flowchart depicting the process of a forward genetic screen and counter screen using a pPrey mutagenesis library (steps 30–43). Step numbers corresponding to the written protocol are shown on the left; descriptions of the steps are shown in the middle, and a schematic representation of the process in terms of both the genotypes and phenotypes of mutants in the library is shown on the right. Three approaches to counter screens to eliminate uninformative mutations, such as premature stop codons, are described in step 37, that allow for identification of mutants that produce general or RNA-specific binding defects.
Another possible screening application is a screen of genomic fragment libraries from organisms with no known RNA-chaperone proteins to identify novel RNA-binding proteins that interact with known sRNAs in the organism. Screening chromosomal fragment libraries has been previously used to detect new RNA-protein interactions in the Y3H assay10,11 and new protein-protein interactions in the B2H assay24. Although the use of the B3H assay in this capacity has not yet been demonstrated, the success of this approach in similar assays suggests that it could also be an application for the B3H assay.
Experimental design
We provide a protocol that details cloning, transformation and growth of reporter cells, and carrying out the B3H assay. The B3H assay protocol itself can be used quantitatively and/or qualitatively, and methods are given detailing both approaches as well as the use of screening for a forward genetics approach. Below, we detail the logic and available choices in experimental design. These options are summarized in Table 2.
Table 2.
Overview of options to consider in B3H experimental design.
| Choice | Options | Recommendations |
|---|---|---|
| Reporter Strain (details in Table 3) |
hfq+ vs. Δhfq | Δhfq behaves best for Hfq and ProQ interactions and is recommended for new interactions if Hfq may compete. |
| F’ episome marked with tetR vs. kanR | Choose based on other antibiotic needs in system (e.g. F’ providing tetracycline resistance is well suited for use with deletion alleles from Keio collection)30 | |
| pAdapter plasmids (details in Table 2) |
IPTG-inducible (pKB989) vs. constitutive expression of DNA-RNA Adapter (pCW17 or p35u4) | pCW17 recommended for ProQ-RNA interactions; p35u4 for Hfq-sRNA interactions |
| Transformation Style | Direct Inoculation from Bulk Transformation | Ideal for 96-well transformations for qualitative readout; able to screen a large number of constructs quickly |
| Inoculation from Single Colonies | Ideal for quantitative readout; recommended for 48 or fewer transformations; provides access to biological replicates from individual colonies from a single transformation | |
| Readout | Qualitative vs. Quantitative | These work well to perform in tandem and have different sensitivities. Since one readout may be ideal for a certain interaction, it is recommended to try both. |
| Counter assay for Mutagenesis Screen | No counter assay | You could choose to sequence all hits from your primary screen. If screening for mutants with a loss-of-interaction from a random mutagenesis library, it is anticipated that many hits will be undesired mutations such as premature stop codons. |
| Secondary Interaction | If a protein-protein B2H interaction is available that would be orthogonal to the RNA interaction being disrupted, this is a straightforward way to ensure functionality of mutants isolated in the primary screen using the same detection strategy. Counter-screening with another RNA bait in a B3H setup can identify mutants with RNA-specific binding defects. |
|
| Dot Blot (Immunodetection) | Only reports on expression levels of a mutant, not functionality; requires low endogenous expression relative to the prey fusion protein and an antibody specific to the prey. |
Choosing a Readout
Qualitative and quantitative methods of the B3H assay can be conducted together to complement each other, or performed on their own. Both methods can be conducted on the same day, saving time and resources relative to conducting separate transformations. Qualitative detection on indicator plates seems to provide more sensitivity than detection with liquid β-gal assays: we have found that interactions with lower signal-to-noise can be detected with more day-to-day consistency on plates. Quantitative experiments allow interactions to be studied during either log or stationary phases of growth, while qualitative experiments require colony growth in which cells are primarily in stationary phase. It is possible that this difference in growth phases will make one method more optimal for detecting certain interactions. For instance, we have found qualitative readouts to be more helpful in dissecting RNA interactions with ProQ compared to Hfq. For protein-RNA pairs that have not yet been tested in the B3H system, we recommend trying both methods in tandem, or using the qualitative method as an initial test for whether an interaction is likely to be detectable and then following up with the quantitative method.
Choosing a reporter strain and pAdapter
Available reporter strains are listed in Table 3. Endogenous Hfq may be a broad competitor for binding with multiple RNAs. Because of this we used a Δhfq strain which improves detection of both Hfq and ProQ interactions, even more so than a ΔproQ strain16. Reporter strains are available that provide either kanamycin (kan) or tetracycline (tet) antibiotic resistance. The protocol below assumes you are using a tetracycline-resistant strain, but this can be substituted with kanamycin throughout if reporter strain FW102 or KB473 is used (Table 3).
Table 3.
Bacterial Reporter Strains for B2H and B3H assays. Each FW102-based strain contains a plac-OL2–62 test promoter fused to lacZ as well as chromosomally encoded StrR31. The protocols in this article assume use of a tetR strain; simply substitute tetracycline with kanamycin if using a kanR strain.
Available adapter plasmids are listed in Table 1. The original publication of this assay used an IPTG-inducible adapter (pKB989)12, but subsequent publications used an adapter with a constitutive promoter (pCW17 or p35u4)15,16. Here, we suggest using pCW17 as it has been established to work well with ProQ or Hfq interactions, though p35u4 may be a slightly better choice for Hfq interactions15,16.
Planning your Transformations
Table 4 provides an example transformation plan. For each RNA-protein interaction being probed, three associated negative controls should be conducted, replacing one hybrid component at a time with a corresponding “empty” vector. The negative controls demonstrate how much β-gal is produced in the absence of an RNA-protein interaction and define the baseline signal. Typically, ɑ-empty (an incomplete pPrey with no protein of interest) has the lowest β-gal signal, while CI-empty (an incomplete pAdapter with no ability to bind the hybrid RNA) and MS2hp-empty (an incomplete pBait with no experimental RNA) produce slightly higher signal. The interaction between Hfq and ChiX (pPrey: pKB817 and pBait: pCH6) is useful as a robust positive control, which produces ~6–10 fold-stimulation in β-gal activity over basal levels (Fig. 4). Additional details about plasmids for negative and positive controls can be found in Table 1.
Table 4.
Layout for B3H transformations. Four transformations provide a full set of negative controls (2–4 below) for each experimental condition (1). In this example the experimental bait is SibB (pSP14) and it is being tested for an interaction with the prey protein ProQ (pKB949). In each of the three negative controls, half of one of the hybrid components is removed (α-empty; CI-empty; MS2hp-empty) to establish basal levels of lacZ transcription.
| Prey | Adapter | Bait | |
|---|---|---|---|
| 1. Experimental | ProQ (pKB949) | pCW17 | SibB (pSP14) |
| 2. Control (α-empty) | α empty (pBR-α) | pCW17 | SibB (pSP14) |
| 3. Control (CI-empty) | ProQ (pKB949) | λCI empty (pAC-λCI) | SibB (pSP14) |
| 4. Control (MS2hp-empty) | ProQ (pKB949) | pCW17 | 1xMS2hp empty (pCH1) |
Figure 4.
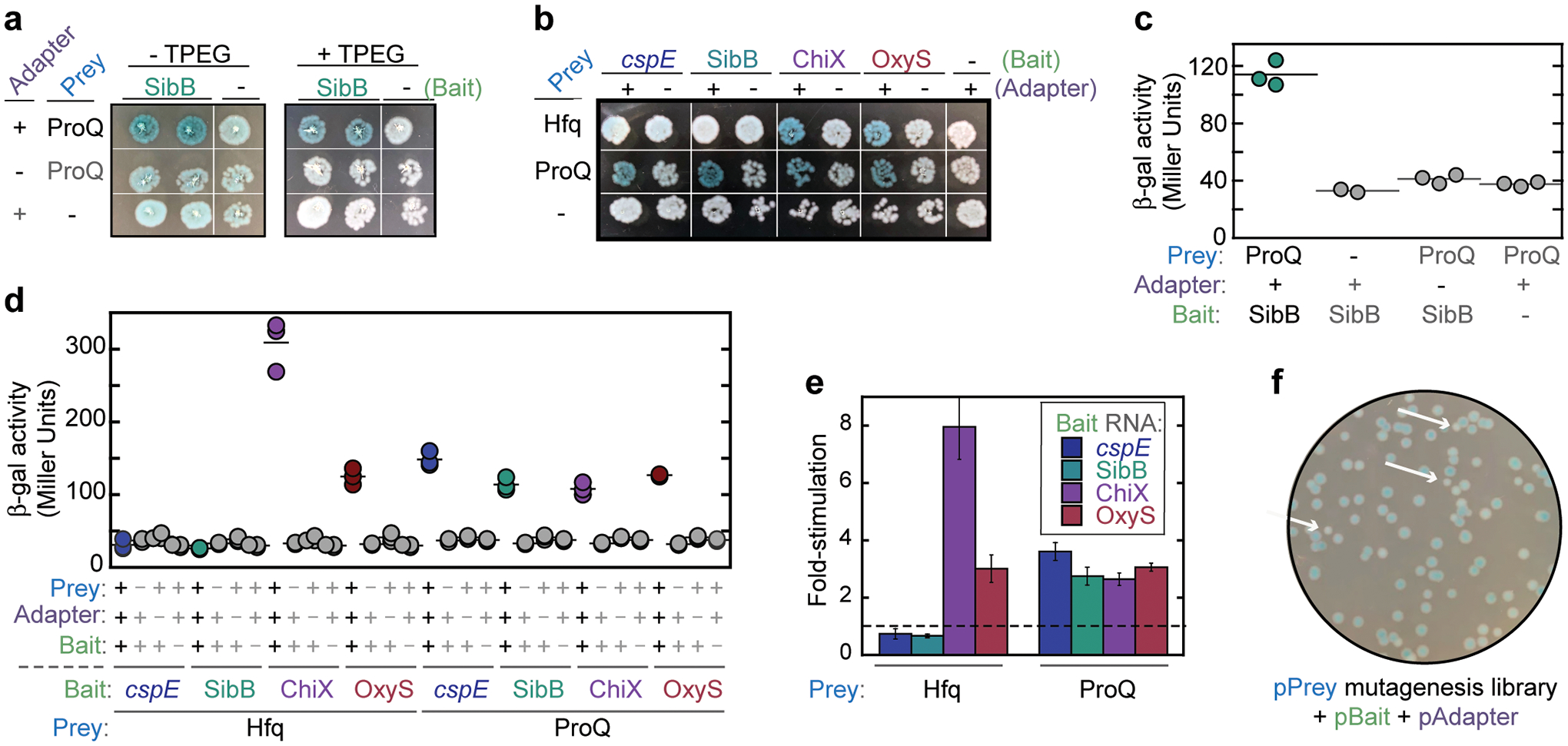
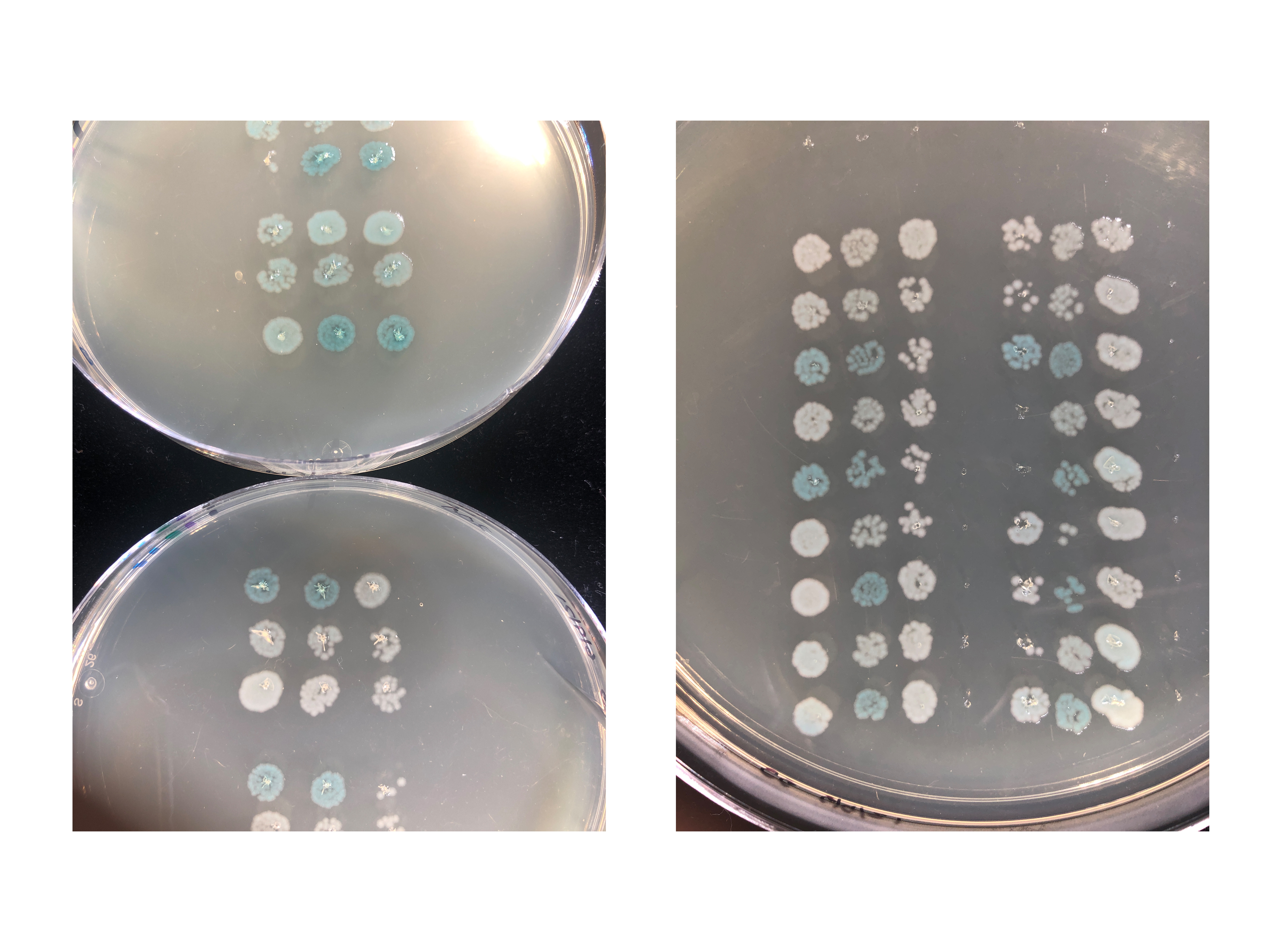
Example B3H data for qualitative readout a-b, quantitative readout c-e, and a forward genetic screen f. B3H data for these RNA-protein interactions have been published in Pandey et al16 and Stein et al17. a, Qualitative plate-based readout of B3H interaction between the RNA SibB in pBait and ProQ in pPrey. Reporter cells (Δhfq) were transformed with a pBait, pPrey and pAdapter plasmid, or one negative control (-) plasmid, as shown in Table 4. The effect of TPEG on qualitative readout is shown. Addition of 200 μM TPEG to X-gal indicator plates reduces the pale blue color of the negative controls, making the bacteria representing the experimental ProQ-SibB condition look more clearly blue than the negative controls. b, Example B3H experiment with qualitative readout of a broader panel of RNA-protein interactions. Reporter cells (Δhfq) were transformed with pPrey (Hfq, ProQ or empty), pBait (cspE 3’UTR, SibB, ChiX, OxyS or empty) and pAdapter (or a CI empty control lacking the MS2CP). Hfq matched with an RNA that is not a known binding partner (cspE, SibB) produces white patches, similar to negative controls. ProQ, which is less specific with its requirements for a binding partner, yields blue patches for all the experimental RNAs. Indicator plates in both (a) and (b) werse supplemented with 40 μg/mL X-gal, 1.5 μM IPTG and 0.2% (wt/vol) arabinose; plates in (b) were additionally supplemented with 200 μM TPEG. c, Quantitative readout of B3H interaction between the RNA SibB in pBait and ProQ in pPrey. Transformations correspond to qualitative readout shown in (a) and cells were grown in the presence of 0.2% (wt/vol) arabinose. Raw β-galactosidase (β-gal) activity is shown in Miller units. Each data point represents the β-gal activity measured from a single well and the horizontal bar represents the average of these replicate values. d, Example B3H experiment with quantitative readout of a broader panel of RNA-protein interactions. Transformations correspond to qualitative readout shown in (b) and raw β-gal activity is shown as in (b). e, Data from (d) plotted as fold-stimulation over basal levels. This is calculated as the β-gal activity of the experimental condition (e.g. pPrey-ProQ + pBait-SibB + pAdapter; teal data points in (c)) divided by the highest β-gal activity of the three negative controls (here, pPrey-ProQ + pBait-SibB + pAdapter-empty). Bar graphs show the average fold-stimulation over basal levels, calculated as described above, from independent measurements of the experimental condition (n=3) and each negative control (n=3; n=3; n=2). Error bars represent the propagated error from each β-gal measurement (see Step 29(B)vii) f, Example plate from a forward genetic screen. A pPrey mutagenesis library was transformed into reporter cells (Δhfq) containing pBait and pAdapter and plated on X-gal-indicator plates (see reagent setup). Example white and pale colonies are indicated with white arrows. Source data available in supplemental information.
Both qualitative and quantitative methods require transformation of the three plasmids, providing the ‘bait’, ‘adapter’, and ‘prey’ components. We share two protocols for growing bacterial cells following transformation, inoculating cultures from either bulk transformations or from single colonies. The single-colony protocol, 26(A), takes two days – one to transform and plate transformants on appropriate antibiotic-containing plates and the second to pick individual colonies and inoculate liquid medium supplemented with antibiotics and arabinose. The bulk-inoculation protocol, 26(B), can be completed on a single day: transformants are inoculated directly into liquid LB without selecting first for individual colonies. Although this shortens the procedure by a day, a single transformation can only provide technical replicates, while the first protocol 26(A) allows for multiple biological replicates to be inoculated from individual colonies from a single transformation.
After growing the inoculated culture overnight, RNA-protein interactions can be analyzed with qualitative and quantitative readouts, step 29(A) and 29(B) respectively. For a qualitative readout on blue-white indicator plates, a strong RNA-protein interaction is indicated by a more blue bacterial patch color and a weak RNA-protein interaction is indicated by a more white bacterial patch color (Fig. 4a, b). For a quantitative readout, β-gal activity is measured from cell lysates taken at mid-log in a microplate spectrophotometer (Fig. 4c,d). Quantitative data from these experiments can be shown over background as fold-stimulation by dividing the experimental β-gal value by the value of the highest of the three negative controls (Fig. 4e).
MATERIALS
REAGENTS
for molecular cloning
2X Phusion PCR Master Mix (New England Biolabs, NEB.com, cat. no. M0532S)
10X CutSmart buffer (New England Biolabs, NEB.com, cat. no. B7204S)
T4 DNA ligase (New England Biolabs, NEB.com, cat. no. M0202S)
Antarctic Phosphatase (New England Biolabs, NEB.com, cat. no. M0289L)
Ultrapure H2O
Genomic or template DNA encoding RNA- and protein-of-interest
B3H plasmids/vectors (see Table 1)
Forward and reverse primers for RNA and protein insert (see Fig. 2d)
Plasmid miniprep kit (e.g. Zyppy Plasmid Miniprep, Zymo, geneseesci.com, cat. no. 11–309B)
Gel purification kit (e.g. Zymoclean Gel DNA Recovery, Zymo, geneseesci.com, cat. no. 11–300)
PCR cleanup kit (e.g. DNA Clean and Concentrator, Zymo, geneseesci.com, cat. no. 11–305)
- LB broth (see reagent setup)
- Tryptone (BD Bacto, thermofisher.com, cat. no. 211699)
- NaCl (EMD Millipore, emdmillipore.com, cat. no. SX0420–3)
- yeast extract (BD Bacto, thermofisher.com, cat. no. 212720)
- LB-agar plates (see reagent setup)
- agar (Genesee Scientific, geneseesci.com, cat. no. 20–248)
for beta-galactosidase assays:
β-mercaptoethanol (Millipore Sigma, sigmaaldrich.com, cat. no. 444203)
O-nitrophenyl-β-D-galactopyranoside (ONPG) (Gold Biotechnology, golbio.com, cat. no. N-275)
PopCulture Reagent (Novagen/Millipore Sigma, emdmillipore.com, cat. no. 71092)
400 U/μL rLysozyme (Novagen/Millipore Sigma, emdmillipore.com, cat. no. 71110)
0.2% (w/v) L-Arabinose, filter sterilize, and stored at room temperature (RT, 20–25°C) protected from light (Gold Biotechnology, goldbio.com, cat. no. A-300)
X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) stock at 40 mg/mL in dimethyl formamide (DMF) stored at −20°C (Gold Biotechnology, goldbio.com, cat. no. X4281)
IPTG (Isopropyl-β-D-thiogalactoside) stock at 1M, filter sterilized, and stored at −20°C protected from light (Gold Biotechnology, goldbio.com, cat. no. I2481)
TPEG (phenylethyl-β-D-thiogalactopyranoside) 250 mM in DMF stored at −20°C protected from light (Gold Biotechnology, goldbio.com, cat. no. P-125)
Glycerol stock at 80% (v/v) stored at RT (Macron Fine Chemicals, vwr.com, cat. no. MK509216)
- Antibiotic stock solutions:
- Carbenicillin (carb): 100 mg/mL in 50% ethanol (v/v) stored at −20°C (Gold Biotech, goldbio.com, cat. no. C-103)
- Chloramphenicol (cm): 25 mg/mL in 100% ethanol stored at −20°C (Gold Biotech, goldbio.com, cat. no. C-105)
- Kanamycin (kan): 50 mg/mL in ultrapure water, filter sterilized, stored at 4°C (Gold Biotech, goldbio.com, cat. no. K-120)
- Spectinomycin (spec): 100 mg/mL in ultrapure water, filter sterilized, stored at −20°C (Gold Biotech, goldbio.com, cat. no. S-140
- Tetracycline (tet): 10 mg/mL in 100% ethanol, store solution at −20°C protected from light (Gold Biotech, goldbio.com, cat. no. T-101)
for PCR mutagenesis:
Quick-Load® Taq 2X Master Mix (New England Biolabs, NEB.com, cat. no. M0271L)
for dot-blot counter assay:
Primary antibody, specific to prey-protein of interest
Appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (e.g. goat anti-rabbit) (Biorad, bio-rad.com, cat. no. 1706515).
Chemiluminescence detection reagent (e.g. BioRad Clarity Western ECL Substrate) (BioRad, bio-rad.com, cat. no. 1705061)
Tween-20 (VWR, vwr.com, cat. no. 97062–332)
TBS (VWR, vwr.com, cat. no. 97064–338)
nonfat powdered milk (store brand)
for competent cells:
EQUIPMENT
PCR tubes
PCR thermocycler
Petri dishes
Microcentrifuge tubes
Heating block at 42°C
Microplate, 96-well flat-bottom clear polystyrene (greiner bio-one, sigmaaldrich.com, cat. no. 655101)
Sterile, flat bottom, suspension culture plate 96-well (Olympus, geneseesci.com, cat. no. 25–104)
2mL 96-well deep well blocks (VWR, vwr.com, cat. no. 10755–248)
96-well non-skirted PCR plate (VWR, vwr.com, cat. no. 82006–636)
Adhesive film for culture plates, porous (VWR, vwr.com, cat. no. 60941–086)
Microplate spectrophotometer (e.g. Molecular Devices SpectraMax190)
Microplate shaker (e.g. VWR 12620–928 or Genesee 31–213) in 37°C incubator
Photography station: Oblique lighting (e.g. 4 Table top portable LED lights), black velvet, ring stand + camera
Chemiluminescence Imaging system (e.g. www.azurebiosystems.com, Azure c600)
Centrifuge (e.g. Beckman J2-MI)
Centrifuge bottles (e.g. Fisher, fishersci.com, cat. no. 05–562-20)
50mL Falcon tubes (VWR, vwr.com, cat. no. 89039–656)
Nitrocellulose Protran membranes (Fisher, fishersci.com, cat. no. 45–004-006)
REAGENT SETUP
E. coli strains
For cloning, use a cloning strain that provides lacIq, e.g. NEB 5-α F’Iq cells (New England Biolabs, NEB.com, cat. no. C2992I). For B3H assay, use an E. coli reporter strain from Table 3. Store competent cells at −80°C for up to a year. This protocol assumes tet resistant strains; replace tet with kan if using kan resistant strains. For forward genetic screen pre-transform reporter cells with two of the three plasmids (pAdapter and pBait for a pPrey/protein screen, or pAdapter and pPrey for a pBait/RNA screen).
Z-buffer
Dissolve 8.0 g Na2HPO4-7H2O, 2.8 g NaH2PO4-H2O, 372.8 mg KCl and 60.18 mg MgSO4 in water to a final volume of 500 mL. Confirm the pH is 7.0 and filter sterilize. Store at room temperature for up to a year.
ONPG Solution, 4 mg/mL
Dissolve 200 mg O-nitrophenyl-β-D-galactopyranoside (ONPG) in a final volume of 50 mL Z-buffer and filter sterilize. Store at −20°C in 2 mL aliquots for up to 6 months.
Z-buffer with β-mercaptoethanol and ONPG
Dilute 4 mL ONPG (4 mg/mL) in 16 mL Z-buffer and 43.2 μL β-mercaptoethanol. CRITICAL Prepare fresh each time. CAUTION: open β-mercaptoethanol in fume hood.
SOC medium
Dissolve 1.47g Tryptone, 0.915g yeast extract, 0.088g NaCl, and 0.028g KCl in 100mL of Ultrapure water. Bring volume up to 150mL and autoclave. Add 0.04g of MgCl2 to 20mL of water and filter sterilize. Add 0.08g of glucose to 20mL of water and filter sterilize. Add 10.13mL of the MgCl2 solution and 10.35mL of the glucose solution to the autoclaved SOC solution. Store long-term at −20°C in 10 mL aliquots and a working stock at 4°C for up to a week.
LB liquid medium
Using a stir bar in 800 mL of ultrapure water, dissolve 10 g Tryptone (ACROS brand), 10 g NaCl (Fisher Scientific), and 5 g of yeast extract (BD Bacto). Bring to a final volume of 1 L and autoclave. Store at room temperature up to six months.
LB-agar plates
Add 1.5% (wt/vol) agar to LB liquid medium, autoclave, after the solution is no longer hot to the touch mix with appropriate antibiotics, and pour the mixture into sterile plates. Allow to dry and store upside down at 4°C up to two weeks until use. Before plating, allow plates to dry at room temperature.
X-gal-(blue/white)-indicator plates
Same protocol as LB-agar plates. Before pouring plates add 0.2% (wt/vol) arabinose, carb (100 μg/mL), cm (25 μg/mL), tet (10 μg/mL), spec (100 μg/mL), 40 μg/mL X-gal, 200 μM TPEG, and 1.5 μM IPTG. CRITICAL Conditions should be optimized to maximize the visual difference between bacterial patches for the experimental condition (+ Bait + Prey + Adapter) bacterial patches representing negative-control conditions (e.g. - Prey) that produce only background levels of β-gal. Recommended concentration ranges: X-gal: 10–80 μg/mL, TPEG: 0–200 μM and IPTG: 0– 50 μM IPTG; see “Anticipated Results” for further information on optimizing this assay.)
Solution A + glycerol
Using a stir bar, mix 150 mL glycerol, 10 mL 1M MnCl2, 50 mL 1M CaCl2, 200 mL 50mM MES pH 6.3, and 590 mL ultrapure water. Filter sterilize and store protected from light at 4°C for up to a year.
PROCEDURE
Molecular Cloning of Bait and Prey Plasmids: Timing 4–6 days
Prepare B3H vector backbones.
- Acquire and digest a B3H vector plasmid (from Table 1). Use appropriate restriction enzymes depending on the vector (Fig. 2).
-
For the pBait vector, use XmaI and HindIII-HF.
-
For the pPrey vector, use NotI-HF and BamHI-HF.
Component Amount Final Vector Plasmid (40–80 ng/μL) 50 μL 2–4 μg 10x CutSmart Buffer 6 μL 1x Restriction Enzyme 1 2μL 4 units Restriction Enzyme 2 2 μL 4 units Total 60 μL -
Incubate 2–16 hr at 37°C
Add 1 μL Antarctic phosphatase and incubate at 37°C for 1 hour.
Purify the digested vector plasmid by gel electrophoresis and gel extraction. Use gel purification kit (e.g. Zymo Gel DNA Recovery Kit) and follow instructions from the manufacturer.
Prepare Bait and Prey inserts
-
3Obtain the DNA sequences encoding the RNA and/or protein of interest. This can be in the form of genomic DNA from a bacterial species of interest. Ensure that your DNA insert does not contain the sequences of the restriction enzymes you are using, and amplify the insert using PCR (see Fig. 2d for primer design).
Component Amount Final 2x Phusion PCR Master Mix 15 μL 1x F primer (10 μM) 1 μL 0.33 μM R primer (10 μM) 1 μL 0.33 μM Template 1 μL ~0.5 ng/uL plasmid or genomic DNA 1 μL 0.5 ng ultra-pure water 12 μL Total 30 μL Step Temperature (°C) Time Initial denaturation 98°C 30 seconds 30 cycles 98°C 5 seconds 50–60°C (annealing) 10 seconds 72°C 15 seconds per kb Final elongation 72°C 5 minutes Hold 10°C -
4
Verify the length of the amplified insert using an agarose gel and conduct PCR cleanup (e.g. Zymo “DNA Clean and Concentrator” Kit), following instructions from the manufacturer.
-
5
Digest PCR products with appropriate restriction enzymes (see step 1). CRITICAL STEP: if your PCR template was a plasmid with the same antibiotic resistance markers as pPrey or pBait, treatment of the PCR product with DpnI is recommended.
-
6
Repeat PCR cleanup as in step 6.
Pause Point Inserts and vectors (digested or undigested) can be stored at −20°C until required.
Ligate vector and insert and identify a correct clone
-
7
Use T4 DNA ligase to ligate insert and vector. Follow protocol provided with the DNA ligase.
-
8
Transform ligation products into a cloning strain that provides lacIq, e.g. NEB 5-α F’Iq, New England Biolabs), following instructions of the manufacturer. Use a positive control with an undigested B3H vector, and negative controls with no insert, or no ligase, to determine efficiency of ligation.
-
9Plate transformation on LB-agar with the appropriate antibiotic.
- pPrey: carb (100 μg/mL).
- pBait: spec (100 μg/mL).
-
10Pick single colonies and inoculate each ~5mL of LB liquid medium with appropriate antibiotic. Grow overnights at 37°C while shaking or spinning.
- pPrey: carb (100 μg/mL).
- pBait: spec (100 μg/mL).
-
11
Isolate plasmid DNA from overnight cultures in the previous step using a plasmid miniprep kit (e.g. Zyppy Plasmid MiniPrep Kit from Zymo), following the manufacturer’s instructions.
-
12
Sequence the plasmids to ensure that your RNA or protein of interest has been properly inserted. Recommended sequencing primers are provided in Table 5.
Pause Point DNA plasmids can be stored at −20°C until required.
Table 5.
Primers for sequencing of B3H vectors. Forward primers bind upstream of XmaI and NotI restriction sites of pBait and pPrey, respectively. Reverse primers bind downstream of HindIII and BamHI sites of pBait and pPrey, respectively.
| Oligo | Sequence (5’-3’) |
|---|---|
| pBait F | CCGGTAACCCCGCTTATTAAAAGC |
| pBait R | TATCAGACCGCTTCTGCGTTC |
| pPrey F | GAACAGCGTACCGACCTGG |
| pPrey R | GGTGATGTCGGCGATATAGG |
Make Competent Cells: Timing 3 days
CRITICAL Prepare competent cells from E. coli reporter strains (see table 4) to prepare for heat-shock transformations.
-
13Streak E. coli reporter strain from frozen glycerol stock on LB agar plate with appropriate antibiotic.
- FW102 or KB473: kan (50 μg/mL)
- KB480 or KB483: tet (10 μg/mL)
-
14
Pick single colonies and grow overnight cultures (~5 mL) at 37°C.
-
15
Dilute 2 mL of the overnight cultures 1:100 into 200mL liquid LB with appropriate antibiotics into 500mL flasks.
-
16
Grow cells in shaking incubator at 37°C to mid-log (OD600 0.5–0.8) by measuring optical density using a spectrophotometer.
-
17
Once cell culture reaches mid-log, add each culture to a previously autoclaved and chilled centrifuge bottle and place on ice. CRITICAL STEP: From this point on, it is important to keep cells on ice.
-
18
Centrifuge for 10 minutes at 4°C at 3800 xg and gently discard supernatant.
-
19
Resuspend cells in 50mL of Solution A + glycerol and then transfer resuspended cell culture to 50 mL Falcon tube.
-
20
Incubate on ice for 30 minutes.
-
21
Centrifuge Falcon tubes at 2000 xg, 4 degrees for 15 minutes. Discard supernatant.
-
22
Resuspend cell pellet in 10mL of Solution A + glycerol.
-
23
Aliquot 500 μL cell culture to microcentrifuge tubes and place the tube in liquid nitrogen to flash freeze. Store at −80°C up to 1 year. CAUTION: use of liquid nitrogen requires adequate ventilation and personal protective equipment such as safety goggles and properly insulated gloves.
Transformation, Growth, and Induction: Timing 2–3 days
-
24Transform plasmids from Step 14 into reporter cells. Option A inoculates from single-colonies over two days and allows for multiple biological replicates from a single transformation, while Option B inoculates from the bulk transformation and can be performed on a single day, though each transformation will only allow for technical – not biological – replicates.
-
Transformations for inoculation from single colonies.CRITICAL Transform competent reporter cells with one plasmid of each type.
- Pipette 1 μL of each of three plasmids (pPrey, pBait, pAdapter) into microcentrifuge tubes (see Table 4 for example transformation plan and negative controls). Plasmid stocks should have concentrations of 40–80 ng/μL in miniprep elution buffer. Chill tubes on ice.
- Add 30 μL of heat-shock competent reporter-strain cells. Mix by gentle flicking and incubate on ice for at least 20 minutes (no more than 1 hour).
Component Amount Final pBait plasmid (40–80 ng/μL) 1 μL 40–80 ng pPrey plasmid (40–80 ng/μL) 1 μL 40–80 ng pAdapter plasmid (40–80 ng/μL) 1 μL 40–80 ng heat-shock competent reporter-strain cells 30 μL Total 33 μL - Heat 45 seconds at 42°C.
- Return to ice for 2 minutes.
- Add 300–500 μL SOC medium and incubate, shaking at 200 rpm, at 37°C for 1 hr.
- Pellet cells by centrifuging at 4000 xg for 3 minutes. Remove supernatant leaving behind about 20 μL. Gently resuspend cells by pipetting up and down.
- Pipette entire transformation onto LB-agar supplemented with carb (100 μg/mL), cm (25 μg/mL), tet (10 μg/mL)), and spec (100 μg/mL). Streak for single colonies. Multiple transformations (4–8) can fit on a single demarcated plate.
- Incubate overnight at 37°C.
-
Select single colonies from transformation plates and grow in 1 mL LB liquid medium supplemented with carb (100 μg/mL), cm (25 μg/mL), tet (10 μg/mL), spec (100 μg/mL) and 0.2% (wt/vol) arabinose in a 2 mL 96-well deep well block (VWR). Pick multiple single colonies per transformation, each in their own well to serve as biological replicates. Be sure to include the three negative controls for each plasmid component (see Table 4 for example negative controls) and to leave blank wells with no colonies as a control for bacterial growth.? Troubleshooting
- Seal with breathable film (VWR) and shake at 900 rpm at 37°C overnight.
-
Transformations for direct inoculation from 96-well plate.CRITICAL Transform competent reporter cells with one plasmid of each type (pAdapter, pBait, and pPrey). To have biological replicates, multiple transformations for each combination of plasmids should be conducted.
- Chill non-skirted 96-well PCR plate on ice or a cold block.
- Pipette 1 μL of each of three plasmids (pPrey, pBait, pAdapter; Table 4) and 30 μL of heat-shock competent reporter-strain cells. Plasmid stocks should have concentrations of 40–80 ng/uL.
- Cover with breathable film and incubate on ice for 30–60 mins.
- Heat shock 42°C for 45 seconds, using a PCR machine.
- Return to ice for 5 minutes.
- Add 70 μL SOC medium, cover with breathable firm and incubate at 37°C for 1 hr (no need to shake).
- Use up to 100 μL of bulk transformation recovery to inoculate 1 mL LB liquid medium supplemented with carb (100 μg/mL), cm (25 μg/mL), tet (10 μg/mL), spec (100 μg/mL) and 0.2% (wt/vol) arabinose in a 2 mL 96-well deep well block (VWR). Be sure to leave blank wells with no transformants.
-
Seal with breathable film (VWR) and shake at 900 rpm at 37°C overnight.? Troubleshooting
-
-
25
The next day, ensure that the blank wells are still clear. Dilute 5 μL of the overnight cultures 1:40 into 200 μL LB liquid medium in optically clear 200 μL flat bottom 96-well plates (Olympus) covered with plastic lids. When taking overnight culture be sure to take cells that are suspended in media and avoid cells that have sedimented at the bottom of the 96-well deep well block. LB should be supplemented as above with appropriate antibiotics and arabinose. During this step adding IPTG is optional. We have found that different interactions are optimized with different IPTG concentrations and recommend testing a concentration range such as 0, 10, 25 and 50 μM. Larger concentrations of IPTG can result in toxicity that leads to inconsistent cell growth.
-
26
Grow cells to mid-log (OD600 0.4–0.8) by measuring optical density using a spectrophotometer. OD600 readings of individual wells should be as close to each other as possible (ideally within 0.2 OD600 units).
? Troubleshooting
Detection of RNA-Protein interactions
-
27Measure RNA-protein interactions using (A) qualitative and/or (B) quantitative readout(s) of the assay. Qualitative readout requires the use of indicator plates and one to two days for the plates to develop. The indicator plate recipe requires optimization depending on the RNA-protein interaction being tested. Quantitative readout requires a microplate reader and produces a numerical value for β-gal production in Miller units. Quantitative readout does not require the same recipe modifications but can be less reliable when trying to detect weaker interactions. Doing both is ideal and both readouts can be performed from the same set of transformations. Note that either transformation method above (Step 26A or 26B) can be used with either detection method (see Experimental Design).
- Qualitative Readout of RNA-Protein interactions (plate-based assay): Timing 2–3 days.
- Dilute mid-log cells (OD600 0.4–0.8) from step 28 1:100.
- Spot 4 μL of this dilution on X-gal indicator plates. We recommend using a multichannel pipette for this step. Allow to dry so as to avoid bleeding together of the patches.
- Incubate overnight at 37°C.
-
Allow to sit at 4°C for 1–2 days to allow bacterial patch color to develop.? Troubleshooting
-
Photograph plates with a black-velvet background and oblique lighting.? Troubleshooting
- Adjust brightness and contrast levels evenly across photographs.
- Quantitative Readout of RNA-Protein interactions: Timing 1 day.
-
Begin lysis of cells grown to mid-log (OD600 0.4–0.8) from step 28 by combining 100 μL of the cells and 10 μL of lysis mixture (1.2 mL PopCulture and 1.2 μL of 400 U/ μL rLysozyme) in optically clear 200 μL flat bottom 96-well plates (Olympus) covered with plastic lids.? Troubleshooting
- Allow to lyse at RT for at least 30 minutes, but not for more than 2–3 hours.
- Freshly prepare Z-buffer with β-mercaptoethanol and ONPG as per “Reagent Setup”. CRITICAL STEP: Z-buffer with β-mercaptoethanol and ONPG must be prepared fresh.CAUTION: open β-mercaptoethanol in fume hood.
- Dilute 30 μL of lysed cells into 150 μL of Z-buffer mixture into optically clear 200 μL flat bottom 96-well plates (Olympus). CRITICAL STEP: immediately go to the next step.
-
Using a microplate spectrophotometer, immediately take OD420 readings at 28°C to measure β-gal activity. We recommend collecting data every minute for 1 hour and extracting the slope of these data for normalization by OD600 measurements to yield β-gal activity in Miller units, according to the formula25:? Troubleshooting
-
β-gal measurements averaged from multiple biological or technical replicates have a standard deviation (σ) associated with them. The estimated error of the fold-stimulation can be calculated by propagating the relative errors of each β-gal value:Pause Point Steps 30–43 are optional and do not have to be performed directly after the previous steps. The protocol can continue using the appropriate plasmids at any time.
-
Forward Genetic Mutagenesis Screen: Timing 11–13 days
CRITICAL As an optional step to complement hypothesis-driven mutagenesis experiments, the forward genetic screen protocol allows for the screening of a large library, first using qualitative screening methods to identify mutant RNA/proteins with desired molecular phenotypes, counter screening to confirm them, and then using the quantitative and/or qualitative method to verify findings. This screening method allows for many protein or RNA mutants to be screened in the B3H assay for changes in RNA-protein interactions. (See Figure 3 for a schematic of the forward genetic mutagenesis screen)
Mutagenesis library
-
28Obtain a mutagenesis library in pPrey or pBait for your protein or RNA of interest. The following protocol assumes you are screening a pPrey library for protein variants with altered phenotypes, but the logic can be applied for screening bait RNA mutant libraries.
- Using a previously created library
- Move directly to step 31.
- Make a pPrey mutagenesis library for a protein of interest Timing: 4 days
- Perform mutagenic PCR amplification on the protein or RNA portion of the wild-type pPrey or pBait plasmid using Taq DNA Polymerase. The protocol below is a straight-forward approach that relies on a large number of rounds of PCR and does not require special buffers; while sufficient for many libraries, this approach may result in bias of what mutations are created. For a more rigorous PCR mutagenesis protocol that results in lower bias, we recommend the protocol from Cadwell and Joyce26.
Component Amount Final 2x Taq Master Mix 90 μL 1x F primer (10 μM) 6 μL 0.33 μM R primer (10 μM) 6 μL 0.33 μM Template (round 1: 1 μL ~50 ng/uL WT plasmid created in step 14; round 2: 1 μL first-round product) 1 μL 50 ng ultra-pure water 72 μL Total 180 μL - Divide 180 μL reaction evenly into 6 PCR tubes (30 μL each) and perform 40 rounds of PCR.
Step Temperature (°C) Time Initial denaturation 95°C 30 seconds 40 cycles 95°C 30 seconds 50–60°C (annealing) 30 seconds 68°C 1 minute per kb Final elongation 68°C 5 minutes Hold 10°C - Recombine the PCR reactions from all 6 tubes and conduct PCR cleanup (e.g. Zymo “DNA Clean and Concentrator” Kit), following instructions from the manufacturer.
- Repeat Steps 30(B)i-iii for a second 40 rounds to obtain an 80-round Taq-amplified PCR product.
- Digest PCR products with Dpnl to remove template (WT) plasmid.
- Digest PCR products with NotI and BamHI in the same way as described in “Cloning for B3H Plasmids”.
- Gel purify the products to ensure they are approximately the same length as WT.
- Ligate (T4 DNA ligase; New England Biolabs) as in “Cloning for B3H Plasmids”.
- Transform the product into a cloning strain that provides lacIq, e.g. NEB 5-α F’Iq, New England Biolabs) as in “Cloning for B3H Plasmids”. Cells can be grown as near-lawns on LB plates with carb (100 μg/mL); dilutions can also be plated to obtain single colonies for sequencing individual clones.
- Miniprep a resuspension of the colonies to yield the plasmid library. To create the suspension, add 5 mL of LB to each plate and gently resuspend with a glass rod. Pool 1–2 mL from each plate to miniprep. Aim for 10,000+ colonies, based on the size of the gene you are mutagenizing.
-
From a plate of single colonies, sequence a subset (10–20) of miniprepped plasmids to check mutagenesis rate. Ideally most plasmids will be wild type and 10–20% will contain single mutations in the gene of interest, as effects of multiple mutations require additional work to disentangle).? Troubleshooting
-
29
Make competent cells from reporter cells pre-transformed with the two B3H plasmids not used for the mutant library (see steps 15–25; if screening pPrey mutations, pre-transform with pBait and pAdapter).
Primary Screen: Identify mutations of interest by screening with an initial partner Timing: 4–6 days.
-
30
Transform the plasmid library into cells with pre-transformed reporter cells (see step 26).
-
31
Alongside transformation of the library, conduct positive and negative control transformations. For example, α-empty would be used as a negative control for a protein prey mutagenesis library and WT pPrey would be used for the positive control. These will assist in assessing baseline colony-color phenotypes, since colonies will become more blue over time.
-
32
Plate on X-gal indicator plates with appropriate antibiotics, inducers and indicators (see reagent setup). It may take some time to find the appropriate X-gal + TPEG concentration combination to best accentuate the white of your negative control and the blue of your positive control. The ideal concentrations can differ between various RNA-protein interactions based on the amount of lacZ being produced.
-
33
Incubate plates overnight at 37°C, then at 4°C for an additional ~24–48 h. This allows the blue color to develop and facilitates distinctions between different levels of interaction.
? Troubleshooting
-
34
Identify and restreak colonies of interest on X-gal indicator plates, alongside negative and positive controls. If looking for interactors with a reduced interaction, restreak pale/white colonies to confirm colors. If looking for increased levels of interaction, restreak blue colonies.
? Troubleshooting
Counter Screen: Perform a counter assay with the mutant plasmids isolated from the primary screen.
-
35Determine whether you are screening for mutations that cause (A, B) general-binding or (C) partner-specific binding effects. If searching for general-binding effects, the goal of the counter screen will be to eliminate the subset of plasmids from the primary screen that contained uninformative mutations such as premature stop codons or substitutions that caused misfolding/degradation of the prey protein; this can be achieved either with (A) a dot blot with immunodetection16 or (B) a B2H assay with a secondary protein interactor12 (Fig. 3). Partner-specific effects can be identified by counter screening with a (C) B3H interaction with a second RNA bait.
-
Counter screen for prey protein stability using immunodetectionCRITICAL A dot-blot counter-screen with anti-prey antibodies can quickly identify mutations that produce protein variants with low expression levels and should be discarded. This approach requires an antibody that is specific to the prey protein and requires that any endogenous expression of the prey protein is substantially lower than overexpression of the α-prey fusion protein. While Western blots could also be helpful to confirm the stability of protein mutants (or Northern blots for RNA mutants), they are not recommended as a counter screen prior to sequencing individual colonies due to their low throughput nature.
- Inoculate a 96-well plate containing 1 mL (per well) LB liquid medium supplemented with antibiotics + arabinose directly from colonies of confirmed hits from the primary screen. No miniprep or retransformation is necessary at this stage. Be sure to include positive controls (WT prey) and negative controls (α empty).
- Follow instructions for bacterial growth and lysis from inoculation in step 26(A) through lysis in step 28(B).
- Using multichannel pipette, transfer cell lysates (3 μL) from β-gal assays directly to nitrocellulose Protran membranes (Amersham). Simply spot lysate onto the membrane and allow to dry.
- Block membrane in blocking solution (e.g., 2% (wt/vol) milk and 0.1% (wt/vol) Tween in TBS) for 30 minutes at RT, then incubate with primary antibody, rocking overnight at 4°C.
- Rinse with 0.1% (wt/vol) Tween in TBS three times for 10 minutes each, then incubate with a secondary antibody rocking at RT for 2 hours, and rinse again with 0.1% (wt/vol) Tween in TBS three times for 10 minutes each.
- Detect chemiluminescent signal from antibody conjugated to horseradish peroxidase using ECL detection reagent and an imaging system according to manufacturer’s instructions.
- Identify the wells that produce comparable prey expression as a WT control and return to the plate of restreaked colonies of primary hits to inoculate individual cultures to miniprep desired mutants.
-
Counter screen for protein stability and function through a B2H protein-protein interaction assayCRITICAL A protein-protein B2H interaction is a straightforward secondary assay that can report on the overall folding and function of variants of prey protein. For instance, counter screening pBrα-Hfq mutants against a B2H interaction with pACλCI-Hfq effectively eliminated mutants containing premature stop codons12. Because this approach does not require an antibody with low background to endogenous proteins and makes use of the same detection method as the primary screen, this is recommended if a B2H interaction with a protein partner can be established with your RNA-binding protein of interest.
- Miniprep to isolate plasmids from colonies from the primary screen (step 36) that produced the phenotype of interest. For >10 primary hits, we recommend pooling all hits by inoculating a single overnight culture with each colony of interest. Bacterial cultures can be grown with only the relevant antibiotic (e.g. carbenicillin for pPrey libraries), but keep in mind that these minipreps will likely still contain the other two plasmids that were present in the reporter cells used for the screen (pAdapter, pBait).
- Transform pooled miniprep of primary screen hits into reporter cells pre-transformed with an empty pAdapter plasmid and a pACI-fusion protein encoding an interacting protein (e.g. CI-Hfq)12. CRITICAL STEP: It is important to use pre-transformed cells in the counter screen in order to out-compete contaminating plasmids (e.g. pAdapter + pBait) present in the miniprep of primary-screen hits.
- Follow instructions as for step 33–35. During the counter screen, aim to screen at least 10 times as many colonies as your primary hits to ensure that each hit from the pooled miniprep is sampled.
- Pick single blue colonies to inoculate an individual culture to miniprep desired mutants.
- Counter screen for RNA-specific binding effects through a B3H assay with a second RNA bait:
- Repeat steps from 37(B)ii-iv but using reporter cells pre-transformed with pAdapter and pBait encoding RNA Bait #2. By screening for the subset of colonies that lost interaction with RNA Bait #1 in the primary screen but retain interaction with RNA Bait #2 in this secondary screen, you can identify the rare but mechanistically interesting candidates that cause RNA-specific binding defects.
-
Miniprep and sequence mutants-of-interest
-
36
From colonies containing plasmids with desired mutants, grow overnight cultures (~5 mL) in LB-carbenicillin at 37°C for minipreps. Keep in mind that these minipreps will likely still contain the other two plasmids that were present in the reporter cells used for the screen (pAdapter, pBait; see Figure 3).
-
37
Sequence mutants-of-interest. Refer to Table 5 for a list of primers for use in sequencing. While it is possible to perform steps 40–43 before sequencing, we have found the minipreps that contain all three plasmids (pPrey, pBait, pAdapter) have sufficient concentration of pPrey for sequencing.
Isolate pPrey plasmid from other two vectors for use in follow-up experiments
-
38
Begin with miniprep from B2H/B3H screen that contains a mixture of mutant pPrey plasmid, pAdapter and pBait. Dilute this miniprep ~1:100 to reduce chances of multiple plasmids being transformed into single cells.
-
39
Transform diluted miniprep into NEB 5-α F’Iq E. coli and plate on LB-carbenicillin to select only for pPrey.
-
40
Patch single colonies on LB-agar plates containing one of the following antibiotics at a time: carb, spec, cm. To isolate pPrey, you will look for a colony that is resistant to carb, but sensitive to spec and cm, indicating the loss of pBait and pAdapter, respectively.
-
41
From a patch that is sensitive to the appropriate antibiotics, streak for single colonies on the appropriate antibiotic, and isolate plasmid from an overnight culture inoculated from a single colony using a miniprep kit. This plasmid prep can now be used in triple transformations into reporter cells lacking any plasmids.
Timing
Steps 1–14, Molecular Cloning: 4–6 days
Step 15–25, Make Competent Cells: 3 days
- Steps 26–28, Transformation, growth, and induction:
- Transform into liquid: 1 day
- Transform onto plates and inoculate: 2 days
- Step 29, Detection of RNA-protein interactions:
- Qualitative: 2–3 days
- Quantitative: 1 day
Steps 30–43, Random-mutagenesis screen: 11–13 days
Troubleshooting
Troubleshooting advice can be found in Table 6.
Table 6.
Troubleshooting
| Step | Problem | Possible Reason | Possible Solution |
|---|---|---|---|
| 26(A)ix | No colonies on plates | Competent cells are not competent enough | Remake competent cells being careful to keep cold throughout. Increase concentration of DNA being transformed; if necessary, pre-transform cells with one of the vectors so that you need only conduct a double transformation. |
| 26(B)viii | No growth in overnight cultures | ||
| 28 | Uneven OD600 readings. | Cells are growing at different speeds. | Plate and/or lyse fast-growing cells when they reach mid-log or late-mid log, allowing the rest of the cells to continue growing, lysing cells as they reach the desired level of growth. Lysed cells can remain at room temperature for up to 3 hrs. |
| Evaporation of media is causing volume to be inconsistent across samples. | Ensure plates are covered. Increase humidity of the incubator you are growing cells in and/or reduce growing time of the cells to reduce evaporation. If the incubator allows it, 70–80% humidity is ideal; if using an incubator without humidity control, warm water in the base of the incubator can help. | ||
| 29(A)iv | Indicator plates: negative controls appear too blue. | The concentrations of [TPEG] and [X-gal] in indicator plates may not be optimal for this interaction. | Try decreasing [TPEG] or increasing [X-gal] and repeat positive and negative controls to identify appropriate screening conditions. |
| Indicator plates: positive controls appear too white. | Try increasing [TPEG] or decreasing [X-gal]. (See Fig. 4b for the effect of TPEG in reducing the blue color of negative controls and accentuating the differences between positives and negatives.) | ||
| Indicator plates have splatter of bacterial colonies surrounding individual bacterial patches. | Fully ejecting the samples onto the plate will push air through the multichannel pipet if you go past the first stop. | When plating samples only go to the first stop. It is okay if some of the patches are smaller, because phenotype is based on color, not size, of the bacterial patch or number of colonies. | |
| High β-gal from a negative control. | Could be a direct interaction between your Bait and Prey and another component of the system. | Perform double-negative controls to better understand what is producing high signal in negative controls. | |
| 29(A)v | Trouble taking photographs of indicator plates | Lighting or set up may make it hard to see color differences on photos. | We have found that the best images were produced by placing the plate on black velvet, using four desk lights to create even, oblique, lighting, and placing the camera on a ring stand to keep it steady. |
| 29(B)i | Cells are growing too slowly. | IPTG levels may be too high and causing toxicity. | Lower IPTG concentration. |
| Δhfq are growing too slowly. | Δhfq allele introduces a growth defect. | Try B3H interaction in hfq+ strain instead. | |
| 29(B)v | Seeing low interactions in hfq+ strain | The endogenous E. coli RNA-binding protein Hfq may be competing with your Prey protein | Try B3H interaction in Δhfq strain or delete your protein-of-interest if encoded in the E. coli genome. |
| New interaction not being detected well. | Different interactors have different ideal conditions. | New interactions should be tested in a variety of IPTG concentrations as well as at a variety of OD readings (see next). | |
| Low β-gal from a WT interaction. | Different proteins seem to have different preferences for bacterial growth for optimal signals. | Try increasing or decreasing the OD at which you lyse your bacterial cultures. The ideal OD value may change depending on the interaction being studied. For us, experiments with Hfq generally had better B3H signal in cells at a lower OD (OD=0.3–0.4), while experiments with ProQ performed better at a slightly higher OD (0.8–0.9). | |
| High β-gal from a negative control. | Could be a direct interaction between your Bait and Prey and another component of the system. | Perform double-negative controls to better understand what is producing high signal in negative controls. | |
| 30(B)xi | Most clones from mutagenesis library contain multiple mutations. | Mutation rate too high. | Fewer cycles of mutagenic PCR. |
| Most clones from mutagenesis library contain no mutations (wild type sequences). | Mutation rate is too low. | More cycles of PCR; or use protocol with varied [dNTP] and addition of 0.1M MnCl2.26 | |
| 35 | Not enough colonies on screening plates to effectively cover the size of the mutagenesis library. | Transformation efficiency is too low when introducing three plasmids simultaneously to reporter cells. | Competent cells of the reporter strain can be made with one of the three plasmids (e.g. pAdapter) pre-transformed so that only two plasmids need to be transformed in at the same time for the screen. |
| See troubleshooting for 29A(iv) regarding indicator plates. | |||
| 36 | Colony color of apparent hits doesn’t repeat upon restreaking. | False positives or negatives. | Revisit plating conditions [X-gal, TPEG, IPTG] with positive and negative controls to maximize blue/white difference; compare colonies that are similar in size as larger colonies will naturally appear more blue in color; avoid picking colonies near the edge of the plate. |
Anticipated Results
Fig. 4a shows an example B3H assay with qualitative detection (step 29(A)) using the RNA-binding protein ProQ as prey and the sRNA SibB as bait; the combinations of plasmids used in transformations for this experiment are shown in Table 4. Bacteria transformed with all three hybrid components (positive interactions) develop blue color while negative controls with one of three plasmids empty stay white, grey, or a lighter blue (Fig. 4a). Duplicate patches for each transformation show good consistency in phenotype between biological replicates. The strength of the B3H interaction is not just the blue color of the experimental patch but the additional blue color above any background produced by the negative controls. Fig. 4a also shows the helpful effects of TPEG — a competitive inhibitor of β-galactosidase — in minimizing the pale blue color of negative-control patches, which facilitates the distinction of color between positive and negative patches. Note that the plate conditions used in this protocol were optimized to show clear data for ProQ/Hfq with these RNA targets. It is likely that a different protein/RNA pair may require different concentrations of each component in the indicator plates (see Troubleshooting). Use of TPEG is strongly recommended when working with relatively weak interactions (less than three-fold stimulation of lacZ).
To demonstrate the potential of this assay to report on the specificity of interactions, we show a second example qualitative experiment in which we use both Hfq and ProQ as prey proteins (Fig. 4b). We chose RNA baits that are known interactors with Hfq (OxyS and ChiX) and with ProQ (SibB and cspE 3’ UTR)27–29. When pPrey-Hfq is co-transformed with pBait-OxyS or pBait-ChiX, blue bacterial patches are seen (Fig. 4b), while SibB-Hfq and cspE-Hfq pairs produce lighter or white patches, consistent with the latter RNA-protein pairs not interacting strongly. In contrast, ProQ appears to be more promiscuous, interacting not only with cspE and SibB, but also with the Hfq-dependent sRNAs OxyS and ChiX (Fig. 4b). These assays were conducted in an Δhfq reporter strain; it is interesting to note that the presence of endogenous Hfq can affect the strength of the observed ProQ-RNA interactions16,17.
When the same experiments are conducted with the quantitative readout (step 29(B)), the β-gal activity measured from liquid cultures corresponds nicely to the blue phenotypes seen on indicator plates (Fig. 4c–e vs. 4a,b). Fig. 4c shows the absolute β-gal activities from transformations to report on the SibB-ProQ interactions. Positive interactions often have β-gal activity at or above 70 Miller units, while negative controls with one of the three plasmids “empty” usually show background signals around 20–50 Miller units (Fig. 4c,d). To quantitatively compare multiple interaction, we often show these results as fold-stimulation above the highest negative control (Fig. 4e); for the SibB-ProQ interaction shown in Fig. 4c, this is calculated as
For genetic screens (Steps 30–43), transformants with a mutagenesis library should be plated at a density that allows for clear distinction between individual white and blue colonies (Fig. 4f). Indicator-plate conditions should be adjusted so that hits in the screen are easily distinguished from controls based on their blue color (see Troubleshooting in Table 6).
Supplementary Material
{kind=link}
Acknowledgements
We thank members of the Berry lab for discussion and suggestions and Ann Hochschild, Padraig Deighan and Danielle Heller for discussions and advice in establishing these protocols. This work was supported with funding from the National Institutes of Health [R15GM135878], the Camille and Henry Dreyfus Foundation, the Henry R. Luce foundation, and Mount Holyoke College.
Footnotes
Competing interests
The authors have no competing interests to report.
Data availability
A dataset used for construction of Fig. 4c–e (Stockert_SourceData_Fig_4cde.xlsx) and raw images used for construction of Fig. 4a–b (Stockert_SourceData_Fig_4ab.jpg) are available in the Supplementary Information.
REFERENCES:
- 1.Wagner EGH & Romby P Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv. Genet 90, 133–208 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Gottesman S & Storz G Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harbor perspectives in biology 3, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng Kwan Lim E, Sasseville C, Carrier M-C & Massé E Keeping Up with RNA-Based Regulation in Bacteria: New Roles for RNA Binding Proteins. Trends in Genetics 37, 86–97 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Quendera AP et al. RNA-Binding Proteins Driving the Regulatory Activity of Small Non-coding RNAs in Bacteria. Front. Mol. Biosci 7, 78 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olejniczak M & Storz G ProQ/FinO-domain proteins: another ubiquitous family of RNA matchmakers?: New family of RNA matchmakers. Molecular Microbiology 104, 905–915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmqvist E & Vogel J RNA-binding proteins in bacteria. Nat Rev Microbiol 16, 601–615 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Licatalosi DD, Ye X & Jankowsky E Approaches for measuring the dynamics of RNA-protein interactions. Wiley Interdiscip Rev RNA 11, e1565 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramanathan M, Porter DF & Khavari PA Methods to study RNA-protein interactions. Nat Methods 16, 225–234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.SenGupta DJ et al. A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A 93, 8496–8501 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koh YY & Wickens M Identifying Proteins that Bind a Known RNA Sequence Using the Yeast Three-Hybrid System - ScienceDirect. Methods in Enzymology 539, 195–214 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Hook B, Bernstein D, Zhang B & Wickens M RNA-protein interactions in the yeast three-hybrid system: affinity, sensitivity, and enhanced library screening. RNA 11, 227–233 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry KE & Hochschild A A bacterial three-hybrid assay detects Escherichia coli Hfq-sRNA interactions in vivo. Nucleic Acids Res. 46, e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dove SL, Joung JK & Hochschild A Activation of prokaryotic transcription through arbitrary protein–protein contacts. Nature 386, 627–630 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Dove SL & Hochschild A Bacterial two-hybrid analysis of interactions between region 4 of the sigma(70) subunit of RNA polymerase and the transcriptional regulators Rsd from Escherichia coli and AlgQ from Pseudomonas aeruginosa. J. Bacteriol 183, 6413–6421 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang CD, Mansky R, LeBlanc H, Gravel CM & Berry KE Optimization of a bacterial three-hybrid assay through in vivo titration of an RNA–DNA adapter protein. RNA 27, 513–526 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandey S et al. Genetic identification of the functional surface for RNA binding by Escherichia coli ProQ. Nucleic Acids Res. 48, 4507–4520 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stein EM et al. Determinants of RNA recognition by the FinO domain of the Escherichia coli ProQ protein. Nucleic Acids Res 48, 7502–7519 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faner MA & Feig AL Identifying and characterizing Hfq–RNA interactions. Methods 63, 144–159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HJ, Chaulk S, Arthur D, Edwards RA & Glover JN M. Biochemical Methods for the Study of the FinO Family of Bacterial RNA Chaperones. in RNA Chaperones (ed. Heise T) vol. 2106 1–18 (Springer US, 2020). [DOI] [PubMed] [Google Scholar]
- 20.Fields S & Song O A novel genetic system to detect protein–protein interactions. Nature 340, 245–246 (1989). [DOI] [PubMed] [Google Scholar]
- 21.Chien CT, Bartel PL, Sternglanz R & Fields S The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. PNAS 88, 9578–9582 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikulecky PJ et al. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nat Struct Mol Biol 11, 1206–1214 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olejniczak M Despite Similar Binding to the Hfq Protein Regulatory RNAs Widely Differ in Their Competition Performance. Biochemistry 50, 4427–4440 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Rao X et al. A regulator from Chlamydia trachomatis modulates the activity of RNA polymerase through direct interaction with the β subunit and the primary σ subunit. Genes Dev. 23, 1818–1829 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thibodeau SA, Fang R & Joung JK High-throughput β-galactosidase assay for bacterial cell-based reporter systems. BioTechniques 36, 410–415 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Cadwell RC & Joyce GF Mutagenic PCR. Cold Spring Harb Protoc, doi: 10.1101/pdb.prot4143 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Smirnov A et al. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc Natl Acad Sci USA 113, 11591–11596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmqvist E, Li L, Bischler T, Barquist L & Vogel J Global Maps of ProQ Binding In Vivo Reveal Target Recognition via RNA Structure and Stability Control at mRNA 3′ Ends. Molecular Cell 70, 971–982.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Melamed S, Adams PP, Zhang A, Zhang H & Storz G RNA-RNA Interactomes of ProQ and Hfq Reveal Overlapping and Competing Roles. Molecular Cell 77, 411–425.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baba T et al. Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol 2, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whipple FW Genetic analysis of prokaryotic and eukaryotic DNA-binding proteins in Escherichia coli. Nucleic Acids Res 26, 3700–3706 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A dataset used for construction of Fig. 4c–e (Stockert_SourceData_Fig_4cde.xlsx) and raw images used for construction of Fig. 4a–b (Stockert_SourceData_Fig_4ab.jpg) are available in the Supplementary Information.