

Summary
Intellectual disability (ID) and multiple congenital anomalies (MCA) are major contributors to infant mortality, childhood morbidity, and long-term disability, with multifactorial aetiology including genetics. We aim to set a diagnostic approach for genetic evaluation of patients with ID and MCA, which can be applied efficiently with a good diagnostic rate in Indonesia or other low resources settings. Out of 131 ID cases, twenty-three individuals with ID/global developmental delay (GDD) and MCA were selected from two-steps of dysmorphology screening and evaluation. Genetic analysis included chromosomal microarray (CMA) analysis, targeted panel gene sequencing, and exome sequencing (ES). CMA revealed conclusive results for seven individuals. Meanwhile, two out of four cases were diagnosed by targeted gene sequencing. Five out of seven individuals were diagnosed using ES testing. Based on the experience, a novel and comprehensive flowchart combining thorough physical and dysmorphology evaluation, followed by suitable genetic tests is proposed as a diagnostic approach to elucidate the genetic factor(s) of ID/GDD and MCA in low resources settings such as Indonesia.
Keywords: intellectual disability, multiple congenital anomalies, genetic testing, diagnostic procedures, low-and middle-income countries
1. Introduction
Intellectual disability (ID) is defined in the latest DSM- 5 as a disorder with onset during a developmental period, that includes both intellectual and adaptive functioning deficits in conceptual, social, and practical domains (1). ID is considered a global lifelong health problem, which affects around 1-3% of the world's population. In Indonesia, the estimated proportion of children aged 5 to 17 years with disability is 3.3 percent (2). Children with special needs, which includes those with ID are estimated at around 1.6 million, or 2% of all children in the population (3,4), but this number is likely to be higher because of the disparity on available data. Genetic ID comprises approximately 50-65% of moderate and severe ID, while only 20% of mild ID cases are of genetic predominant aetiology (5). Understanding the aetiology of ID, as well as multiple congenital anomalies (MCA) will help the parents, family, and health care providers to give more appropriate medical and supportive care.
Genetic testing methods have improved significantly from conventional cytogenetic analysis to next-generation sequencing (NGS), which can identify pathogenic variants from the whole human exome or genome (6). However, the availability of genetic testing varies across countries, and in Indonesia, it is only available in some research-based institutions. Other challenges include the lack of health professional and public awareness, medical genetic infrastructure including the recognition of its profession, law and regulation, limitation of national health insurance coverage, minimum government support, and lack of expertise and interests from researchers in genetic diseases (7,8). Availability of diagnosis will give better comprehension of genetic counselling and family understanding to facilitate autonomous decision making, avoid unnecessary testing or medical treatment, and eventually increase patient's quality of life (9,10).
To evaluate and diagnose patients with MCA, global developmental delay (GDD), and ID, several strategies have been established. First-line genetic testing, such as routine karyotyping and/or chromosomal microarray, is generally applied for patients with noticeable dysmorphic features (11). When there is evidence for autistic features or a family history with ID, FMR1 gene analysis is warranted to elucidate Fragile X syndrome (FXS). Then, second-tier testing is performed to find common monogenic disorders (12). Further analysis by next-generation sequencing (NGS), a high-throughput technology, is warranted for unexplained ID and MCA after conventional testing. Although still deemed costly, especially in developing countries, the use of exome sequencing (ES) and whole genome sequencing (WGS) increase the diagnostic yield of individuals with ID and MCA and may end a diagnostic odyssey (13-15). There are two approaches applied to discover pathogenic variants, which are the phenotype-first approach which investigates patients based on the clinical features, while the genotype-first approach offers unbiased examination to find causative genetic variants by simultaneously analyzing sequences of the patient's and the respective parents' DNA (trio approach) (16). These strategies are believed cost-effective in most countries, and therefore even some developed countries are implementing a genotype-first approach in genetic diagnosis (13,17). Still, it is not feasible to have this approach put into practice in Indonesia. In most developing countries where advanced molecular facilities are limited, a phenotype-first approach is more favorable. For example, in Morocco, genetic testing methods such as cytogenetic, molecular cytogenetic, and several molecular diagnostics are available for several conditions such as constitutional chromosomal abnormalities, inborn error of metabolisms, chronic myeloid leukemia, and thalassemia (18). Similarly in Pakistan, the number of genetic diagnostic laboratories are limited, and no newborn screening program is conducted at the national level (19). Meanwhile in Sri Lanka, aside from cytogenetics service, NGS is available for university-based cancer genetic diagnosis (20).
We aim to set a systematic flowchart for the genetic evaluation of patients with ID and MCA, which can be applied efficiently in Indonesia or other low resources settings, while attempting to achieve a good diagnostic yield.
2. Materials and Methods
2.1. Patients
Our study population consisted of 155 individuals, of which 133 individuals were included from an institution for ID individuals, and 22 MCA patients with global developmental delay (GDD) were referred from clinicians to our laboratory of Center for Biomedical Research through Diponegoro National University Hospital, from 2016 to 2018. We excluded 24 individuals with clinical features suggestive of Down syndrome (DS) for further analysis (21). Blood sampling from all individuals were performed for chromosomal and molecular analyses. We obtained blood from the index cases, and follow-up when necessary for trio blood collection. Patients with GDD and MCA with unexplained aetiology were referred by clinicians/pediatricians for genetic testing. All individuals were investigated through physical and dysmorphology examination, and the clinical data was recorded using a standardized form. Clinical photographs were taken from all examined individuals.
All parents/legal guardians signed a consent form, including for publication of photographs prior to study. The study complied with the Declaration of Helsinki. Ethical clearance was obtained from Health Research Ethic Committee, Faculty of Medicine, Universitas Diponegoro, Semarang (No. 1.032/EC/FK-RSDK/ XII/2016). We applied the study procedure as followed.
2.2. Study procedure
Patient inclusion and procedure of this study are shown on Figure 1. Routine cytogenetic analysis using G-banding was performed on all individuals with ID/ DD and dysmorphic features (except one patient due to the patient's poor condition) to exclude the possibility of a chromosomal abnormality. Individuals with mild to moderate ID from institutions were screened for FXS, using FastFrax™ Identification, Sizing, and Methylation Status kits (The Biofactory Pte Ltd, Singapore) as previously described (22).
Figure 1.
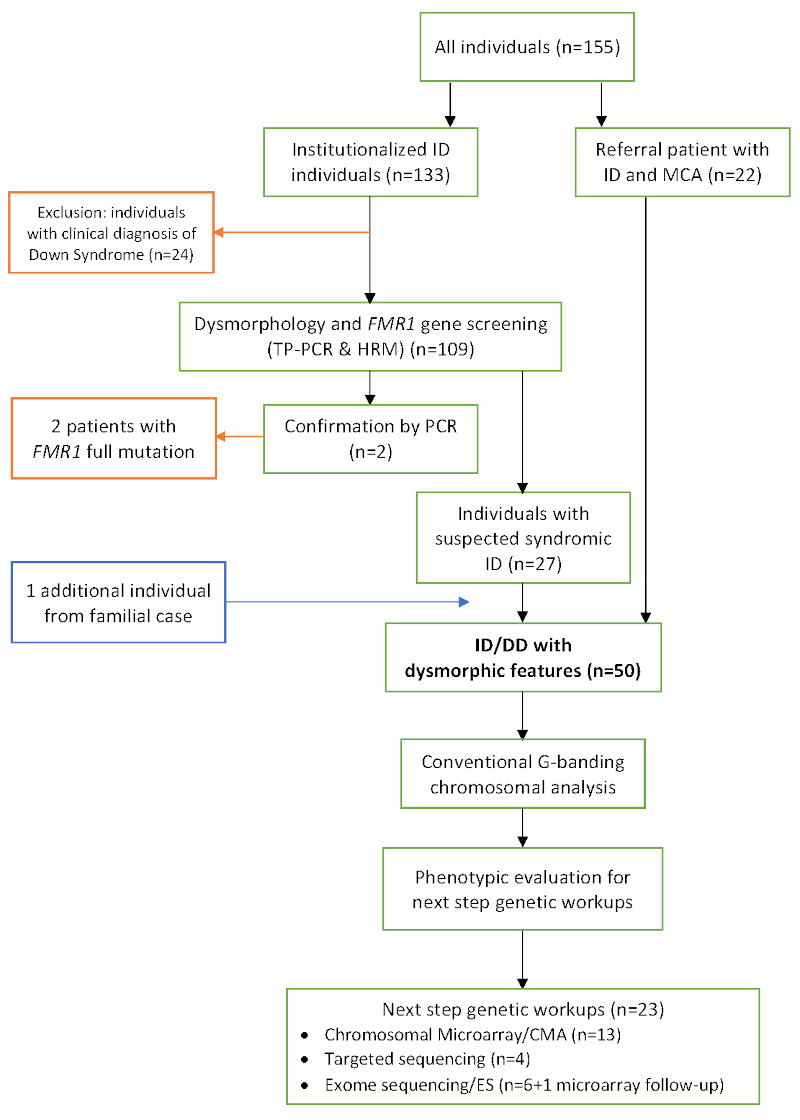
Flowchart and patient selection for the research. Red boxes and arrows indicate excluded individuals, blue box and arrows indicate additional individual from a familial case (hereafter referred to as P6).
We performed dysmorphology evaluation in two steps. The first step of screening aimed to identify individuals with syndromic ID/GDD, by evaluating facial dysmorphic features and existing comorbidities (e.g., congenital heart abnormalities). Clinical and dysmorphic features were analyzed using the London Dysmorphology Database which is available online from the Face2Gene system software (FDNA Inc, Boston, MA, USA). From this screening, we selected 50 individuals for further evaluation. The second step of phenotypic evaluation was performed by researchers and experienced clinical geneticists (IvB and BvB), in order to determine accurate genetic testing for each individual based on its disease mechanism and mode of inheritance. From the comprehensive clinical screening, we selected 23 patients for further analysis, i.e., chromosomal microarray (CMA) for 13 individuals, targeted gene sequencing (panels) for 4 individuals and exome sequencing (ES) for 6 individuals. One individual was added to ES analysis as a follow-up of CMA examination.
2.3. Chromosomal microarray (CMA)
CMA was performed at the Division of Genome Diagnostics of the Radboud university medical center (Nijmegen, Netherlands) for 13 individuals using the CytoScan HD array platform (Thermo Fisher Scientific Inc., Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. Interpretation was made according to the following categories, based on the ACMG standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants (23).
2.4. Next generation sequencing (NGS)
Exome sequencing (ES) was performed on DNA from seven individuals at the Division of Genome Diagnostics (Radboudumc, Netherlands). Exome capturing was carried out using Agilent SureSelect Target Enrichment V5 (Agilent Technologies, Santa Clara, CA, USA) as described previously (24). Next, sequencing was performed using the Illumina HiSeq 2000 platform (Illumina, Inc. San Diego, CA). Illumina base calling software v1.7 was performed using the Roche Newbler software (v2.3) using human genome build hg19/ GRCH37.
Seven major steps were taken to select all high-quality potentially pathogenic variants, as previously described. The exome sequencing results were confirmed by Sanger sequencing. Primers for the amplification of the exons carrying variants were designed using Primer3. PCR reactions were performed on 50 ng of genomic DNA with Taq DNA polymerase (Invitrogen, Carlsbad, CA). PCR amplicons were purified with NucleoFast 96 PCR plates (Clontech Lab, Mountain View, CA), according to the manufacturers protocol. ABI PRISM Big Dye Terminator Cycle Sequencing V3.1 Ready Reaction Kit and the ABI PRISM 3730 DNA Analyzer were used to perform sequencing (Applied Biosystems, Foster City, CA, USA) (25).
Targeted gene sequencing was performed on DNA from four individuals with suggestive clinical features of syndromic ID. Targeted gene sequencing was done on BCOR and NAA11 genes for one individual and on the KMT2A gene on another. Panel sequencing was done on DNA from two patients for Stickler syndrome and Noonan syndrome, respectively. The Stickler syndrome panel consisted of COL11A1, COL11A2, COL2A1, COL9A1, COL9A2, COL9A3, SLC26A2, VCAN genes, and the Noonan syndrome panel included 17 genes (BRAF, CBL, HRAS, KRAS, LZTR1, MAP2K1, MAP2K2, NRAS, PPP1CB, PTPN11, RAF1, RIT1, RREB1, SHOC2, SOS1, SOS2, SPRED1).
3. Results
We examined individuals with ID/GDD and MCA by combining conventional analysis, stringent patient selection from two-steps evaluation, and advanced genetic testing. Out of 109 individuals from the institution who underwent FMR1 gene analysis, two individuals were found to have a full mutation of the FMR1 gene with a methylated repeat allele (22). Subsequently, based upon first dysmorphology evaluation using the dysmorphology database and facial analysis software of Face2Gene (Facial Dysmorphology Novel Analysis, FDNA), 27 (out of the 107) individuals with the most prominent dysmorphisms and/or congenital anomalies and comorbidities were selected for further genetic testing.
The second group of samples came from referred patients (n = 22) with ID/GDD and MCA. All but one patient underwent routine cytogenetic analysis with no visible aberration. Finally, one individual was added from a cascade testing of a family with ID and MCA.
Following two steps of dysmorphology evaluation, 23 individuals were included to next step genetic analysis. The main clinical findings and the genetic test results are summarized in Table 1 (Online Data, http:// www.irdrjournal.com/action/getSupplementalData. php?ID=145). The frontal facial photographs of each diagnosed individuals are shown in Figure 2.
Figure 2.
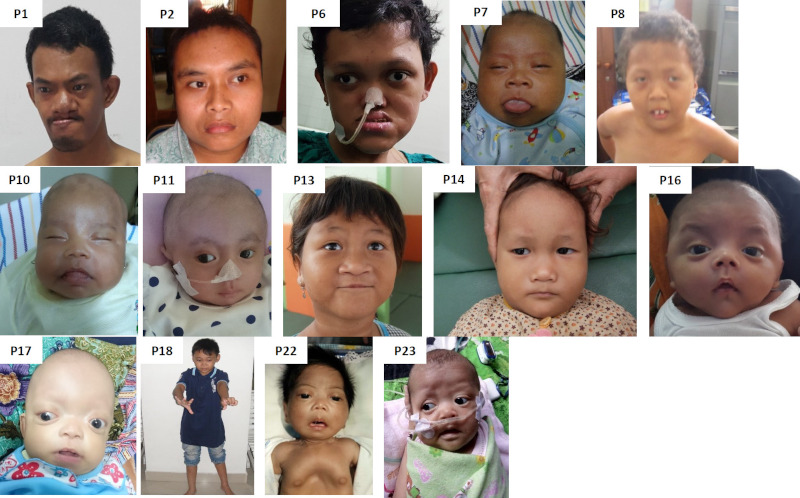
Frontal documentation of diagnosed individuals with ID/GDD and MCA. Supplementary data on P16 is provided.
3.1. Chromosomal microarray (CMA)
CMA was performed on DNA from 13 individuals (Table 1). Of these 13 cases, seven individuals received conclusive results, yielding a diagnostic rate of 54% in this selected group of patients.
From thirteen cases who underwent microarray, P4, P5, and P6 were siblings with ID and similar dysmorphic features. Individuals P5 and P6 were twins, with P6 having more severe clinical features compared to the twin sister. Subject P6 was included from the family cascade testing prior to further dysmorphology evaluation. Since no genomic aberrations were found, P6 was subjected to further analysis using exome sequencing to search for possible pathogenic variants (26).
Subject P11 was included for array analysis due to the suggestive features of split hand foot malformation (SHFM). Routine cytogenetic analysis was not performed due to the patient's poor condition (i.e., insufficient blood collection) and high suspicion of this syndrome. Array analysis revealed a complete trisomy 18, as reported earlier (27).
3.2. Next generation sequencing (NGS)
Seven individuals were subjected to ES, while some other patients underwent targeted sequencing due to high suspicion of a specific syndrome diagnosis, as shown in Table 1.
In this cohort, P13 and P14 were two sisters with developmental delay and similar clinical features, who were first suspected of having Angelman syndrome. The older sister (P13) was sent in for array analysis and P14 was subjected to exome sequencing. Both results showed an interstitial gain in band q31.3q41 of chromosome 1, as reported previously (28). Further analysis using exome sequencing on P6 detected a nonsense pathogenic variant in the NFIX gene, which was also found in the two other affected sisters. A clinical report of this family has been reported in detail elsewhere (26). Taken together, the diagnostic rate of ES was 71% (5 out of 7 individuals diagnosed) in this selected group of individuals. Meanwhile in targeted gene sequencing, 2 out of 4 cases were concluded, yielding a 50% diagnostic rate.
4. Discussion
Our study applied a stepwise phenotype-first approach to elucidate the aetiology of ID/GDD and MCA patients. The first step was performing thorough physical examination to identify dysmorphic features in each individual. The existing physical characteristic found was defined using standardized terms from The Elements of Morphology (29). Additionally, facial recognition software was utilized to aid phenotyping towards a presumptive syndrome diagnosis (30). The purpose of this step is to recognize syndromic ID. Meanwhile, the second step applied thorough evaluation and reassessment, which was conducted by experienced clinical geneticists, aimed to determine appropriate advanced genetic tests. Although genetic testing in recent years has rapidly advanced and genotype-first approach has progressed, dysmorphology examination still remains an important component to make a presumptive diagnosis (31). Evidently, the phenotype-first approach is still widely used and obligatory, especially in developing countries with limited access or resources to be able to perform next generation sequencing in individuals with ID/GDD and MCA (32). Many different diagnostic approaches on individuals with ID/GDD and MCA individuals have been established within different settings (32-34). We described our experience in Indonesia, a low-middle income country setting without the possibility to perform MCA and ES in all individuals with ID/GDD without performing patient selection. Hence, we propose a genetic diagnostic approach for individuals with ID/ GDD and MCA, which is applicable to developing countries setting as shown in Figure 3.
Figure 3.
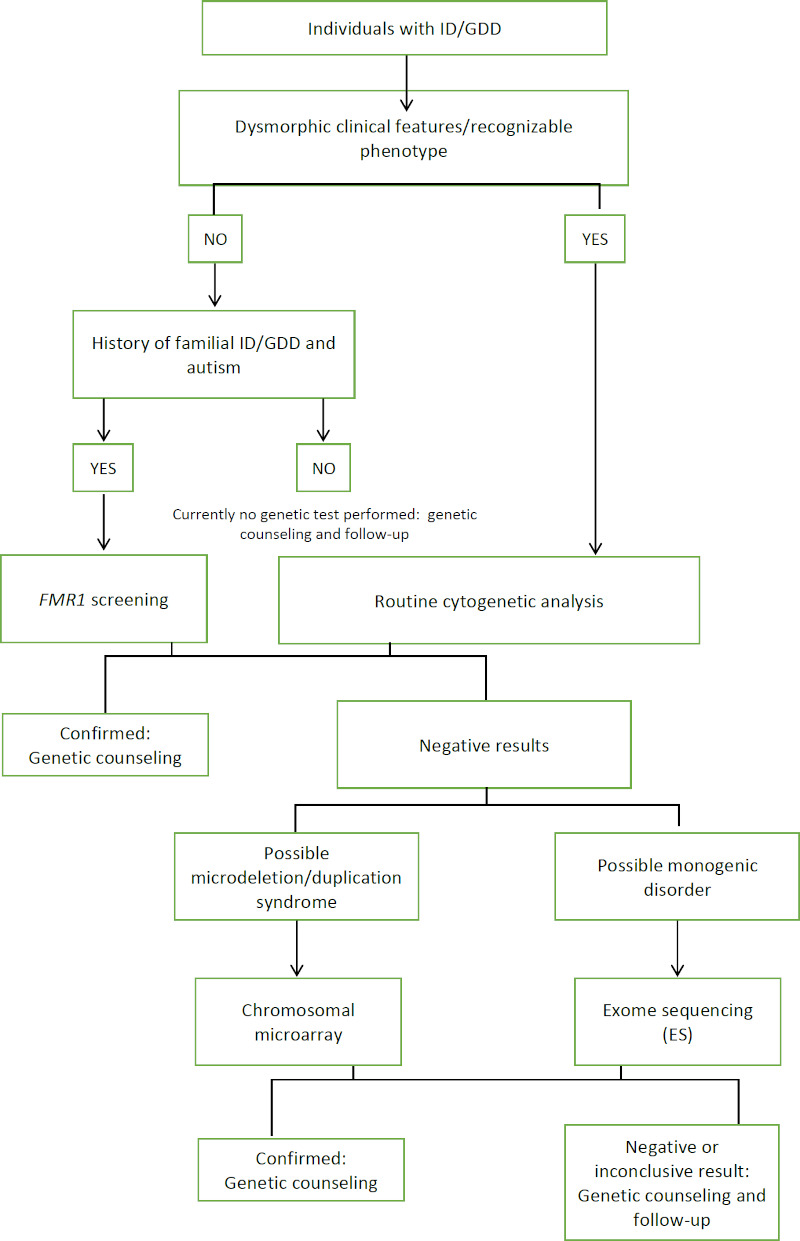
Proposed diagnostic approach for individuals with ID/GDD and MCA in Indonesia. Stepwise phenotype-first approach starts from identifying dysmorphic features, conducting routine cytogenetic analysis, and follow-up with CMA/ES according to possible clinical diagnosis.
In countries with limited resources setting such as Indonesia, cytogenetic analysis is still deemed useful in evaluating individuals with ID/GDD and MCA. Routine cytogenetic analysis with G-banded karyotyping has been applied for more than three decades in Indonesia. A study by Mundhofir et al. suggested that cytogenetic analysis could detect chromosomal aberrations in 16.5% of ID population including 14% of trisomy 21. A similar study was done in a Rwandan ID/GDD and MCA population, resulting in a 39% diagnostic yield, including 30% of individuals with Down syndrome (35,36). However, due to the low sensitivity, most individuals with smaller chromosomal abnormalities or microdeletion/ monogenic disorders remain undiagnosed. Here, we were able to use CMA as the next approach for a selected group of individuals with a normal karyotype result.
CMA analysis provided a diagnosis in seven out of 13 individuals. From seven individuals, there were two individuals with deletions, two cases with a copy number gain, two cases with large regions of homozygosity, and one trisomy 18 case in whom no karyotyping was done prior to array analysis because of his poor condition and unsuccessful karyotyping. Meanwhile, from the six undiagnosed cases, we found one case where the genome contained large homozygous regions, confirming parental consanguinity. Since 2010, CMA has been recommended as the first-tier diagnostic test for individuals with ID/GDD and MCA, instead of G-banded karyotyping (11). CMA can detect copy number variations (CNVs), as well as regions of homozygosity if a SNP-based array platform is used to reveal conditions such as uniparental isodisomy (UPD) and parental consanguinity, as shown in our results. One of the limitations of CMA is the inability to detect balanced chromosomal rearrangements, which was apparent in case P9 with a 46,XY, (t1;2)(q43;q10) karyotype. No genomic imbalances were observed with CMA, even at the translocation breakpoints in the long arms of chromosomes 1 and 2, respectively.
In the CMA study, we obtained a diagnosis in 4 out of 7 cases after finding relatively large chromosomal imbalances (ranging from 16.71 Mb-20.48 Mb), including two cases in whom the imbalance was not previously identified upon routine cytogenetic analysis. Several factors may have led to these missed diagnoses in the first-tier karyotype test. First, in these cases the resolution was low, the band size was approximately 400-450 bands, and as a result, structural chromosomal abnormalities such as deletions or duplications of up to 20 Mb were not resolvable. For postnatal indications karyotyping using a banding technique such as G-banding requires a minimum banding quality of 550 bands per haploid set (37), but preferably the chromosome resolution is at or above the 650-band stage (resolution at the 850-band level may be necessary) for structural abnormalities (38). Second, initial clinical suspicion may hinder the unbiased decision to evaluate the karyotype or to enroll the patient in a specific diagnostic test. These potential pitfalls have been discussed in our previous reports (27,28).
We performed ES and targeted sequencing as the next approach in undiagnosed cases. From seven individuals who underwent ES, pathogenic variants were obtained in four cases, and one individual (P14) yielded a large CNV similar to the sister's (P13) CMA results. The availability of exome data in this familial case could further delineate possible breakpoints, whether it occurred in a functional gene region, and whether any pathogenic nucleotide variants were found in the patients. The use of ES is highly efficient, since it can detect both single nucleotide variants as well as CNVs at the sequence-level in the protein-coding exome and at the intron-exon boundaries (28,39,40).
By utilizing ES, a diagnosis was finally established in a male baby (P6) after more than one year in which he underwent several evaluations and follow-ups with various differential diagnoses, from Robinow syndrome on the first examination to Antley-Bixler syndrome on the second evaluation, due to clinical characteristics and dysmorphic features alteration. When all genetic testing results came back with no pathogenic variant found, a trio ES was performed to elucidate the causative pathogenic variant in the patient, which revealed a de novo, pathogenic variant in the Filamin-A (FLNA) gene (NM_001456.3:c.3425A>T; p(Asp1142Val)). This variant has been described as a gain-of-function pathogenic variant, which causes frontometaphyseal dysplasia type 1 (FMD1, OMIM #305620), a spectrum of otopalatodigital syndrome (see Supplementary File, http://www.irdrjournal.com/ action/getSupplementalData.php?ID=146) (41). The accessibility and availability of a diagnosis in this patient has finally put an end to his diagnostic odyssey, which is described as a condition where a strong diagnostic hypothesis is absent even after clinical evaluation, or a negative diagnostic work-up including array analysis, FXS screening, and targeted testing for monogenic disorders (42). In a study from Brazil, ES identified underlying pathogenic variants in almost half (9 out of 19) of individuals with undiagnosed ID/GDD with MCA (43). ES is beneficial compared to conventional approaches in terms of diagnosing atypical forms of known syndromes, recently described genes and/or syndromes, and ultra-rare conditions (42). Moreover, although the utility of ES did not directly change the treatment, therapy or prognosis, the results are important to improve the family members' understanding of the psychological condition, especially because the diagnostic odyssey could be ended. In addition, genetic counselling can be done when the disease is inherited to improve the understanding of the disease, to identify the family member(s) who may be at risk to be a carrier or affected (adult onset disease), calculating the recurrence risk, and to provide continuing support to the family member who needs more information in the future (44,45). When the recurrence risk can be calculated, it helps the family, especially young couples, to plan future reproductive options or to consider invasive prenatal diagnosis or pre-implantation genetic testing (PGT) when available.
By using targeted sequencing (single-gene sequencing and panel sequencing), we obtained a genetic diagnosis in 2 out of 4 individuals. Both solved cases were diagnosed with panel sequencing on specific syndromes, i.e., Stickler syndrome and Noonan syndrome, respectively. Meanwhile, in the other two individuals, we only attempted sequencing of one or two genes. It is important to notice that targeted Sanger sequencing will be most beneficial for individuals with recognizable syndromes (46).
The detection rate of each genetic test in our study varied between 50% and 71%, with the highest rate using ES. Current evidence suggests that for ES, the diagnostic yield is around 34% for patients with ID/ GDD (14). The high detection rate in our relatively small cohort was due to selection bias, because stringent selection was made by experienced clinicians beforehand (32,47,48).
There are some limitations to our study. Although the aetiology on half of the cases evaluated using exome sequencing have been found, some cases remained elusive, thus further follow-up is needed for possible retesting or reanalysis. This could be due to the unavailability of a trio de novo analysis. In addition, other genetic factors, such as methylation abnormalities, repeat expansion disorders or intronic variants are not possible to detect by ES. Finally, non-genetic factors will not be detected using this diagnostic strategy. This study involved individuals from an institutionalized intellectually disabled population as well as referred patients from clinicians. Some clinical data was incomplete, for example on severe cases of MCA, information on clinical characteristics including dysmorphic features were limited at times, and patients had passed away after only a few hours or days of life, thus making diagnostic workups difficult. To deal with this issue, it is important for the referring clinicians to document comprehensive notes on patients, including good photographs, which include a frontal facial photo and detailed pictures of dysmorphic features.
Although the advancement of ES technology is promising, there are some challenges in applying ES in a routine clinical setting. For instance, while ES can facilitate the diagnosis in atypical and heterogeneous cases, it should not replace the need for thorough clinical evaluation by the clinician to narrow down the clinical diagnosis and select the appropriate panel testing, if available (49). A good diagnostic approach includes awareness of positive signs during history taking, pedigree construction, physical and dysmorphology evaluations, which prompt further genetic testing, and performing comprehensive analysis based on suspected conditions or syndromes, starting from routine cytogenetic analysis to NGS. The presence of recognizable dysmorphic features, growth abnormalities or peculiar comorbidities should prompt clinicians on the possibility of a genetic origin. Additionally, parental reproductive issues and family history of ID/GDD and MCA are considered as a "warning sign" to initiate genetic evaluation.
When no conclusive results are obtained in routine cytogenetic analysis, further genetic testing should be done together with parental samples, in order to do trio analysis. Conducting the flowchart does not warrant conclusive results in all patients. In patients with moderate to severe ID/GDD, accumulated diagnostic yield from cytogenetic analysis to whole genome sequencing can be achieved in mostly around 55-70% of all cases (50). Thus, pre-test and post-test genetic counselling is important and take a longer time in order to plan for follow-up or re-testing when more causative pathogenic variants are elucidated. Genetic counselling in most families has been done in the clinic or by conducting home visits. However, several others were done virtually by video call or phone call, since patients would have to come from various parts of Java Island, including remote areas. Virtual counselling may hinder full comprehension about the patient's condition and diagnosis, consequently, patients or family may decide for a less suitable option for (follow-up) testing.
Recently, the Indonesia Health Ministry took a step forward by launching the Biomedical and Genome Science Initiative (BGSi), which is aimed to integrate genomics capacity into health services for rare diseases, metabolic syndrome, infectious diseases, cancer, and wellness (51). In the future, NGS will become widely available for genetic diagnosis services in Indonesia. However, considering the current policy national healthcare insurance, which does not cover genetic testing, a phenotype-first approach will remain more cost-effective compared to a genotype-first approach.
In conclusion, our flowchart is applicable for the genetic diagnosis of ID/GDD and MCA in Indonesia and similar countries with limited resources available for genetic services. Here we established a diagnosis in 17 of 23 patients. By recognizing the phenotype and categorizing syndromic ID, followed by conducting appropriate genetic testing, most syndromes are explained, and diagnostic odysseys have been solved using this comprehensive approach. While some laboratory facilities such as array analysis and exome sequencing are not widely available in Indonesia, close collaboration between clinical and laboratory centers with a research institution, both nationwide and international, government, and stakeholders will improve the possibility of providing a genetic diagnosis for individuals with ID/GDD and MCA.
Acknowledgements
We would like to thank Dr. Ineke van der Burgt, Dr. Helger Ijntema, and Dr. Dominique Smeets, from the Radboud University Medical Center in Nijmegen, the Netherlands, for their support and expertise towards this study.
Funding:
The study was supported by the Indonesia Ministry of Education, Culture, Research, and Technology grant of the Program Pendidikan Magister menuju Doktor untuk Sarjana Unggul (PMDSU, grant No. 102-02/UN7.P4.3/PP/2018).
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. American Psychiatric Association. Highlights of Changes from DSM-IV-TR to DSM-5. https://www.psychiatry.org/File%20Library/Psychiatrists/Practice/DSM/APA_DSM_Changes_from_DSM-IV-TR_-to_DSM-5.pdf (accessed October 5, 2022).
- 2. Ministry of Health Republic Indonesia. Laporan Nasional Riset Kesehatan Dasar 2018. http://repository.bkpk.kemkes.go.id/3514/1/Laporan%20Riskesdas%202018%20Nasional.pdf (accessed October 5, 2022). (in Indonesian).
- 3. Ministry of Education and Culture Republic Indonesia. Sekolah Inklusi dan Pembangunan SLB Dukung Pendidikan Inklusi. https://www.kemdikbud.go.id/main/blog/2017/02/sekolah-inklusi-dan-pembangunan-slb-dukung-pendidikan-inklusi (accessed October 5, 2022). (in Indonesian).
- 4. Kementerian Pemberdayaan Perempuan dan Perlindungan Anak, Badan Pusat Statistik. Profil Anak Indonesia 2018. https://www.kemenpppa.go.id/lib/uploads/list/74d38-buku-pai-2018.pdf (accessed October 5, 2022). (in Indonesian).
- 5. van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011; 45:81-104. [DOI] [PubMed] [Google Scholar]
- 6. Yim SY. Diagnostic approach for genetic causes of intellectual disability. Journal of Genetic Medicine. 2015; 12:6-11. [Google Scholar]
- 7. Ariani Y, Soeharso P, Sjarif DR. Genetics and genomic medicine in Indonesia. Mol Genet Genomic Med. 2017; 5:103-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hetu M, Koutouki K, Joly Y. Genomics for all: International open science projects and capacity building in the developing world. Front Genet. 2019; 10:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Polla DL, Cardoso MTO, Silva MCB, Cardoso ICC, Medina CTN, Araujo R, Fernandes CC, Reis AMM, de Andrade R V, Pereira RW, Pogue R. Use of targeted exome sequencing for molecular diagnosis of skeletal disorders. PLoS One. 2015; 10:e0138314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lay-Son G, Espinoza K, Vial C, Rivera JC, Guzmán ML, Repetto GM. Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J Pediatr (Rio J). 2015; 91:189-195. [DOI] [PubMed] [Google Scholar]
- 11. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010; 86:749-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han JY, Lee IG. Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Clin Exp Pediatr. 2020; 63:195-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vissers LELM, Van Nimwegen KJM, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, Pfundt R, Van Der Wilt GJ, Krabbenborg L, Brunner HG, Van Der Burg S, Grutters J, Veltman JA, Willemsen MAAP. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017; 19:1055-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, Massingham LJ, Miller D, Yu TW, Hisama FM. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021; 23:2029-2037. [DOI] [PubMed] [Google Scholar]
- 15. de Ligt J, Willemsen MH, van Bon BWM, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012; 367:1921-1929. [DOI] [PubMed] [Google Scholar]
- 16. Carvill G, Mefford H. Next-Generation Sequencing in Intellectual Disability. J Pediatr Genet. 2015; 4:128-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jansen S, Hoischen A, Coe BP, et al. A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur J Hum Genet. 2018; 26:54-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Belhassan K, Ouldim K, Sefiani AA. Genetics and genomic medicine in Morocco: The present hope can make the future bright. Mol Genet Genomic Med. 2016; 4:588-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Riaz M, Tiller J, Ajmal M, Azam M, Qamar R, Lacaze P. Implementation of public health genomics in Pakistan. Eur J Hum Genet. 2019; 27:1485-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sirisena ND, Dissanayake VHW. Genetics and genomic medicine in Sri Lanka. Mol Genet Genomic Med. 2019; 7:e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Devlin L, Morrison PJ. Accuracy of the clinical diagnosis of Down syndrome. Ulster Med J. 2004; 73:4-12. [PMC free article] [PubMed] [Google Scholar]
- 22. Sihombing NRB, Cai S, Wong DPW, Guan M, Chong SS-C, Faradz SM, Winarni TI. Repeat expansion and methylation-sensitive triplet-primed polymerase chain reaction for fragile X mental retardation 1 gene screening in institutionalised intellectually disabled individuals. Singapore Med J. 2021; 62:143-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. South ST, Lee C, Lamb AN, Higgins AW, Kearney HM; Working Group for the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet Med. 2013; 15:901-909. [DOI] [PubMed] [Google Scholar]
- 24. Lelieveld SH, Reijnders MRF, Pfundt R, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016; 19:1194-1196. [DOI] [PubMed] [Google Scholar]
- 25. Riazuddin S, Hussain M, Razzaq A, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. 2017; 22:1604-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sihombing NRB, Winarni TI, van Bokhoven H, van der Burgt I, de Leeuw N, Faradz SMH. Pathogenic variant in NFIX gene affecting three sisters due to paternal mosaicism. Am J Med Genet A. 2020; 182:2731-2736. [DOI] [PubMed] [Google Scholar]
- 27. Sihombing NRB, Purwanti A, Utari A. A rare case of trisomy 18 with split-hand/split-foot malformation (SHFM). Journal of Biomedicine and Translational Research. 2018; 4:41-44. [Google Scholar]
- 28. Sihombing NRB, de Leeuw N, van Bokhoven H, Faradz SMH. Duplication of 1q31.3q41 in two affected siblings due to paternal insertional translocation. BMJ Case Rep. 2019; 12:e230941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allanson JE, Biesecker LG, Carey JC, Hennekam RCM. Elements of morphology: Introduction. Am J Med Genet A. 2009; 149:2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, Basel-Salmon L, Krawitz PM, Kamphausen SB, Zenker M, Bird LM, Gripp KW. Identifying facial phenotypes of genetic disorders using deep learning. Nature Med. 2019; 25:60-64. [DOI] [PubMed] [Google Scholar]
- 31. Hurst ACE. Facial recognition software in clinical dysmorphology. Curr Opin Pediatr. 2018; 30:701-706. [DOI] [PubMed] [Google Scholar]
- 32. Altlner Ş, Yürür Kutlay N. Importance of patient selection criteria in determining diagnostic copy number variations in patients with multiple congenital anomaly/mental retardation. Mol Cytogenet. 2019; 12:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol. 2012; 19:173-180. [DOI] [PubMed] [Google Scholar]
- 34. Muhle RA, Reed HE, Vo LC, Mehta S, McGuire K, Veenstra-VanderWeele J, Pedapati E. Clinical diagnostic genetic testing for individuals with developmental disorders. J Am Acad Child Adolesc Psychiatry. 2017; 56:910-913. [DOI] [PubMed] [Google Scholar]
- 35. Mundhofir FEP, Winarni TI, van Bon BW, Aminah S, Nillesen WM, Merkx G, Smeets D, Hamel BCJ, Faradz SMH, Yntema HG. A Cytogenetic study in a large population of intellectually disabled Indonesians. Genet Test Mol Biomarkers. 2012; 16:412-417. [DOI] [PubMed] [Google Scholar]
- 36. Uwineza A, Hitayezu J, Jamar M, Caberg JH, Murorunkwere S, Janvier N, Bours V, Mutesa L. Cytogenetic studies of Rwandan pediatric patients presenting with global developmental delay, intellectual disability and/or multiple congenital anomalies. J Trop Pediatr. 2016; 62:38-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Silva M, de Leeuw N, Mann K, Schuring-Blom H, Morgan S, Giardino D, Rack K, Hastings R. European guidelines for constitutional cytogenomic analysis. Eur J Hum Genet. 2019; 27:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaffer LG; American College of Medical Genetics Professional Practice and Guidelines Committee. American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation. Genet Med. 2005; 7:650-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pfundt R, Del Rosario M, Vissers L, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Me. 2017; 19:667-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, Firth H V. , Frazier T, Hansen RL, Prock L, Brunner H, Hoang N, Scherer SW, Sahin M, Miller DT. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019; 21:2413-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moutton S, Fergelot P, Naudion S, et al. Otopalatodigital spectrum disorders: refinement of the phenotypic and mutational spectrum. J Hum Genet. 2016; 61:693-699. [DOI] [PubMed] [Google Scholar]
- 42. Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, St-Onge J, Steinmetz A, Huet F, Chouchane M, Darmency-Stamboul V, Callier P, Thauvin-Robinet C, Faivre L, Riviere JB. Diagnostic odyssey in severe neurodevelopmental disorders: Toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. 2016; 89:700-707. [DOI] [PubMed] [Google Scholar]
- 43. da Cunha Leite AJ, Pinto IP, Leijsten N, Ruiterkamp- Versteeg M, Pfundt R, de Leeuw N, da Cruz LAD, Minasi B. Diagnostic yield of patients with undiagnosed intellectual disability, global developmental delay and multiple congenital anomalies using karyotype, microarray analysis, whole exome sequencing from Central Brazil. PLoS One. 2022; 17:e0266493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Basel D, McCarrier J. Ending a diagnostic odyssey: Family education, counseling, and response to eventual diagnosis. Pediatr Clin North Am. 2017; 64:265-272. [DOI] [PubMed] [Google Scholar]
- 45. Dave UP. Genetic counseling in developmental disability. Int J Hum Genet. 2016; 16:89-97. [Google Scholar]
- 46. Saudi Mendeliome Group. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015; 16:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iourov IY, Vorsanova SG, Kurinnaia OS, Zelenova M a, Silvanovich AP, Yurov YB. Molecular karyotyping by array CGH in a Russian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogenet. 2012; 5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roselló M, Martínez F, Monfort S, Mayo S, Oltra S, Orellana C. Phenotype profiling of patients with intellectual disability and copy number variations. Eur J Paediatr Neurol. 2014; 18:558-566. [DOI] [PubMed] [Google Scholar]
- 49. Volk A, Conboy E, Wical B, Patterson M, Kirmani S. Whole-exome sequencing in the clinic: Lessons from six consecutive cases from the clinician's perspective. Mol Syndromol. 2015; 6:23-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vissers LELM, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016; 17:9-18. [DOI] [PubMed] [Google Scholar]
- 51. Kementerian Kesehatan Republik Indonesia. About BGSi. https://bgsi.kemkes.go.id/about-bgsi/about-us (accessed August 19, 2022).