

Summary
Bacterial vaginosis (BV) is characterized by depletion of Lactobacillus and overgrowth of anaerobic and facultative bacteria, leading to increased mucosal inflammation, epithelial disruption, and poor reproductive health outcomes. However, the molecular mediators contributing to vaginal epithelial dysfunction are poorly understood. Here we utilize proteomic, transcriptomic, and metabolomic analyses to characterize biological features underlying BV in 405 African women and explore functional mechanisms in vitro. We identify five major vaginal microbiome groups: L. crispatus (21%), L. iners (18%), Lactobacillus (9%), Gardnerella (30%), and polymicrobial (22%). Using multi-omics we show that BV-associated epithelial disruption and mucosal inflammation link to the mammalian target of rapamycin (mTOR) pathway and associate with Gardnerella, M. mulieris, and specific metabolites including imidazole propionate. Experiments in vitro confirm that type strain G. vaginalis and M. mulieris supernatants and imidazole propionate directly affect epithelial barrier function and activation of mTOR pathways. These results find that the microbiome-mTOR axis is a central feature of epithelial dysfunction in BV.
Keywords: vaginal microbiome, metabolomics, proteomics, transcriptomics, bacterial vaginosis, BV, vaginal dysbiosis, mTOR signaling, in vitro models, epithelial barrier, inflammation, imidazolepropionic acid
Graphical abstract
Highlights
-
•
Dysbiotic vaginal bacteria modify key signaling networks in the host including mTOR
-
•
mTOR signaling activation associated with both clinical Nugent BV and molecular BV
-
•
Epithelial barrier disruption is modulated through metabolites and bacterial products
-
•
The host-microbiome axis involving mTOR links to inflammation and barrier disruption
Berard et al. find a key cellular central signaling network, mTOR, that associates with host inflammation and epithelial barrier disruption in women with a dysbiotic vaginal microbiome and bacterial vaginosis (BV). This network, as well as barrier disruption, is directly modulated by bacterial products and BV-associated metabolites in vitro.
Introduction
The female genital tract (FGT) mucosal microenvironment comprises host immunological features as well as a resident microbiome which together maintain mucosal homeostasis and immunity against pathogenic infections. The vaginal microbiome is commonly dominated by Lactobacillus bacteria (including L. crispatus, L. gasseri, L. iners, and L. jensenii).1 The combination of both anaerobic and aerobic bacteria can lead to a condition termed vaginal microbial dysbiosis or bacterial vaginosis (BV). A Lactobacillus-dominant (LD) vaginal profile is considered an optimal microbiome because of the protective characteristics these bacteria have on the vaginal environment. These protective characteristics include, through the fermentation of glucose, the production of lactic acid, which is the primary acidifier of the vagina and has immunomodulatory and antimicrobial properties.2 Lactobacillus produces bacteriocins and hydrogen peroxide, which also help to keep the environment unfavorable for invading pathogens, although levels of these antimicrobials produced play a partial, not crucial, role of inhibition.3,4,5,6 In contrast to an optimal Lactobacillus-dominated microbiome, some women have a vaginal microbiome harboring a diverse community of anaerobic bacteria.1 This is often termed vaginal microbial dysbiosis or BV, although other etiologies such as aerobic vaginitis can occur that cause non-optimal vaginal microbiome environments. Microbiologically, BV is specifically defined by the loss of Lactobacillus as a dominant species, and an overgrowth (though not necessarily dominant presence) of obligate and facultative anaerobes such as Gardnerella, Prevotella, Atopobium, and Mobiluncus.7,8,9,10 This is accompanied by changes to the vaginal microenvironment including an increase in pH, the induction of inflammatory cytokines, breakdown of mucins by sialidases, and the production of immunoglobulin A proteases and short-chain fatty acids leading to immunomodulation.3,4 The production of amines (cadaverine, N-acetylputrescine) leads to the characteristic foul odor associated with clinically diagnosed BV.4 Traditionally, BV is diagnosed in the clinic using either multiple clinical characteristics defined by Amsel’s criteria (“Amsel BV”) or microscopic characteristics defined by Nugent score (“Nugent BV”).11 Advanced molecular tools, including 16S rRNA sequencing, metagenomics, and metaproteomics, have helped refine classifications of suboptimal vaginal microbiomes that are relevant to adverse reproductive health outcomes, termed “molecular BV.”11 BV represents the most common vaginal disorder among women of reproductive age, with clinical symptoms consisting of increased vaginal discharge, vaginal discomfort/itching, pain during urination or sex, and a strong fish-like odor.12 BV can significantly affect reproductive health by increasing the risk of sexually transmitted diseases13,14,15 and decreasing the chances for a healthy pregnancy.16,17 Although BV is normally treated with metronidazole or clindamycin, these treatments are associated with a 50% recurrence rate within 1 year after treatment,18,19,20,21 indicating the need for better treatment options.
Vaginal dysbiosis is linked to increased mucosal inflammation, the activation of immune cells,3,22,23 and reduced mucosal wound-healing ability within the cervicovaginal compartment.24 Various short-chain fatty acids are increased in BV and are thought to be involved in the recruitment and activation of innate immune cells.25 Recently, studies have shown that the presence of a dysbiotic vaginal microbiome is associated with antigen-presenting cells expressing inflammatory genes and pathways including those involved in the interferon response, self/non-self recognition, innate immunity, and T cell-mediated adaptive immunity.26 However, the molecular mediators and host pathways governing epithelial barrier disruption and inflammation in BV are not completely understood.
Colonization of a variety of bacteria on a 3D vaginal epithelial cell in vitro model showed species-specific immune signatures, such as the change of mucin expression, pattern-recognition receptor signaling, or proinflammatory cytokine expression that may play a role in susceptibility to sexually transmitted diseases.27 Fluctuations in epithelial barrier proteins are amplified in non-Lactobacillus-dominant (nLD) women.28 Other mucosal environmental factors are known to also affect inflammation and barrier function, including bacterial-derived metabolites such as lactic acid,2 short-chain fatty acids, organic acids, and biogenic amines.29,30 Inoculation of BV bacteria into the in vitro 3D model has identified certain metabolites, including amino acids and nucleotides, that have been used to predict the presence of these bacteria.31 The presence of amines, such as tyramine, trimethylamine, and cadaverine, are largely responsible for the fishy odor of the vaginal discharge in women with BV.30 Short-chain fatty acids are also increased in vaginal secretions during BV and may help recruit and activate innate immune cells.25
In this study, we used an integrated multi-omics approach to identify microbial mediators and host pathways in the FGT that underlie BV-associated epithelial dysfunction and tested these observations in bacterial co-culture models.
Results
Cohort description
Cervicovaginal vaginal swab samples collected from 405 women from Kenya and Uganda enrolled in the Partners PrEP Study32 were used to generate microbiome and proteomic data. The samples were processed and analyzed from two different participant groups in the same cohort, the first group of 315 women ranging in age from 18 to 51 years and the second group of 90 women ranging in age from 25 to 51 years. The larger participant group was selected to include individuals with exposures to a diverse set of HIV risk factors, whereas the second participant group was identified through participant willingness to consent to multiple genital sample collections. The second group of women had vaginal biopsies collected and processed for transcriptomic data, and the proteomic data from vaginal swabs collected from this group was used as a validation for proteomic signatures identified in the first participant group. Both groups included women from Kenya and Uganda, with some women in the pre-exposure prophylaxis (PrEP) treatment arm. Information was collected on depot medroxyprogesterone acetate (DMPA) usage, protected and unprotected sex frequency, and history of sexually transmitted infections. Vaginal microbiome data were generated using vaginal swab eluate samples to obtain microbiome composition and functional information. Clinical, demographic, and behavioral information for study participants is shown in Table 1, comparing different microbiome groups, and Table S1, comparing LD and nLD groups.
Table 1.
Clinical, demographic, and behavioral epidemiological information for both participant groups (n = 315 and n = 90 for participant groups 1 and 2, respectively)
| Participant group |
Epidemiological variable |
Group 0: Lactobacillus unspecified |
Group 1: L. crispatus dominant |
Group 2: L. iners dominant |
Group 3: G. vaginalis dominant |
Group 4: polymicrobial/heterogeneous |
p value |
Statistical test |
|
|---|---|---|---|---|---|---|---|---|---|
| Participant group 1 |
total n = 315 |
n = 4 |
n = 52 |
n = 84 |
n = 93 |
n = 82 |
|||
| Participant group 2 | total n = 90 | n = 32 | n = 16 | n = 1 | n = 31 | n = 10 | |||
| Participant group 1 | Nugent score | 0 | 0 (0–2) | 0 (0–8) | 8 (0–10) | 8 (0–10) | 1.00E−15 | Kruskal-Wallis | |
| Participant group 2 | 0 (0–3) | 0 (0–2) | 0 | 3 (0–8) | 3 (0–7) | 2.26E−07 | |||
| Participant group 1 | BV diagnosis | positive (Nugent score 7–10) | 0 (0%) | 0 (0%) | 1 (1%) | 63 (68%) | 70 (85%) | 1.00E−15 | chi-squared |
| negative (Nugent score 0–3) | 4 (100%) | 52 (100%) | 80 (95%) | 21 (22%) | 11 (14%) | ||||
| intermediate (Nugent score 4–6) | 0 (0%) | 0 (0%) | 3 (4%) | 9 (10%) | 1 (1%) | ||||
| Participant group 2 | positive (Nugent score 7–10) | 0 | 0 | 0 | 6 (22%) | 1 (11%) | 5.50E−04 | ||
| negative (Nugent score 0–3) | 32 | 14 (100%) | 1 (100%) | 15 (56%) | 5 (45%) | ||||
| intermediate (Nugent score 4–6) | 0 | 0 | 0 | 6 (22%) | 3 (33%) | ||||
| Participant group 1 | age | 37.4 (33.3–41.3) | 35.1 (24.0–50.1) | 34.6 (19.8–47.1) | 34.1 (20.6–51.5) | 34.3 (18.3–48.9) | 0.8 | Kruskal-Wallis | |
| Participant group 2 | 37.8 (25.9–49.9) | 38.2 (28.1–45.7) | 25.4 | 36.6 (26.1–50.1) | 36.3 (27.0–51.6) | 0.33 | |||
| Participant group 1 | proportion of visits with unprotected sex | 0.011 (0–0.90) | 0.021 (0–0.95) | 0.066 (0–0.97) | 0.068 (0–0.95) | 0.048 (0–0.97) | 0.0408 | Kruskal-Wallis | |
| Participant group 2 | 0 (0–0.93) | 0.026 (0–0.52) | 0.057 | 0.027 (0–0.70) | 0.080 (0–0.45) | 0.90 | |||
| Participant group 1 | number of unprotected sex acts at visit | 0.5 (0–35) | 1 (0–41) | 2 (0–36) | 2 (0–37) | 1.5 (0–41) | 0.0432 | Kruskal-Wallis | |
| Participant group 2 | 0 (0–27) | 1 (0–14) | 2 | 1 (0–19) | 3 (0–15) | 0.84 | |||
| Participant group 1 | ever unprotected sex | no | 2 (50%) | 24 (46%) | 21 (25%) | 27 (29%) | 25 (30%) | 0.1006 | chi-squared |
| yes | 2 (50%) | 28 (54%) | 63 (75%) | 66 (71%) | 57 (70%) | ||||
| Participant group 2 | no | 17 (53%) | 8 (50%) | 0 (0%) | 14 (45%) | 4 (40%) | 0.8014 | ||
| yes | 15 (47%) | 8 (50%) | 1 (100%) | 17 (55%) | 6 (60%) | ||||
| Participant group 1 | HSV-2 at enrollment | positive | 2 (50%) | 41 (79%) | 67 (80%) | 78 (84%) | 71 (87%) | 0.5511 | chi-squared |
| negative | 2 (50%) | 8 (15%) | 13 (15%) | 11 (12%) | 9 (11%) | ||||
| indeterminate/unavailable | 0 (0%) | 3 (6%) | 4 (5%) | 4 (4%) | 2 (2%) | ||||
| Participant group 2 | positive | 23 (72%) | 9 (56%) | 1 (100%) | 19 (70%) | 7 (70%) | 0.52 | ||
| negative | 6 (19%) | 6 (38%) | 0 (0%) | 12 (30%) | 3 (30%) | ||||
| indeterminate/unavailable | 3 (9%) | 1 (6%) | 0 (0%) | 0 (0%) | 0 (0%) | ||||
| Participant group 1 | HSV-2 at visit | positive | 2 (50%) | 41 (79%) | 68 (81%) | 81 (87%) | 73 (89%) | 0.0456 | chi-squared |
| negative | 0 (0%) | 4 (8%) | 10 (12%) | 6 (6%) | 3 (4%) | ||||
| unknown | 2 (50%) | 7 (13%) | 6 (7%) | 6 (6%) | 6 (7%) | ||||
| Participant group 2 | positive | 24 (75%) | 11 (69%) | 1 (100%) | 22 (71%) | 7 (70%) | 0.957 | ||
| negative | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||||
| unknown | 8 (25%) | 5 (31%) | 0 (0%) | 9 (29%) | 3 (30%) | ||||
| Participant group 1 | clinical site | Kenya | 2 (50%) | 15 (29%) | 20 (24%) | 17 (18%) | 19 (23%) | 0.4299 | chi-squared |
| Uganda | 2 (50%) | 37 (71%) | 64 (76%) | 76 (82%) | 63 (77%) | ||||
| Participant group 2 | Kenya | 22 (69%) | 13 (81%) | 0 (0%) | 21 (68%) | 7 (70%) | 0.49 | ||
| Uganda | 10 (31%) | 3 (19%) | 1 (100%) | 10 (32%) | 3 (30%) | ||||
| Participant group 1 | study arm (PrEP use) | treatment | 2 (50%) | 16 (31%) | 34 (40%) | 47 (51%) | 24 (29%) | 0.0368 | chi-squared |
| placebo | 2 (50%) | 36 (69%) | 50 (60%) | 46 (49%) | 58 (71%) | ||||
| Participant group 2 | treatment | 21 (66%) | 15 (94%) | 1 (100%) | 24 (77%) | 8 (80%) | 0.27 | ||
| placebo | 11 (34%) | 1 (6%) | 0 (0%) | 7 (23%) | 2 (20%) | ||||
| Participant group 1 | reported DMPA use | positive | 2 (50%) | 26 (50%) | 50 (60%) | 41 (44%) | 21 (26%) | 0.0002 | chi-squared |
| negative | 2 (50%) | 26 (50%) | 31 (37%) | 40 (43%) | 48 (58%) | ||||
| indeterminate | 0 (0%) | 0 (0%) | 3 (3%) | 12 (13%) | 13 (16%) | ||||
| Participant group 2 | positive | 5 (16%) | 1 (6%) | 1 (100%) | 1 (3%) | 1 (10%) | 9.80E−02 | ||
| negative | 17 (53%) | 8 (50%) | 0 (0%) | 19 (61%) | 7 (70%) | ||||
| indeterminate | 10 (31%) | 7 (44%) | 0 (0%) | 11 (36%) | 2 (20%) | ||||
| Participant group 1 | measured MPA levels | 0.024 (0–1.03) | 0.066 (0–2.10) | 0.041 (0–3.17) | 0.025 (0–2.24) | 0 (0–2.77) | 0.1056 | Kruskal-Wallis | |
| Participant group 2 | 0 (0–0.407) | 1.175 | 0 (0–0.104) | 0 (0–0.817) | 0 (0–0.826) | 1.70E−10 | |||
| Participant group 1 | last known partner plasma HIV-1 RNA level | 4.1 (0–5.4) | 3.9 (0–6.0) | 4.1 (0–6.2) | 4.0 (0–6.6) | 4.2 (0–6.2) | 0.95 | Kruskal-Wallis | |
| Participant group 2 | 2.8 (0–5.1) | 4.5 | 3.5 (0–5.1) | 3.2 (0–5.9) | 2.7 (0–5.5) | 0.62 | |||
| Participant group 1 | gonorrhea at enrollment | no | 4 (100%) | 45 (87%) | 76 (91%) | 87 (94%) | 74 (90%) | 0.7226 | chi-squared |
| yes | 0 (0%) | 1 (2%) | 2 (2%) | 0 (0%) | 3 (4%) | ||||
| not done | 0 (0%) | 6 (12%) | 6 (7%) | 6 (6%) | 5 (6%) | ||||
| Participant group 2 | no | 32 (100%) | 16 (100%) | 1 (100%) | 30 (97%) | 10 (100%) | 0.7496 | ||
| yes | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||||
| not done | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) | 0 (0%) | ||||
| Participant group 1 | Chlamydia at enrollment | no | 4 (100%) | 45 (87%) | 76 (91%) | 86 (92%) | 77 (94%) | 0.8404 | chi-squared |
| yes | 0 (0%) | 1 (2%) | 2 (2%) | 1 (1%) | 0 (0%) | ||||
| not done | 0 (0%) | 6 (12%) | 6 (7%) | 6 (6%) | 5 (6%) | ||||
| Participant group 2 | no | 30 (94%) | 15 (94%) | 1 (100%) | 28 (90%) | 10 | 0.9526 | ||
| yes | 2 (6%) | 1 (6%) | 0 (0%) | 2 (6.5%) | 0 (0%) | ||||
| not done | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3.2%) | 0 (0%) | ||||
| Participant group 1 | Trichomonas at enrollment | no | 4 (100%) | 48 (92%) | 72 (86%) | 83 (89%) | 71 (87%) | 0.864 | chi-squared |
| yes | 0 (0%) | 2 (4%) | 7 (8%) | 8 (9%) | 8 (10%) | ||||
| not done | 0 (0%) | 2 (4%) | 5 (6%) | 2 (2%) | 3 (4%) | ||||
| Participant group 2 | no | 30 (94%) | 16 (100%) | 1 (100%) | 29 (94%) | 10 (100%) | 0.8831 | ||
| yes | 2 (6%) | 0 (0%) | 0 (0%) | 1 (3%) | 0 (0%) | ||||
| not done | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) | 0 (0%) | ||||
| Participant group 1 | gonorrhea at visit | no | 4 (100%) | 44 (85%) | 66 (79%) | 72 (77%) | 63 (77%) | 0.8968 | chi-squared |
| yes | 0 (0%) | 0 (0%) | 2 (2%) | 2 (2%) | 1 (1%) | ||||
| not done | 0 (0%) | 8 (15%) | 16 (19%) | 19 (20%) | 18 (22%) | ||||
| Participant group 2 | no | 11 (34%) | 5 (31%) | 0 (0%) | 11 (35%) | 4 (40%) | 0.7493 | ||
| yes | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||||
| not done | 21 (66%) | 11 (69%) | 1 (100%) | 20 (65%) | 6 (60%) | ||||
| Participant group 1 | Chlamydia at visit | no | 4 (100%) | 44 (85%) | 67 (80%) | 74 (80%) | 63 (77%) | 0.8762 | chi-squared |
| yes | 0 (0%) | 0 (0%) | 1 (1%) | 0 (0%) | 1 (1%) | ||||
| not done | 0 (0%) | 8 (15%) | 16 (19%) | 19 (20%) | 18 (22%) | ||||
| Participant group 2 | no | 11 (34%) | 5 (31%) | 0 (0%) | 11 (35%) | 4 (40%) | 0.7493 | ||
| yes | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||||
| not done | 21 (66%) | 11 (69%) | 1 (100%) | 20 (65%) | 6 (60%) | ||||
| Participant group 1 | Trichomonas at visit | no | 4 (100%) | 41 (79%) | 65 (77%) | 70 (75%) | 60 (73%) | 0.9733 | chi-squared |
| yes | 0 (0%) | 2 (4%) | 3 (4%) | 5 (5%) | 4 (5%) | ||||
| not done | 0 (0%) | 9 (17%) | 16 (19%) | 18 (19%) | 18 (22%) | ||||
| Participant group 2 | no | 11 (34%) | 5 (31%) | 0 (0%) | 11 (35%) | 4 (40%) | 0.7493 | ||
| yes | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||||
| not done | 21 (66%) | 11 (69%) | 1 (100%) | 20 (65%) | 6 (60%) | ||||
Comparisons were made between women who had one of five different vaginal microbiome groups. p value statistical results and tests performed are indicated in the right-hand columns. See Table S1 for LD vs. nLD comparisons.
Vaginal microbiome
We identified 3,280 bacterial proteins from 20 unique genera from 315 women in the first participant group, and 522 bacterial proteins from 15 unique genera from 90 women in the second participant group, using vaginal mucosal fluid samples analyzed by mass spectrometry (Figure 1 and Table S2). Five major microbiome groups from all 405 women were identified using unsupervised hierarchical clustering with average Euclidean linkage, and belonged to either LD or nLD groups. The microbiome group nomenclatures were designated based on whether the microbiome profiles had a dominant, or more than 50%, bacterial genera or species. If no dominant bacterial genera was observed (i.e., bacterial species made up less than 50% of the bacteria within the vaginal microbiome), the microbiome group was designated as a polymicrobial profile. The groups identified included an LD profile not species specific (could not be resolved to species level) (9%, group 0, average Shannon’s diversity index 1.494), Lactobacillus crispatus dominant (21%, group 1, average Shannon’s diversity index 1.009), Lactobacillus iners dominant (18%, group 2, average Shannon’s diversity index 1.278), Gardnerella dominant (30%, group 3, average Shannon’s diversity index 1.066), and a polymicrobial group which was a heterogeneous community containing Gardnerella, Prevotella species, Megasphaera, and Mobiluncus mulieris in higher proportions as well as numerous other anaerobic bacterial species in generally lower amounts (22%, group 4, average Shannon’s diversity index 1.908). Participant groups are indicated in Figure 1 (participant group 1 = dark blue; participant group 2 = light blue), and bacterial functions were also annotated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database on the identified bacterial proteins. Clinical, demographic, and behavioral information of women belonging to these microbiome groups, including both participant groups, are shown in Table 1. Women with non-Lactobacillus microbiomes (identified as having “molecular BV”) were significantly more likely to have Nugent BV in both groups (p values 1.0E−15 and 2.26E−07 for cohort groups 1 and 2, respectively). This was determined by using the Nugent score as a continuous variable using Kruskal-Wallis analysis. Using the categorical Nugent scoring nomenclature for BV diagnosis of positive (Nugent score 7–10), intermediate (Nugent score 4–6), and negative (Nugent score 0–3), we also showed a significant association between Nugent BV and molecular BV using chi-squared analysis. Herpes simplex virus (HSV), gonorrhea, Chlamydia, and Trichomonas infections at enrollment were not different between microbiome groups. In participant group 1, some factors such as the proportion or number of sex acts and the study arm were slightly different (p < 0.05) between microbiome groups.
Figure 1.

Vaginal microbiome composition
Vaginal swabs collected from 315 women were analyzed by mass spectrometry-based metaproteomics and passed quality control criteria (dark-blue participant group). 3,280 bacterial proteins from 20 unique genera and 142 species were identified in this group. Vaginal swab and vaginal tissues collected from another 90 HIV-negative women were analyzed by mass spectrometry and passed quality control criteria (light-blue participant group). 522 bacterial proteins from 15 unique genera were identified in this second group. All 405 samples were clustered and designated microbiome profile groups 0–4. Bacterial functions were also annotated using the KEGG database and are shown in the bottom panel in the same order as in the microbiome grouping. See also Table S2.
Cervicovaginal proteomic and transcriptomic differences between Lactobacillus-dominant and non-Lactobacillus-dominant vaginal microbiomes
We performed proteomic and transcriptomic analysis of cervicovaginal mucosal specimens to better understand host immunological differences between major microbiome groups. In the mucosal proteome of the first group, there were a total of 353 out of 434 (81% having <5% false discovery rate [FDR]) host proteins significantly differentially abundant between LD and nLD microbiomes (Figure 2A and Table S3), whereas the second participant group had 274 out of 454 (60% having <5% FDR) host proteins significantly different between LD and nLD microbiomes (Table S4 and Figure S1A). Hierarchical clustering of these significantly different proteins between LD and nLD groups in the first participant group, and annotation of pathways by DAVID gene ontology (GO),33 showed a general upregulation of proteins involved in the innate immune response, small-molecule metabolism, antigen presentation, and nuclear factor κB (NF-κB) signaling (p < 0.0001), suggesting an overall increase in immune activation. Downregulated proteins in nLD microbiomes were involved in cell-cell adhesion, differentiation, peptide crosslinking, and keratinization functions (p < 0.0001), suggesting epithelial barrier disruption. Many of these pathways were also highlighted in pathway analysis of the second participant group (Figure S1B), indicating that these two groups had similar protein profile differences between microbiome groups and validated these pathways using two separate cohort groups. Transcriptomic analysis of vaginal biopsy samples of a subset of women in the second participant group (n = 80 tissues) identified 1,435 genes to be differentially expressed between LD and nLD women (<5% FDR) (Figure 2B and Table S5), providing a deeper analysis of cellular signaling pathways in the tissue that associates with microbiome profiles. Hierarchical clustering of differentially expressed genes identified two major clusters based on up- and downregulated genes, including an upregulation of genes in nLD women involved in the innate immune response, antigen presentation, and cell cycle and division (p < 0.0001), and decreased expression of genes involved in tissue development, cell differentiation, Wnt signaling, and cell mobility (p = 0.003). Gene set enrichment analysis (GSEA)34 of proteomic and transcriptomic datasets identified many differentially expressed pathways between nLD and LD microbiomes with a pathway score representing the scale of the entire pathway, rather than the individual proteins (Figures 2C and 2D). In general, nLD microbiomes showed enrichment for pathways involved in interferon-γ (IFNγ), tumor necrosis factor-α (TNFα), inflammation, metabolism, and cell division. A key observation in both the proteomic and transcriptomic datasets was enrichment for the mammalian target of rapamycin (mTOR) and MYC targets signaling pathways in multiple comparisons between nLD and LD microbiomes. For the second participant group, GSEA proteomic analysis identified mTOR signaling as the only pathway significantly different between nLD and LD microbiome groups (Figure S1B). It has been established in the literature that mTOR signaling activates MYC targets,35,36 linking these signaling pathways. In addition, many other pathways related to mTOR activation and signaling were associated with nLD microbiomes, including cell division (G2M, E2F),37 metabolism (glycolysis, fatty acid metabolism),38,39 and immune response (IFNγ, TNFα/NF-κB, complement)40,41,42 (p < 0.0001, Figure 2D). These collective data suggested a linkage between the vaginal microbiome and epithelial dysfunction, inflammation, and mTOR signaling.
Figure 2.

Proteomic and transcriptomic differences in cervicovaginal mucosal and tissue samples between vaginal microbiome groups
(A) Heatmap showing differentially abundant proteins (353, 5% FDR) between vaginal microbiome groups, with functional annotation of biological pathways (p < 0.0001) shown on the right, for participant group 1. See Figure S1A for proteomics on participant group 2.
(B) Gene set enrichment analysis (GSEA) of vaginal mucosal proteome differences between microbiome groups for participant group 1. See Figure S1B for pathway analysis of participant group 2.
(C) Heatmap of differentially expressed genes by transcriptomic analysis of vaginal biopsy samples from a subset of 80 women (1,435 genes [<5% FDR]) between vaginal microbiome groups. Functional annotated pathways are indicated with major gene clusters.
(D) GSEA of differentially expressed genes in vaginal tissue between microbiome groups.
(E) Heatmap of vaginal mucosal metabolites significantly different between vaginal groups (27, 5% family-wise error rate).
See Figure S5 for a summary of these interactions. See also Table S3. Protein expression from mucosal fluid taken by a vaginal swab in women from the first participant group, related to Figure 2, Table S4. Protein expression from mucosal fluid taken by a vaginal swab in women from the second participant group, related to Figures 2 and S1, Table S5. Gene (transcriptomics) expression from vaginal biopsies in women from the second participant group, related to Figure 2 for protein and gene expression.
Metabolites in vaginal fluid were also measured in a subset of women from both participant groups (n = 142). There was a distinct separation of vaginal metabolites by microbiome groups (Figure 2E), where nLD microbiomes showed an expected decrease of lactate as well as asparagine, L-acetyl carnitine, and glycerophosphocholine, and increases in N-acetylputrescine, 5-aminovalerate, and imidazolepropionic acid (ImPA).
Generation of a multi-omics host immune signature underlying mucosal dysfunction
To better understand microbiome features that underlie epithelial dysfunction and inflammation in the cervicovaginal mucosa, we utilized a multi-omics latent variable meta-model approach to evaluate host-microbiome interactions. One of the observations from the univariate proteomic and transcriptomic analysis above was that mTOR signaling is a common feature of vaginal microbial dysbiosis. A recent review of host and microbiome interactions in the gut suggested that numerous studies have given evidence to support a pathophysiological interaction through the master cell-regulatory signaling pathway: mTOR.43 Since mTOR is a master signaling network coordinating immune response, antigen presentation, cell growth and differentiation, and nutrient availability,44,45 we hypothesized that this signaling network would also associate with many different and important elements of microbial dysbiosis, inflammation, and epithelial dysfunction in the vaginal environment. To test this hypothesis, we constructed an mTOR activity score in each sample. To create a representation of microbiome-associated mTOR activity, we performed partial least-squares discriminant analysis (PLS-DA) to extract a one-dimensional latent variable (LV), an “mTOR score” that captured variation in the host mTOR pathway proteins (identified with GSEA), relative to a binary outcome of LD or nLD microbiome. To create the mTOR activation score, we used protein expression values of the leading-edge proteins (those contributing the most to the pathway enrichment score in GSEA) (Figures 3A and 3B). Samples with high expression of mTOR pathway proteins have a positive mTOR score, while samples with low expression have a negative mTOR score. We investigated whether this mTOR score was also representative of clinical BV measured by Nugent score. To accomplish this, we recalculated the mTOR score using a continuous partial least-squares regression (PLSR) of mTOR pathway proteins against the Nugent score. We found that the correlation between the mTOR score derived by the Nugent-score and LD-status approaches were nearly identical (Spearman rho = 0.995, Figure S2A), indicating that both the binary LD-status and continuous Nugent-score approaches to deriving the mTOR score give similarly predictive downstream models with similar biological inferences drawn.
Figure 3.

Relationship between the mTOR activity signature in the vaginal microenvironment
(A) Heatmap of the leading mTOR edge proteins identified by GSEA and relationship to vaginal microbiome.
(B) Core mTOR proteins identified using the Pathway Interaction Database were used to identify mTOR expression. An mTOR activity score was determined using partial least-squares discriminant analysis (PLS-DA) inference of the latent variable combining pathway proteins or transcripts to describe mTOR pathway activity relative to LD status (“mTOR Score”). This numeric score was then used to determine both host and bacterial factors that associate with a high mTOR score. See Figure S2A for mTOR score analysis performed using Nugent score.
(C) Vaginal tissue gene expression for immune response pathways (inflammation, TNFα, IFNγ) overlapped with genes associated with mTOR and LPS-stimulated genes identified using significant pathways identified GSEA when comparing Lactobacillus-dominant (LD) with non-dominant (nLD) groups.
(D) mTOR score is significantly correlated with measured epithelial barrier protein expression in the mucosal fluid (Spearman’s correlations, p < 0.0001) showing key distinctions in barrier function and structural changes that were clearly separated by cluster analysis.
To evaluate the utility of the microbiome-associated mTOR score for mucosal dysfunction, we compared the proteome signature with respect to host immune parameters associated with microbial dysbiosis, including lipopolysaccharide (LPS) response, inflammation, and epithelial barrier disruption. As mTOR is known to be activated by bacterial infection and play a functional role in the inflammatory response,45,46 we compared LPS signaling genes from the smaller subset of vaginal tissue transcriptomic data with mTOR activation in the proteome. Focusing on proteins and genes that functionally overlap between different inflammatory pathways, Figure 3C shows a clear relationship between increased mTOR activity, activation of LPS-stimulated genes, TNFα and IFNγ responses, and nLD microbiomes. Performing a correlation analysis between individual mTOR activity scores and protein expression identified in the samples, there were numerous epithelial proteins significantly associated (Spearman’s correlation, p < 0.05) with mTOR score. These epithelial proteins were involved in cell-cell adherence, keratinization, and differentiation (Figure 3D), showing downregulation of proteins involved in basal membrane integrity (LAD1) and differentiation (IVL). This identified a clear relationship between mTOR activity, mucosal dysfunction, and vaginal microbial dysbiosis.
mTOR activity is linked to specific bacterial taxa and functional properties of the vaginal microbiome
Once we were able to designate per-sample host mTOR activity scores, we developed a partial least-squares path model (PLS-PM) approach47 to integrate metabolomics and microbiome composition data from the vaginal mucosal specimens (Figure 4A). In brief, we trained PLSR models to predict host mTOR activity from the metabolomics, microbiome composition, or bacterial protein data individually, and identified predictive features for each model by calculating variable importance of projection (VIP) scores. VIP scores calculate the relative importance of a feature for predicting the outcome variable by averaging the weighted score of the feature by the percent variance explained by latent variables across the model, where average VIP values across the model are 1. Therefore, features that have VIP scores >1 are considered significantly contributing to the model prediction relative to all other features in the model (Table S6). Bacterial taxa that negatively associated with mTOR score included L. crispatus (VIP score 2.2) and L. iners (VIP score 2.2), while positive mTOR-associated bacteria included Gardnerella (VIP score 2.3), M. mulieris (VIP score 1.5), and Prevotella amnii (VIP score 1.2). Top bacterial proteins associated with mTOR score were all negatively associated and included ribosome-recycling factor (VIP score 2.1), glutamine synthetase (VIP score 1.9), 60 kDa chaperonin (VIP score 1.6), and triosephosphate isomerase (VIP score 1.6). Bacterial functions identified using KEGG GO databases were also analyzed, and the top microbiome functions that associate with host mTOR score were negatively associated and include starch and sucrose metabolism (VIP score 2.7), glycolysis/gluconeogenesis (VIP score 2.7), pentose phosphate pathway (VIP score 2.2), and amino acid sugar and nucleotide sugar metabolism (VIP score 1.39). As energy levels and nutrient availability in the surrounding environment are directly linked to mTOR function, this suggested that functional products of bacteria affect mTOR activation and, potentially, the resulting epithelial dysfunction.
Figure 4.

Host mTOR activity associates with bacterial variables and host immune and barrier functions in the mucosal environment
Construction of multi-omics meta-model for microbiome-to-host regulation of mTOR activity.
(A) Latent variables were computed for each data stream (microbiome composition, bacterial proteins, and metabolites) using partial least-squares regression (PLSR) to predict mTOR score before integration via a generalized linear model with interaction effects.
(B–E) The mTOR activity score was then regressed against (B) bacterial taxa, (C) bacterial proteins, (D) bacterial GO functions, and (E) metabolites measured in the vaginal fluid.
(F) Multi-omics integration via latent variable interaction effects linear modeling identified top variables that associate with an activated mTOR pathway. The significant interactions between mTOR pathway and latent variables, bacterial composition LV1, and metabolome LV2 are shown here. Bacterial taxa positively associated with an activated mTOR score include Gardnerella, Prevotella amnii, and Mobiluncus mulieris. Vaginal metabolites associated with activated mTOR expression include 5-aminovalerate, N-acetylputrescine, imidazolepropionic acid, xanthine, and tyramine.
See also Figures S2B–S2D and S3; Table S6.
We also trained single-omic PLSR models (metabolomics, bacterial taxa, bacterial proteins, bacterial functions) to predict host mTOR activity, where host proteomics was used to define the mTOR activity score (Figures 4B–4E). The single-omic vaginal microbiome composition and function models showed that the activation of the host mTOR pathway could separate microbiome composition in women and that mTOR activation can be predicted on the basis of microbiome composition. Individual microbiota features across different cohort groups and different sample types are predictive of higher vaginal mTOR activity including the bacterial taxa Gardnerella and M. mulieris (Figures S2B and S2E), and the bacterial functional molecules phosphoglycerate kinase and chaperonin (Figures S2D and S2G).
The single-omic LVs were then integrated into a multi-omics meta-model in the form of a generalized linear model with interaction effects between microbiome LVs to predict host mTOR activity. We included three computational controls for these models. For the first model, we trained single-omic models using the host vaginal transcriptomics data to define the mTOR score using the GSEA Hallmark Pathways MTORC1 gene set (Figure 4). Top bacteria that predicted mTOR activation included L. crispatus, Gardnerella, P. amnii, and M. mulieris (Figure 4F). Mucosal metabolites positively associated with mTOR activity included 5-aminovalerate, N-acetylputrescine, ImPA, xanthine, and tyramine, whereas asparagine, arginine, and uric acid were negatively associated with mTOR activity.
For the other computational controls, we validated the previous one by using a different gene set, the Pathway Interaction Database (PID), rather than GSEA to define the host mTOR score using the vaginal transcriptomics or host proteomics. We identified the same top associations of bacterial factors with these mTOR scores using the PID protein list (Figure S3) on vaginal mucosal proteins as in our GSEA, indicating a high reproducibility of our analysis. We saw similar reproducibility in predictive microbiome factors with either proteomics- or transcriptomics-defined host mTOR activity.
Bacterial products linked to mTOR signaling directly affect vaginal epithelial barrier integrity in vitro
The association between bacterial products and species with mTOR activation led us to hypothesize that these bacteria could modulate epithelial function and stimulate these pathways. We performed bacterial supernatant treated cell-culture experiments in vaginal epithelial cells and mapped intracellular pathways. We chose four type strain bacterial species purchased through the American Type Culture Collection for performance of these experiments, namely L. crispatus, Gardnerella vaginalis, M. mulieris, and P. amnii, because they were the top species associated with mTOR score and were also found to have the greatest number of unique proteins and functional pathways from the identified bacterial proteins in the clinical cohorts. To test the direct impact of vaginal bacterial species on the epithelial barrier, we employed a reductionist model of inoculating vaginal cells with bacterial culture supernatants. This allowed us to observe the effects of secreted bacterial products on the epithelial cells without numerous variables that have to be accounted for in animal models or more complex multi-cellular model systems. The in vitro vaginal VK2 E6/E7 cells in our air-to-liquid model systems resemble the upper layers of the squamous epithelial layer known as the stratum corneum in vivo, with little to no tight junctions present and multiple cell layers thick.48 These cells were therefore used to assess epithelial thickness and overall host protein responses after treatment with bacterial products. The bottom-most layer of the squamous epithelial layer, known as the parabasal epithelium,49 as well as the single polarized columnar epithelial cell barrier of the upper FGT,50 have the most robust tight junctions in the FGT. Hec1A cells are endometrial cells that are known to be one of the only FGT cell lines available producing robust tight junctions in vitro51 and were therefore used in separate assays to assess junctions (transepithelial electrical resistance [TEER]), porosity, and wound-healing capabilities in our model systems.
VK2 were treated with cell-culture media controls (cell-culture media alone or with bacterial media: NYC and NF) as well as bacterial products collected and filtered from cultures of L. crispatus, G. vaginalis, P. amnii, and M. mulieris for 24 h (bacterial supernatants). The VK2 cell lysates were measured for proteome changes by mass spectrometry. In comparison with L. crispatus supernatant treated cells, the addition of BV-associated bacterial supernatants resulted in significant epithelial proteome changes, including those treated with G. vaginalis (187 proteins, 8.4%), M. mulieris (642 proteins, 28.7%), and P. amnii (660 proteins, 29.5%). Hierarchal clustering of differentially abundant proteins identified three clusters that separated each bacterial supernatant treatment (Figure 5A). These proteins were searched against the literature for experimentally determined links between mTOR signaling or epithelial barrier function and structure. Proteins upregulated with BV-associated bacteria treatments included mTOR signaling and related pathways, including remodeling of epithelial adherens junction,52,53 Rho signaling,54 Cdc42 signaling,55 PI3K signaling,56 and others (adjusted p < 0.05) (Figure 5B).
Figure 5.

BV-associated bacterial supernatants affect vaginal epithelial barrier function in vitro and affect pathways involved in mTOR activity and epithelial function
(A) Heatmap showing differentially abundant proteins in VK2 cells treated with different bacterial culture supernatants for 24 h, as observed by mass spectrometry. Proteins that activate mTOR cluster in the group that is upregulated in BV-associated bacterial treated cells (red box). Numerous structural and signaling adherens proteins were also differentially regulated by BV-associated bacteria (highlighted in green).
(B) Pathway analysis showing top pathways including wound healing, tissue remodeling, and mTOR activation.
(C) G. vaginalis inhibited VK2 cell differentiation and thickness compared with L. crispatus (n = 9).
(D) G. vaginalis inhibited wound-healing ability as measured by scratch assay in Hec1a cells (n = 9).
(E) M. mulieris decreased barrier integrity as measured by electrical resistance (TEER) on transwell membranes of Hec1a cells (n = 12).
(F) M. mulieris induced a leaky barrier with protein-sized particles (dextran-FITC, 70 kDa) able to translocate across the membrane, but not larger-sized particles (n = 6).
All error bars represent standard deviation from the mean, p values represent unpaired t test results. See also Figure S4.
To evaluate the physiological consequences of molecular changes induced by BV-associated bacteria on epithelial barriers, we measured several physical parameters of vaginal and endometrial epithelial layer function, including proteomic pathway characterization (VK2 cells), cell differentiation using an air-to-liquid in vitro VK2 cell model system,57 a transwell barrier model to assess porosity and electrical resistance (Hec1a cells),58 and a wound-healing model (Hec1a cells).59 We characterized wound-healing capabilities by scratch-test assay, tight junction formation by using TEER, and epithelial layer porosity by using fluorescently tagged protein-sized dextran-fluorescein isothiocyanate (FITC) molecules (∼70 kDa) and fluorescently tagged virus-particle-sized FluoSpheres (0.1 μm). G. vaginalis supernatants significantly inhibited multi-cellular epithelial barrier growth and differentiation (Figure 5C) (p = 0.0005), similar to the positive control 0.1% nonoxynol-9 (p = 0.0002), and this was visibly evident by microscopy (Figure S4A). In addition, G. vaginalis decreased wound-healing capacity at 48 h post scratch, in comparison with the cell-culture and bacterial culture media controls (p = 0.0011 and p = 0.149, respectively) (Figures 5D and S4B). Cell growth and differentiation was not affected by L. crispatus, M. mulieris, or P. amnii (p > 0.05). M. mulieris decreased barrier integrity as measured by TEER 24 h post treatment, compared with cell-culture media and NF bacterial media controls (p = 0.0029 and p < 0.0001, respectively), similar to the positive control nonoxynol-9 (p < 0.0001) (Figure 5E). M. mulieris treatment also affected epithelial porosity, leading to a significant translocation of the smaller-sized dextran-FITC particles compared with both the cell-culture and bacterial media controls (p = 0.0003 and p = 0.0005, respectively). However, translocation of the larger FluoSpheres was not affected, indicating that M. mulieris induces a semi-leaky barrier. The positive control nonoxynol-9 treatment showed a significant translocation of both the dextran-FITC (Figure 5F, p = 0.0022) and FluoSpheres (Figure S4C, p = 0.0043). None of the other bacterial supernatants tested induced significantly affected TEER measurements or translocation of either size of fluorescent particles (p > 0.05). Collectively, these results indicate that products from dysbiotic bacteria linked to mTOR activity in vivo can modulate these pathways in vitro, resulting in physiological effects on epithelial function.
Vaginal metabolites cause epithelial dysfunction in vitro
In our meta-model analysis, we identified metabolites that were associated with mTOR activity and microbiome taxa, including imidazolepropionic acid (ImPA), an amino-acid-derived metabolite produced by certain gut microbiota that impairs insulin signaling through mTOR, contributing to type 2 diabetes.60 In addition, we found that ImPA is present in cultures of vaginal bacterial species in vitro (Figure 6A). Therefore, we hypothesized that ImPA may affect vaginal epithelial barrier function. To test this, we co-cultured vaginal epithelial cells with 1 mM pure cell-culture sterilized ImPA (from Sigma) in cell medium and monitored the molecular and physiological effects. This dose was used because it is on the higher end of ImPA concentration that was generated in bacterial cultures of people with diabetes,60 although whether this concentration is physiologically relevant in the vaginal microenvironment needs to be determined. After 24 h, ImPA-treated Hec1a epithelial cells showed significant changes to the proteome (Figure 6B), including upregulation of carbohydrate metabolic processes (p = 0.004), proteins that can activate mTOR (CUL4B, HDAC2, MAPK1, ARHGAP4), and decreased proteins involved in cell adhesion (PARVA). Ingenuity Pathway Analysis61 associated integrin, ILK, AMPK, and leukocyte extravasation signaling with these proteome changes (p < 0.05) (Figure 6C). While mTOR signaling did not reach statistical significance (p = 0.276), Integrin and ILK are shown to activate mTOR,62 AMPK negatively regulates mTOR,63 and leukocyte extravasation occurs through mTOR activation.64 Using GSEA on these samples, transforming growth factor-β and apical junction pathways are highlighted as positively associated (p < 0.05) with ImPA treatment, and others including mTOR signaling trending. For negative associations, KRAS signaling and hypoxia (p < 0.05) were identified. ImPA-treated cells showed a significant, dose-dependent decrease in TEER (p < 0.05) (Figure 6D). These results suggest that bacterial metabolites identified in vivo as linked to mTOR activity are contributing to epithelial dysfunction.
Figure 6.

Bacterial and mTOR-associated metabolite imidazolepropionic acid leads to epithelial barrier disruption
(A–C) (A) One of the metabolites identified in our multi-omics model (imidazolepropionic acid) to be significantly correlated with mTOR activation and BV-associated bacteria was found to be produced by G. vaginalis in supernatants of pure cultures measured against negative control bacterial media samples. Imidazolepropionic acid purchased from Sigma was used to treat Hec1a cells and proteomics, analysis of which showed both (B) protein expression and (C) pathway-level disruption of mTOR activity.
(D) We next showed that imidazolepropionic acid disrupted barrier integrity in a dose-dependent manner using transepithelial electrical resistance of Hec1a cells on transwell membranes. The error bar represents the standard deviation from the mean; n = 12, p value represents unpaired t test results.
Discussion
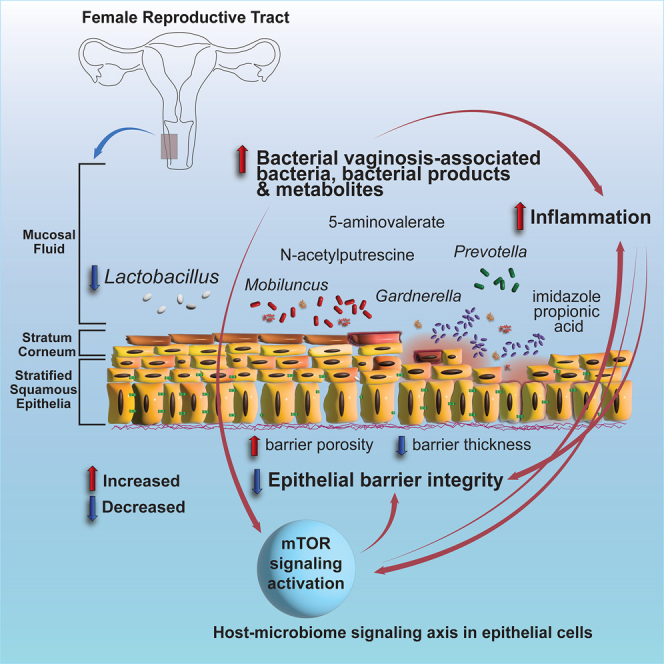
In this study we identified key relationships between host immunity and specific functional properties of the vaginal microbiome. We show that mTOR is a key feature underlying epithelial dysfunction and inflammation related to molecular BV. We show that metabolic products from Gardnerella and M. mulieris, as well as ImPA, contribute to vaginal epithelial dysfunction in vitro and associate with mTOR signaling. Therefore, these data suggest that the microbiome-mTOR axis, driven by molecular mediators that are derived from the microbiota, is a key component of epithelial dysfunction and may contribute to the pathogenesis of molecular BV.
BV is associated with inflammation of the FGT.11,65 Vaginal microbial communities of high diversity are strongly correlated with genital proinflammatory cytokines,66 which has previously been linked to Toll-like receptor 4 (TLR4) signaling by antigen-presenting cells.3 Previous literature has shown a link between mTOR activation45 and LPS stimulation through TLR4, activating either NF-κB or PI3K67,68 during other bacterial infections, including lung69 and kidney70 injury. A previous study has also shown that Lactobacillus casei extract inhibits NF-κB and mTOR signaling in the gastrointestinal tract.71 A central control response for metabolism,44,72 mTOR is tightly regulated by the presence of nutrients and growth factors, such as amino acids, ATP, oxygen levels, glucose, and insulin.73,74,75 The mTOR pathway plays a large role in immune regulation45 including regulation of CD8 T cell responses,76 differentiation of regulatory and T cell subsets,77 development and maturation of B cells,78 generation of functional dendritic cells,79,80 and the production of cytokines.45,81 This central regulator is activated by numerous signaling pathways,44,72,74 such as upstream Wnt75 and LPS induction of NF-κB, as well as downstream signaling of RhoA,82 integrin,83 and ARP2/3,84,85 all of which were significantly associated with the vaginal microbiome through pathway analysis in our study (Figure S4). In addition, cytokines can activate mTOR expression, such as induction of T helper cell differentiation via interleukin-2 (IL-2).86 TNFα signaling can regulate epithelial cell proliferation through the activation of mTOR87 and induce IL-10 production through mTOR-dependent activity in macrophages during chronic inflammatory conditions.88
One of the defining pathologies of BV is a breakdown of epithelial barrier integrity.89,90 This barrier disruption is thought to increase inflammation through a leaky membrane, allowing epithelial and resident immune cells to be exposed to microbial products within the tissues.91 In our study, gene and protein expression analysis showed a clear difference in the cell-cell adherens, keratinization, and cell differentiation expression patterns in women with nLD microbiomes. Our experiments showed that G. vaginalis inhibited cell growth, including a decrease of wound healing, and cell differentiation, in line with previous observations.24 Cell differentiation has been shown to be regulated by bacteria in the gut both in vitro and in vivo,92 but it has not yet been described in the FGT. M. mulieris caused decreased epithelial barrier integrity, which is associated with reductions of adherens junction remodeling. The mTOR network plays a major role in cell growth, proliferation, and differentiation75,93 as well as functions in wound healing and autophagy,44,94 and was activated in the presence of P. amnii and M. mulieris bacterial growth culture supernatants containing by-products of bacterial metabolic processes. Therefore, epithelial dysfunction during BV may, in part, be mediated through mTOR signaling, and further studies evaluating how this pathway modifies vaginal epithelial function are an important area of investigation. As our experiments show, there is diversity in barrier changes and mTOR activation in the presence of products from different bacterial species, indicating that all BV-associated bacteria are not equal with respect to BV pathology. More studies that determine whether the barrier damage is exacerbated in the presence of multiple bacterial species, such as a combination of Gardnerella and M. mulieris, or other BV-associated bacteria, also needs to be determined.
Bacterially produced metabolites play a role in epithelial barrier function and mucosal immunity such as lactic acid95 and short-chain fatty acids.96 In the vaginal tract, lactic acid maintains homeostasis by attenuating inflammation, and short-chain fatty acids alter innate mucosal immunity, leading to reduced barrier integrity.5 A proinflammatory response in the vagina is elicited by BV-associated bacterial species through TLR signaling, promoting production of IL-8, TNFα, and IL-1β.5 Other metabolites may also play a role in vaginal barrier function. We identified metabolites that predict mTOR activation, such as 5-aminovalerate, N-acetylputrescine, xanthine, xanthosine, lactate, and ImPA. Using in vitro models, we showed that ImPA directly inhibited FGT barrier function. ImPA has been shown to affect insulin signaling through mTOR, but this is the first study showing that direct treatment of epithelial cells with this metabolite affects barrier function. The effect of barrier damage from the bacteria was greater than that of ImPA, likely because ImPA is only one of numerous factors that affect the vaginal barrier. A more comprehensive investigation using the other metabolites identified and clinical isolates may be beneficial in future studies. Different growth conditions of bacteria, including the growth medium in culture compared with in the vaginal environment, whether these bacteria are in their log or stationary phase, and whether they are growing in pure cultures or as part of a mixed colony, may change the metabolites produced and the effects on vaginal epithelial cells. In addition, it needs to be determined whether the bacteria would produce similarly damaging products when growing on vaginal cells. A more detailed investigation of the growing conditions that can most damage the vaginal environment, by either bacterial cells directly growing in the model or different culture conditions prior to bacterial supernatant harvest, is an avenue of future studies.
The vaginal microbiome profiles in this study were consistent with other studies of African women.1,3,97,98,99 A total of 53% of the women in our study had an nLD microbiome (molecular BV), with 35% having a positive Nugent-BV diagnosis and 5% with an indeterminate Nugent-BV diagnosis. These numbers agree with previous literature that describes greater microbiome diversity for African women in comparison with Caucasian women, ranging from 21% to 59% based on Nugent score.100 Other studies in Kenya have observed a Nugent-BV prevalence of 36.7%–55.9% for women aged 15–49 years.15 Molecular analysis (molecular BV) of vaginal microbiome groups in African women also show a large percentage (∼40%) of women with a non-Lactobacillus microbiome profile.101
The interaction between the microbiome and mucosa of the FGT is very complex, and the epithelial layer plays a first-line defense from invading pathogens. While nLD microbiomes may affect other aspects of genital immunity, here we show that dysbiotic bacteria directly modify key signaling networks in epithelial cells through soluble products and metabolites, which associates with decreased epithelial function and may contribute to the pathogenesis of BV through increased inflammation. A better understanding of the host-microbiome axis involving mTOR in the FGT is important for devising new interventions to prevent BV and/or its deleterious effects on vaginal health and risk of sexually transmitted infections.
Limitations of the study
There are several limitations of this study. First, we did not obtain information on immune cell subsets, which limited our ability to evaluate the role of cellular inflammation in epithelial dysfunction. It is likely that epithelial disruption independent of microbiome-epithelial interactions through inflammatory signaling by resident immune cells could also contribute to our observations. Also, while we chose mTOR as an endpoint in our data-driven models of BV-associated inflammation, it is possible that other pathways may be important for the pathogenesis of BV. Investigating the pathways found to be significantly different between microbiome groups indicated that many of these pathways connected to the central mTOR signaling cascade. As such, this pathway was the focus of our analysis. However, a similar analysis could be performed on other pathways, which could identify biomarkers and metabolites of interest. In addition, the modulation of epithelial dysfunction by bacteria may include other inflammatory metabolites that we did not test in our assays, and thus imidazole propionate likely represents one of many metabolite contributors to in vivo epithelial dysfunction. Finally, the in vitro assays we utilized to test epithelial dysfunction do not represent the complex in vivo mucosal microenvironment and, thus, these effects by bacteria and metabolites may be modulated, either positively or negatively, by other components such as resident immune cells. Further studies that employ more complex model systems that incorporate microbiota, epithelial cells, and immune cells may better recapitulate these processes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Involucrin | Novus Biologicals | cat# NBP2-16981; RRID:AB_2936950 |
| DAPI | Novus Biologicals | cat# NBP2-31156; RRID:DAPI Solution |
| Alexa Fluor® 647 Phalloidin | ThermoFisher | cat# A22287; RRID:AB_2620155 |
| Goat anti-Rabbit IgG (H + L) Secondary Antibody, Alexa Fluor® 488 conjugate | ThermoFisher | cat# A11008; RRID:AB_143165 |
| Bacterial and Virus Strains | ||
| Lactobacillus crispatus bacteria type strain | DSMZ | DSM20584 |
| Prevotella amnii bacteria type strain | DSMZ | DSM23384 |
| Gardnerella vaginalis bacteria type strain, strain designation 594 | ATCC | ATCC14018 |
| Mobiliuncus mulieris bacteria type strain, strain designation SV175, SV17J | DSMZ | DSM25311 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imidazolepropionic acid | Millipore Sigma | 77951-250MG |
| Critical Commercial Assays | ||
| LavaPep’s Fluroescent Peptide and Protein Quantification Kit | Gel Company, CA, USA | LP022010 |
| RNeasy Fibrous Tissue Mini Kit | Qiagen | Cat. No. ID: 74704 |
| Cell Proliferation Reagent WST-1 | Millipore Sigma | 5015944001 |
| Deposited Data | ||
| Transcriptomics of vaginal biopsies | GEO | Accession number: GSE139655 |
| Metabolomics of vaginal swab elutions | MetaboLights | Accession number: MTBLS7087 |
| Proteomics/metaproteomics of vaginal swab elutions | PRIDE | Accession numbers: PXD040895 and PXD040280 |
| Experimental Models: Cell Lines | ||
| VK2/E6E7 immortalized human epithelial vaginal cell line | ATCC | Cat# CRL-2616; RRID: CVCL_6471 |
| HEC-1-A immortalized human epithelial endometrium cells | ATCC | Cat# HTB-112; RRID: CVCL_0293 |
| Software and Algorithms | ||
| Mascot v2.4.0 | Matrix Science | RRID:SCR_014322 |
| Progenesis | Nonlinear Dynamics | RRID:SCR_018923 |
| R programming software | R Foundation for Statistical Computing | RRID:SCR_001905 |
| R Studio | Posit Software, PBC | Version 2022.12.0 + 353 |
| R NMF package | BMC Bioinformatics | RRID:SCR_023124 |
| R ColorBrewer package | Erich Neuwirth | RRID:SCR_016697 |
| R ggplot2 | Springer-Verlag | RRID:SCR_014601 |
| R COMBAT | Minghui Wang and Yiyuan Liu and Shizhong Han | RRID:SCR_010974 |
| R reshape2 package | Hadley Wickham | RRID:SCR_022679 |
| R plyr | Hadley Wickham | https://www.jstatsoft.org/v40/i01/ |
| R ggfortify package | Yuan Tang and Masaaki Horikoshi and Wenxuan Li | https://doi.org/10.32614/RJ-2016-060 |
| R survival package | Terry M Therneau | RRID:SCR_021137 |
| R vegan package | Jari Oksanen | RRID:SCR_011950 |
| DAVID | DAVID bioinformatics team (LHRI/ADRD at Frederick National Laboratory) | RRID:SCR_001881 |
| Ingenuity Pathway Analysis | Ingenuity | RRID:SCR_008653 |
| Gene Set Enrichment Analysis | Broad Institute | RRID:SCR_003199 |
| FlowJo | LLC | RRID:SCR_008520 |
| Adobe Photoshop CC | Adobe | RRID:SCR_014199 |
| MATLAB | Mathworks | R2021a |
| Partial Least Squares Path Modeling | This Work | https://doi.org/10.5281/zenodo.7810345 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Adam Burgnener (adam.burgener@case.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant detail
Clinical sample collection
Ethics statement
All women who participated in this study provided written informed consent. This study was approved by the University of Washington Human Subjects Review Committee, the Kenya Medical Research Institute, and the Research Ethics Board of the University of Manitoba.
Study population
This study included cervicovaginal samples and data collected from Ugandan and Kenyan female partners of HIV-1 serodifferent couples enrolled in the Partners PrEP study103 – a randomized, placebo-controlled trial of daily tenofovir-based pre-exposure prophylaxis (PrEP) to prevent HIV-1 infection. HIV-1 seronegative partners were followed at monthly visits that included detailed assessments of behavioral and medical histories. For this study, we included 405 women between the ages of 18 and 52, which were separated into participant groups: participant group 1, n = 315 vaginal swabs for MS/MS analysis; participant group 2, n = 90 vaginal swabs for MS/MS analysis and n = 80 vaginal biopsies for transcriptomics analysis. The first group included 192 women who were enrolled in the Partners PrEP Study placebo arm while 123 received either tenofovir or tenofovir-emtricitabine, with a p = 0.03 significant difference between microbiome groups in this participant group analysis. The second participant group was used to validate functional pathways observed in the first group without any treatment (placebo vs. treatment p = 0.27), behavioral, clinical, or demographic differences between non-Lactobacillus dominant and Lactobacillus dominant women, which were observed in the larger, first participant group. In addition, the matched vaginal tissue biopsies allowed for complimentary transcriptomic analysis of the immunological, cell signaling, and barrier structure analysis at the tissue level. These groups were analyzed independently, for validation of molecular analysis of pathways associated with BV. All women from both analysis sets remained HIV-1 negative throughout the study. Women were also evaluated at baseline for bacterial STIs, Trichomonas vaginalis, Neisseria gonorrhoeae, and Chlamydia trachomatis and treated if found to be positive.
Cell lines
VK2/E6E7 cells (Vagina, mucosa; E6/E7 transformed, ATCC CRL-2616) were grown in Keratinocyte-Serum Free medium (ThermoFisher), supplemented with 0.1 ng/mL human recombinant EGF, 0.05 mg/mL bovine pituitary extract, and 0.4 mM calcium chloride, in incubators set at 37°C and 5% CO2 concentration. Cells were subcultured at 75–85% (unless left to full confluence for cell differentiation purposes), using DMEM:F12 transfer medium (ThermoFisher) supplemented with 10% heat-inactivated FBS (Sigma-Aldrich) after dissociation using Trypsin-EDTA (0.25%, phenol red, ThermoFisher). Hec1A cells (uterus, endometrium; adenocarcinoma, ATCC HTB-112) were grown in McCoy’s 5A (Modified) Medium (ThermoFisher), supplemented with 10% heat-inactivated FBS. Cell were subcultured at 75–85% after dissociation using Trypsin-EDTA. Cell viability was monitored using a microscope and colored media indicators, as per usually laboratory practices.
Bacterial cultures
Lactobacillus crispatus type strain DSM 20584 (Leibniz DSMZ) was grown on De Man, Rogosa, and Sharpe agar (MRS; National Microbiology Lab media prep). To prepare liquid culture, 1–2 colonies were suspended in 2 mL NYC III broth (ATCC 1685). Gardnerella vaginalis type strain ATCC 14018 (ATCC Cedarlane) was grown on tryptic soy agar with 5% sheep’s blood (TSAB; National Microbiology Lab media prep), and loopful of growth was added to 2 mL NYC III broth to prepare liquid culture. Mobiluncus mulieris type strain DSM 25311 (Leibniz DSMZ) was grown on tryptic soy agar with 5% sheep’s blood (TSAB; National Microbiology Lab media prep), and plate growth was harvested into 2 mL of NF broth, an in-house mixture of NYC III and Fastidious Anaerobe Broth (1:1), pre-reduced anaerobically in previous 24 h. A 20% inoculum of this bacterial suspension was used to inoculate multiple tubes of 2 mL pre-reduced NF broth. Prevotella amnii type strain DSM 23384 (Leibniz DSMZ) was grown on chocolate agar (CA; National Microbiology Lab media prep) and plate growth was harvested into 2 mL NF broth, pre-reduced anaerobically in previous 24 h. A 10% inoculum of this bacterial suspension was used to inoculate multiple tubes of 2 mL pre-reduced NF broth. All cultures were inoculated at 37ᵒC for 24 h under anaerobic conditions, achieved using BD GasPak Anaerobic Sachets (Becton-Dickinson).
Aliquots of bacterial cultures were taken for cell enumeration via optical density reading and flow cytometry cell counting. Remaining volume of culture was spun down at 3,000 rpm for 15 min, cells discarded, and supernatant filter-sterilized using 0.22 μm Millex-GV Syringe Filter (Merck). Preparation of cell free supernatants was performed on bacterial cultures grown to approximately 109 cells/ml.
Method details
Sample collection
Copan Floqswabs (Copan Diagnostics, Murrieta CA, USA) were used to collect vaginal secretions. A vaginal swab was collected by a physician and assisted by another clinician or nurse, after participants provided consent. The swab was placed with the long edge along the lateral vaginal mucosal then gently rotated while moving the swab in a circular motion. The swab was then snipped with cleaned scissors into an empty cryovial to freeze at −80°C for future mass spectrometry processing and analysis.
A vaginal biopsy was collected by a physician from an area of the vaginal mucosa that can be visualized on the lateral vaginal wall adjacent to (not overlapping with) the sites where the swab samples were collected. Approximately 4mm biopsies were collected using forceps and placed into a cryovial with 0.5mL RNAlater. Samples were stored at −80°C until processed for transcriptomic analysis.
Mass spectrometry analysis
A total of 405 vaginal copan swab samples were analyzed by tandem mass spectrometry. Vaginal swabs were eluted with PBS (pH 7.0). Swabs were then centrifuged in SpinX tubes with a bonded fritted bottom (Corning, Corning, NY) and eluate was collected. Eluates were centrifuged at 23,000g for 30 min at 4°C and 100μL of supernatant per sample was then denatured, reduced, alkylated and digested into peptides. Peptides were cleaned of salt and detergents by reverse-phase liquid chromatography using a step-function gradient. Cleaned peptides were quantified using LavaPep’s Fluroescent Peptide and Protein Quantification Kit (Gel Company, CA, USA) according to the manufacturer’s protocol. One microgram of peptide per sample was then prepared for mass spectrometry as previously described.104 Peptide samples were injected into a nano-flow LC system (Easy nLC, Thermo Fisher) connected inline to an LTQ Orbitrap Velos (Thermo Fisher) mass spectrometer and analyzed in a label-free manner as described previously.104 All samples were analyzed by mass spectrometry (MS) in batches of ∼50 samples (6 batches in total). Within each batch run were replicates of a technical mix (pooled swab eluate from 50 random different samples) which was used to evaluate the technical variance between batches allowing for batch effect corrections. Human peptide identity searching was performed with Mascot v2.4.0 (Matrix Science) against the human SwissProt database (June 2015). Bacterial peptide identity searches were performed using a manually curated TrEMBL database containing the major genera identified from an initial search and taxa described by 16S rRNA studies (16 genera total).3 A decoy database was included to determine the false discovery rate. Search results were imported into Scaffold to validate the protein identifications, using the following criteria: ≤0.1% False Discovery Rate (FDR) for peptide identification, ≤1% FDR for protein identification and at least 2 unique peptides identified per protein. Host proteome results were imported into Progenesis LC-Mass Spectrometry software to perform label-free differential protein expression analysis based on MS peak intensities. Feature detection, normalization and quantification were all performed using default settings from the software. Microbial abundance was calculated by summing normalized total spectral counts from Scaffold for all proteins associated with each genus. Database construction and analysis details are described in the Supplementary Methods. For the initial group, a total of 1174 host proteins were identified (passing quality assurance) across all experimental batches, 434 of which were identified consistently in every batch and therefore used in downstream analysis. The data were uploaded to PRIDE: PXD040895 (participant group 1) and PRIDE: PXD040280 (participant group 2).
Metaproteomic data analysis
Unsupervised hierarchical clustering with average Euclidean linkage using bacterial proteins identified in our mass spectrometry analysis was performed using binned genus or species level microbial abundance proportions for each sample and stacked bar charts were generated in RStudio (1.1.453) using ggplot2 and NML packages.105 Shannon’s H index diversity scores were calculated and compared between groups using Mann-Whitney U tests in GraphPad Prism. Women were defined as having a species-specific dominance if they had >50% of their total bacterial protein abundance represented by that bacterial species’ proteins,106 if no particular species was dominant, then they were considered to have a polymicrobial profile.
Most of the Gardnerella dominant proteins identified as Gardnerella vaginalis in our protein database, however immerging research indicates that Gardnerella can now be resolved into numerous different species, such as G. leopoldii, G. piotii, and G. swidsinkii.107 NCBI currently has 3 new species in their refSeq protein database, but there are very few peptide sequences (ie. 285 vs. 136,000 vaginalis sequences) that map to other Gardnerella species. These new species are not yet included in Uniprot. To investigate our current data, we blasted the top 5 Gardnerella protein hits against the NCBI sequences. This showed that though all the proteins’ top hits were 100% similar to G. vaginalis, 3 of the 5 proteins also showed 100% similarity to one of the other new Gardnerella species, indicating that our current proteomic database may not confidently resolve Gardnerella to species specificity. As such, we labeled any identified Gardnerella proteins as non-species specific.
Metabolomic analysis
Metabolomics procedures were adapted from Srinivasan et al.108 Metabolites were extracted from cervicovaginal lavages (CVL) using a 1:4 ratio of CVL to methanol, followed by vortexing and centrifugation at 16,000 × g for 30 min at 4°C. The supernatants are then transferred to new tubes and dried using a Speed Vac system. For MS analysis, dried metabolite samples are resuspended in 200 μL of sample buffer (a mixture of 30% Buffer A: 70% Buffer B containing 53.1 μM 13C5-15N1-glutamic acid and 58.2 μM 13C2-succinic acid. Buffer A = 5 mM ammonium acetate in 0.1% acetic acid, Buffer B = 0.1% acetic acid in acetonitrile) vortexed for 15 min and then centrifuged at 16,000 g for 30 min to remove particulate matter. A standard sample containing a mixture of 10 known metabolites along with a mix sample consisting of extracted metabolites from a mixture of CVL from all samples was injected periodically throughout each batch run for QC and monitoring LC-MS/MS conditions. For LC-MS/MS analysis, metabolites are then separated using an Agilent 1200 binary pump HPLC system equipped with a ZIC-cHILIC column (150 × 2.1 mm, 3.0 μm particle size) (Millipore) for HILIC mode analysis of polar metabolites. The flow rate was set to 200 μL/min and the column temperature is kept at 40°C. The separation gradient was set as follows: 0–3 min: 70% B, 3–7.5 min: 70-30% B, 7.5–13.5 min: 30% B, 13.5–16.5 min: 30–70% B, 16.5–27 min: 70% B. 8 μL of each sample was injected for simultaneous positive and negative mode analysis on a Fusion Lumos Tribrid mass spectrometer. Targeted quantification of metabolites was accomplished in parallel reaction monitoring (PRM) mode, targeting 121 different metabolites (66 in positive mode, 55 in negative mode). Source conditions for positive mode were set as follows: Spray Voltage: 3500 V, Sheath Gas: 3 (Arbitrary Units), Aux Gas: 1.2 (Arbitrary Units), Ion Transfer Tube Temp: 275°C. Source conditions for negative mode are the same as for positive except the Spray Voltage is set to 2100 V. Mass spectrometry data is acquired by alternating MS1 and MS2 scans in both positive and negative mode. MS1 scans are collected with the following parameters (the same for positive and negative unless listed otherwise): Scan range: 55–280 m/z (positive), 65–500 (negative), Orbitrap resolution: 30000, RF Lens (%): 30, AGC Target: 4.0e5, Max Injection Time: 50 ms. MS2 scans are collected with the following parameters (the same for positive and negative mode unless listed otherwise): Isolation Window: 1.6 m/z, HCD Collision Energy: 30%, Stepped Collision Energy: +/− 10%, Orbitrap Resolution: 15000, Maximum Injection Time 22 ms. For MS2 scans the mass range was set from 50 m/z to the mass of the targeted metabolite +10 m/z. Raw files obtained are subsequently uploaded into Skyline for metabolite peak integration and quantification using a custom method. The data were uploaded to Metabolights: MTBLS7087.
Transcriptomic analysis
Vaginal biopsies were processed as previously described.109 Briefly, vaginal biopsies were stored at −80C in RNALater Stabilization Solution (Thermo Fisher Scientific, Waltham, MA, USA). Biopsies were thawed and homogenized in 600 μL of Buffer RLT (Qiagen, Hilden, Germany) using a Bio-Gen PRO200 homogenizer (PRO Scientific, Oxford, CT, USA). RNA was extracted on a QIAcube using the Rneasy fibrous tissue mini kit (Qiagen). RNA quality was determined using the TapeStation R6K assay (Agilent, Santa Clara, CA, USA). Samples were prepared for microarray using the Ovation PicoSL WTA System V2 (NuGEN, San Carlos, CA, USA) and Encore BiotinIL kits (NuGEN). Labeled cDNA was hybridized to HumanHT-12 v4 Expression BeadChips (Illumina, San Diego, CA, USA) and images were converted to expression data using GenomeStudio (Illumina). The data were uploaded to GEO with accession number GSE139655.
Cell viability assays
Cells were tested for viability in different bacterial supernatant treatment conditions by trypan blue exclusion assay (# live cells/200 total cells counted), as well as WST-1 (Sigma) as per manufacturer’s protocol. Briefly, 40μL of WST-1 reagent was added to each well in a 24-well plate 24 h post-treatment. The plate was incubated for 2 h at 37°C at 5% CO2 then measured using a spectrometer at both 690nm and 450nm; values at the 690nm wavelength were subtracted from the 450nm values.
Optical density readings
Aliquots of 100 μL were taken from undiluted 24-h cultures in triplicate, and optical density measured at wavelength of 600 nm using Synergy H1 Microplate Reader (BioTek). Optical density data for each culture was matched with corresponding bacterial cell count data obtained from flow cytometry.
Flow cytometry – Bacterial cell quantification
A 100 μL aliquot of each undiluted 24-h culture was combined with 100 μL of 4% paraformaldehyde (Sigma-Aldrich) to fix cells, then samples were stored at 4ᵒC for at least 24 h. To prepare samples for flow cytometry, a 10 μL aliquot of fixed bacteria was added to 200 μL of 0.001% propidium iodide (Sigma-Aldrich) and 100 μL of fluorescent yellow-green microparticles, at known concentration of 106/mL (FluoSpheres Polystyrene Microspheres, 1.0 μm; ThermoFisher Scientific). Samples were run on BD LSR II flow cytometer (Becton-Dickinson), with total of 5000 events recorded per sample. Data was analyzed using FlowJo v.10 (FlowJo, LLC).
Scratch test wound healing assay
A scratch test to determine wound healing ability was performed as previously described.106 Briefly, 100% confluent and differentiated (≥7 days post-seeding) Hec1A cells seeded onto 12-well tissue culture plates were scratched using sterile P20 pipet tips. Cells were then fed with cell media or a mixture of fresh, filtered bacterial supernatants diluted in cell media at a 1:5 ratio. Negative controls were performed using cell media only as well as a mix of 1:5 ratio of bacterial growth media in cell media, using either the NYCII broth (Lactobacillus and Gardnerella controls) or the half and half NYC III and Fastidious Anaerobe Broth media (Mobiluncus and Prevotella controls). Microscopy images (EVOS digital inverted microscope) were used to visualize the wound closure, and images were taken at 0-, 24-, and 48-h post-scratch. Wound size area was calculated using the measurements tool in Adobe Photoshop CC, then % healed wound sizes were calculated based on time 0 measurements. The statistical significance of results was calculated using ANOVA with Kruskal-Wallis correction, and unpaired t-tests. Each wound healing experiment was performed independently 3 times. For each experiment, 3 separate sections (marked with a permanent marker) were captured for each biological replicate (each well). The size of the wounds was measured by tracing the area of the scratch (where there were no cells present) in the series of images (taken at the same magnification) using the Adobe CC measurement tool. An example of this time course is shown in Figure S4B. In addition, when measuring our wound sizes throughout the time course, our images were blinded and randomized prior to measuring the wound area to prevent any biased reporting. After measurements were collected, the samples were unblinded and analysis of wound closures were calculated as a percentage based on the original size of the scratch to normalize the assay.
Transepithelial electrical resistance
Transepithelial electrical resistance was used to measure the integrity of tight junction formation as previously described.110 To test the tight junction integrity of the epithelial barrier, Hec1A cells were used to form tight junctions using transepithelial electrical resistance (TEER). A previous study51 showed that this cell type is the best FGT cell line to test junction integrity, as other cell lines including VK2 cells do not form tight junctions. The ability/inability to form tight junctions of Hec1A and VK2 cells was confirmed in our lab with Western blot and imaging (data not shown). Briefly, Hec1A cells were seeded on polyethylene-terephtalate (PET) filter inserts (Millicell Hanging Cell Culture Insert, 0.1 μm pore diameter, area 0.3cm2, used for 24-well plate) at a density of 1 × 105 cells/insert and maintained for approximately 7 days for complete cell differentiation, with ∼80% media changes to both the apical and basolateral sides every 2–3 days. Cell resistance, based on tight junction formation, was measured using TEER (trans epithelial electric resistance) using a Millicell ERS-2 Voltohmmeter (Sigma) per manufacturer’s protocol. The final resistance values are expressed as Ωcm2 using the following equation: TEER = (R-Rb) x A, where R is the measured resistance of the cells, Rb is the resistance of the filter alone in media (no cells), and A is the area of the filter (0.3cm2). Once TEER measurements stably reached ∼190 Ωcm2 indicating tight junction formation, fresh, filtered bacterial supernatants were diluted in cell media at a 1:5 ratio and added to the apical side. Negative controls were performed using cell media only as well as a mix of 1:5 ratio of bacterial growth media in cell media, using either the NYCII broth (Lactobacillus and Gardnerella controls) or the half and half NYC III and Fastidious Anaerobe Broth media (Mobiluncus and Prevotella controls). For the imidazole propionic acid (ImPA) assay, ImPA purchased from Sigma (Sigma 77951-250MG) was dissolved at a high concentration in PBS, filter sterilized, then diluted in cell media at the indicated concentrations. TEER measurements were then taken immediately following treatment and 24 h post-treatment. The statistical significance of results was calculated using ANOVA with Kruskal-Wallis correction, and unpaired t-tests. Each TEER experiment was performed independently 4 times in triplicate for a total of 12 replicates.
Porosity assays
The porosity, or leakiness, of the epithelial barrier in the presence of different bacterial supernatants were measured by determining the amount of translocation occurring across the barrier using different sized fluorescently labeled particles (Fluorescein isothiocyanate (FITC)–dextran, Sigma Aldrich; FluoSpheres Carboxylate-Modified Microspheres, 0.1 μm, yellow-green fluorescent (505/515), ThermoFisher) as previously described.51,111 After seeding Hec1A cells on filter inserts and left to form tight junctions as described above, fresh, filtered bacterial supernatants diluted in cell media at a 1:5 ratio were added to the apical side of the inserts. Negative controls were performed using cell media only as well as a mix of 1:5 ratio of bacterial growth media in cell media, using either the NYCII broth (Lactobacillus and Gardnerella controls) or the half and half NYC III and Fastidious Anaerobe Broth media (Mobiluncus and Prevotella controls). Cell were treated for 24 h then either dextran-FITC (at 1 mg/mL) or 0.1 μm FluoSpheres (at 1:20 dilution into cell media, filter sterilized) were added to the apical side. Controlling for volume, media from the basal side was tested for the presence of fluorescence at 488mm to indicate translocation of fluorescent particles across the membrane. After 1 h (dextran-FITC) or 24 h (FluoSpheres), the amount of fluorescence in the basal side of the membrane was measured using a Synergy H1 hybrid reader (BioTek). The statistical significance of results was calculated using ANOVA with Kruskal-Wallis correction, and unpaired t-tests. Each porosity experiment was performed independently 3 times in triplicate.
Epithelial barrier thickness assay
VK2 cells were used in an air-to-liquid interface culture method, as previously described,57 to determine whether bacterial products could prevent the formation of a multicellular layer similar to the stratified squamous epithelium found in the lower vaginal tract. Briefly, VK2 cells were seeded on polyethylene-terephtalate (PET) filter inserts (Millicell Hanging Cell Culture Insert, 0.1 μm pore diameter, area 0.3cm2, used for 24-well plate) at a density of 1 × 105 cells/insert. After 24 h, the media from the apical side was removed and the media in the basal side was maintained with media changes every 2–3 days for approximately 14 days to ensure complete cell differentiation and multilayer production. For cells treated with bacterial supernatant, fresh, filtered supernatant from either of the 4 bacterial species tested was diluted at a 1:5 ratio then added to the basal sides of the membranes. Negative controls were performed using cell media only as well as a mix of 1:5 ratio of bacterial growth media in cell media, using either the NYCII broth (Lactobacillus and Gardnerella controls) or the half and half NYC III and Fastidious Anaerobe Broth media (Mobiluncus and Prevotella controls). After 14 days of treatment, the cells were washed 3× with warm PBS, and fixed with 4% formaldehyde (Pierce 16% Formaldehyde (w/v), Methanol-free, ThermoFisher, cat# 28908; diluted in PBS) for 15 min, washed again 3× with PBST, then blocked using blocking buffer (PBS with 0.1% Tween 20 (PBST), 5% skim milk and 0.1% saponin) for 1 h. Cells were then probed with primary antibodies for 1 h in blocking buffer (Rabbit anti-Involucrin, Novus Biologicals, cat# NBP2-16981), washed 3× with PBST and probed with secondary antibodies in PBST with 0.1% saponin for 1 h (DAPI, Novus Biologicals, cat# NBP2-31156; Alexa Fluor 647 Phalloidin, ThermoFisher, cat# A22287; Goat anti-Rabbit IgG (H + L) Secondary Antibody, Alexa Fluor 488 conjugate, ThermoFisher, cat# A11008). Membranes were then removed from the inserts using a scalpel and placed cell-side up onto microscope slides. ProLong Diamond Antifade Mountant was added to the membrane, then coverslips (Cover glasses, high performance, D = 0.17mm, 18 × 18mm, type 1 ½, Zeiss) were placed on top and sealed with clear nail polish.
Images were taken using an inverted Zeiss confocal microscope LSM700 (Laser Scanning Microscope) with 4 lasers (405, 488, 555, 633). A 40× objective was used to take z stack images and the ZEN 2 software program was used for 3D image rendering. To take area measurements of the barrier thickness, the measurement tool in Adobe Photoshop CC (2017) was used, with the scale manually set to the image axis scale (μm) and the selection highlighted where the fluorescent staining marked the cells. The statistical significance of results was calculated using ANOVA with Kruskal-Wallis correction, and unpaired t-tests. Each barrier thickness experiment was performed independently 3 times in triplicate.
In-vitro epithelial cell assay with vaginal bacteria supernatants and metabolites
VK2 cells were grown in T75 flasks to full confluency then continued for 14 days post-seeding to promote differentiation with ∼80% media changes every 2–3 days. Markers of differentiation (SPINK5) were tested by Western blot to confirm differentiation occurs by 14 days (data not shown). Differentiated cells were used for bacterial supernatant exposure assay. All used media was discarded, then fresh cell media mixed with fresh, filtered bacteria supernatant from either Lactobacillus crispatus, Gardnerella vaginalis, Mobiluncus mulieris or Prevotella amnii (diluted at a 1:5 ratio of cell media to bacterial supernatant), or bacterial media and cell media (for negative controls) was added to the flask. Three different negative controls were used in the experiment and included cell media only, as well as a mix of 1:5 ratio of bacterial growth media in cell media, using either the NYCII broth (Lactobacillus and Gardnerella controls) or the half and half NYC III and Fastidious Anaerobe Broth media (Mobiluncus and Prevotella controls). For the imidazole propionic acid (ImPA) assay, ImPA purchased from Sigma (Sigma 77951-250MG) was dissolved at a high concentration in PBS, filter sterilized, then diluted in cell media at 1 mM. At 24 h post-bacterial supernatant or metabolite exposure, cells were harvested for mass spectrometry analysis. Briefly, cells were washed 3× with PBS after dissociation using Trypsin-EDTA, then 150μL SDS buffer (4% SDS, 50mM HEPES pH 8.8, 100mM DTT) was added to the cells. Samples were heated at 95°C for 5 min then run through QIAshredder tubes (Qiagen) at 15,000rpm for 2 min. The samples were then frozen at −80°C for mass spectrometry analysis similar to above. Proteomic data from each bacterial supernatant experiment was normalized to media control followed by median centering and log2 transformation for downstream pathway analysis. The statistical significance of results was calculated using ANOVA with Kruskal-Wallis correction, and unpaired t-tests. Each experiment was performed independently 3 times in triplicate.
Quantification and statistical analysis
Statistical analysis
Host proteome data was batch corrected in R using the combat package.112 Both proteome and transcriptome datasets were normalized via median centering and log 2 data transformation. Independent group comparisons of continuous variables were conducted using Student’s t tests or Mann-Whitney U tests where appropriate. Categorical variable comparisons were conducted via Fisher’s Exact Test or Chi-square where appropriate. Local false discovery rate (Benjamini-Hochberg) threshold of 5 percent was applied as a multiple hypothesis test correction where applicable.
Pathway analysis
Hierarchical clustering was performed using complete linkage and Pearson correlation distance metric in R using the NMF and RColorBrewer packages. Functional analysis was performed using DAVID Functional Annotation Bioinformatics Microarray Analysis Resource 6.8 and Ingenuity Pathway Analysis (QIAGEN Inc., https://digitalinsights.qiagen.com/IPA),61 with homo sapiens as the comparison dataset. Right-tailed Fisher’s exact tests (Benjamini-Hochberg corrected) were used to calculate the probability that the association between each protein in the dataset and the biological function or pathway was random.
Microbiome multi-‘omics partial least squares path model
mTOR score calculation
Gene Set Enrichment Analysis (GSEA) of the host proteomics and transcriptomics data implicated the MTORC1 Hallmarks pathway in bacterial vaginosis. GSEA Leading Edge analysis identified the members of the MTORC1 pathway most significantly driving the enrichment score in each data type, these proteins or genes constituting the leading edge subset of the pathway. We used these leading edge features to construct per-sample mTOR activity scores in the host proteomics and transcriptomics data via a partial least squares discriminant analysis (PLS-DA) model that collapsed protein or transcriptomic values to a single latent variable maximally explaining BV-associated mTOR activity. For a given data type, we constructed a PLSR X-block by filtering the dataset to just those leading-edge features identified in GSEA. We then trained a PLS-DA model using a single protein or RNA latent variable to predict BV status (Y-block) from the mTOR pathway members. The vector of coefficients describing how a given patient sample projected on this single latent variable, constructed from RNA or protein data, was extracted as the BV-associated mTOR score for downstream modeling.
Single-omic PLSR models and variable importance of projection (VIP) analysis
We trained single-omic PLSR models using either the metabolomics, bacterial taxa, bacterial proteins, or bacterial functions to individually predict the BV-associated host mTOR score. Models underwent 10-fold cross validation during construction to infer the latent variable coefficients and number. We visualized the projection scores of patient samples on the first two latent variables and extracted the loading coefficients of metabolites, microbial taxa, bacterial proteins, and bacterial functions predictive of host mTOR score. The predictive power of these features on the mTOR score, in the models overall and on individual latent variables, was inferred by variable importance of projection (VIP) analysis of the feature loadings coefficients. In brief, VIP scores calculate the relative importance of a feature for predicting the outcome variable by averaging the weighted score of the feature by the percent variance explained by latent variables across the model using the following equation:
| (Equation 1) |
where m is the number of features, i is the index of the LV, L is the total number of model LVs, wij is the weight coefficient of feature j on latent variable i and is the variance in Y (mTOR score) explained by the LV i. By normalization, the average value of VIPs across the model is 1, therefore, features that have VIP scores >1 are considered significantly contributing to the model prediction relative to all other features in the model.
Multi-‘omic meta-model
Direct merging of raw multi-‘omics data by constructing a single unified matrix fails to account for differences in data distributions, scales, and measurement technologies. Therefore, we combined our metabolomics, bacterial taxa, and bacterial protein data in a meta-model comprised of single-omic LVs that maintained within-data type properties while allowing causal prediction of the host mTOR response. This took the form of a generalized linear model with mTOR score as the Y variable and the patient scores on single-omic LV’s as the predictors X. The first two LVs from each single-omic PLSR model were extracted and used to define the model main effects and then interaction terms were constructed for each pair of LVs from a different molecular data type. In total, each meta-model contained 19 terms, an intercept (1-term), single-omic LV main effects (6-terms) and multi-‘omic LV interaction effects (12-terms). Models were interpreted by examining the coefficient values and p values on the regression terms to identify LV’s significantly predictive of mTOR score. VIP scores were used to identify significant features on predictive LVs by adapting Equation 1 for a single latent variable.
Acknowledgments
Study medication in the Partners PrEP Study was donated by Gilead Sciences. This work would not have been possible without the study participants for the Partner’s PrEP study, and we would like to thank them for their participation. This work was supported by the Canadian Institutes of Health Research Team grant (A.D.B.: HB3-164066), the National Institutes of Health (J.L.: MEHP R01 Al 111738), the Bill and Melinda Gates Foundation Partners PrEP study (J.L.: 47674), the Army Institute for Collaborative Biotechnologies UARC contract (D.A.L.: W911NF-19-D-0001), and Cooperative Agreement from the Army Research Office (D.A.L.: W911NF-19-2-0026).
Author contributions
Conceptualization, A.D.B., J.L., and F.H.; methodology, A.D.B., J.L., F.H., A.R.B., and D.K.B.; software, D.K.B., L.N.-R., S.K., A.R.B., A.D.B., and D.A.L.; investigation, A.R.B., K.B., A.L., R.D.M., X.H., E.I., N.M., T.R.M., E.K., S.M.H., C.L., J.M.B., C.C., and F.L.C.; validation, A.R.B. and M.P.; resources, A.R.B., A.L., and M.P.; data curation, L.N.-R., A.R.B., S.K., and D.K.B.; visualization, A.R.B., D.K.B., and S.K.; writing – original draft, A.R.B.; writing – review & editing, D.K.B., A.D.B., J.M.B., C.C., and A.R.B.; supervision, A.D.B., D.A.L., J.L., and F.H.; funding acquisition, A.D.B. and J.L.
Declaration of interests
J.M.B. is an employee of Gilead Sciences, outside of the submitted work.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 6, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112474.
Supplemental information
Clinical, demographic, and behavioral epidemiological information for both participant groups (n = 315, n = 90 for participant groups 1 and 2, respectively). Comparisons were made between women who had either a Lactobacillus-dominant or a non-Lactobacillus-dominant vaginal microbiome. p-value statistical results and tests performed are indicated in the table.
Data has been normalized to 1, and proportions are indicated in the table. See Figure 1 for bar graph of these data.
Expression values for Lactobacillus-dominant (LD, yellow) and non-Lactobacillus-dominant (nLD, purple) microbiome groups. t test p values, FDR values, and average log2 fold change (FC) for protein comparison between LD and nLD groups.
Expression values for Lactobacillus-dominant (LD, yellow) and non-Lactobacillus-dominant (nLD, purple) microbiome groups. t test p values, FDR values, and average log2 fold change (FC) for protein comparison between LD and nLD groups.
Expression values for Lactobacillus-dominant (LD, yellow) and non-Lactobacillus-dominant (nLD, purple) microbiome groups. t test p values, FDR values, and average log2 fold change (FC) for protein comparison between LD and nLD groups.
VIP scores calculate the relative importance of a feature for predicting the outcome variable by averaging the weighted score of the feature by the percent variance explained by latent variables across the model, where average VIP values across the model are 1. Therefore, features that have VIP scores >1 are considered significantly contributing to the model prediction relative to all other features in the model.
Data and code availability
All data has been supplies in Supplementary tables including corresponding Uniprot accession numbers (proteomics), ProbeIDs (transcriptomics) and Gene names. Transcriptomics data has been uploaded to GEO with accession number GSE139655. Metabolomics data has been uploaded to MetaboLights with accession number MTBLS7087. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE102 partner repository with the accession numbers PXD040895 (participant group 1) and PXD040280 (participant group 2).
All original code has been deposited at Zenodo (10.5281/zenodo.7810345) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ravel J., Gajer P., Abdo Z., Schneider G.M., Koenig S.S.K., McCulle S.L., Karlebach S., Gorle R., Russell J., Tacket C.O., et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA. 2011;108:4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tachedjian G., Aldunate M., Bradshaw C.S., Cone R.A. The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 2017;168:782–792. doi: 10.1016/j.resmic.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Anahtar M.N., Byrne E.H., Doherty K.E., Bowman B.A., Yamamoto H.S., Soumillon M., Padavattan N., Ismail N., Moodley A., Sabatini M.E., et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. 2015;42:965–976. doi: 10.1016/j.immuni.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anahtar M.N., Gootenberg D.B., Mitchell C.M., Kwon D.S. Cervicovaginal microbiota and reproductive health: the virtue of simplicity. Cell Host Microbe. 2018;23:159–168. doi: 10.1016/j.chom.2018.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Aldunate M., Srbinovski D., Hearps A.C., Latham C.F., Ramsland P.A., Gugasyan R., Cone R.A., Tachedjian G. Antimicrobial and immune modulatory effects of lactic acid and short chain fatty acids produced by vaginal microbiota associated with eubiosis and bacterial vaginosis. Front. Physiol. 2015;6:164. doi: 10.3389/fphys.2015.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strus M., Brzychczy-Włoch M., Gosiewski T., Kochan P., Heczko P.B. The in vitro effect of hydrogen peroxide on vaginal microbial communities. FEMS Immunol. Med. Microbiol. 2006;48:56–63. doi: 10.1111/j.1574-695X.2006.00120.x. [DOI] [PubMed] [Google Scholar]
- 7.Kenyon C.R., Osbak K. Recent progress in understanding the epidemiology of bacterial vaginosis. Curr. Opin. Obstet. Gynecol. 2014;26:448–454. doi: 10.1097/GCO.0000000000000112. [DOI] [PubMed] [Google Scholar]
- 8.Ling Z., Kong J., Liu F., Zhu H., Chen X., Wang Y., Li L., Nelson K.E., Xia Y., Xiang C. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genom. 2010;11:488. doi: 10.1186/1471-2164-11-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coudray M.S., Madhivanan P. Bacterial vaginosis-A brief synopsis of the literature. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020;245:143–148. doi: 10.1016/j.ejogrb.2019.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng L., Norenhag J., Hu Y.O.O., Brusselaers N., Fransson E., Ährlund-Richter A., Guðnadóttir U., Angelidou P., Zha Y., Hamsten M., et al. Vaginal microbiota and human papillomavirus infection among young Swedish women. NPJ Biofilms Microbiomes. 2020;6:39. doi: 10.1038/s41522-020-00146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKinnon L.R., Achilles S.L., Bradshaw C.S., Burgener A., Crucitti T., Fredricks D.N., Jaspan H.B., Kaul R., Kaushic C., Klatt N., et al. The evolving facets of bacterial vaginosis: implications for HIV transmission. AIDS Res. Hum. Retroviruses. 2019;35:219–228. doi: 10.1089/AID.2018.0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paladine H.L., Desai U.A. Vaginitis: diagnosis and treatment. Am. Fam. Physician. 2018;97:321–329. [PubMed] [Google Scholar]
- 13.Allsworth J.E., Peipert J.F. Severity of bacterial vaginosis and the risk of sexually transmitted infection. Am. J. Obstet. Gynecol. 2011;205:113–113.e6. doi: 10.1016/j.ajog.2011.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atashili J., Poole C., Ndumbe P.M., Adimora A.A., Smith J.S. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS. 2008;22:1493–1501. doi: 10.1097/QAD.0b013e3283021a37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torrone E.A., Morrison C.S., Chen P.L., Kwok C., Francis S.C., Hayes R.J., Looker K.J., McCormack S., McGrath N., van de Wijgert J.H.H.M., et al. Prevalence of sexually transmitted infections and bacterial vaginosis among women in sub-Saharan Africa: an individual participant data meta-analysis of 18 HIV prevention studies. PLoS Med. 2018;15 doi: 10.1371/journal.pmed.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koedooder R., Singer M., Schoenmakers S., Savelkoul P.H.M., Morré S.A., de Jonge J.D., Poort L., Cuypers W.J.S.S., Beckers N.G.M., Broekmans F.J.M., et al. The vaginal microbiome as a predictor for outcome of in vitro fertilization with or without intracytoplasmic sperm injection: a prospective study. Hum. Reprod. 2019;34:1042–1054. doi: 10.1093/humrep/dez065. [DOI] [PubMed] [Google Scholar]
- 17.Kroon S.J., Ravel J., Huston W.M. Cervicovaginal microbiota, women's health, and reproductive outcomes. Fertil. Steril. 2018;110:327–336. doi: 10.1016/j.fertnstert.2018.06.036. [DOI] [PubMed] [Google Scholar]
- 18.Bagnall P., Rizzolo D. Bacterial vaginosis: a practical review. JAAPA. 2017;30:15–21. doi: 10.1097/01.JAA.0000526770.60197. [DOI] [PubMed] [Google Scholar]
- 19.Koumans E.H., Sternberg M., Bruce C., McQuillan G., Kendrick J., Sutton M., Markowitz L.E. The prevalence of bacterial vaginosis in the United States, 2001-2004; associations with symptoms, sexual behaviors, and reproductive health. Sex. Transm. Dis. 2007;34:864–869. doi: 10.1097/OLQ.0b013e318074e565. [DOI] [PubMed] [Google Scholar]
- 20.Bradshaw C.S., Morton A.N., Hocking J., Garland S.M., Morris M.B., Moss L.M., Horvath L.B., Kuzevska I., Fairley C.K. High recurrence rates of bacterial vaginosis over the course of 12 months after oral metronidazole therapy and factors associated with recurrence. J. Infect. Dis. 2006;193:1478–1486. doi: 10.1086/503780. [DOI] [PubMed] [Google Scholar]
- 21.Sobel J.D., Kaur N., Woznicki N.A., Boikov D., Aguin T., Gill G., Akins R.A. Conventional oral and secondary high dose vaginal metronidazole therapy for recurrent bacterial vaginosis: clinical outcomes, impacts of sex and menses. Infect. Drug Resist. 2019;12:2297–2307. doi: 10.2147/IDR.S213853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchell C., Marrazzo J. Bacterial vaginosis and the cervicovaginal immune response. Am. J. Reprod. Immunol. 2014;71:555–563. doi: 10.1111/aji.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thurman A.R., Doncel G.F. Innate immunity and inflammatory response to Trichomonas vaginalis and bacterial vaginosis: relationship to HIV acquisition. Am. J. Reprod. Immunol. 2011;65:89–98. doi: 10.1111/j.1600-0897.2010.00902.x. [DOI] [PubMed] [Google Scholar]
- 24.Zevin A.S., Xie I.Y., Birse K., Arnold K., Romas L., Westmacott G., Novak R.M., McCorrister S., McKinnon L.R., Cohen C.R., et al. Microbiome composition and function drives wound-healing impairment in the female genital tract. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirmonsef P., Zariffard M.R., Gilbert D., Makinde H., Landay A.L., Spear G.T. Short-chain fatty acids induce pro-inflammatory cytokine production alone and in combination with toll-like receptor ligands. Am. J. Reprod. Immunol. 2012;67:391–400. doi: 10.1111/j.1600-0897.2011.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrne E.H., Farcasanu M., Bloom S.M., Xulu N., Xu J., Hykes B.L., Mafunda N.A., Hayward M.R., Dong M., Dong K.L., et al. Antigen presenting cells link the female genital tract microbiome to mucosal inflammation, with hormonal contraception as an additional modulator of inflammatory signatures. Front. Cell. Infect. Microbiol. 2021;11 doi: 10.3389/fcimb.2021.733619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doerflinger S.Y., Throop A.L., Herbst-Kralovetz M.M. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J. Infect. Dis. 2014;209:1989–1999. doi: 10.1093/infdis/jiu004. [DOI] [PubMed] [Google Scholar]
- 28.Bradley F., Birse K., Hasselrot K., Noël-Romas L., Introini A., Wefer H., Seifert M., Engstrand L., Tjernlund A., Broliden K., Burgener A.D. The vaginal microbiome amplifies sex hormone-associated cyclic changes in cervicovaginal inflammation and epithelial barrier disruption. Am. J. Reprod. Immunol. 2018;80 doi: 10.1111/aji.12863. [DOI] [PubMed] [Google Scholar]
- 29.Ceccarani C., Foschi C., Parolin C., D'Antuono A., Gaspari V., Consolandi C., Laghi L., Camboni T., Vitali B., Severgnini M., Marangoni A. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-50410-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vitali B., Cruciani F., Picone G., Parolin C., Donders G., Laghi L. Vaginal microbiome and metabolome highlight specific signatures of bacterial vaginosis. Eur. J. Clin. Microbiol. Infect. Dis. 2015;34:2367–2376. doi: 10.1007/s10096-015-2490-y. [DOI] [PubMed] [Google Scholar]
- 31.Łaniewski P., Herbst-Kralovetz M.M. Bacterial vaginosis and health-associated bacteria modulate the immunometabolic landscape in 3D model of human cervix. NPJ Biofilms Microbiomes. 2021;7:88. doi: 10.1038/s41522-021-00259-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heffron R., McClelland R.S., Balkus J.E., Celum C., Cohen C.R., Mugo N., Bukusi E., Donnell D., Lingappa J., Kiarie J., et al. Efficacy of oral pre-exposure prophylaxis (PrEP) for HIV among women with abnormal vaginal microbiota: a post-hoc analysis of the randomised, placebo-controlled Partners PrEP Study. Lancet. HIV. 2017;4:e449–e456. doi: 10.1016/S2352-3018(17)30110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., Imamichi T., Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu P., Ge M., Hu J., Li X., Che L., Sun K., Cheng L., Huang Y., Pilo M.G., Cigliano A., et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology. 2017;66:167–181. doi: 10.1002/hep.29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pourdehnad M., Truitt M.L., Siddiqi I.N., Ducker G.S., Shokat K.M., Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michaloglou C., Crafter C., Siersbaek R., Delpuech O., Curwen J.O., Carnevalli L.S., Staniszewska A.D., Polanska U.M., Cheraghchi-Bashi A., Lawson M., et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol. Cancer Ther. 2018;17:908–920. doi: 10.1158/1535-7163.MCT-17-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu G.Y., Sabatini D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao Z., Zhang W. Role of mTOR in glucose and lipid metabolism. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19072043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su X., Yu Y., Zhong Y., Giannopoulou E.G., Hu X., Liu H., Cross J.R., Rätsch G., Rice C.M., Ivashkiv L.B. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dan H.C., Cooper M.J., Cogswell P.C., Duncan J.A., Ting J.P.Y., Baldwin A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hess C., Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity. 2016;45:240–254. doi: 10.1016/j.immuni.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noureldein M.H., Eid A.A. Gut microbiota and mTOR signaling: insight on a new pathophysiological interaction. Microb. Pathog. 2018;118:98–104. doi: 10.1016/j.micpath.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 44.Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 45.Weichhart T., Hengstschläger M., Linke M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015;15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribeiro M.C., Peruchetti D.B., Silva L.S., Silva-Filho J.L., Souza M.C., Henriques M.D.G., Caruso-Neves C., Pinheiro A.A.S. LPS induces mTORC1 and mTORC2 activation during monocyte adhesion. Front. Mol. Biosci. 2018;5:67. doi: 10.3389/fmolb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu D.Y., Gorman M.J., Yuan D., Yu J., Mercado N.B., McMahan K., Borducchi E.N., Lifton M., Liu J., Nampanya F., et al. Defining the determinants of protection against SARS-CoV-2 infection and viral control in a dose-down Ad26.CoV2.S vaccine study in nonhuman primates. PLoS Biol. 2022;20 doi: 10.1371/journal.pbio.3001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson D.J., Marathe J., Pudney J. The structure of the human vaginal stratum corneum and its role in immune defense. Am. J. Reprod. Immunol. 2014;71:618–623. doi: 10.1111/aji.12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blaskewicz C.D., Pudney J., Anderson D.J. Structure and function of intercellular junctions in human cervical and vaginal mucosal epithelia. Biol. Reprod. 2011;85:97–104. doi: 10.1095/biolreprod.110.090423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berard A.R., Perner M., Mutch S., Farr Zuend C., McQueen P., Burgener A.D. Understanding mucosal and microbial functionality of the female reproductive tract by metaproteomics: implications for HIV transmission. Am. J. Reprod. Immunol. 2018;80 doi: 10.1111/aji.12977. [DOI] [PubMed] [Google Scholar]
- 51.Gali Y., Ariën K.K., Praet M., Van den Bergh R., Temmerman M., Delezay O., Vanham G. Development of an in vitro dual-chamber model of the female genital tract as a screening tool for epithelial toxicity. J. Virol. Methods. 2010;165:186–197. doi: 10.1016/j.jviromet.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Mok K.W., Mruk D.D., Lee W.M., Cheng C.Y. Rictor/mTORC2 regulates blood-testis barrier dynamics via its effects on gap junction communications and actin filament network. Faseb. J. 2013;27:1137–1152. doi: 10.1096/fj.12-212977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sato T., Ishii J., Ota Y., Sasaki E., Shibagaki Y., Hattori S. Mammalian target of rapamycin (mTOR) complex 2 regulates filamin A-dependent focal adhesion dynamics and cell migration. Gene Cell. 2016;21:579–593. doi: 10.1111/gtc.12366. [DOI] [PubMed] [Google Scholar]
- 54.Gordon B.S., Kazi A.A., Coleman C.S., Dennis M.D., Chau V., Jefferson L.S., Kimball S.R. RhoA modulates signaling through the mechanistic target of rapamycin complex 1 (mTORC1) in mammalian cells. Cell. Signal. 2014;26:461–467. doi: 10.1016/j.cellsig.2013.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang Y., Park I.H., Wu A.L., Du G., Huang P., Frohman M.A., Walker S.J., Brown H.A., Chen J. PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr. Biol. 2003;13:2037–2044. doi: 10.1016/j.cub.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 56.Dibble C.C., Cantley L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee Y., Dizzell S.E., Leung V., Nazli A., Zahoor M.A., Fichorova R.N., Kaushic C. Effects of female sex hormones on susceptibility to HSV-2 in vaginal cells grown in air-liquid interface. Viruses. 2016;8 doi: 10.3390/v8090241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeste J., Illa X., Alvarez M., Villa R. Engineering and monitoring cellular barrier models. J. Biol. Eng. 2018;12:18. doi: 10.1186/s13036-018-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinotti S., Ranzato E. Scratch wound healing assay. Methods Mol. Biol. 2020;2109:225–229. doi: 10.1007/7651_2019_259. [DOI] [PubMed] [Google Scholar]
- 60.Koh A., Molinaro A., Ståhlman M., Khan M.T., Schmidt C., Mannerås-Holm L., Wu H., Carreras A., Jeong H., Olofsson L.E., et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175:947–961.e17. doi: 10.1016/j.cell.2018.09.055. [DOI] [PubMed] [Google Scholar]
- 61.Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mousavizadeh R., Hojabrpour P., Eltit F., McDonald P.C., Dedhar S., McCormack R.G., Duronio V., Jafarnejad S.M., Scott A. β1 integrin, ILK and mTOR regulate collagen synthesis in mechanically loaded tendon cells. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-69267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cork G.K., Thompson J., Slawson C. Real talk: the inter-play between the mTOR, AMPK, and hexosamine biosynthetic pathways in cell signaling. Front. Endocrinol. 2018;9:522. doi: 10.3389/fendo.2018.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farkas S., Hornung M., Sattler C., Guba M., Steinbauer M., Anthuber M., Herfarth H., Schlitt H.J., Geissler E.K. Rapamycin decreases leukocyte migration in vivo and effectively reduces experimentally induced chronic colitis. Int. J. Colorectal Dis. 2006;21:747–753. doi: 10.1007/s00384-005-0793-7. [DOI] [PubMed] [Google Scholar]
- 65.Ravel J., Moreno I., Simón C. Bacterial vaginosis and its association with infertility, endometritis, and pelvic inflammatory disease. Am. J. Obstet. Gynecol. 2021;224:251–257. doi: 10.1016/j.ajog.2020.10.019. [DOI] [PubMed] [Google Scholar]
- 66.Mtshali A., San J.E., Osman F., Garrett N., Balle C., Giandhari J., Onywera H., Mngomezulu K., Mzobe G., de Oliveira T., et al. Temporal changes in vaginal microbiota and genital tract cytokines among South African women treated for bacterial vaginosis. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.730986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abdel-Nour M., Tsalikis J., Kleinman D., Girardin S.E. The emerging role of mTOR signalling in antibacterial immunity. Immunol. Cell Biol. 2014;92:346–353. doi: 10.1038/icb.2014.3. [DOI] [PubMed] [Google Scholar]
- 68.Luo L., Wall A.A., Yeo J.C., Condon N.D., Norwood S.J., Schoenwaelder S., Chen K.W., Jackson S., Jenkins B.J., Hartland E.L., et al. Rab8a interacts directly with PI3Kgamma to modulate TLR4-driven PI3K and mTOR signalling. Nat. Commun. 2014;5:4407. doi: 10.1038/ncomms5407. [DOI] [PubMed] [Google Scholar]
- 69.Hu Y., Lou J., Mao Y.Y., Lai T.W., Liu L.Y., Zhu C., Zhang C., Liu J., Li Y.Y., Zhang F., et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy. 2016;12:2286–2299. doi: 10.1080/15548627.2016.1230584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang X., Howell G.M., Guo L., Collage R.D., Loughran P.A., Zuckerbraun B.S., Rosengart M.R. CaMKIV-dependent preservation of mTOR expression is required for autophagy during lipopolysaccharide-induced inflammation and acute kidney injury. J. Immunol. 2014;193:2405–2415. doi: 10.4049/jimmunol.1302798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hwang J.W., Baek Y.M., Yang K.E., Yoo H.S., Cho C.K., Lee Y.W., Park J., Eom C.Y., Lee Z.W., Choi J.S., Jang I.S. Lactobacillus casei extract induces apoptosis in gastric cancer by inhibiting NF-kappaB and mTOR-mediated signaling. Integr. Cancer Ther. 2013;12:165–173. doi: 10.1177/1534735412442380. [DOI] [PubMed] [Google Scholar]
- 72.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonvini A., Coqueiro A.Y., Tirapegui J., Calder P.C., Rogero M.M. Immunomodulatory role of branched-chain amino acids. Nutr. Rev. 2018;76:840–856. doi: 10.1093/nutrit/nuy037. [DOI] [PubMed] [Google Scholar]
- 74.Laplante M., Sabatini D.M. mTOR signaling at a glance. J. Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shimobayashi M., Hall M.N. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014;15:155–162. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 76.Araki K., Turner A.P., Shaffer V.O., Gangappa S., Keller S.A., Bachmann M.F., Larsen C.P., Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeng H., Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr. Opin. Immunol. 2017;46:103–111. doi: 10.1016/j.coi.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iwata T.N., Ramírez-Komo J.A., Park H., Iritani B.M. Control of B lymphocyte development and functions by the mTOR signaling pathways. Cytokine Growth Factor Rev. 2017;35:47–62. doi: 10.1016/j.cytogfr.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Linke M., Fritsch S.D., Sukhbaatar N., Hengstschläger M., Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017;591:3089–3103. doi: 10.1002/1873-3468.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sukhbaatar N., Hengstschläger M., Weichhart T. mTOR-mediated regulation of dendritic cell differentiation and function. Trends Immunol. 2016;37:778–789. doi: 10.1016/j.it.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Araki K., Ellebedy A.H., Ahmed R. TOR in the immune system. Curr. Opin. Cell Biol. 2011;23:707–715. doi: 10.1016/j.ceb.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gulhati P., Bowen K.A., Liu J., Stevens P.D., Rychahou P.G., Chen M., Lee E.Y., Weiss H.L., O'Connor K.L., Gao T., Evers B.M. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011;71:3246–3256. doi: 10.1158/0008-5472.CAN-10-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ata R., Antonescu C.N. Integrins and cell metabolism: an intimate relationship impacting cancer. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou K., Muroyama A., Underwood J., Leylek R., Ray S., Soderling S.H., Lechler T. Actin-related protein2/3 complex regulates tight junctions and terminal differentiation to promote epidermal barrier formation. Proc. Natl. Acad. Sci. USA. 2013;110:E3820–E3829. doi: 10.1073/pnas.1308419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.DeMali K.A., Barlow C.A., Burridge K. Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J. Cell Biol. 2002;159:881–891. doi: 10.1083/jcb.200206043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ray J.P., Staron M.M., Shyer J.A., Ho P.C., Marshall H.D., Gray S.M., Laidlaw B.J., Araki K., Ahmed R., Kaech S.M., Craft J. The interleukin-2-mTORc1 kinase Axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity. 2015;43:690–702. doi: 10.1016/j.immuni.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou J.X., Fan L.X., Li X., Calvet J.P., Li X. TNFα signaling regulates cystic epithelial cell proliferation through Akt/mTOR and ERK/MAPK/Cdk2 mediated Id2 signaling. PLoS One. 2015;10 doi: 10.1371/journal.pone.0131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huynh L., Kusnadi A., Park S.H., Murata K., Park-Min K.H., Ivashkiv L.B. Opposing regulation of the late phase TNF response by mTORC1-IL-10 signaling and hypoxia in human macrophages. Sci. Rep. 2016;6 doi: 10.1038/srep31959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Borgdorff H., Gautam R., Armstrong S.D., Xia D., Ndayisaba G.F., van Teijlingen N.H., Geijtenbeek T.B.H., Wastling J.M., van de Wijgert J.H.H.M. Cervicovaginal microbiome dysbiosis is associated with proteome changes related to alterations of the cervicovaginal mucosal barrier. Mucosal Immunol. 2016;9:621–633. doi: 10.1038/mi.2015.86. [DOI] [PubMed] [Google Scholar]
- 90.Plesniarski A., Siddik A.B., Su R.C. The microbiome as a key regulator of female genital tract barrier function. Front. Cell. Infect. Microbiol. 2021;11 doi: 10.3389/fcimb.2021.790627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burgener A., McGowan I., Klatt N.R. HIV and mucosal barrier interactions: consequences for transmission and pathogenesis. Curr. Opin. Immunol. 2015;36:22–30. doi: 10.1016/j.coi.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 92.Becker S., Oelschlaeger T.A., Wullaert A., Vlantis K., Pasparakis M., Wehkamp J., Stange E.F., Gersemann M. Bacteria regulate intestinal epithelial cell differentiation factors both in vitro and in vivo. PLoS One. 2013;8 doi: 10.1371/journal.pone.0055620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ikenoue T., Inoki K., Yang Q., Zhou X., Guan K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ren C., Zhang Q., de Haan B.J., Faas M.M., Zhang H., de Vos P. Protective effects of lactic acid bacteria on gut epithelial barrier dysfunction are Toll like receptor 2 and protein kinase C dependent. Food Funct. 2020;11:1230–1234. doi: 10.1039/c9fo02933h. [DOI] [PubMed] [Google Scholar]
- 96.Parada Venegas D., De la Fuente M.K., Landskron G., González M.J., Quera R., Dijkstra G., Harmsen H.J.M., Faber K.N., Hermoso M.A. Short chain fatty acids (SCFAs)-Mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019;10:277. doi: 10.3389/fimmu.2019.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hummelen R., Fernandes A.D., Macklaim J.M., Dickson R.J., Changalucha J., Gloor G.B., Reid G. Deep sequencing of the vaginal microbiota of women with HIV. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Borgdorff H., Tsivtsivadze E., Verhelst R., Marzorati M., Jurriaans S., Ndayisaba G.F., Schuren F.H., van de Wijgert J.H.H.M. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 2014;8:1781–1793. doi: 10.1038/ismej.2014.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Albert A.Y.K., Chaban B., Wagner E.C., Schellenberg J.J., Links M.G., van Schalkwyk J., Reid G., Hemmingsen S.M., Hill J.E., Money D., VOGUE Research Group A study of the vaginal microbiome in healthy Canadian women utilizing cpn60-based molecular profiling reveals distinct Gardnerella subgroup community state types. PLoS One. 2015;10 doi: 10.1371/journal.pone.0135620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jespers V., Crucitti T., Menten J., Verhelst R., Mwaura M., Mandaliya K., Ndayisaba G.F., Delany-Moretlwe S., Verstraelen H., Hardy L., et al. Prevalence and correlates of bacterial vaginosis in different sub-populations of women in sub-Saharan Africa: a cross-sectional study. PLoS One. 2014;9 doi: 10.1371/journal.pone.0109670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Noël-Romas L., Perner M., Molatlhegi R., Farr Zuend C., Mabhula A., Hoger S., Lamont A., Birse K., Berard A., McCorrister S., et al. Vaginal microbiome-hormonal contraceptive interactions associate with the mucosal proteome and HIV acquisition. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1009097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baeten J.M., Donnell D., Ndase P., Mugo N.R., Campbell J.D., Wangisi J., Tappero J.W., Bukusi E.A., Cohen C.R., Katabira E., et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N. Engl. J. Med. 2012;367:399–410. doi: 10.1056/NEJMoa1108524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Birse K.D., Romas L.M., Guthrie B.L., Nilsson P., Bosire R., Kiarie J., Farquhar C., Broliden K., Burgener A.D. Genital injury signatures and microbiome alterations associated with depot medroxyprogesterone acetate usage and intravaginal drying practices. J. Infect. Dis. 2017;215:590–598. doi: 10.1093/infdis/jiw590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gaujoux R., Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinf. 2010;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Klatt N.R., Cheu R., Birse K., Zevin A.S., Perner M., Noël-Romas L., Grobler A., Westmacott G., Xie I.Y., Butler J., et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science. 2017;356:938–945. doi: 10.1126/science.aai9383. [DOI] [PubMed] [Google Scholar]
- 107.Castro J., Jefferson K.K., Cerca N. Genetic heterogeneity and taxonomic diversity among Gardnerella species. Trends Microbiol. 2020;28:202–211. doi: 10.1016/j.tim.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 108.Srinivasan S., Morgan M.T., Fiedler T.L., Djukovic D., Hoffman N.G., Raftery D., Marrazzo J.M., Fredricks D.N. Metabolic signatures of bacterial vaginosis. mBio. 2015;6 doi: 10.1128/mBio.00204-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hughes S.M., Levy C.N., Calienes F.L., Stekler J.D., Pandey U., Vojtech L., Berard A.R., Birse K., Noël-Romas L., Richardson B., et al. Treatment with commonly used antiretroviral drugs induces a type I/III interferon signature in the gut in the absence of HIV infection. Cell Rep. Med. 2020;1 doi: 10.1016/j.xcrm.2020.100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Srinivasan B., Kolli A.R., Esch M.B., Abaci H.E., Shuler M.L., Hickman J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015;20:107–126. doi: 10.1177/2211068214561025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ariën K.K., Vanham G., Gali Y. A dual-chamber model of the female genital tract to evaluate epithelial toxicity of candidate anti-HIV microbicides. Curr. Protoc. Cell Biol. 2011 doi: 10.1002/0471143030.cb2613s52. Chapter 26, Unit26 13. [DOI] [PubMed] [Google Scholar]
- 112.Leek J.T., Storey J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical, demographic, and behavioral epidemiological information for both participant groups (n = 315, n = 90 for participant groups 1 and 2, respectively). Comparisons were made between women who had either a Lactobacillus-dominant or a non-Lactobacillus-dominant vaginal microbiome. p-value statistical results and tests performed are indicated in the table.
Data has been normalized to 1, and proportions are indicated in the table. See Figure 1 for bar graph of these data.
Expression values for Lactobacillus-dominant (LD, yellow) and non-Lactobacillus-dominant (nLD, purple) microbiome groups. t test p values, FDR values, and average log2 fold change (FC) for protein comparison between LD and nLD groups.
Expression values for Lactobacillus-dominant (LD, yellow) and non-Lactobacillus-dominant (nLD, purple) microbiome groups. t test p values, FDR values, and average log2 fold change (FC) for protein comparison between LD and nLD groups.
Expression values for Lactobacillus-dominant (LD, yellow) and non-Lactobacillus-dominant (nLD, purple) microbiome groups. t test p values, FDR values, and average log2 fold change (FC) for protein comparison between LD and nLD groups.
VIP scores calculate the relative importance of a feature for predicting the outcome variable by averaging the weighted score of the feature by the percent variance explained by latent variables across the model, where average VIP values across the model are 1. Therefore, features that have VIP scores >1 are considered significantly contributing to the model prediction relative to all other features in the model.
Data Availability Statement
All data has been supplies in Supplementary tables including corresponding Uniprot accession numbers (proteomics), ProbeIDs (transcriptomics) and Gene names. Transcriptomics data has been uploaded to GEO with accession number GSE139655. Metabolomics data has been uploaded to MetaboLights with accession number MTBLS7087. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE102 partner repository with the accession numbers PXD040895 (participant group 1) and PXD040280 (participant group 2).
All original code has been deposited at Zenodo (10.5281/zenodo.7810345) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.