

Abstract
Pfam is a large collection of protein multiple sequence alignments and profile hidden Markov models. Pfam is available on the WWW in the UK at http://www.sanger.ac.uk/Software/Pfam/ , in Sweden at http://www.cgr.ki.se/Pfam/ and in the US at http://pfam.wustl.edu/ . The latest version (4.3) of Pfam contains 1815 families. These Pfam families match 63% of proteins in SWISS-PROT 37 and TrEMBL 9. For complete genomes Pfam currently matches up to half of the proteins. Genomic DNA can be directly searched against the Pfam library using the Wise2 package.
INTRODUCTION
Pfam is a database of protein domain families. Pfam contains curated multiple sequence alignments for each family, as well as profile hidden Markov models (profile HMMs) for finding these domains in new sequences. Pfam contains functional annotation, literature references and database links for each family. There are two multiple alignments for each Pfam family, the seed alignment that contains a relatively small number of representative members of the family and the full alignment that contains all members in the database that can be detected. All alignments use sequences taken from pfamseq, which is a non-redundant protein set composed of SWISS-PROT and SP-TrEMBL. The profile HMM is built from the seed alignment using the HMMER package (see http://hmmer.wustl.edu/ ), which is then used to search the pfamseq sequence database. All the matches found above the curated thresholds are aligned using the profile HMM to make the full alignment. The largest full alignment in Pfam, for the HIV GP120 glycoprotein, has >16 000 members, yet the seed alignment only has 24 representative members. The latest version of Pfam (4.3) contains 1815 families that have matches to 63% of sequences, covering 45% of residues in the sequence database.
One of the main goals of Pfam was to aid the annotation of the Caenorhabditis elegans genome (1). Traditional approaches to large scale sequence annotation use a pairwise sequence comparison method such as BLAST (2) to find similarity to proteins of known function. Annotations are then transferred from the protein of known function to the predicted protein. The pairwise similarity search does not give a clear indication of the domain structure of the proteins. Mistakes in annotation can result from not considering the domain organisation of proteins (3). For example a protein may be misannotated as an enzyme when the similarity is only to a regulatory domain. Since its inception, Pfam has been developed to provide broad support for automated protein sequence classification and annotation. During the last year there have been significant changes and extensions to Pfam, which further this role.
Pfam WEBSITES
There are currently three Pfam websites that are maintained independently. All of the sites contain core functionality, including searching the Pfam library of HMMs, searching the text annotation of Pfam and viewing the multiple alignments for each family. A few new features are not yet implemented on all sites.
The Pfam WWW servers can present the domain architecture of a protein graphically as ‘beads on a string’ with a colour-coded and hyperlinked bead for each domain (4). To get an overview of the different domains involved in a family, it is possible to list graphical schematics for all family members in one view. By browsing the sequence annotation together with these schematics, one can get a rough idea of the evolution and functional implications of domain combinations. For instance, if a certain combination is uniquely associated with proteins of a distinct functional class, this would suggest that other proteins with this combination have the same function. Likewise, if a certain combination is present in a certain taxonomic group only, it may confer a function that is specific for those organisms. If a combination is found scattered over a range of taxa, this might suggest that it arose multiple times independently.
For a more fine-grained analysis of the evolution of domain architectures, we have developed a novel tool that displays the graphical domain schematics of each sequence connected in an evolutionary tree. This tool is implemented as a Java applet, NIFAS, which at present is available from the Pfam servers in Sweden and the UK. It requires Netscape 4.5 or Internet Explorer 4.0. An example of a NIFAS view is shown in Figure 1. Trees are calculated from Pfam seed or full multiple alignments. We are currently using the neighbour-joining tree construction method in Clustalw (5). NIFAS can be used to analyse whether two or more domains have co-evolved or have recombined recently. For instance, the bacterial sugar transferase proteins PTF1_RHOCA (P23388) and PTF1_XANCP (P45597) are clustered together in Figure 1, in which the tree was calculated for the enzymatic domain. The NIFAS view based on the EIIA_2 domain shows the same two sequences grouped, and based on the HPR domain they are grouped too, although not as reliably (data not shown). This analysis thus suggests that an ancestral protein existed with all three domains, and the two present proteins are its direct descendants.
Figure 1.

NIFAS view of the Pfam family Pep-utilizers (PF00391). Only members in the seed alignment are shown. Each sequence in the tree is shown with name/organism and Pfam domains as coloured boxes. Large boxes are Pfam-A domains while thin multi-coloured boxes are Pfam-B domains. The domains that were used to calculate the tree are marked with a small tree icon (green domains). The tree was calculated by Clustalw with bootstrapping. If a node has >90% bootstrap support it is marked with a green box; 75–90% with a yellow box; 50–75% with a white box; and 0–50% with no box.
Pfam-A is supplemented by Pfam-B, however it has previously not been possible to annotate new proteins with matches to Pfam-B families. Protein sequence submitted to the UK Pfam search server is now automatically searched for Pfam-B domains (as well as the standard search for Pfam-A domains). This is performed by using BLAST2 to search against a database of the sequence fragments that form Pfam-B, with some post-processing of the results. Sequence segments matching a Pfam-B family can then be aligned against the family using a profile HMM. These profile HMMs are built on-the-fly; profile HMMs for Pfam-B families are not currently part of the Pfam distribution.
A further enhancement of Pfam’s utility is the addition of structural information to alignments with members of known 3D-structure. Secondary structure and relative solvent accessibility values extracted from the DSSP database (6) are included as alignment markups (labels ‘#=GR.. SS’ and ‘#=GR.. SA’) as of Pfam 4.3. Furthermore, the corresponding entries in the PDB database (7) are referenced with residue coordinates. These references are linked to rasmol (8) for visualisation of the structural entity that corresponds to the Pfam domain.
CHANGES TO Pfam-B
Pfam-B is an automatically generated supplement to Pfam-A, that provides completeness in terms of coverage. Pfam-B has also provided a useful resource for new Pfam-A families. Pfam version 4 has seen a marked change in the way that Pfam-B is constructed. Up to and including the 3.4 release of Pfam, Pfam-B was constructed using the Domainer algorithm (9). The basis for this algorithm was a computationally expensive all-against-all BLAST comparison of the subsequences not found in Pfam-A. As a result it became infeasible to re-construct Pfam-B at every monthly release.
Since the 4.0 release of Pfam, Pfam-B has been constructed using the ProDom database of protein domain families (10), which is a high quality automatically generated protein families database constructed over the same underlying sequence database as Pfam (SWISS-PROT and TrEMBL). The new construction process for Pfam-B is fast, and as a result Pfam-B is now re-built at every point monthly release. Pfam-B in principle is made from the parts of ProDom not covered by Pfam-A. The Pfam-B construction process is conceptually a function taking a ProDom alignment as input and giving between zero and three Pfam-B families as output. The function is applied to all families in ProDom to form Pfam-B. In some cases, a ProDom family is effectively subsumed by one or more Pfam-A families. These ProDom families are ignored. In other cases, a ProDom family has no overlap with any Pfam-A family. These alignments become Pfam-B families with no alteration. More interesting are cases where the ProDom alignment is truncated or bisected by a Pfam-A family, as displayed pictorially in Figure 2. In these cases, the ProDom alignment is cut at the maximal extent of the intruding Pfam-A family to form one (Fig. 2a), or in the case of bisection, two or three Pfam-B families (Fig. 2b). Here, the domain boundaries of Pfam-A are used to infer domain boundaries for Pfam-B families. New Pfam-B alignments are only included if they are wider than 20 columns. Cases such as the ‘bisection’ example shown in Figure 2 are particularly useful for Pfam curation. In such cases the ProDom family has more members than the Pfam-A family it subsumes, and this implies that perhaps the Pfam-A family is missing some members. By adding a link from the new Pfam-B family 2 to the Pfam-A family, this potential deficit is flagged for future consideration.
Figure 2.
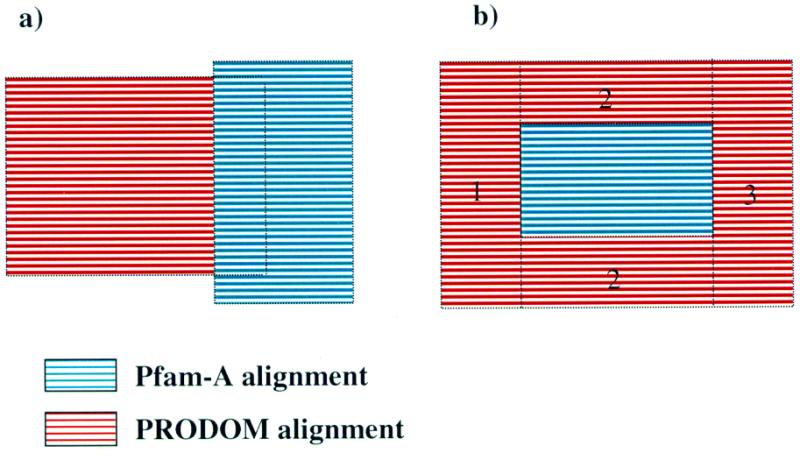
Two possible types of overlap between Pfam-A and ProDom families are shown. (a) A partial overlap that gives one Pfam-B family. (b) A case where the Pfam-A family is subsumed by a ProDom family to create three numbered Pfam-B families.
QUALITY CONTROL
Curating a large number of families presents many challenges for quality control, both for annotation and family membership. We have recently added a spell checking functionality to Pfam, allowing us to store a dictionary of words that are allowed in the free text lines of Pfam.
Pfam-B is now providing useful quality control for Pfam-A that was not present before. The comparison of Pfam-A and ProDom that occurs during Pfam-B construction has provided Pfam with an excellent way to detect missing members of families. This has led to large increases in membership for some families. For example, in Pfam version 4.1 the rieske domain family (PF00355) had 51 members. This was found to be related to Pfam-B family 31 by ProDom. By including some of the related rieske domains from Pfam-B 31 in the seed alignment the new Pfam-A profile HMM found 192 rieske domains.
One of the most important quality controls is the overlap check. This states that no residue of any protein can belong to more than one family. As new families are added to Pfam an overlap to an existing family may signify that the new family is related to a preexisting family. In this case we can extend the existing family to include the members of the new family. The overlap could also be due to incorrectly choosing domain boundaries for a family, which can be easily fixed by trimming the seed alignment. As Pfam’s residue coverage increases this control becomes more stringent and therefore more useful.
SEARCHING GENOMES WITH Pfam
An important goal of Pfam is to enable rapid automatic classification of predicted proteins into protein domain families. Pfam is used around the world as an aid to genomic annotation in one of two ways: (i) Pfam can be used to annotate protein translations using the HMMer software; or (ii) Pfam can be used to predict genes and annotate genomic DNA using the Wise2 package.
Although Pfam’s coverage across the sequence databases is high (63%), we know that these databases are biased towards some protein families and organisms. Therefore it is useful to know what fraction of protein sequences in whole genome sequencing projects are annotated by Pfam analysis. Table 1 shows a summary of a Pfam/HMMER analysis of the predicted proteins from five representative genomes: the bacteria Escherichia coli and Rickettsia prowazekii, the nematode Caenorhabditis elegans, the yeast Saccharomyces cerevisiae and the archaeon Methanococcus jannaschii. Pfam identifies domains in 40–50% of the proteins in each genome, except for the archaeal M.jannaschii genome where the fraction is somewhat lower (33%). This compares favourably to the fraction of proteins that can be annotated by standard pair-wise BLAST analysis: for example, the worm genome project reported that ~42% of worm proteins had an informative BLAST similarity to a non-nematode protein (11).
Table 1. The fraction of proteins and fraction of residues hit by a Pfam analysis in each of five genomes.
| E.coli | R.prowazekii | C.elegans | S.cerevisiae | M.jannaschii | |
|---|---|---|---|---|---|
| Total no. proteins | 4290 | 837 | 16 332 | 6305 | 1771 |
| No. proteins hit | 2020 | 421 | 6344 | 2542 | 582 |
| Protein coverage (%) | 47 | 50 | 39 | 40 | 33 |
| Total no. residues | 1 363 501 | 280 233 | 7 120 115 | 2 983 822 | 501 797 |
| No. residues hit | 493 103 | 105 338 | 1 515 030 | 652 191 | 128 469 |
| Residue coverage (%) | 36 | 38 | 21 | 22 | 26 |
1664 Pfam 4.2 HMMs were searched (using hmmsearch 2.1.1 on a Linux PVM cluster) against each genome’s protein database. Every protein domain satisfying the curated Pfam GA (gathering threshold) score cutoffs was tabulated as a hit. Sources of the five genome protein databases were as follows: E.coli, release M52, http://www.genetics.wisc.edu/ ; R. prowazekii, release 11/12/98, http://evolution.bmc.uu.se/~siv/gnomics/Rickettsia.html ; C.elegans, release wormpep-16, http://www.sanger.ac.uk/Projects/C_elegans/wormpep/ ; S.cerevisiae, release 6/21/99, ftp://genome-ftp.stanford.edu/pub/yeast/yeast_ORFs/ ; M.jannaschii, release 9/29/98, http://www.tigr.org/tdb/CMR/arg/htmls/SplashPage.html
Increasing the number of models in Pfam will increase the hit rate, of course. However, the expected return on such an effort is less than one might guess, as illustrated in Table 2. As a rough rule of thumb, the 10–20 largest protein families can account for ~10% of each genome. To cover 20%, it takes ~50–100 families; to get 30%, it takes ~150–300 families; and to get our full current coverage in each genome, it takes ~500–1000 families. The representation of a given protein family varies substantially from genome to genome. The top 10 families that account for 10% of one genome are not the same as the top 10 families in another genome. For example, the largest bacterial protein family is the ABC transporter family; in the two eukaryotes, the protein kinases are the most numerous. The last line in Table 2 shows the number of Pfam families that show one or more hits in one genome but no hits in any of the other genomes, showing that there is substantial non-overlap in the representation of Pfam families in various genomes. Also, 471 of the 1664 Pfam 4.2 models showed no hits to any proteins in these five genomes; many of these models cover protein families specific to vertebrates or viruses.
Table 2. The numbers of matching Pfam families in five complete genomes.
| E.coli | R.prowazekii | C.elegans | S.cerevisiae | M.jannaschii | |
|---|---|---|---|---|---|
| No. families to cover 10% | 14 | 17 | 9 | 13 | 23 |
| No. families to cover 20% | 61 | 59 | 40 | 69 | 98 |
| No. families to cover 30% | 151 | 139 | 156 | 224 | 282 |
| No. Pfam families with a hit | 694 | 337 | 815 | 717 | 339 |
| No. families ‘unique’ to genome | 105 | 1 | 185 | 26 | 8 |
A considerable amount of sequence data is released as raw genomic sequence. Analysis of this sequence is greatly hampered by the presence of (i) introns and (ii) frameshifting sequencing errors in the DNA sequence, which makes deducing the protein sequence of genes contained in the genomic DNA sequence difficult. It is estimated that ~50% of metazoan exons are predicted correctly when standard gene programs are run (T.Hubbard, personal communication). If Pfam is searched against protein translations of genomic DNA, in many cases valid protein domains are missed due to the inaccuracy of gene prediction. The algorithm GeneWise (12) allows a protein profile HMM to be compared directly to genomic DNA, without the need for any gene prediction and allowing for potential frameshifting sequencing errors. GeneWise contains a gene prediction method which it integrates with the profile HMM during the comparison. Tests of GeneWise show that it produces 98% accurate gene predictions in the region of the homology (R.Guigo, personal communication). Unfortunately, GeneWise is a very CPU expensive program, and comparing 100 kb of DNA sequence to the entire Pfam library takes ~30 h on a Unix server machine (Compaq Alpha).
To allow the large scale application of Pfam to genomic DNA we used a pre-filter that incorporated a Perl script called halfwise based on BLASTX (2) to cut the running time down to an average of 2 h. The BLAST search is of the DNA sequence against a constructed protein database which attempts to represent Pfam hits sensibly. This is made by taking the Pfam full alignments and making them non-redundant to a maximum pairwise identity of 75%. This pre-filter is run with a low threshold to select candidate profile HMMs to be compared to the DNA sequence using GeneWise. In tests, the sensitivity loss of using this pre-filter was ~10%, and it also showed greater robustness towards low complexity regions in the genomic data, such as unmasked microsatellite repeats. Halfwise is part of the Wise2 package that provides access to the GeneWise algorithm in a number of different forms (see http://www.sanger.ac.uk/Software/Wise2/ ).
AVAILABILITY OF Pfam
Pfam is available on the WWW in Europe at http://www.sanger.ac.uk/Software/Pfam/ and http://www.cgr.ki.se/Pfam/ , and in the US at http://pfam.wustl.edu/ . The Pfam distribution contains a number of files: Pfam-A.seed and Pfam-A.full contain the seed and full alignments with annotation in Stockholm format; Pfam is a file containing the library of Pfam profile HMMs; PfamFrag is a library of profile HMMs designed specifically to find matches to protein fragments; SwissPfam is a file containing the domain organisation for each protein in the database; Pfam-B contains the data for Pfam-B families in Stockholm format; diff is a file containing the changes between release to allow incremental updates of Pfam derived data; pfamseq contains the underlying sequence database, in fasta format, that all sequences in Pfam are taken from.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to the many people who have submitted data to Pfam. In particular we thank Matthew Bashton who added many of the new families in Pfam, Christian Storm for writing NIFAS, and Michael Åsman and Mats Jonsson for adding new features to the websites.
REFERENCES
- 1.Sonnhammer E.L.L., Eddy,S.R. and Durbin,R. (1997) Proteins, 28, 405–420. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 3.Galperin M.Y. and Koonin,E.V. (1998) In Silico Biol., 1, 55–67. [PubMed] [Google Scholar]
- 4.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Finn,R.D. and Sonnhammer,E.L.L. (1999) Nucleic Acids Res., 27, 260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabsch W. and Sander,C. (1983) Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein F.C., Koetzle,T.F., Williams,G.J.B., Meyer,E.F., Brice,M.D., Rodgers,J.R., Kennard,O., Shimanouchi,T. and Tasumi,M. (1977) J. Mol. Biol., 112, 535–542. [DOI] [PubMed] [Google Scholar]
- 8.Sayle R.A. and Milner-White,E.J. (1995) Trends Biochem. Sci., 20, 374. [DOI] [PubMed] [Google Scholar]
- 9.Sonnhammer E.L.L. and Kahn,D. (1994) Protein Sci., 3, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corpet F., Gouzy,J. and Kahn,D. (1999) Nucleic Acids Res., 27, 263–267. Updated article in this issue: Nucleic Acids Res. (2000), 28, 267–269.9847197 [Google Scholar]
- 11.The C.elegans Sequencing Consortium (1998) Science, 282, 2012–2018.9851916 [Google Scholar]
- 12.Birney E. and Durbin,R. (1997) ISMB, 5, 56–64. [PubMed] [Google Scholar]