

Abstract
The efficacy of treating sarcoidosis‐associated pulmonary hypertension (SAPH) with pulmonary vasodilator therapy is unclear. The INCREASE trial showed improvement in 6‐minute walk distance (6MWD) and in decline in functional vital capacity (FVC) in patients with interstitial lung disease and pulmonary hypertension. We hypothesize that patients with SAPH treated with pulmonary vasodilators have reduced decline in FVC. We retrospectively analyzed patients with SAPH who underwent lung transplantation evaluation. The primary objective was to compare change in FVC between patients with SAPH who received pulmonary vasodilators (treated) and those who did not (untreated). Secondary objectives were to compare the change in 6MWD, change in oxygen requirement, transplant rates, and mortality between treated and untreated SAPH patients. We identified 58 patients with SAPH; 38 patients received pulmonary vasodilator therapy, and 20 patients did not. Treated SAPH patients had significantly less decline in FVC than untreated SAPH patients (+54 mL vs. −357 mL, p < 0.01). Treated SAPH patients had significantly higher survival than untreated SAPH patients. Receiving PH therapy was significantly associated with a change in FVC (estimate 0.36 ± 0.07, p < 0.01) and decreased mortality (hazard ratio 0.29, confidence interval 0.12–0.67, p < 0.01). Among patients with SAPH, those who received pulmonary vasodilator therapy had significantly less decline in FVC and increased survival. Receiving pulmonary vasodilator therapy was significantly associated with FVC change and decreased mortality. These study findings point towards potential benefit of pulmonary vasodilator therapy in SAPH patients. Further prospective studies are required to fully elucidate the benefits of pulmonary vasodilator therapy in SAPH.
Keywords: functional vital capacity, lung transplant, pulmonary hypertension, pulmonary vasodilator therapy, sarcoidosis
Abbreviations
- 6MWD
6‐minute walk distance
- APS
advanced pulmonary sarcoidosis
- CI
cardiac index
- COPD
chronic obstructive pulmonary disease
- CPFE
combine pulmonary fibrosis and emphysema
- DLCO
diffusing capacity of the lung for carbon monoxide
- ERA
endothelin receptor antagonist
- FEV1
forced expiratory volume in 1 s
- FVC
functional vital capacity
- ILD
interstitial lung disease
- IRB
Institutional Review Board
- LAS
lung allocation score
- mPAP
mean pulmonary artery pressure
- NYHA
New York Heart Association
- PA
pulmonary artery
- PCWP
pulmonary capillary wedge pressure
- PDE
phosphodiesterase
- PH
pulmonary hypertension
- PVR
pulmonary vascular resistance
- RA
right atrial
- RHC
right heart catheterization
- RV
right ventricle
- SAPH
sarcoidosis‐associated pulmonary hypertension
- STROBE
Strengthening the Reporting of Observational Studies in Epidemiology
- TLC
total lung capacity
- WASOG
World Association of Sarcoidosis and Other Granulomatous Diseases
- WU
Woods units
INTRODUCTION
Sarcoidosis is a multisystem inflammatory disease characterized by respiratory involvement in the vast majority of patients. 1 A small percentage of patients with pulmonary sarcoidosis develop advanced pulmonary sarcoidosis (APS) despite optimal pharmacologic management, which includes sarcoidosis‐associated pulmonary hypertension (SAPH). 2 Anywhere from 5.7% to 28.3% of all sarcoidosis patients develop SAPH, with pulmonary hypertension (PH) most recently defined as mean pulmonary artery pressure (mPAP) > 20 mmHg. 3 , 4 SAPH occurs more so within patients with interstitial lung disease and pulmonary fibrosis secondary to sarcoidosis, but it can occur even in the absence of significant lung disease. Most patients with APS referred for lung transplantation have SAPH. 5 Among patients with sarcoidosis, those with SAPH have increased morbidity and mortality. 3
There is limited and mixed data on the benefit of treating SAPH with pulmonary vasodilator therapies. To our knowledge, there have been two prospective randomized trials evaluating pulmonary vasodilator therapy in SAPH, specifically bosentan, and riociguat. Bosentan, an endothelin‐receptor antagonist (ERA), was shown to improve pulmonary hemodynamics but not 6‐minute walk distance (6MWD) or quality of life, while riociguat, a soluble guanylate cyclase stimulator, showed improved 6MWD and prolonged time to clinical worsening. These studies did not show a benefit in lung function or mortality with treatment of SAPH. 6 , 7 However, a post hoc analysis of the INCREASE trial, which evaluated the efficacy of inhaled treprostinil, a prostacyclin analog, in patients with PH and interstitial lung disease (ILD), showed a significant improvement in placebo‐corrected difference in percent‐predicted functional vital capacity (FVC) over 16 weeks and a trend towards in improvement in actual FVC. 8 Notably, the INCREASE trial did not include patients with sarcoidosis.
We hypothesize that patients with SAPH who receive pulmonary vasodilator therapy have decreased lung function decline. Our primary objective was to compare the change in FVC between patients with SAPH who received PH treatment and those who did not. Our secondary objectives were to compare change in 6MWD, change in oxygen requirement, transplant rates, and mortality before transplant between patients with SAPH who received PH treatment and those who did not.
STUDY DESIGN AND METHODS
Study design and definitions
This was a retrospective cohort study of patients with SAPH who underwent lung transplantation evaluation at our institution, Temple University Hospital, from 2008 to 2021. SAPH was defined as mPAP ≥ 25 mmHg on right heart catheterization (RHC) in patients with biopsy‐proven sarcoidosis, as 25 mmHg was the cut‐off for pulmonary hypertension for the majority of the time period of the study. We excluded patients evaluated/listed for combined heart–lung transplantation. Patient characteristics, comorbidities, anti‐inflammatory treatment, and right heart catheterization values at time of PH diagnosis were collected. Pre‐capillary pulmonary hypertension was defined as mPAP ≥ 25 mmHg, pulmonary capillary wedge pressure (PCWP) ≤ 15 mmHg, and pulmonary vascular resistance (PVR) ≥ 3 Woods units (WU). Spirometry, 6MWD, and oxygen requirement were collected at time of PH diagnosis and at 12 months after initial PH diagnosis; as part of the lung transplant evaluation at our institution, patients undergoing evaluation perform routine spirometry and 6‐min walk testing every 4–8 weeks. If patients were deceased or underwent lung transplantation before 12 months after PH diagnosis, values at last follow‐up before death or transplant were collected. Mortality before transplantation, lung transplantation rates, and time to death were collected as well. These variables were compared between patients with SAPH who received pulmonary vasodilator therapy and patients with SAPH who did not receive pulmonary vasodilator therapy. The decision to treat PH was at the discretion of the individual pulmonary physicians providing care for these patients. Our study met approval for waiver of informed consent by the Western Institutional Review Board (IRB, Protocol # 29957). Procedures were followed in accordance with the ethical standards of the Western IRB and the Helsinki Declaration of 1975. The authors used the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist in the preparation of this manuscript. Patients or the public were involved in the design, or conduct, or reporting, or dissemination plans of our research.
Statistical analysis
Statistical analysis was performed comparing treated SAPH to untreated SAPH. All continuous variables were presented as mean + standard deviation unless otherwise stated. Categorical variables were compared using Pearson chi‐squared test or Fisher exact test where applicable. Continuous variables were compared between groups using Mann–Whitney U test. Univariate analysis was performed to identify significant associations with the outcomes of change in FVC and mortality. For a change in FVC, univariate analysis consisted of one‐way analysis of variance if the independent variable was a categorical variable or linear regression if the independent variable was a continuous variable. For mortality, univariate analysis was performed with cox proportional hazards analysis. Variables with significant association with the outcomes (p < 0.05) were subsequently utilized in multivariable regression models for change in FVC and for mortality. Kaplan‐Meier analysis was performed to compare survival before transplant between patients with SAPH who received pulmonary vasodilator treatment and patients with SAPH who did not receive pulmonary vasodilator treatment. Statistical analysis was performed with the use of Stata 14.0 (StataCorp LP).
RESULTS
We identified 58 patients with SAPH who underwent lung transplant evaluation at our institution between 2008 and 2021 (Figure 1). Of these, 38 patients received pulmonary vasodilator treatment, and 20 patients did not. Baseline patient characteristics, comorbidities, and oxygen requirements were statistically similar between the two groups (Table 1). Twenty‐three patients in the cohort had additional lung disease, all of which was chronic obstructive pulmonary disease (COPD). 18 patients with SAPH and concurrent COPD received pulmonary vasodilator therapy, and 5 did not. Most patients identified as black race. There were similar rates of extra‐pulmonary sarcoidosis involvement between the two groups. Prednisone dose, rates of non‐corticosteroid anti‐inflammatory treatment, and rates of anti‐fibrotic treatment were similar between the two groups as well (Table 1).
Figure 1.
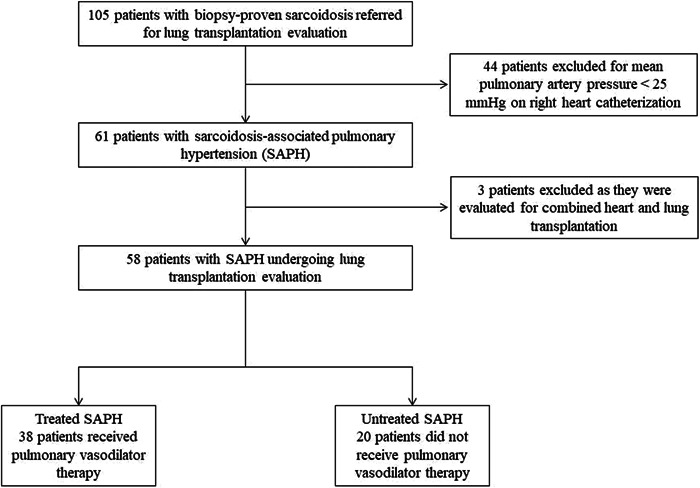
Inclusion and exclusion of patients with sarcoidosis‐associated pulmonary hypertension. Patients with sarcoidosis referred for lung transplantation were excluded if they did not have pulmonary hypertension on right heart catheterization, defined as mean pulmonary artery pressure ≥ 25 mmHg, and if they were evaluated for combined heart and lung transplantation. 58 total patients with sarcoidosis‐associated pulmonary hypertension evaluated for lung transplantation were included in the study.
Table 1.
Baseline and clinical characteristics at time of PH diagnosis.
| Treated SAPH (n = 38) | Untreated SAPH (n = 20) | |
|---|---|---|
| Age (years) | 57.9 ± 9.4 | 55.0 ± 6.8 |
| Gender | ||
| Female, n (%) | 23 (60.5) | 8 (40.0) |
| Male, n (%) | 15 (39.5) | 12 (60.0) |
| Weight (kg) | 77.3 ± 20.9 | 79.8 ± 11.6 |
| Race | ||
| Black, n (%) | 32 (84.2) | 14 (70.0) |
| White, n (%) | 4 (10.5) | 3 (15.0) |
| Other, n (%) | 2 (5.3) | 3 (15.0) |
| Smoking history, n (%) | 20 (52.6) | 11 (55.0) |
| Diastolic dysfunction, n (%) | 12 (31.6) | 4 (20.0) |
| Systolic dysfunction, n (%) | 3 (7.9) | 2 (10.0) |
| Kidney disease, n (%) | 21 (55.2) | 9 (45.0) |
| Liver disease, n (%) | 5 (13.1) | 1 (5.0) |
| Diabetes, n (%) | 10 (26.3) | 3 (15.0) |
| Additional lung disease/concurrent COPD, n (%) | 18 (47.4) | 5 (25.0) |
| Extrapulmonary sarcoidosis, n (%) | 9 (23.7) | 5 (25.0) |
| Oxygen requirement (L/min) | 3.3 ± 2.9 | 2.8 ± 2.7 |
| Prednisone dose (mg) | 9.5 ± 8.6 | 12.1 ± 6.2 |
| Additional anti‐inflammatory treatment | ||
| Methotrexate, n (%) | 13 (34.0) | 9 (45.0) |
| Other agents, n (%) | 5 (13.0) | 4 (20.0) |
| Anti‐fibrotic Therapy, n (%) | 1 (2.6) | 0 (0.0) |
| RA Pressure (mmHg) | 8.4 ± 4.4 | 6.9 ± 2.7 |
| PA systolic (mmHg)* | 58.5 ± 15.5 | 49.3 ± 10.5 |
| mPAP (mmHg)* | 36.3 ± 8.7 | 31.4 ± 5.9 |
| PCWP (mmHg) | 9.5 ± 5.5 | 11.0 ± 4.1 |
| PVR (Woods units)* | 6.6 ± 3.2 | 3.8 ± 1.3 |
| CI (L/min/m2) | 2.5 ± 0.6 | 2.9 ± 0.9 |
Note: Continuous variables are presented as mean and standard deviation. Statistical comparison of each variable between treated SAPH and untreated SAPH groups yielded p > 0.05. All patients with additional lung disease had chronic obstructive pulmonary disease (COPD). All patients were on prednisone.
Abbreviations: CI, Cardiac Index; COPD, chronic obstructive pulmonary disease; mPAP, mean pulmonary artery pressure; PA, pulmonary artery; PH, pulmonary hypertension; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RHC, right‐heart catheterization; SAPH, sarcoidosis‐associated pulmonary hypertension.
*p < 0.05.
Pulmonary hemodynamics determined by RHC showed that patients with SAPH who received PH treatment had higher mPAP (36.3 mmHg vs. 31.4 mmHg) and PVR (6.6 WU vs. 3.8 WU) than patients with SAPH who did not receive PH treatment (Table 1). Among both groups, the mean PCWP was ≤ 15 mmHg and mean PVR was > 3 WU, suggesting that the majority of patients had precapillary SAPH.
Pulmonary vasodilator therapy prescribed among treated SAPH patients is seen in Table 2. All therapy was prescribed after RHC was performed and demonstrated PH. Most patients were prescribed oral agents, typically either phosphodiesterase‐5 inhibitors (sildenafil or tadalafil) or one of the endothelin‐receptor antagonists (ambrisentan, bosentan, or macitentan). The majority of patients were prescribed single‐agent therapy, and only one patient was prescribed triple‐agent therapy.
Table 2.
Pulmonary vasodilator treatment among treated SAPH Group.
| Pulmonary vasodilator treatment | Prescribed treatment (n = 38) |
|---|---|
| Sildenafil or Tadalafil (PDE‐5 inhibitor), n (%) | 23 (60.5) |
| Ambrisentan (ERA), n (%) | 9 (23.6) |
| Bosentan (ERA), n (%) | 9 (23.6) |
| Macitentan (ERA), n (%) | 8 (21) |
| Inhaled prostacyclin, n (%) | 3 (7.9) |
| Subcutaneous prostacyclin, n (%) | 3 (7.9) |
Abbreviations: ERA, endothelin receptor antagonist; PDE, phosphodiesterase; PH, pulmonary hypertension; SAPH, sarcoidosis‐associated pulmonary hypertension.
Mean spirometry, 6MWD, oxygen requirement, and New York Heart Association (NYHA) functional class at time of PH diagnosis were statistically similar between the treated SAPH and untreated SAPH groups (Table 3). However, a higher proportion of patients in the untreated SAPH group had a preserved FEV1/FVC ratio than in the treated SAPH group (75.0% vs. 47.3%, p = 0.04). All patients had Scadding Stage 4 disease on chest radiograph, indicating that all patients in the cohort had pulmonary fibrosis secondary to sarcoidosis. The mean percent‐predicted diffusing capacity of the lung for carbon monoxide (DLCO) was similar between the two groups as well (<30% predicted).
Table 3.
Spirometry, 6MWD, oxygen requirement, NYHA functional class, and scadding stage at time of PH diagnosis.
| Treated SAPH (n = 38) | Untreated SAPH (n = 20) | |
|---|---|---|
| FEV1 (L) | 1.2 ± 0.5 | 1.4 ± 0.6 |
| % Predicted FEV1 (%) | 45.7 ± 15.6 | 49.8 ± 17.9 |
| FVC (L) | 1.7 ± 0.6 | 2.0 ± 0.8 |
| % Predicted FVC (%) | 52.3 ± 13.6 | 54.6 ± 18.3 |
| FEV1/FVC > 0.7, n (%)* | 18 (47.3) | 15 (75.0) |
| Percent‐predicted DLCO | 26.7 ± 12.3 | 29.6 ± 11.8 |
| 6MWD (m) | 224.4 ± 89.0 | 267.7 ± 78.3 |
| NYHA functional class | ||
| II, n (%) | 0 (0) | 1 (5) |
| III, n (%) | 16 (42.1) | 13 (65) |
| IV, n (%) | 22 (57.9) | 6 (30) |
| Scadding stage | ||
| 0, n (%) | 0 (0) | 0 (0) |
| I, n (%) | 0 (0) | 0 (0) |
| II, n (%) | 0 (0) | 0 (0) |
| III, n (%) | 0 (0) | 0 (0) |
| IV, n (%) | 38 (100) | 20 (100) |
Abbreviations: 6MWD, 6‐minute walk distance; DLCO, diffusing capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, functional vital capacity; NYHA, New York Heart Association; PH, pulmonary hypertension; SAPH, sarcoidosis‐associated pulmonary hypertension.
p < 0.05.
Table 4 shows change in spirometry, 6MWD, and oxygen requirement at 12‐month follow‐up (or last follow‐up before death or lung transplant if either occurred before 12 months since PH diagnosis). Patients with SAPH who did not receive pulmonary vasodilators had a statistically significant decrease in FEV1 (p = 0.0001) and FVC (p < 0.0001) when compared with patients with SAPH who did. Among the treated SAPH group, FEV1 was stable, and FVC increased by 54 mL from time of PH diagnosis to the 12‐month follow‐up (or sooner if death or lung transplantation occurred before 12 months). Among the untreated SAPH group, FEV1 decreased by 257 mL, and FVC decreased by 357 mL. Both groups had a decrease in 6MWD to a similar degree. Supplemental oxygen requirement was higher in the untreated SAPH group than the treated SAPH group, but not significantly so. Additionally, while there was a trend towards weight loss in the treated SAPH group as compared to the untreated group, this difference was not statistically significant.
Table 4.
Outcomes 12 months (or last follow‐up) after PH diagnosis.
| Treated SAPH (n = 38) | Untreated SAPH (n = 20) | |
|---|---|---|
| Change in FEV1 (mL)* | +13 ± 146 | −257 ± 237 |
| Change in FVC (mL)* | +54 ± 229 | −357 ± 332 |
| Change in 6MWD (m) | −24.5 ± 80.1 | −32.9 ± 52.9 |
| Change in weight (kg) | −0.44 ± 5.5 | 0.95 ± 4.1 |
| Oxygen requirement (L/min) | 4.4 ± 3.4 | 5.4 ± 3.7 |
| Lung transplant recipient* | ||
| Yes, n (%) | 22 (57.9) | 4 (20.0) |
| No, n (%) | 16 (42.1) | 16 (80.0) |
| Death before transplant | ||
| Yes, n (%) | 13 (34.2) | 11 (55.0) |
| No, n (%) | 25 (65.8) | 9 (45.0) |
| Median time to death after PH diagnosis, months (IQR)* | 50 (30–70) | 24 (14–34) |
| Death or lung transplant within 12 months of PH diagnosis, n (%)** | 5 (13.1) | 7 (35.0) |
Note: Patients with SAPH who received PH treatment had stable FEV1 and mildly improved FVC, while those who did not receive PH treatment had a decrease in FEV1 and FVC after 12 months, or the last follow‐up before death or transplant. The values listed for oxygen requirement are the new oxygen requirement at the time of follow‐up, not the change in oxygen requirement from baseline.
Abbreviations: 6MWD, 6‐minute walk distance; FEV1, forced expiratory volume in 1 s; FVC, functional vital capacity; IQR, interquartile range; PH, pulmonary hypertension; SAPH, sarcoidosis‐associated pulmonary hypertension.
p < 0.01
p < 0.05.
There was a significantly higher rate of lung transplantation in the treated SAPH group as compared to the untreated group (57.9% vs. 42.1%, p < 0.01), as well as a trend towards a higher rate of death before transplant among the untreated group (Table 4). A higher proportion of patients with untreated SAPH died or received lung transplants within 12 months of PH diagnosis than patients with treated SAPH. Eight patients (three in treated SAPH, five in untreated SAPH) were still alive and had not yet received a lung transplant. Kaplan–Meier survival analysis demonstrated decreased survival probability among untreated SAPH patients as compared with treated SAPH patients (longrank p = 0.002, Figure 2). Median survival was 50 months among treated SAPH patients and 24 months among untreated SAPH patients.
Figure 2.
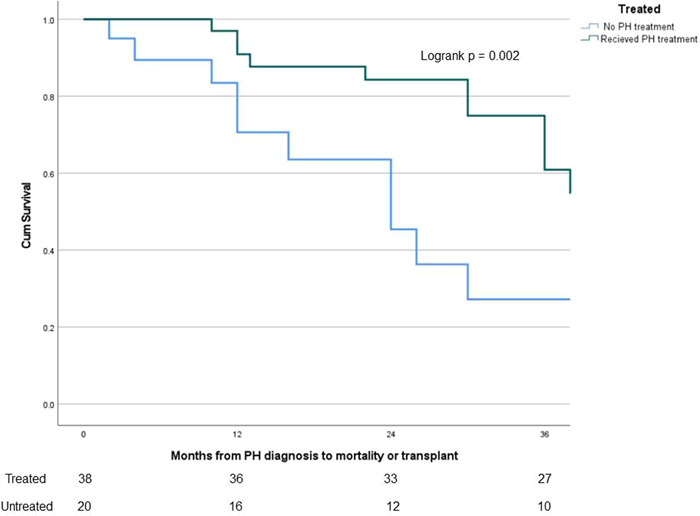
Survival analysis of treated SAPH versus untreated SAPH since PH diagnosis. Comparison of survival probability between treated SAPH and untreated SAPH patients from time of PH diagnosis. The number of patients at risk in each group displayed on bottom. Changes in the graph due to death before transplant while also censoring those who received lung transplant, as they would no longer be at risk of death before transplant. Lung transplantation is not reflected in the actual Kaplan–Meier curves. A significant difference seen, with lower survival probability demonstrated in the untreated SAPH group (logrank p = 0.002). PH, pulmonary hypertension; SAPH, sarcoidosis‐associated pulmonary hypertension.
Univariate analysis found initial FEV1 (β = −0.27 ± 0.08, p = 0.001), initial FVC (β = −0.22 ± 0.06, p = 0.0005), PVR at time of PH diagnosis (β = 0.04 ± 0.01, p = 0.012), and whether pulmonary vasodilator treatment was received (F(1, 56) = 29.44, p < 0.0001) to be significantly associated with change in FVC (Table 5). Subsequent multivariable analysis with these variables found only receiving pulmonary vasodilator therapy (β = 0.35 ± 0.08, p < 0.0001) to be independently and significantly associated with change in FVC (Table 5). With regard to mortality before transplantation, univariate analysis found that receiving pulmonary vasodilator treatment (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.12–0.67, p = 0.004) was significantly associated with decreased mortality before lung transplant. None of the other variables tested were found to have an association with mortality, so multivariable analysis was not performed (Table 5).
Table 5.
Univariate and multivariable analysis for change in FVC and for mortality.
| Variable | Univariate analysis for association with change in FVC | Multivariable analysis for association with change in FVC | Univariate analysis for association with mortality before transplant |
|---|---|---|---|
| Age | β = 0.0002 ± 0.005, p = 0.96 | HR 0.90, 95% CI 0.58–1.42, p = 0.66 | |
| Change in Weight | Β = −0.004 ± 0.009, p = 0.69 | HR 1.02, 95% CI 0.95–1.10, p = 0.59 | |
| Gender | F(1, 56) = 0.86, p = 0.36 | Female: HR 0.61, 95% CI 0.27–1.39, p = 0.24 | |
| Race | F(2, 55) = 0.08, p = 0.92 | Black as reference; White: HR 2.01, 95% CI 0.45–8.88; Other: HR 2.07, 95% CI 0.47–9.07; p = 0.45 | |
| FEV1 at time of PH diagnosis | β = −0.27 ± 0.08, p = 0.001* | β = −0.05 ± 0.10, p = 0.62 | HR 1.04, 95% CI 0.82–1.32, p = 0.75 |
| FVC at time of PH diagnosis | β = −0.22 ± 0.06, p = 0.0005* | β = −0.13 ± 0.08, p = 0.09 | HR 1.05, 95% CI 0.53–2.06, p = 0.89 |
| FEV1/FVC > 0.70 | F(1, 56) = 2.09, p = 0.15 | HR 1.07, 95% CI 0.47–2.43, p = 0.86 | |
| DLCO at time of PH diagnosis | β = −0.007 ± 0.006, p = 0.26 | HR 1.00, 95% CI 0.96–1.05, p = 0.92 | |
| Change in 6MWD | β = 0.0003 ± 0.0006, p = 0.69 | HR 1.02, 95% CI 0.82–1.28, p = 0.84 | |
| Prednisone dose at PH diagnosis | β = −0.009 ± 0.006, p = 0.10 | HR 1.12, 95% CI 0.97–1.28, p = 0.12 | |
| Additional anti‐inflammatory treatment | F(1, 56) = 0.01, p = 0.92 | HR 1.25, 95% CI 0.55–2.83, p = 0.59 | |
| Treatment with pulmonary vasodilator therapy | F(1, 56) = 29.44, p < 0.0001* | β = 0.35 ± 0.08, p < 0.0001** | HR 0.286, 95% CI 0.12–0.67, p = 0.004* |
| Anti‐fibrotic therapy | F(1, 56) = 0.79, p = 0.38 | HR 0.05, 95% CI 0.0–35.5, p = 0.59 | |
| History of smoking | F(1, 56) = 0.02, p = 0.90 | HR 1.20, 95% CI 0.53–2.70, p = 0.66 | |
| History of diastolic dysfunction | HR 1.09, 95% CI 0.45–2.62, p = 0.86 | ||
| History of systolic dysfunction | HR 1.05, 95% CI 0.24–4.49, p = 0.95 | ||
| History of additional lung disease/concurrent COPD | F(1, 56) = 0.28, p = 0.60 | HR 0.89, 95% CI 0.38–2.09, p = 0.80 | |
| History of diabetes | HR 0.90, 95% CI 0.33–2.41, p = 0.83 | ||
| History of kidney disease | HR 0.64, 95% CI 0.28–1.47, p = 0.29 | ||
| History of liver disease | HR 0.83, 95% CI 0.24–2.82, p = 0.77 | ||
| Extrapulmonary sarcoidosis | F(1, 56) = 0.02, p = 0.90 | HR 0.90, 95% CI 0.34–2.43, p = 0.84 | |
| RA pressure at PH diagnosis | β = −0.007 ± 0.01, p = 0.55 | HR 0.89, 95% CI 0.65–1.21, p = 0.44 | |
| mPAP at PH diagnosis | β = 0.005 ± 0.006, p = 0.37 | HR 1.11, 95% CI 0.90–1.38, p = 0.32 | |
| PCWP at PH diagnosis | β = −0.01 ± 0.009, p = 0.24 | HR 0.97, 95% CI 0.71–1.32, p = 0.85 | |
| CI at PH diagnosis | β = −0.11 ± 0.06, p = 0.076 | HR 1.11, 95% CI 0.89–1.39, p = 0.34 | |
| PVR at PH Diagnosis | β = 0.04 ± 0.01, p = 0.012* | β = 0.003 ± 0.01, p = 0.81 | HR 1.03, 95% CI 0.80–1.33, p = 0.81 |
Note: For the outcome change in FVC, univariate analysis was performed with either linear regression or one‐way ANOVA. For the outcome of mortality, cox proportional hazards analysis was performed. Univariate analysis for mortality only showed treatment with pulmonary vasodilator therapy to be associated with decreased mortality. No other variables had a significant association, so multivariable analysis was not performed.
Abbreviations: 6MWD, 6‐minute walk distance; CI, Cardiac index; CI, confidence interval; COPD, chronic obstructive pulmonary disease; DLCO, diffusing capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, functional vital capacity; HR, hazard ratio; mPAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; RA, right atrial.
*Variable had significant association (p < 0.05) with outcome in univariate analysis. For change in FVC, these were included in multivariable regression analysis.
**Variable had a significant, independent association with change in FVC in multivariable regression.
DISCUSSION
Among patients with SAPH who underwent lung transplant evaluation at our institution, those who received pulmonary vasodilator therapy had significantly less decline in FVC over the course of 12 months, higher rates of lung transplantation, and increased survival as compared to those who did not receive pulmonary vasodilator therapy. Receiving pulmonary vasodilator therapy was independently and significantly associated with improvement in FVC and decreased mortality before lung transplantation. These findings point towards a potential benefit of pulmonary vasodilator therapy in properly selected SAPH patients.
To our knowledge, this is the first study to demonstrate a beneficial association between pulmonary vasodilator therapy and lung function and mortality among patients with SAPH. Prior studies have shown mixed results with regard to the benefit of pulmonary vasodilator therapy in SAPH. Some studies have shown an increase in 6MWD and quality of life, 7 , 9 , 10 yet other studies did not show such improvement in 6MWD. 6 , 11 Improvement in pulmonary hemodynamics with pulmonary vasodilator therapy among SAPH patients has been seen as well. 6 , 12 , 13 However, no study has shown an improvement in lung function decline or association with decreased mortality. The endpoint of time to clinical worsening in the RCT evaluating riociguat included all‐cause mortality and was prolonged in the riociguat‐treated group, but this was driven by decrease in 6MWD rather than mortality. 7
As mentioned previously, the post‐hoc analysis of the INCREASE trial showed significant improvement in placebo‐corrected difference in percent‐predicted FVC and a trend towards in improvement in actual FVC in patients with ILD and PH, but it did not include patients with SAPH. 7 Notably, the difference in change in FVC between treated and untreated SAPH that we found (~400 mL at 1 year) was larger than the difference in FVC change between placebo and treprostinil group in the INCREASE trial (44.4 mL at 16 weeks). 7 Such difference may be attributable to the longer observed time period in our study as well as the inclusion of all pulmonary vasodilators, rather than focusing on inhaled treprostinil alone, as was done in the INCREASE trial.
The reasons for improvement in the decline in FVC among our patient cohort are unclear yet interesting to speculate. In the post hoc analysis of the INCREASE trial, the authors found that when stratifying by PVR value, those with a higher PVR demonstrated improvement in FVC with inhaled treprostinil therapy. The authors posited that pulmonary vessels may contribute to lung restriction through vascular compliance, which would improve with pulmonary vasodilator therapy, and that inhaled treprostinil could lead to improved right ventricle (RV) function and/or cardiac output, leading to improved respiratory muscle strength and improved FVC. 8 In chronic heart failure, an increase in pulmonary congestion may lead to blood flow back up into bronchial circulation and subsequently influence airway caliber, resulting in airflow limitation. 14 Cardiac enlargement may also pose constraints on the lungs and play a role in restrictive breathing patterns. 15 Increased pulmonary congestion and cardiac size, namely right ventricle, can be seen in pulmonary hypertension and improved by pulmonary vasodilator therapy. Also, the INCREASE trial did show an improvement in 6MWD in those treated with inhaled treprostinil. 16 Additionally, certain pulmonary vasodilators may have anti‐fibrotic properties. The authors of the INCREASE trial noted that treprostinil has anti‐fibrotic effects. 8 Riociguat has demonstrated anti‐fibrotic properties in animal models and was found to stabilize percent‐predicted FVC in patients with systemic sclerosis and associated pulmonary fibrosis. 17
Some of these reasons could certainly apply to our SAPH patient cohort; for example, the treated SAPH group had a higher mean PVR at time of PH diagnosis than the untreated group, and PVR was significantly associated with change in FVC in univariate analysis. However, the significance of PVR did not carry through when accounting for other variables in the multivariable analysis. The treated SAPH cohort may have benefitted from improvements in pulmonary congestion and cardiac size associated with pulmonary vasodilator therapy; such evaluation is limited as there was no consistent follow‐up RHC or echocardiogram. All patients in our cohort had pulmonary fibrosis secondary to their sarcoidosis, so they may have benefitted from the anti‐fibrotic properties of pulmonary vasodilator therapy. We notably did not find an increase in 6MWD among this cohort, in contrast to the INCREASE trial. However, it is important to note that 6MWD is affected by multiple factors in patients with sarcoidosis aside from pulmonary vasodilator therapy, including gender, pulmonary function parameters, dyspnea scores, and partial pressure of oxygen. 18 We also did not find baseline lung function, change in weight, anti‐inflammatory treatment, anti‐fibrotic therapy, presence of additional lung disease, or smoking to have significant, independent associations with change in FVC.
Although FVC is slightly reduced in the setting of obesity, and weight gain is associated with accelerated decline in lung function, 19 , 20 change in weight was not a significant contributor to change in FVC among our cohort. Anti‐inflammatory therapy with prednisone and infliximab has been found to increase FVC among patients with sarcoidosis. 21 , 22 This effect is not reflected in our study, likely due to the end‐stage and “burnt‐out” nature of APS in this cohort undergoing lung transplantation evaluation. Anti‐inflammatory treatment was similar between the treated and untreated SAPH groups. The effect of anti‐fibrotic therapy was likely not reflected in our study as only one patient in the cohort was prescribed anti‐fibrotic therapy. These findings further highlight the significance of the potential effect of pulmonary vasodilator therapy on FVC and lung function decline among this cohort of patients with APS and SAPH undergoing lung transplant evaluation.
With regard to mortality, PH is an independent risk factor for mortality among sarcoidosis patients. 23 Mortality is increased among patients with SAPH with pulmonary vascular resistance (PVR) ≥ 3 mmHg. 24 Other predictors of reduced transplant‐free survival among patients with SAPH include a 6MWD < 300 meters and DLCO < 35% predicted. 25 , 26 However, we did not find any pulmonary hemodynamic values or 6MWD to be associated with increased mortality in our cohort, which is comprised of patients with end‐stage APS. Comorbid conditions, baseline spirometry values, and anti‐inflammatory treatment were not associated with mortality either. All patients in both groups had pulmonary fibrosis, suggesting against pulmonary fibrosis contributing to the differences in mortality between the treated and untreated SAPH groups. We only found pulmonary vasodilator therapy to be associated with decreased mortality. Our findings could be explained by the severity of disease in our patient population; mean 6MWD across this patient cohort was already less than 300 meters. Also, over 1/3rd of our patient cohort was NYHA Functional Class IV; in the prospective randomized trial evaluating bosentan, NYHA Functional Class IV patients were excluded. 6 Unfortunately, we did not have reliable follow‐up DLCO or total lung capacity (TLC) data as much of the pulmonary function testing for these patients during the specified time period (date of RHC to 1 year after RHC) was simple spirometry.
It is important to note that the majority of patients with SAPH and concurrent COPD did receive pulmonary vasodilator therapy. The presence of COPD was not associated with worse outcomes, including mortality, suggesting tolerance of and potential benefit of pulmonary vasodilator therapy in patients with SAPH and concurrent COPD. To our knowledge, such a benefit in patients with ILD and concurrent COPD has not been seen. Patients with combined pulmonary fibrosis and emphysema (CPFE) in the INCREASE trial did not have the same benefit in terms of lung function as patients with other ILDs. 8 Additionally, a post‐hoc analysis of the RISE‐IIP study, which examined riociguat in patients with PH and idiopathic interstitial pneumonia, actually found that patients with high parenchymal disease burden and the presence of more emphysema than fibrosis may have predisposed patients in the study to worse outcomes. 27 The contrast of our findings highlights the variability in efficacy and safety of pulmonary vasodilator therapy in patients with PH related to lung disease, specifically those with a component of COPD, and the need for further studies to evaluate pulmonary vasodilator therapy in these patients.
There has long been concern and uncertainty with regard to the utility of pulmonary vasodilator therapy in patients with SAPH. The most recent World Association of Sarcoidosis and Other Granulomatous Diseases (WASOG) statement on the diagnosis and management of SAPH did not provide definitive recommendations on the use of pulmonary vasodilator therapy due to lack of sufficient evidence, but advocated for an individualized approach with a discussion between a pulmonologist and pulmonary hypertension specialist. 4 One of the concerns of treating PH in patients with lung disease, including pulmonary sarcoidosis, is the worsening of ventilation‐perfusion mismatch and hypoxemia. 28 This concern was not replicated in our study. In fact, while treated SAPH patients had a higher oxygen requirement at the time of PH diagnosis, the oxygen requirement in the untreated SAPH group was higher at the 12‐month follow‐up (although statistically similar). We feel that this finding, along with an associated reduced decline in FVC and improved survival, can provide further evidence to the potential benefits of the use of pulmonary vasodilator therapy in patients with SAPH.
Limitations of our study include its retrospective, single‐center nature, and small sample size. Given that this study focused on patients with SAPH undergoing lung transplantation evaluation, these findings may not be generalizable to those with less severe lung disease, such as those with sarcoidosis presenting as mediastinal lymphadenopathy or less severe ILD, or those who may not be candidates for lung transplantation evaluation, such as older patients with significant comorbid conditions. There is also a subset of patients in our cohort with concurrent COPD; while PH is likely a result of the underlying sarcoidosis, given the pulmonary fibrosis present, we cannot know for certain if PH is at least in part a result of COPD. However, this does not take away from the significance of the potential benefits of pulmonary vasodilator therapy seen in this study. Treatment with pulmonary vasodilator therapy was not standardized and was at the discretion of individual providers; there may have been a bias to treat PH in the treated SAPH group due to higher mPAP and PVR in these patients. However, SAPH patients treated with pulmonary vasodilators did have less decline in FVC and lived longer. We also did not have consistent follow‐up RHC data and were unable to assess the effect of treatment on pulmonary hemodynamics. The higher rate of lung transplantation among the treated SAPH group suggests there may be factors that we were unable to and did not capture in our analysis, namely those that contribute to the Lung Allocation Score (LAS) and organ distribution aside from mPAP; higher mPAP values contribute to a higher LAS score.
CONCLUSION
Among patients with SAPH, we demonstrated that those who received pulmonary vasodilator therapy had a significantly higher change in FVC and survival over 12 months from the time of PH diagnosis as compared to those who did not receive pulmonary vasodilator therapy. Receiving pulmonary vasodilator therapy was significantly associated with a decreased decline in FVC and decreased mortality. Patients who received pulmonary vasodilator therapy did not have worsening hypoxemia either. We believe these findings can provide evidence for the benefits of pulmonary vasodilator therapy among patients with SAPH. Given the limitations of our study, further prospective, randomized studies are required to fully elucidate the benefits of treating SAPH with pulmonary vasodilator therapy.
AUTHOR CONTRIBUTIONS
Authors Shameek Gayen, Sohaib Ansari, and Bilal H. Lashari contributed to the data analysis and manuscript writing/editing. Shameek Gayen and Albert James Mamary formulated the study design. Huaqing Zhao contributed to statistical analysis. Huaqing Zhao, Albert James Mamary, Gerard J. Criner, and Rohit Gupta revised and reviewed the manuscript.
CONFLICT OF INTEREST STATEMENT
Each author declares that he or she has no commercial associations (e.g. consultancies, stock ownership, equity interest, patent/licensing arrangement etc.) that might pose a conflict of interest in connection with the submitted article.
ETHICS STATEMENT
This study was performed in accordance with the ethical standards of the Western IRB and the Helsinki Declaration of 1975.
ACKNOWLEDGMENTS
Shameek Gayen is the guarantor of the content of the manuscript, including the data and analysis. This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
Gayen S, Ansari S, Lashari BH, Zhao H, Criner GJ, Gupta R, James Mamary A. Pulmonary vasodilator therapy in sarcoidosis‐associated pulmonary hypertension may decrease lung function decline and mortality. Pulm Circ. 2023;13:e12245. 10.1002/pul2.12245
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Crouser ED, Maier LA, Wilson KC, Bonham CA, Morgenthau AS, Patterson KC, Abston E, Bernstein RC, Blankstein R, Chen ES, Culver DA, Drake W, Drent M, Gerke AK, Ghobrial M, Govender P, Hamzeh N, James WE, Judson MA, Kellermeyer L, Knight S, Koth LL, Poletti V, Raman SV, Tukey MH, Westney GE, Baughman RP. Diagnosis and detection of sarcoidosis. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):e26–51. 10.1164/rccm.202002-0251ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gupta R, Judson MA, Baughman RP. Management of advanced pulmonary sarcoidosis. Am J Respir Crit Care Med. 2022;205(5):495–506. 10.1164/rccm.202106-1366CI [DOI] [PubMed] [Google Scholar]
- 3. Duong HT, Bonham CA. Sarcoidosis‐associated pulmonary hypertension: pathophysiology, diagnosis, and treatment. Clin Pulmon Med. 2018;25(2):52–60. 10.1097/CPM.0000000000000252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savale L, Huitema M, Shlobin O, Kouranos V, Nathan SD, Nunes H, Gupta R, Grutters JC, Culver DA, Post MC, Ouellette D, Lower EE, Al‐Hakim T, Wells AU, Humbert M, Baughman RP. WASOG statement on the diagnosis and management of sarcoidosis‐associated pulmonary hypertension. Eur Respir Rev. 2022;31(163):210165. 10.1183/16000617.0165-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shorr AF, et al. Pulmonary hypertension in advanced sarcoidosis: epidemiology and clinical characteristics. Eur Respir J. 2005;25(5):783–8. 10.1183/09031936.05.00083404 [DOI] [PubMed] [Google Scholar]
- 6. Baughman RP, Culver DA, Cordova FC, Padilla M, Gibson KF, Lower EE, Engel PJ. Bosentan for sarcoidosis‐associated pulmonary hypertension. Chest. 2014;145(4):810–7. 10.1378/chest.13-1766 [DOI] [PubMed] [Google Scholar]
- 7. Baughman RP, Shlobin OA, Gupta R, Engel PJ, Stewart JI, Lower EE, Rahaghi FF, Zeigler J, Nathan SD. Riociguat for sarcoidosis‐associated pulmonary hypertension. Chest. 2022;161(2):448–57. 10.1016/j.chest.2021.07.2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nathan SD, Waxman A, Rajagopal S, Case A, Johri S, DuBrock H, De La Zerda DJ, Sahay S, King C, Melendres‐Groves L, Smith P, Shen E, Edwards LD, Nelsen A, Tapson VF. Inhaled treprostinil and forced vital capacity in patients with interstitial lung disease and associated pulmonary hypertension: a post‐hoc analysis of the INCREASE study. Lancet Resp Med. 2021;9(11):1266–74. 10.1016/S2213-2600(21)00165-X [DOI] [PubMed] [Google Scholar]
- 9. Barnett CF, Bonura EJ, Nathan SD, Ahmad S, Shlobin OA, Osei K, Zaiman AL, Hassoun PM, Moller DR, Barnett SD, Girgis RE. Treatment of sarcoidosis‐associated pulmonary hypertension. Chest. 2009;135(6):1455–61. 10.1378/chest.08-1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keir GJ, Walsh SL, Gatzoulis MA, Marino PS, Dimopoulos K, Alonso R, Raposeiras‐Roubin S, Renzoni EA, Maher TM, Wells AU, Wort SJ. Treatment of sarcoidosis‐associated pulmonary hypertension: a single centre retrospective experience using targeted therapies. Sarcoidosis, Vasc Diffuse Lung Dis. 2014;31(2):82–90. https://www.ncbi.nlm.nih.gov/pubmed/25078636 [PubMed] [Google Scholar]
- 11. Judson MA, Highland KB, Kwon S, Donohue JF, Aris R, Craft N, Burt S, Ford HJ. Ambrisentan for sarcoidosis associated pulmonary hypertension. Sarcoidosis, Vasc Diffuse Lung Dis. 2011;28(2):139–45. https://www.ncbi.nlm.nih.gov/pubmed/22117505 [PubMed] [Google Scholar]
- 12. Bonham CA, Oldham JM, Gomberg‐Maitland M, Vij R. Prostacyclin and oral vasodilator therapy in sarcoidosis‐associated pulmonary hypertension. Chest. 2015;148(4):1055–62. 10.1378/chest.14-2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boucly A, Cottin V, Nunes H, Jaïs X, Tazi A, Prévôt G, Reynaud‐Gaubert M, Dromer C, Viacroze C, Horeau‐Langlard D, Pison C, Bergot E, Traclet J, Weatherald J, Simonneau G, Valeyre D, Montani D, Humbert M, Sitbon O, Savale L. Management and long‐term outcomes of sarcoidosis‐associated pulmonary hypertension. Eur Respir J. 2017;50(4):1700465. 10.1183/13993003.00465-2017 [DOI] [PubMed] [Google Scholar]
- 14. Petermann W, Barth J, Entzian P. Heart failure and airway obstruction. Int J Cardiol. 1987;17(2):207–9. 10.1016/0167-5273(87)90132-x [DOI] [PubMed] [Google Scholar]
- 15. Olson TP, Beck KC, Johnson BD. Pulmonary function changes associated with cardiomegaly in chronic heart failure. J Card Failure. 2007;13(2):100–7. 10.1016/j.cardfail.2006.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Waxman A, Restrepo‐Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–34. 10.1056/NEJMoa2008470 [DOI] [PubMed] [Google Scholar]
- 17. Khanna D, Allanore Y, Denton CP, Kuwana M, Matucci‐Cerinic M, Pope JE, Atsumi T, Bečvář R, Czirják L, Hachulla E, Ishii T, Ishikawa O, Johnson SR, De Langhe E, Stagnaro C, Riccieri V, Schiopu E, Silver RM, Smith V, Steen V, Stevens W, Szücs G, Truchetet ME, Wosnitza M, Laapas K, de Oliveira Pena J, Yao Z, Kramer F, Distler O. Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE‐SSc): randomised, double‐blind, placebo‐controlled multicentre trial. Ann Rheum Dis. 2020;79(5):618–25. 10.1136/annrheumdis-2019-216823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alhamad EH, Shaik SA, Idrees MM, Alanezi MO, Isnani AC. Outcome measures of the 6 minute walk test: relationships with physiologic and computed tomography findings in patients with sarcoidosis. BMC Pulm Med. 2010;10:42. 10.1186/1471-2466-10-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dixon AE, Peters U. The effect of obesity on lung function. Expert Rev Resp Med. 2018;12(9):755–67. 10.1080/17476348.2018.1506331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peralta GP, Marcon A, Carsin AE, Abramson MJ, Accordini S, Amaral AF, Antó JM, Bowatte G, Burney P, Corsico A, Demoly P, Dharmage S, Forsberg B, Fuertes E, Garcia‐Larsen V, Gíslason T, Gullón JA, Heinrich J, Holm M, Jarvis DL, Janson C, Jogi R, Johannessen A, Leynaert B, Rovira JMM, Nowak D, Probst‐Hensch N, Raherison C, Sánchez‐Ramos JL, Sigsgaard T, Siroux V, Squillacioti G, Urrutia I, Weyler J, Zock JP, Garcia‐Aymerich J. Body mass index and weight change are associated with adult lung function trajectories: the prospective ECRHS study. Thorax. 2020;75(4):313–20. 10.1136/thoraxjnl-2019-213880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ungprasert P, Ryu JH, Matteson EL. Clinical manifestations, diagnosis, and treatment of sarcoidosis. Mayo Clin Proc: Innov, Qual Outcomes. 2019;3(3):358–75. 10.1016/j.mayocpiqo.2019.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Broos CE, Wapenaar M, Looman CWN, in 't Veen JCCM, van den Toorn LM, Overbeek MJ, Grootenboers MJJH, Heller R, Mostard RL, Poell LHC, Hoogsteden HC, Kool M, Wijsenbeek MS, van den Blink B. Daily home spirometry to detect early steroid treatment effects in newly treated pulmonary sarcoidosis. Eur Respir J. 2018;51(1):1702089. 10.1183/13993003.02089-2017 [DOI] [PubMed] [Google Scholar]
- 23. Kirkil G, Lower EE, Baughman RP. Predictors of mortality in pulmonary sarcoidosis. Chest. 2018;153(1):105–13. 10.1016/j.chest.2017.07.008 [DOI] [PubMed] [Google Scholar]
- 24. Baughman RP, Engel PJ, Taylor L, Lower EE. Survival in sarcoidosis‐associated pulmonary hypertension. Chest. 2010;138(5):1078–85. 10.1378/chest.09-2002 [DOI] [PubMed] [Google Scholar]
- 25. Shlobin OA, Kouranos V, Barnett SD, Alhamad EH, Culver DA, Barney J, Cordova FC, Carmona EM, Scholand MB, Wijsenbeek M, Ganesh S, Lower EE, Engel PJ, Wort J, Price L, Wells AU, Nathan SD, Baughman RP. Physiological predictors of survival in patients with sarcoidosis‐associated pulmonary hypertension: results from an international registry. Eur Respir J. 2020;55(5):1901747. 10.1183/13993003.01747-2019 [DOI] [PubMed] [Google Scholar]
- 26. Gupta R, Baughman RP, Nathan SD, Wells AU, Kouranos V, Alhamad EH, Culver DA, Barney J, Carmona EM, Cordova FC, Huitema M, Scholand MB, Wijsenbeek M, Ganesh S, Birring SS, Price LC, Wort SJ, Shlobin OA. The six‐minute walk test in sarcoidosis associated pulmonary hypertension: results from an international registry. Respir Med. 2022;196:106801. 10.1016/j.rmed.2022.106801 [DOI] [PubMed] [Google Scholar]
- 27. Nathan SD, Cottin V, Behr J, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H, Mogulkoc N, Ulrich S, Wuyts WA, Yao Z, Ley‐Zaporozhan J, Müller‐Lisse UG, Scholle FD, Brüggenwerth G, Busse D, Nikkho S, Wells AU. Impact of lung morphology on clinical outcomes with riociguat in patients with pulmonary hypertension and idiopathic interstitial pneumonia: a post hoc subgroup analysis of the RISE‐IIP study. J Heart Lung Transplant. 2021;40(6):494–503. 10.1016/j.healun.2021.02.006 [DOI] [PubMed] [Google Scholar]
- 28. Ahmad K, Khangoora V, Nathan SD. Lung disease‐related pulmonary hypertension. Cardiol Clin. 2022;40(1):77–88. 10.1016/j.ccl.2021.08.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.