

Summary
Androgen insensitivity syndrome (AIS) is a rare genetic disorder that affects the development of the male reproductive system in individuals with a 46,XY karyotype. In addition to physical impacts, patients with AIS may face psychological distress and social challenges related to gender identity and acceptance. The major molecular etiology of AIS results from hormone resistance caused by mutations in the X-linked androgen receptor (AR) gene. Depending on the severity of androgen resistance, the wide spectrum of AIS can be divided into complete AIS (CAIS), partial AIS (PAIS), or mild AIS (MAIS). Open issues in the treatment and management of AIS include decisions about reconstructive surgery, genetic counseling, gender assignment, timing of gonadectomy, fertility and physiological outcomes. Although new genomic approaches have improved understanding of the molecular causes of AIS, identification of individuals with AIS can be challenging, and molecular genetic diagnosis is often not achievable. The relationship between AIS genotype and phenotype is not well established. Therefore, the optimal management remains uncertain. The objective of this review is to outline the recent progress and promote understanding of AIS related to the clinical manifestation, molecular genetics and expert multidisciplinary approach, with an emphasis on genetic etiology.
Keywords: AIS, androgen receptor, disorders of sex development (DSD), genetics
1. Introduction
The first case of androgen insensitivity syndrome (AIS; OMIM#300068) was reported in 1953 by Dr. John Morris, who called it testicular feminization, a phenomenon that causes feminization effects and involves the presence of testes observed in the body (1). AIS is a heterogeneous disease of hormone resistance (2) characterized by mutations of the androgen receptor (AR) gene. The incidence of AIS varies from 1:40,800 to 1:99,000 (3). AIS is a common disorder of sex development (DSD) with a 46,XY karyotype (4). It is distinguished by a variety of clinical features, including undermasculinization of the external genitalia at birth, abnormal secondary sexual development at puberty, and infertility. Depending on the degree of androgen resistance and the resulting physical characteristics, AIS is classified as complete (CAIS), partial (PAIS) or mild androgen insensitivity (MAIS). Although AIS mainly results from a loss-of-function in the AR gene (4), only approximately 85% of patients with a clinical diagnosis of CAIS and less than 30% with PAIS can be attributed to inactivating mutations in the AR gene (5). Not all individuals with clinical AIS exhibit mutations in the AR gene (5). The clinical diagnosis and optimal management remain challenging. The biology of masculinization depends on coordination among several signaling networks, such as androgen-dependent signals and downstream events (6).
This review provides an overview of the current research on AIS, covering its clinical manifestations, molecular genetics, and the importance of a multidisciplinary approach, with a particular focus on genetic etiology.
2. Clinical manifestation of AIS
AIS is classically characterized as an X-linked recessive genetic disorder due to absence or reduced functionality of the AR protein, which prevents the body from responding to androgens. People with AIS are born with testes that produce androgens, but their bodies are unable to respond to these hormones. As a result, individuals with AIS may develop female-like physical traits such as breast development and lack of pubic hair.
Depending on the degree of androgen insensitivity (5,7), AIS manifests a broad spectrum of phenotypes from mild to partial or complete androgen insensitivity (8) (Table 1). CAIS occurs when an individual with the XY chromosome has a complete inability to respond to androgens, resulting in a typically female phenotype. The incidence of CAIS is estimated between 1 in 20,000 and 1 in 64,000 individuals with a 46,XY karyotype (9). PAIS occurs when there is some residual androgen receptor activity, leading to varying degrees of undervirilized male external genitalia or partially virilized. The incidence of PAIS is approximately 1 in 130,000 individuals with a 46,XY karyotype (10). PAIS patients usually present with clinical features such as micropenis, hypospadias and cryptorchidism. Individuals with PAIS may also have external genital anomalies such as bifid scrotum and penoscrotal transposition. MAIS is characterized by complete masculinization of the external genitalia, but individuals with MAIS typically show signs of incomplete masculinization, including gynecomastia at puberty and impaired spermatogenesis. Although the incidence of MAIS is much less than CAIS or PAIS, it has not been exactly measured.
Table 1. Clinical manifestation of different AIS phenotypes.
| Phenotypes | Prevalence | AIS With AR mutation | External Genitalia | Clinical characterization | LH (U/L) |
FSH (U/L) |
Testosterone (ng/dl) |
|---|---|---|---|---|---|---|---|
 CAIS |
1:20,000 to 1:64,000 (9) | 85% (5) | Female | Absent or rudimentary Wolffian duct derivatives; Absence or presence of epididymides and/or vas deferens; Inguinal or labial testes; Short blind-ending vagina; Scant or absent pubic and/or axillary hair (8). |
14-43 (15) | 3.5-16 (15) | 186-1,033 (15) |
 PAIS |
1:130,000 (10) | < 30% (5) | Predominantly female | Clitoromegaly and labial fusion, sinus urogenitalis with a wide opening, short, blind-ending vagina; Slight signs of androgen effects: slight clitoromegaly or partial labial fusion, distinct urethral and vaginal opening (8). | 9-32 (15) | 1.1-34 (15) | 157-1,592 (15) |
| Ambiguous | Microphallus with clitoris-like underdeveloped glans, labia majora like bifid scrotum, perineoscrotal hypospadias; Additional sinus urogenitalis with a short, blind ending vagina (8). | ||||||
| Predominantly male | Clitoromegaly and labial fusion, sinus urogenitalis with a wide opening, short, blind-ending vagina; Slight signs of androgen effects: slight clitoromegaly or partial labial fusion, distinct urethral and vaginal opening (8). | ||||||
 MAIS |
NA | 18% (7) | Male | Impaired spermatogenesis and/or impaired pubertal virilization (8). | 2.7-25 (14) | 0.6-50 (14) | 141-2,047 (14) |
AIS: androgen insensitivity syndrome. PAIS: partial androgen insensitivity syndrome. MAIS: mild androgen insensitivity syndrome. CAIS: complete androgen insensitivity syndrome. LH: luteinizing hormone. FSH: follicle-stimulating hormone. NA: not available.
The endocrine profile is responsible for producing and regulating hormones in the body. Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) play critical roles in the regulation of male reproductive function. LH is a gonadotropic hormone and stimulates the production of testosterone in testes (11), while FSH stimulates the production of sperm cells through its synergistic action with testosterone (12). In AIS, the characteristic feature of hormone spectrum is elevated levels of testosterone or normal basal testosterone levels that are associated with high serum LH, indicating impaired androgen negative feedback on the anterior pituitary (13). Despite the differences in the severity of androgen resistance among different types of AIS, there is no difference in hormonal levels (testosterone and LH) between them (14). Serum FSH levels were not different in individuals with MAIS, PAIS and CAIS (14,15). Therefore, hormone screening for AIS would be helpful, but lack specificity.
3. Molecular genetics of AIS
Androgens bind to the AR and activate its signaling pathway, which is essential for male sexual differentiation. The AR protein consists of 919 amino acid residues and is composed of four functional domains (Figure 1A): i) the N-terminal domain (NTD), which is encoded by exon 1 and initiates transcription of target genes; ii) the central DNA-binding domain (DBD) encoded by exons 2 and 3, which interacts with DNA and is critical for binding to hormone response elements (HREs); iii) the Hinge domain encoded by the proximal portion of exon 4, containing the phosphorylation site, which controls the AR activity; iv) the ligand-binding domain (LBD) encoded by the remaining exons 4 and exons 5-8, which first facilitates interaction of AR with heat shock proteins (HSPs) in the cytoplasm, and then interacts with androgens, leading to translocation of the AR to the nucleus. In the androgen reporter databases (www.mcgill.ca/androgendb) (7), approximately 600 mutations in the AR gene were described in AIS. Mutations in the NTD domain are more common in patients with CAIS, whereas variants in LBD (exons 5 and 6) are more common in individuals with PAIS (7). Although almost all AR mutations associated with MAIS have been identified in the NTD domain, the number of AR mutations related to this phenotype remains relatively low (15). In the androgen receptor database, most AR variants identified in patients with AIS are missense mutations. Compared to missense variants, variants with larger effect size (stop codon, insertion, deletion and duplication) are more frequently reported in individuals with CAIS (Figure 1B).
Figure 1.
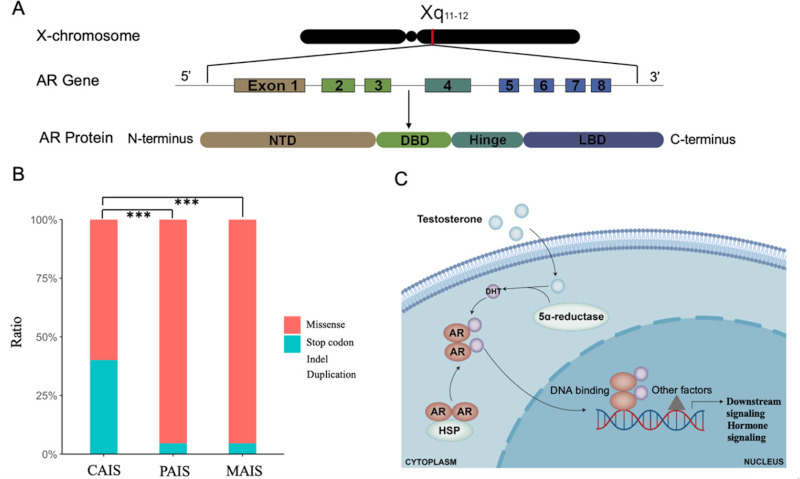
Structure and function of the androgen receptor (AR) gene, distribution of AR mutations in patients with AIS and androgen action. (A). A schematic representation of AR gene and AR protein. (B). Distribution of different types of AR mutations in AIS showed that mutations with larger effect size (stop codon, insertion, deletion and duplication) are more frequently reported in individuals with CAIS in the androgen receptor mutations database. The asterisks indicate significant excess of mutations with larger effect size (***p < 0.001). (C). Mechanism of AR activation through DHT binding.
Although the AIS diagnosis is characterized by the identification of mutations in the AR gene (16), there is not a robust correlation between AIS genotype and phenotype. The incidence of AR gene mutations tends to decrease progressively from CAIS to PAIS to MAIS. While mutations in AR gene were identified in more than 85% of individuals with CAIS, less than 30% of patients with PAIS (5) and about 18% of individuals with MAIS (7) are associated with a genetic abnormality in the AR gene. Especially in PAIS, partial loss of androgen action results in various phenotypes that depend on the overall fetal exposure to androgens. A new class of AIS group, AIS type II or AR-mutation negative AIS was proposed to better understand this type of AIS (5). Gene mutations outside the AR coding sequence have been discovered in patients with AIS, which may influence their response to androgens. For instance, a recurrent germline mutation in the 5'UTR of the AR resulted in aberrant translation in CAIS (17). In addition, epigenetic repression of AR transcription would contribute to AR-mutation negative class of AIS (AIS type II) (18). The aberrant methylation of CpG sites within the proximal AR promoter has been demonstrated to contribute to AIS (19). Furthermore, disruption of AR-dependent cofactors, such as APOD, would cause the AR-mutation negative class of AIS (AIS type II) (5). The duplication of an enhancer upstream of the AR gene leads to an increase in the expression of AR (20). This suggests that while AR is crucial for masculinization, multiple other components of the AR complex, including coactivators (e.g., SRC and p300/ CBP) (21), corepressors (SMRT, NCoR) (22), cofactors (e.g., HSP56, HSP70, HSP90, β-catenin) (23,24) and androgen metabolism related genes (25) might be required for full masculinization.
The genetic etiology and underlying mechanisms leading to androgen resistance need to be further elucidated. Androgen-AR signaling plays a fundamental role in masculinization which involves many precisely regulated steps and biochemical interactions that are modulated by various types of cofactors (Figure 1C). The networks governed by core genes that interact with each other may offer valuable insights into comprehending the etiology of a disease (26). Dysregulation of any step regulated by androgen-AR signaling can potentially result in AIS. Hypospadias is a common feature in individuals with AIS. In hypospadias, more than 70% of the proteins encoded by hypospadias risk genes that interact directly or indirectly with AR, thereby influencing androgen production and signaling (27). A recent study further demonstrated that triple compound rare damaging variants (one variant from AR and two variants from SLC25A5) rather than a single mutation yielded severe hypospadias (25). A following study demonstrated that hypospadias risk associated genes may influence AR expression through the AR-centered network (27) and genetic risk-associated transcription factors (TFs) (28). It has been indicated that AR may play a direct role in the genetic etiology of AIS, while genes that interact with AR and genes related to androgen-AR signaling have a minor impact on the etiology of AIS.
4. General management
Correct diagnosis of AIS patients is the basis for subsequent personalized treatment. In previous disease classifications, AIS was classified as intersex, while the current classification follows the DSD classification standards formulated by the Chicago consensus, which classifies AIS as 46, XY DSD (29). Therefore, the diagnosis of AIS patients may be confused due to the use of different disease classification standards. Since different types of AIS will be treated based on different strategies, it is recommended to re-diagnose some patients who were diagnosed using the old classification method according to the current standard before treatment (2). In addition, since other types of DSD may also exhibit phenotypes similar to AIS (29), it is necessary to perform a differential diagnosis through further endocrine evaluation and genetic sequencing.
The main principles of treatment for AIS focus on three main areas: performing surgery to reconstruct the external genitalia, removing abdominal gonads (such as undescended testes) to reduce the risk of cancer, and selecting the appropriate hormone therapy for the individual. To reduce the risk of testicular malignancy in individuals with CAIS, treatment options include prepubertal removal of the testes and provision for estrogen replacement therapy. Due to the ambiguous or indeterminate features of the external genitalia in PAIS individuals, surgical reconstruction requires more complex and extensive procedures compared to other types of AIS. Treatment for PAIS often involves surgery to reconstruct the external genitalia, removal of abdominal gonads to reduce the risk of cancer, as well as selection of appropriate hormone therapy for the individual. For PAIS, combined therapy is usually required. For example, endocrine and genetic evaluation should be considered for severe hypospadias (30). Additionally, reconstructive surgery can be performed to correct hypospadias, and testosterone therapy can be used to promote male secondary sexual differentiation. In MAIS, there is generally no need for surgical intervention, as affected individuals have fully developed male genitalia. However, counseling and hormonal therapy may be advised to those who experience gender dysphoria or other psychological effects.
Given the long-term consequences associated with the diagnosis of AIS, a collaborative approach involving physicians, patients and parents is crucial for making decisions. The formation of the external genitalia is one of the earliest and most visible signs of sex differentiation that occurs in the uterus during fetal development. Therefore, it may not be possible to initiate normal differentiation of external genitalia in individuals with AIS after birth without medical intervention. Due to androgen resistance, there are a variety of outcomes, including malignant disease (31-35), low bone mineral density and fractures (36-38), infertility (14,39), hypospadias and short vagina (40,41), impaired metabolism and cardiovascular disease (42), and mental disorders (43-47) (Table 2). The future aim of follow-up for AIS individuals is to identify the potential long-term health issues that may arise as a result of the condition, such as cancer risk, metabolic problems, cardiovascular disease, or mental health disorders.
Table 2. Summary of the long-term consequences of AIS.
| Long-term outcomes | CAIS | PAIS | MAIS |
|---|---|---|---|
| Malignant disease | Incidence rate (31,34): 1-2% in CAIS. | Incidence rate (32): > 15% in PAIS. | NA |
| Cause (33): the low rate of germ cell tumor in CAIS could be attributed to the rapid reduction of the germ cell population after the first year of life. | Cause (35): untreated patients have a risk of up to 50% for undescended gonads, while the risk of scrotal testes remains unknown. | NA | |
| Low bone mineral density and fractures | Outcome (37): the final height of CAIS and PAIS individuals was greater than the average height of women and lower than the average height of men. | NA | |
| Fracture rates among patients with CAIS who underwent gonadectomy ranged from 2% to 27% (36,38). | NA | ||
| Infertility, subfertility | No reported cases of biological fertility (14,39). | Usually infertility (14). | Fertility ay occur spontaneously or be induced through androgen treatment (14). |
| Gynaecomastia, hypospadias and short vagina | Patients with CAIS and phenotypically female individuals with PAIS have a short, blind-ending vagina (40). | All individuals with AR mutation develop gynecomastia; hypospadias cases with AR mutation are more likely to require additional surgical treatment than hypospadias cases without an AR mutation (41). | NA |
| Impaired metabolism and cardiovascular disease | Higher prevalence of obesity (16.7% vs. 3.6%), 56% higher total cholesterol, 33% higher low-density lipoprotein-cholesterol, 16% higher triglycerides, and 47% higher HOMA-Index (42). | ||
| Mental disorder | Both PAIS and CAIS patients had lower quality of sexual life (43,46). | NA | |
| Patients with CAIS had a 5-fold higher risk of psychiatric disorders than the general population, a 3-fold higher risk of mood disorders, a 4-fold higher risk of anxiety disorders, and a 20-fold higher risk of obsessive-compulsive disorder (44). | Male PAIS patients often present with psychiatric and psychological problems due to clinical symptoms of poor virilization (45,47). | NA | |
NA: not available.
5. Conclusions
Individuals with AIS may have impaired development of external genitalia and reproductive organs, leading to infertility and other consequences. Defining the genetic etiology for AIS is the foundation for understanding the pathogenesis of AIS and for long-term management of patients. The severity of AIS can vary widely and can be considered as an AR dosage-dependent condition. The largest-effect variants in AR play direct roles in AIS. Although molecular genetic diagnosis is achieved in almost all individuals with CAIS, identifying the genetic etiology of PAIS and MAIS is challenging and molecular genetic diagnosis is often not achieved. With the discovery of new genes contributing to androgen action and mechanisms involved in sexual differentiation, new concepts of AIS are emerging, especially for the AR-mutation negative class of AIS (AIS type II). We proposed that genetic contribution to PAIS is heavily concentrated in genes related to androgen-AR signaling or AR-centered network that are transcribed or expressed in relevant tissues. Rare damaging variants are likely to be causative than other classes of variants and hence explain some of the unknown genetic causes of birth defects, such as hypospadias (48,49). Furthermore, new genomic approaches for identifying non-coding, mosaic, structural or epigenetic variants will improve the understanding of the molecular causes (50). As additional genetic and epigenetic causes of AIS are identified, the diagnosis of these conditions will become more precise.
Patients with AIS, their parents, and healthcare providers face challenging decisions regarding gender assignment, genital surgery, and lifelong care. A multidisciplinary team, such as geneticists, urologists, endocrinologists, and psychologists, can provide expertise in different areas of AIS diagnosis and treatment, and help facilitate shared decision-making. These advances are systematically improving the prediction of prognosis and improving the diagnosis and long-term management of patients with AIS.
Funding:
This work was supported by grants from the National Natural Science Foundation of China (81970572, 82270702), Shanghai Shenkang Hospital Development Center (SHDC2020CR2058B), and Shanghai Commission of Science and Technology (21Y21901000).
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol. 1953; 65:1192-1211. [DOI] [PubMed] [Google Scholar]
- 2. Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J. Androgen insensitivity syndrome. Lancet. 2012; 380:1419-1428. [DOI] [PubMed] [Google Scholar]
- 3. Boehmer AL, Brinkmann O, Brüggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MC, Niermeijer MF, Brunner HG, Rouwé CW, Waelkens JJ, Oostdijk W, Kleijer WJ, van der Kwast TH, de Vroede MA, Drop SL. Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab. 2001; 86:4151-4160. [DOI] [PubMed] [Google Scholar]
- 4. Singh S, Ilyayeva S. Androgen Insensitivity Syndrome. In: StatPearls,Treasure Island (FL). 2023. [Google Scholar]
- 5. Hornig NC, Ukat M, Schweikert HU, et al. Identification of an AR mutation-negative class of androgen insensitivity by determining endogenous AR activity. J Clin Endocrinol Metab. 2016; 101:4468-4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsushita S, Suzuki K, Murashima A, Kajioka D, Acebedo AR, Miyagawa S, Haraguchi R, Ogino Y, Yamada G. Regulation of masculinization: androgen signalling for external genitalia development. Nat Rev Urol. 2018; 15:358-368. [DOI] [PubMed] [Google Scholar]
- 7. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012; 33:887-894. [DOI] [PubMed] [Google Scholar]
- 8. Sinnecker GH, Hiort O, Nitsche EM, Holterhus PM, Kruse K. Functional assessment and clinical classification of androgen sensitivity in patients with mutations of the androgen receptor gene. German Collaborative Intersex Study Group. Eur J Pediatr. 1997; 156:7-14. [DOI] [PubMed] [Google Scholar]
- 9. Mendoza N, Motos MA. Androgen insensitivity syndrome. Gynecol Endocrinol. 2013; 29:1-5. [DOI] [PubMed] [Google Scholar]
- 10. Blackless M, Charuvastra A, Derryck A, Fausto-Sterling A, Lauzanne K, Lee E. How sexually dimorphic are we? Review and synthesis. Am J Hum Biol. 2000; 12:151-166. [DOI] [PubMed] [Google Scholar]
- 11. Nedresky D, Singh G. Physiology, Luteinizing Hormone. In: StatPearls, Treasure Island (FL). 2023. [PubMed] [Google Scholar]
- 12. Santi D, Crepieux P, Reiter E, Spaggiari G, Brigante G, Casarini L, Rochira V, Simoni M. Follicle-stimulating hormone (FSH) action on spermatogenesis: a focus on physiological and therapeutic roles. J Clin Med. 2020; 9:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Melo KF, Mendonca BB, Billerbeck AE, Costa EM, Inacio M, Silva FA, Leal AM, Latronico AC, Arnhold IJ. Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: five novel mutations in the androgen receptor gene. J Clin Endocrinol Metab. 2003; 88:3241-3250. [DOI] [PubMed] [Google Scholar]
- 14. Batista RL, Craveiro FL, Ramos RM, Mendonca BB. Mild androgen insensitivity syndrome: the current landscape. Endocr Pract. 2022; 28:911-917. [DOI] [PubMed] [Google Scholar]
- 15. Batista RL, Costa EMF, Rodrigues AS, Gomes NL, Faria JA, Jr., Nishi MY, Arnhold IJP, Domenice S, Mendonca BB. Androgen insensitivity syndrome: a review. Arch Endocrinol Metab. 2018; 62:227-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Achermann JC, Domenice S, Bachega TA, Nishi MY, Mendonca BB. Disorders of sex development: effect of molecular diagnostics. Nat Rev Endocrinol. 2015; 11:478-488. [DOI] [PubMed] [Google Scholar]
- 17. Hornig NC, de Beaufort C, Denzer F, Cools M, Wabitsch M, Ukat M, Kulle AE, Schweikert HU, Werner R, Hiort O, Audi L, Siebert R, Ammerpohl O, Holterhus PM. A recurrent germline mutation in the 5'UTR of the androgen receptor causes complete androgen insensitivity by activating aberrant uORF translation. PLoS One. 2016; 11:e0154158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornig NC, Rodens P, Dörr H, et al. Epigenetic repression of androgen receptor transcription in mutation-negative androgen insensitivity syndrome (AIS type II). J Clin Endocrinol Metab. 2018; 103:4617-4627. [DOI] [PubMed] [Google Scholar]
- 19. Ammerpohl O, Bens S, Appari M, Werner R, Korn B, Drop SL, Verheijen F, van der Zwan Y, Bunch T, Hughes I, Cools M, Riepe FG, Hiort O, Siebert R, Holterhus PM. Androgen receptor function links human sexual dimorphism to DNA methylation. PLoS One. 2013; 8:e73288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho SS, Urban AE, Mills RE. Structural variation in the sequencing era. Nat Rev Genet. 2020; 21:171-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Waddell AR, Huang H, Liao D. CBP/p300: critical co-activators for nuclear steroid hormone receptors and emerging therapeutic targets in prostate and breast cancers. Cancers (Basel). 2021; 13:2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wong MM, Guo C, Zhang J. Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol. 2014; 2:169-187. [PMC free article] [PubMed] [Google Scholar]
- 23. Mulholland DJ, Dedhar S, Coetzee GA, Nelson CC. Interaction of nuclear receptors with the Wnt/beta-catenin/ Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005; 26:898-915. [DOI] [PubMed] [Google Scholar]
- 24. Tyutyusheva N, Mancini I, Baroncelli GI, D'Elios S, Peroni D, Meriggiola MC, Bertelloni S. Complete androgen insensitivity syndrome: From bench to bed. Int J Mol Sci. 2021; 22:1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z, Lin X, Wang Y, Xie H, Chen F. Dysregulated expression of androgen metabolism genes and genetic analysis in hypospadias. Mol Genet Genomic Med. 2020; 8:e1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Z, Zhang T, Lin J, Yan Z, Wang Y, Zheng W, Weng KC. GeneSense: A new approach for human gene annotation integrated with protein-protein interaction networks. Sci Rep. 2014; 4:4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Z, Lin X, Lei Y, Chen H, Finnell RH, Wang Y, Xu J, Lu D, Xie H, Chen F. Genome-wide association study in Chinese cohort identifies one novel hypospadias risk associated locus at 12q13.13. BMC Med Genomics. 2019; 12:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Z-Z, Gao Y-Q, Xie H, Huang Y-C, Chen F, Lei Y-P. Transcription factors dysregulated in three complex birth defects datasets. Reproductive and Developmental Medicine. 2022; 6:79-85. [Google Scholar]
- 29. Hughes IA, Houk C, Ahmed SF, Lee PA, LWPES Consensus Group, ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child. 2006; 91:554-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson EK, Jacobson DL, Finlayson C, Yerkes EB, Goetsch AL, Leeth EA, Cheng EY. Proximal hypospadias-isolated genital condition or marker of more? J Urol. 2020; 204:345-352. [DOI] [PubMed] [Google Scholar]
- 31. Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006; 27:468-484. [DOI] [PubMed] [Google Scholar]
- 32. Looijenga LH, Hersmus R, Oosterhuis JW, Cools M, Drop SL, Wolffenbuttel KP. Tumor risk in disorders of sex development (DSD). Best Pract Res Clin Endocrinol Metab. 2007; 21:480-495. [DOI] [PubMed] [Google Scholar]
- 33. Kaprova-Pleskacova J, Stoop H, Brüggenwirth H, Cools M, Wolffenbuttel KP, Drop SL, Snajderova M, Lebl J, Oosterhuis JW, Looijenga LH. Complete androgen insensitivity syndrome: Factors influencing gonadal histology including germ cell pathology. Mod Pathol. 2014; 27:721-730. [DOI] [PubMed] [Google Scholar]
- 34. Patel V, Casey RK, Gomez-Lobo V. Timing of gonadectomy in patients with complete androgen insensitivity syndrome-current recommendations and future directions. J Pediatr Adolesc Gynecol. 2016; 29:320-325. [DOI] [PubMed] [Google Scholar]
- 35. Ovidiu B, Marcu DR, Mischianu DLD, Poiana C, Diaconu CC, Bungau SG, Tit DM, Cumpanas A, Bohiltea R. The challenges of androgen insensitivity syndrome. Arch Med Sci. 2021; 18:881-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marcus R, Leary D, Schneider DL, Shane E, Favus M, Quigley CA. The contribution of testosterone to skeletal development and maintenance: Lessons from the androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000; 85:1032-1037. [DOI] [PubMed] [Google Scholar]
- 37. Danilovic DL, Correa PH, Costa EM, Melo KF, Mendonca BB, Arnhold IJ. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos Int. 2007; 18:369-374. [DOI] [PubMed] [Google Scholar]
- 38. Gava G, Mancini I, Orsili I, Bertelloni S, Alvisi S, Seracchioli R, Meriggiola MC. Bone mineral density, body composition and metabolic profiles in adult women with complete androgen insensitivity syndrome and removed gonads using oral or transdermal estrogens. Eur J Endocrinol. 2019; 181:711-718. [DOI] [PubMed] [Google Scholar]
- 39. Hughes IA, Werner R, Bunch T, Hiort O. Androgen insensitivity syndrome. Semin Reprod Med. 2012; 30:432-442. [DOI] [PubMed] [Google Scholar]
- 40. Gomez-Lobo V, Amies Oelschlager AM, North American Society for Pediatric and Adolescent Gynecology. Disorders of sexual development in adult women. Obstet Gynecol. 2016; 128:1162-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lucas-Herald A, Bertelloni S, Juul A, et al. The long-term outcome of boys with partial androgen insensitivity syndrome and a mutation in the androgen receptor gene. J Clin Endocrinol Metab. 2016; 101:3959-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang C, Jackson G, Jones TH, Matsumoto AM, Nehra A, Perelman MA, Swerdloff RS, Traish A, Zitzmann M, Cunningham G. Low testosterone associated with obesity and the metabolic syndrome contributes to sexual dysfunction and cardiovascular disease risk in men with type 2 diabetes. Diabetes Care. 2011; 34:1669-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ranke MB, Saenger P. Turner's syndrome. Lancet. 2001; 358:309-314. [DOI] [PubMed] [Google Scholar]
- 44. Engberg H, Strandqvist A, Nordenström A, Butwicka A, Nordenskjöld A, Hirschberg AL, Frisén L. Increased psychiatric morbidity in women with complete androgen insensitivity syndrome or complete gonadal dysgenesis. J Psychosom Res. 2017; 101:122-127. [DOI] [PubMed] [Google Scholar]
- 45. Kreukels BPC, Köhler B, Nordenström A, Roehle R, Thyen U, Bouvattier C, de Vries ALC, Cohen-Kettenis PT, dsd-LIFE group. Gender dysphoria and gender change in disorders of sex development/intersex conditions: Results from the dsd-LIFE study. J Sex Med. 2018; 15:777-785. [DOI] [PubMed] [Google Scholar]
- 46. Loch Batista R, Inácio M, Prado Arnhold IJ, Gomes NL, Diniz Faria JA, Rodrigues de Moraes D, Frade Costa EM, Domenice S, Bilharinho Mendonca B. Psychosexual aspects, effects of prenatal androgen exposure, and gender change in 46,XY disorders of sex development. J Clin Endocrinol Metab. 2019; 104:1160-1170. [DOI] [PubMed] [Google Scholar]
- 47. Bridi Filho CA, Cardoso SB, Soll BMB, Noal MF, Schwarz K, Rosito TE, Rosito NC, Lobato MIR. Descriptive study of gender dysphoria and sexual behavior in a disorder of sex development group. Front Psychol. 2022; 13:652030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Z, Xie H, Chen F. Re: Vuthy Ea, Anne Bergougnoux, Pascal Philibert, et al. How far should we explore hypospadias? Next-generation sequencing applied to a large cohort of hypospadiac patients. Eur Urol 2021;79:507-515. Eur Urol. 2021; 80:e10-e11. [DOI] [PubMed] [Google Scholar]
- 49. Chen Z, Lei Y, Finnell R, Su Z, Wang Y, Xie H, Chen F. Whole-exome sequencing study of hypospadias. medRxiv. 2022; doi: https://doi.org/10.1101/2022.01.19.22269564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Délot EC, Vilain E. Towards improved genetic diagnosis of human differences of sex development. Nat Rev Genet. 2021; 22:588-602. [DOI] [PMC free article] [PubMed] [Google Scholar]