

Abstract
The current Medicaid system is ill equipped to handle the anticipated approvals of new gene and cell therapy products. These advanced therapies tend to be single-dose, potentially durable options for a variety of indications spanning oncology, rare disease, and more. The up-front cost of these therapies contrasts with chronic care treatment, which may incur cost over the life of a patient. The cost of these innovative treatments, along with the anticipated larger patient pools, can limit patient access as Medicaid programs operate on limited or fixed budgets. Given the value of these therapies for diseases that may have large Medicaid populations, the system will need to grapple with the existing barriers to access to ensure equitable patient care. This review focuses on one such barrier, discrepancies between product indications and state Medicaid and Medicaid Managed Care Organization coverage policies, and it proposes federal policy solutions to this barrier to better accommodate the exponential growth of the gene and cell therapy pipeline.
Keywords: Medicaid, payment policy, CMS, cell therapy, gene therapy, value-based payment, coverage to label, time to treatment, development pipeline, patient experience
Graphical abstract
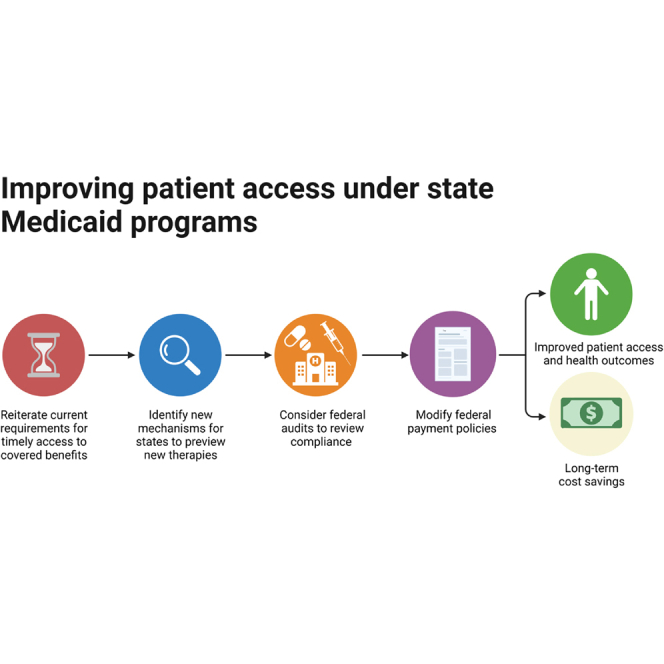
McCombs and colleagues believe improving equitable patient access to gene and cell therapies is critical for the Medicaid system, which faces challenges accommodating approvals of new products and their high, front-loaded costs. The review from McCombs and colleagues highlights the current barriers to access and proposes federal policy solutions to ensure access to this new class of therapeutics.
Introduction
Since the first gene and cell therapies were approved by the FDA in 2017, the Medicaid system has grappled with how to manage patient access to this new class of therapeutics. Gene and cell therapies are durable, potentially curative treatments for a variety of conditions spanning oncology, rare disease, and more. Because of the often-individualized manufacturing process and often single-dose administration, gene and cell therapies carry high front-loaded costs in exchange for long-term health benefits. For example, one-time treatment with ZOLGENSMA for spinal muscular atrophy has a wholesale acquisition cost of $2.125 million.1 This presents a paradigm for which the existing US healthcare system was never built to accommodate.
Oncological indications dominate the gene and cell therapy pipeline.2 However, many gene and cell treatments in development target patient populations with rare, inherited diseases, many of which do not have available treatment options. These conditions, such as hemophilia A and B and Duchenne muscular dystrophy, disproportionately affect children. Rare diseases affecting adult populations often severely impact individuals’ ability to complete education and participate or remain in the workforce. Thus, many rare disease patients, including those with disabilities, are enrolled in state Medicaid programs.3,4 New cell and gene therapy approvals are imminent for such conditions, including sickle cell disease. Sickle cell disease is associated with a high economic burden for state Medicaid programs in the United States, which can be $1.7 million over a lifetime,5 and it presents a health equity issue as the disease largely impacts communities of color. This is on top of the high economic and quality of life burden for patients and their families. To that end, individuals with sickle cell disease enrolled in Medicaid have been found to have significantly higher healthcare utilization and costs compared with individuals without sickle cell disease.6 These therapies offer hope and the possibility to meaningfully transform the lives of patients and their families. As the primary insurer for vulnerable populations, it is crucial for Medicaid to address coverage and reimbursement barriers to ensure timely patient access to these long-awaited innovative gene and cell therapies.
Since gene and cell therapies are potentially curative and intended for one-time administration, payment for the therapies exists as an up-front payment at time of administration. This differs from therapeutics used for symptom and disease management that are administered over a lifetime for conditions that have no disease-modifying alternative and are reimbursed following each administration. Therefore, gene therapies that hold a high price tag that in many cases will rival that current total lifetime care cost create a logistical and budgetary hurdle for state Medicaid programs, especially as the eligible populations for these treatments grow. State budgeting processes can occur sometimes 2 years out in advance of emerging FDA approvals, and state budgets must be balanced each year. This combined with other fiscal pressures—such as pandemic response, rising education costs, and infrastructure—make it especially difficult to accommodate these costs, even if the costs are recouped over a patient’s lifetime.
Federal law sets overarching requirements for state Medicaid programs, which mandate coverage of certain medical benefits. However, the bulk of the operational decisions are left at the discretion of each state, including enrollment eligibility, reimbursement methodology, and service coverage.7 Once approved by the Centers for Medicare and Medicaid Services (CMS), state Medicaid programs may draw down federal funds based on the federal medical assistance percentage (FMAP).8 Under the Medicaid Drug Rebate Program (MDRP),9 states that include prescription drug coverage in their Medicaid programs—which all states do—must cover all drugs approved by the Federal Drug Administration (with limited statutory exceptions) according to their “medically accepted indications,” and in return manufacturers provide rebates on their products to the states, which are then shared between the states and the federal government.10
Under Section 1927 of the Social Security Act (SSA),11 which authorizes the MDRP, the “medically accepted indication” is defined as “any (emphasis added) use of a covered outpatient drug which is approved under the Federal Food Drug And Cosmetic Act (FFDCA),12 or the use of which is supported by one or more citations included or approved for inclusion in any of the compendia described in section 1927(g) (1) (B) (i),” which include the American Hospital Formulary Service Drug Information, United States Pharmacopeia-Drug Information (or its successor publications), and the DRUGDEX Information System. This means the drugs administered under the MDRP, whether by states directly or by Medicaid Managed Care Organizations (MCOs) contracted by the states to administer and run their benefit, are required by federal law to be covered for FDA-approved “indications and usage”13,14 (referred to hereafter as the “labeled indication”).
Despite general coverage to the labeled indication being mandated by federal law, variations in state plans, policies, and practices have created anecdotal inconsistencies15,16,17 regarding how therapies are covered, which populations they cover, and how quickly coverage is approved for individual patients. This leads to patient access issues, as denials or delays in coverage lead to delays in treatment. These disparities and delays all culminate into an unsustainable public payor system that undermines Medicaid’s objectives to improve the care and health of its beneficiaries.
The goal of this article is to concretely identify gaps and discrepancies in Medicaid coverage of gene and cell therapies and to discuss federal policy options that can improve coverage to ensure equitable patient access in the Medicaid program. The authors have noted patient organizations and their companies frequently report issues obtaining comprehensive data on state coverage policies. Not every state posts its coverage policies publicly, and some states make decisions on a case-by-case basis. Thus, ASGCT conducted a study of 16 states and three large MCOs to assess the coverage policies for three gene and cell therapy products. An opinion piece18 was published using these data to provide a broad overview of the issues. In contrast, this review will elaborate on the coverage barriers identified and federal solutions proposed, looking at the data through a more policy-focused lens. The authors acknowledge that inadequate reimbursement may also hinder patient access but have focused this article on coverage considerations and solutions.
Results
Using the parameters for defining “coverage to label” in Tables 1 and 2, the assessment found that coverage varied between types of therapies, with overall fewer restrictions placed on KYMRIAH (Table 3; Figure 1).
Table 1.
FDA-approved indications and usage for KYMRIAH, LUXTURNA, and ZOLGENSMA
| KYMRIAH Labeled indication | LUXTURNA Labeled indication | ZOLGENSMA Labeled indication |
|---|---|---|
| KYMRIAH is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of patients up to 25 years of age with B cell precursor acute lymphoblastic leukemia (ALL) that is refractory or in second or later relapse. | LUXTURNA is an adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Patients must have viable retinal cells as determined by the treating physician(s). | ZOLGENSMA is an adeno-associated virus vector-based gene therapy indicated for the treatment of pediatric patients less than 2 years of age with spinal muscular atrophy (SMA) with biallelic mutations in the survival motor neuron 1 (SMN1) gene. |
| Adult patients with relapsed or refractory (r/r) large B cell lymphoma after two or more lines of systemic therapy, including diffuse large B cell lymphoma (DLBCL) not otherwise specified, high grade B cell lymphoma and DLBCL arising from follicular lymphoma. | Limitations of use: The safety and effectiveness of repeat administration of ZOLGENSMA have not been evaluated. The use of ZOLGENSMA in patients with advanced SMA (e.g., complete paralysis of limbs, permanent ventilator dependence) has not been evaluated. | |
| Limitations of use: KYMRIAH is not indicated for treatment of patients with primary central nervous system lymphoma. |
Table 2.
Summary of determining factors in the categorization of policies as in accordance with the labeled indication vs. not in accordance with the labeled indication
| Payor requirements determined to be | |
|---|---|
| In accordance with the labeled indication | Not in accordance with the labeled indication |
| confirmation of diagnosis or genetic mutation(s) for which the product is approved | age limitations narrower than the label (unless the payor does not cover such ages) |
| confirmation of physical attributes required for the product to work (e.g., confirmation of viable retinal cells to be transduced, tumor marker expression required for drug mechanism of action, etc.) | severity of condition thresholds (visual acuity, advanced disease, physical performance scores, expectation of outcomes) |
| requirements to monitor or assess physiological markers or functionality recommended in the label | limitations on use based on pregnancy or being of childbearing age, even if not recommended in pregnancy in section 8.1 of the FDA package insert |
| requirements to have failed previous lines of therapy consistent with the indications and usage section of the label | limiting use in populations not included in the clinical trial, even if the lack of data from such populations is noted in the indications and usage section of the label |
| limitations that match the contraindications included in the indications and usage section of the label | |
Table 3.
Summary of “to labeled indication” coverage determinations for 16 states and 3 MCOs
| KYMRIAH | LUXTURNA | ZOLGENSMA | |
|---|---|---|---|
| Arizona | N/A | N/A | N/A |
| Arkansas | N/A | N/A | N/A |
| California | to label | more restrictivea,d | more restrictiveb,d |
| Colorado | to label | N/A | more restrictiveb,d |
| Florida | to label | more restrictivea,d | more restrictivea,b,d |
| Georgia | N/A | N/A | N/A |
| Illinois | N/A | N/A | N/A |
| Indiana | N/A | more restrictivea | more restrictivea,b,d |
| Massachusetts | to label | to label | more restrictiveb |
| Michigan | N/A | N/A | N/A |
| Mississippi | more restrictivec,d | more restrictivea,b | more restrictivea |
| New York | to label | to label | more restrictivea,b,d |
| North Carolina | to label | more restrictivea | more restrictiveb,d |
| Oklahoma | to label | more restrictivea,b,c | more restrictiveb |
| Oregon | more permissivee | more restrictiveb | more restrictiveb,d |
| Texas | to label | more restrictivea,b,d | more restrictiveb |
| United Healthcare (MCO Policy) | more restrictivec,d | to label | more restrictiveb |
| Anthem (MCO Policy) | more restrictiveb | to label | more restrictiveb |
| Centene (MCO Policy) | more restrictiveb | more restrictivea | more restrictiveb |
| Covered to, or beyond labeled indication | 9 | 4 | 0 |
| Total policies available | 13/19 | 13/19 | 14/19 |
N/A: no policy publicly available.
Age limitations are narrower than the label (unless the payor does not cover such ages).
Severity of condition thresholds (visual acuity, advanced disease, physical performance scores, expectation of outcomes).
Limitations on use based on pregnancy or being of childbearing age, even if not recommended in pregnancy in section 8.1 of the FDA package insert.
Limiting use in populations not included in the clinical trial, even if the lack of data from such populations is noted in the indications and usage section of the label.
Allowing compendia diagnoses.
Figure 1.

Summary of “to labeled indication” coverage determinations for 16 states and three
MCOs.
KYMRIAH
Published coverage policies assessed showed that nine payors covered KYMRIAH to its labeled indication. Four other payors placed greater restrictions on use of the product, two of which were for pregnancy, one of which required patients to pass performance scores, and one of which required documentation of lymphocyte counts. One state covered beyond the labeled indication and included compendia diagnoses.
LUXTURNA
Published coverage policies assessed found that two states and two MCOs covered the product to the approved labeled indication. Two states and one MCO limited coverage to ages ≥ 3 or 4 rather than the labeled ≥12 months. Five states limited use in the 65 and up population due to the lack of inclusion of this age group in the clinical trials leading to approval. Several states also required additional visual acuity measures not included in the labeled indication.
ZOLGENSMA
Published coverage policies assessed found that no states or MCOs covered the product to the approved labeled indication. Nine payors were determined to be more restrictive based on the exclusion of “advanced” or ventilator-dependent spinal muscular atrophy (SMA) alone. Others required minimum ages and performance scores not noted in the drug’s labeled indication.
Discussion
Our data show that state coverage policies vary widely between products, and for certain products, there are frequently additional exclusionary criteria and/or requests for additional clinical information or assessments beyond the labeled indication. These overly restrictive criteria can result in treatment denials or delays in time to treatment. Moreover, various discrepancies exist between states or MCOs in the coverage and reimbursement of specific products that can limit the ability of providers to treat Medicaid patients.
With over 3,600 gene and cell therapies in the development pipeline, Medicaid and state payors need to consider these issues given the existing disparities with less than 30 globally approved gene therapy products on the market today.2 Many of these therapies in development will have significantly larger overall Medicaid populations, as they aim to address diseases that disproportionately affect certain patients. The current patient access issues within the Medicaid system identified by our results will be exponentially compounded as more therapies transition toward FDA approval and as the Medicaid patient pool expands. This discussion illustrates the prevailing coverage issues, the upstream causes of states’ move to limit coverage, the downstream impacts of such coverage limits, and provides potential federal policy solutions to overcome access barriers.
State Medicaid programs face significant challenges accommodating the high up-front costs of cell and gene therapies
While Medicaid has a duty to provide care to the patients it serves, as a government-funded program, it also has a duty to maintain fiscal integrity to protect both patients and taxpayers. Medicaid programs must be accountable for the funds they receive, ensuring they are used effectively and efficiently with every dollar spent in accordance with federal and state law while supporting the interests of Medicaid patients. States must balance their budgets each year, and their Medicaid programs must operate within the available funding allocated to them by the state and federal government. This requires careful budgeting and cost management to ensure that funds are used to provide high-quality care to eligible patients in a fair and equitable manner, while minimizing waste and inefficiencies.19
Given this, one of the challenges for state Medicaid programs is determining which patients are eligible for high-cost therapies. Cell and gene therapies are often highly targeted to specific populations and clinical trials are smaller than predecessor biologics and small molecules given the large treatment effects. States may be reluctant to cover populations outside of the clinical trial criteria of age and disease progression where the greatest amount of data exist supporting the product’s effectiveness, even though genotyping can identify the targeted population.
In addition to determining eligibility, Medicaid programs must also consider the cost of providing ongoing care for patients who receive high-cost therapies. While these treatments can be highly effective and greatly reduce the overall economic burden associated with a disease, they can also require ongoing monitoring and follow-up care to ensure that patients continue to receive the full benefits of treatment.
Overall, state budgetary pressures are a reality that challenge patient access to cell and gene therapies with high up-front costs. Unnecessarily restricting patient access as a means to balance budgets and manage costs would undermine Medicaid’s responsibility to provide care that is in the best interest of its patients. Innovative solutions, such as value-based pricing models, may help to alleviate some of these financial pressures and ensure that Medicaid programs can continue to provide high-quality care to patients in need.
States and MCOs are not always adhering to the requirements to provide coverage for products to their “medically accepted” indication
A state’s determination of medical necessity does not substitute for the federal definition of medically accepted indication for coverage (expounded upon previously) nor the judgment of the treating physician who determines medical necessity. As seen in the labeled indication for LUXTURNA, “sufficient viable retina cells” are required for administration. Regulators left this term undefined and at the discretion of the treating physician. Payors that required “sufficient viable retina cells” for coverage were not considered more restrictive as outlined in Table 2. Although each state may have a different definition of medical necessity or apply its own parameters for medical necessity decisions for procedures in other medical areas, those parameters for drug products may not be more restrictive than the federal statute. Furthermore, federal regulations mandate that MCOs follow the same obligations applied to state Medicaid programs.20
SSA §1927 does not reference clinical trial criteria or other patient populations that are described in an FDA-approved drug label, information that is designed to inform prescribers about the development process and data supporting FDA’s decision-making.8 To demonstrate product benefit, clinical trial eligibility criteria may be much narrower than the population FDA approves for use in the indication. For instance, ZOLGENSMA’s labeled indication was broadened by FDA upon approval to include patients up to 2 years of age, despite this population not being represented in clinical trials.21 The FDA has wide scientific and legal latitude to establish the labeled indication based on “substantial evidence,” and it does not obligate a clinical trial population to match the patient population once approved.14 Gene and cell therapies frequently have small clinical trial populations by the nature of the rare diseases they aim to treat. In addition, by the nature of the therapies, large treatment effects that demonstrate clinical benefit may be observed in small clinical trial populations. There are no limitations of coverage defined within SSA §1927 that would justify restricting coverage to clinical trial populations for a therapy that is FDA approved and prescribed for use according to its medically accepted indication.
Unfortunately, our study suggests that state coverage policies, including those of MCOs, are not always aligned with the spirit of the law. Despite Medicaid patients meeting the “medically accepted indication” standard, authors have anecdotal experience with many of these patients receiving a denial of insurance approval. This denial can be accompanied by requests for additional information, likened to “eligibility criteria,” that is above and beyond simply requesting confirmation that a patient’s condition corresponds to the labeled indication of the commercial product.
Appeals processes for coverage are slow and burdensome and may delay or prohibit access for the patients most in need
In the case of initial Medicaid denials, SSA §1932(b) (4) requires Medicaid MCOs to establish internal grievance procedures under which Medicaid enrollees may challenge denial of coverage or payment for medical assistance.11,20 For Medicaid patients not enrolled under an MCO, state Medicaid programs are required to offer a hearing process, frequently called the “fair hearing” process, a path also available to MCO enrollees following the initial grievance decision.22 However, these processes can take months to resolve.
Beyond these appeals processes, litigation remains an option for beneficiaries, albeit a time-consuming and expensive one. Litigation processes are complex and therefore not generally a feasible option for patients and their families. For companies, litigation is not often a desirable avenue and is used as a last recourse to resolve compliance issues as it is contentious and can take years to reach a resolution. In addition, litigation initiated by product sponsors regarding coverage of an existing product can hamper the relationship with the state and limit the ability of the manufacturer and state to engage in productive discussions regarding additional products coming to market. Taken together, the lack of reasonable processes compounds the issues with time-to-treatment delays.
Even if Medicaid insurers ultimately approve the coverage of the therapy, their requests for additional clinical information increase the time to treatment, leading to negative clinical outcomes for patients that cannot be reversed.23 Delays to treatment can be as harmful as outright denial of coverage. Long wait times can lead to the progression of a patient’s condition to a point where treatment is no longer effective, and despite being eligible at the time the treatment is prescribed, they may develop comorbidities that make treatment more challenging, or they may “age out” of the labeled population.24 In some rapidly progressing conditions, a patient may die during this waiting period. Adverse impacts to patients’ clinical outcomes due to treatment delays may also end up costing Medicaid more money in the long run than if the therapy were promptly covered following FDA approval.25
Medicaid patients are not receiving timely access to care
In addition to delays for prior authorization, timely access to care is also limited by coverage disputes and delays. Historically, state officials have described difficulties in anticipating the timing of new drug product approvals26 and the patient populations for these therapies. Without the lead time to budget for these products, states may deny coverage of newly approved products with front-loaded costs. However, the authors have anecdotally experienced states limiting the number of meetings a manufacturer may have with the state to discuss their pipeline. One state reportedly gives only one 30-min meeting per year to each manufacturer to discuss all access issues and drugs in clinical development, while some states are not willing to meet with manufacturers at all. There are many resources that states can turn to better understand the product pipeline and projected timelines, including groups like ASGCT that compile this information from members.2
Many gene and cell therapies are targeting rare pediatric diseases for which there are no existing therapies, and timeliness of treatment is critical to maximizing favorable outcomes. Because the volume of cases is often small and complex, gene and cell therapies are often administered in a few centers of excellence (COE) around the country. While Medicaid and Medicaid MCOs are required to cover out-of-state care if not available in state, negotiating payment to a limited number of COEs where these therapies are available can be resource-intensive and time-consuming for states, ultimately delaying Medicaid patients’ access to newly approved products. Medicaid beneficiaries with out-of-state coverage may wait longer for care than patients with commercial insurance or in-state fee for service Medicaid coverage for some gene therapies. Medicaid programs may need to streamline their agreements with COEs to ensure timely access to care across state lines.
Policy solutions
Federal policy changes could significantly impact some of the findings associated with this report. While Medicaid is jointly managed by federal and state governments, Congress and federal agencies have clear legislative authority to make program-wide directives.
In assessing potential policy solutions, we considered two roles for the federal government: first, as a regulator and enforcer and, second, as a critical support mechanism for the states (Table 4).
Table 4.
Summary of proposed federal policy solutions
| Improve federal guidance and transparency efforts supporting implementation of statutory requirements | 1. Issue additional guidance to states outlining current federal requirements for coverage to FDA labeled indications |
| 2. Reiterate current requirements for timely access to covered benefits | |
| 3. Establish clearer channels for stakeholders to report non-compliance with federal coverage rules | |
| 4. Create a public dashboard to track policies, denials, complaints | |
| 5. Consider federal audits to review compliance with federal coverage rules | |
| Support states in integrating new gene and cell therapies into Medicaid benefits | 1. Modify federal payment policies to support states in bearing the up-front costs of gene and cell therapies (VBP/OBA agreements, enhanced federal support) |
| 2. Establish new mechanisms for states to preview gene and cell therapies coming to market | |
| 3. Provide states with a “best practices” guide for coverage of gene and cell therapy and payment arrangements |
Improve federal guidance and transparency efforts supporting implementation of statutory requirements
Congress and CMS can and should play a more active role in ensuring that states are compliant with federal requirements for coverage of gene and cell therapies, particularly as new products come to market with potential to significantly impact Medicaid populations. Recommendations include the following.
Issue additional guidance to states outlining current federal requirements for coverage to label
CMS regularly issues bulletins and other guidance to states that outline their obligations under federal statute and regulations. To ensure that state compliance issues are not a product of misunderstanding of coverage to label, CMS should issue additional guidance to states reiterating the expectations of current requirements relating to coverage to label. This guidance could also identify products that recently received FDA approval, ensuring states are aware of potential impactful products coming to market.
In July 2022, CMS issued an informational bulletin to states and other stakeholders outlining the beneficiary protections included in current Medicaid statute and policy.27 That memorandum outlines some of the coverage requirements associated with outpatient drugs. CMS would be well suited to provide a revised memorandum that offers greater specificity around coverage expectations for gene and cell therapies, particularly as that coverage relates to the description on the label.
Reiterate current requirements for timely access to covered benefits
In the case of gene and cell therapies with time-sensitive applicability, the time required to establish coverage can be equal in importance to the final coverage decision. Initial coverage denials, requirements for additional evidence, and prior authorization policies more broadly can all significantly delay a patient’s access to treatments.
Congress should work with CMS to reinforce timely coverage of gene and cell therapy, which is expected when prescribed according to its medically accepted indication, and limit the unnecessary and time-consuming demands for additional criteria to be met beyond the FDA-approved label. States should be required to issue clear timelines and escalation processes for partial denials or programs’ requests for additional unnecessary information such as prior authorization, single-case agreements, or requiring the primary care physician as the source of the request. CMS should also consider reforms to prior authorization and pre-certification policies to minimize the burden on providers and patients and expedite decisions.
Establish clearer channels for stakeholders to report non-compliance with federal coverage rules
The federal government is only able to respond to issues of non-compliance when it is made aware of potential violations. Given the breadth of programs and requirements CMS is left to manage, stakeholders can play a key role in highlighting state policies in need of review. To that end, CMS should establish a clear method for patients, providers, manufacturers, and other members of the public to identify instances in which state policies relating to coverage of gene and cell therapy fall short of federal expectations. For example, Health and Human Services (HHS) has existing public reporting portals for individuals to flag potential violations that could be used as a model for this case.28,29
Create a public dashboard
CMS should establish a public dashboard tracking coverage policies, denials, complaints, and discrepancies in coverage and reimbursement for each product across states. The information would be useful in quantifying the true scope of the problem and provide a forum to assess claims of overly restricted coverage.
Consider federal audits
Congress could direct federal investigative agencies to conduct regular reviews of compliance with federal coverage rules as new gene and cell therapies come to market. This review could be conducted by the Office of the Inspector General, the Government Accountability Office, or other federally supported entities tasked with tracking compliance with federal regulations.
Support states in integrating new gene and cell therapies into Medicaid benefits
Modify federal payment policies to support states in bearing the up-front costs of gene and cell therapies
The up-front costs for potentially curative therapies are difficult for Medicaid programs to manage in the current system. While the overall costs of chronic care may be more expensive, state systems are able to accommodate those costs as they are predictable and paid out over time.
Congress and the federal government should consider policies that would alleviate the financial strain on states associated with the arrival of gene and cell therapies. Examples of support are as follows.
-
•
Alternative payment options – Payment methodologies such as value-based payment (VBP) or outcomes-based arrangements (OBAs) that spread the cost of therapies over time or tie reimbursement to outcomes hold significant potential to support states in providing access to certain gene and cell therapies. CMS has already modified current rules for the MDRP to better support these arrangements.30 In addition, following President Biden’s executive order on drug pricing,31 the Centers for Medicare and Medicaid Innovation announced a new Cell and Gene Therapy Access Model32 to be tested in the coming years. This model will allow manufacturers the option to negotiate OBAs with CMS, rather than negotiating individually with each state. While the model announcement contained little detail, CMS would be responsible for reconciling the financial and clinical outcomes of the outcome-based agreements. This type of federal support could help spread risk pooling and ensure equitable access to cell and gene therapies regardless of individual states’ programs. However, there are concerns over the “price of admission” to this program and, though voluntary, what the implications would be for manufacturers who opt out. The authors encourage Congress and the Administration to continue to work with stakeholders to identify any additional policy barriers to adoption.
-
•
Enhanced federal support – Congress may consider policies that would enhance the federal government’s role in covering Medicaid costs of these therapies. The federal government generally has greater budget flexibility than states, putting the federal government in a more comfortable position to bear anticipated costs and spread risks. For instance, the federal government could increase its share of payments to states (the federal match percentage; FMAP) for new gene and cell therapies. Alternatively, the Medicaid and CHIP Payment and Access Commission considered establishing gene and cell therapy as a separate Medicaid benefit, with more generous federal financing in the 2021 June Report to Congress.33
Establish new mechanisms for states to preview gene and cell therapies coming to market
It is crucial for states and manufacturers to have productive pre-approval conversations to best anticipate budgeting for these therapies. As it currently stands, the systems that direct these channels of communications vary by state and by year, making it difficult for manufacturers to communicate about the pipeline of soon-to-be-approved therapies and difficult for states to preemptively budget for these therapies with front-loaded costs. The authors suggest creating more standard processes for meetings, akin to the standard processes, timing, and rigor required for FDA meetings. Having two standard meeting types, one for newly approved products and one for soon-to-be approved products in development, would help increase visibility on both sides.
Provide states with a “best practices” guide for coverage of gene and cell therapy
CMS periodically provides states with guides outlining best practices relating to coverage or administration of particular benefits. For example, during the COVID-19 Public Health Emergency, CMS provided states with a “State Medicaid & CHIP Telehealth Toolkit”34 that outlined key policy considerations for states with respect to telehealth. As more gene and cell therapies come to market, CMS should provide states with comparable resources that can help states more seamlessly integrate these therapies into their benefit offerings.
Future investigations should examine the impact of Medicaid reimbursement levels
This paper focuses primarily on coverage decisions of Medicaid programs. Payment policy can play an equally impactful role in coverage decisions, as insufficient payment levels can jeopardize the willingness of providers to offer novel products to Medicaid beneficiaries.
Consider the scenario created by bundled payments. Payment for the administration of gene and cell therapies and the accompanying clinical care, including most often in the inpatient setting, can be made through bundled payments that include the cost of the drug and the cost to treat the patient. The bundled payment amounts are set prospectively and for that reason may not include the cost of new therapies. Because the bundled payment is often significantly less than the cost of the drug itself, providers and hospitals may be less likely to offer advanced gene and cell therapies. For example, when CAR T’s KYMRIAH35 and YESCARTA36 launched in 2017, initial reimbursement was $43,094 (DRG 016),37 substantially less than the total cost of CAR T administration, creating a situation where hospitals had a disincentive to provide access to these lifesaving therapies.
Reimbursement policies pegged to site of care may also be a limiting factor to patient access to innovative gene and cell therapies. For example, Medicaid policies for gene-modified cell therapies targeting B cell malignancies commonly limit reimbursement to outpatient administration, denying inpatient coverage and reimbursement to patients who require hospitalization, despite both inpatient and outpatient administration studied in clinical trials.38 Limiting reimbursement to a specific site of care is inconsistent with products’ labeled indications and disincentivizes provider adoption of novel gene and cell therapies due to the uncertainty in payment if a patient moves between sites of care.
Materials and Methods
The 16 states included in the survey are Arizona, Arkansas, California, Colorado, Florida, Georgia, Illinois, Indiana, Massachusetts, Michigan, Mississippi, New York, North Carolina, Oklahoma, Oregon, and Texas. The three national Medicaid MCOs included in the survey are United Healthcare, Anthem, and Centene. The states and MCOs chosen are representative of over 46 million out of nearly 90 million Medicaid-covered individuals across the country and provide diversity in state geography, population size, political leadership, and Medicaid program structure. These states were selected by the authors’ work group based on a variety of factors impacting Medicaid programs to ensure diversity. Texas, California, Florida, New York, and Illinois were included for their diverse populations and geographic spread. Some states, like Massachusetts, were selected due to their notable biotechnology ecosystems and innovative reimbursement policies adopted to date for novel therapies. Other states, such as Arizona, were chosen to include the perspective of large states with reinsurance funds. The states’ economic capacity—the funding levels available for public payor programs—was also considered in the selection process to ensure diversity.
Policies for the selected states were collected from public internet sources; MCO policies were collected from their respective websites. The authors did not work to privately obtain policies or policy decisions from the insurers in order to most accurately capture the provider and patient journey. The authors of this article reviewed the dataset and identified which publicly available policies were consistent with the product’s FDA labeled indication. Coverage policies were considered to be “more restrictive” than the labeled indication if payors covered a population more narrow than what is outlined in the “indications and usage” section of the FDA label. For example, denying coverage based on pregnancy, age, or defined functional or laboratory criteria not included in the indications and usage section would be considered “more restrictive.” The review of coverage policies considered the products’ FDA-approved indications and the factors listed in Table 2 in determining if states were overly restrictive, or inconsistent, in their administration of three key products.
-
•
LUXTURNA (voretigene-neparvovec), a gene therapy for children and adults with an inherited retinal dystrophy caused by mutations in the RPE65 gene and sufficient viable retinal cells as determined by the treating physician39
-
•
ZOLGENSMA (onasemnogene abeparvovec-xioi), a gene therapy to treat children under 2 years old with SMA21
-
•
KYMRIAH (tisagenlecleucel), a CAR T cell therapy to treat adult relapsed/refractory diffuse large B cell lymphoma (DLBCL) as well as pediatric and young adult relapsed/refractory B-acute lymphoblastic leukemia (B-ALL)35
While patient monitoring requirements, provider reimbursement, and prior authorization processes can also impact access, which is addressed in the discussion, these factors were not considered when assessing whether a state covers a product to the FDA-approved labeled indication.
The proposed policy solutions identified in the discussion section were selected based on feasibility, similar efforts already in progress, and the scope of the federal government authorities. The goal was to provide actionable recommendations based on the current state of coverage for cell and gene therapies. Each proposed solution aims to resolve one or more of the barriers to coverage illustrated by the results.
Conclusions
Medicaid coverage for some of the first gene and cell therapies on the market has sometimes been inconsistent with federal legal requirements, with delayed and narrowly defined coverage. Even when covered by a Medicaid program, various factors in the appeals and administration process cause delays in time to treatment. For gene and cell therapies specifically, this delay can have a negative impact on patient health outcomes and often leads to disqualification from treatment, despite being eligible at the time of prescription, and it could deny patients the only or last option for treating their disease.
Understanding the impact of existing arrangements, along with vital implementation details, is critical in having a complete understanding of the gene and cell therapy marketplace. This understanding can help policymakers identify the need for additional federal policy changes. Streamlining the bureaucratic process, increasing transparency and accountability for state programs, and supporting CMS enforcement mechanisms to help reduce time to treatment will result in improved patient outcomes and equity.
Acknowledgments
Conceptualization and review support was provided by the ASGCT Government Relations committee. Manuscript preparation support was provided by Erin Frey and Lisa Kahlman, employees of Ultragenyx Pharmaceuticals, and Margarita Valdez-Martinez, Christina Mayer, and Chiagbanwe Enwere, employees of ASGCT.
Author contributions
Project administration, conceptualization, writing, review, and editing, J.A., D.B., F.C., J.W., and C.M.; writing support, review, and editing A.H., R.R., and A.S.
Declaration of interests
J.W. is an Independent Director on the Board of Atsena Therapeutics, Inc., and sits on the Board of Directors at ASGCT. R.R. has consulting agreements with and receives honoraria from Pfizer Inc., sits on ASGCT’s Board of Directors, receives research funding from Tessa Therapeutics, and receives honoraria from Kite-Gilead Sciences. The content of this article represents the authors’ opinions and may not necessarily represent the views of their employers.
References
- 1.Novartis . 2019. AveXis Announces Innovative Zolgensma® Gene Therapy Access Programs for US Payers and Families.https://www.novartis.com/news/media-releases/avexis-announces-innovative-zolgensma-gene-therapy-access-programs-us-payers-and-families [Google Scholar]
- 2.ASGCT, Citeline . 2022. Gene, Cell, & RNA Therapy Landscape: Q4 2022 Quarterly Data Report.https://asgct.org/global/documents/asgct_citeline-q4-2022-report_final.aspx [Google Scholar]
- 3.Centers for Medicare & Medicaid Services Who Enrolls in Medicaid and CHIP? https://www.medicaid.gov/state-overviews/scorecard/who-enrolls-medicaid-chip/index.html
- 4.Medicaid and CHIP Payment and Access Commission . 2021. Racial and Ethnic Disparities in Medicaid: An Annotated Bibliography.https://www.macpac.gov/wp-content/uploads/2021/04/Racial-and-Ethnic-Disparities-in-Medicaid-An-Annotated-Bibliography.pdf [Google Scholar]
- 5.Kauf T.L., Coates T.D., Huazhi L., Mody-Patel N., Hartzema A.G. The cost of health care for children and adults with sickle cell disease. Am. J. Hematol. 2009;84:323–327. doi: 10.1002/ajh.21408. [DOI] [PubMed] [Google Scholar]
- 6.Grady A., Fiori A., Patel D., Nysenbaum J. Profile of Medicaid enrollees with sickle cell disease: a high need, high cost population. PLoS One. 2021;16:e0257796. doi: 10.1371/journal.pone.0257796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medicaid and CHIP Payment and Access Commission State Plans. https://www.macpac.gov/subtopic/state-plan/
- 8.Medicaid and CHIP Payment and Access Commission Matching Rates. https://www.macpac.gov/subtopic/matching-rates/
- 9.Center for Medicare & Medicaid Services . 2021. Medicaid Drug Rebate Program (MDRP)https://www.medicaid.gov/medicaid/prescription-drugs/medicaid-drug-rebate-program/index.html [Google Scholar]
- 10.Medicaid and CHIP Payment and Access Commission . 2022. Trends in Medicaid Drug Spending and Rebates.https://www.macpac.gov/wp-content/uploads/2022/10/07_Trends-in-Medicaid-Drug-Spending-and-Rebates-Chris.pdf [Google Scholar]
- 11.U.S. Congress . 1994. Social Security Act. 42 U.S.C. § 1396(r-8)https://www.ssa.gov/OP_Home/ssact/title19/1927.htm [Google Scholar]
- 12.U.S. Congress . 2010. Federal Food, Drug, and Cosmetics Act. 21 U.S.C. § 355(d)https://www.govinfo.gov/app/details/USCODE-2021-title21/USCODE-2021-title21-chap9-subchapV-partA-sec355 [Google Scholar]
- 13.U.S. Congress . 1994. Social Security Act. 42 U.S.C. § 1396(a)https://www.ssa.gov/OP_Home/ssact/title19/1902.htm [Google Scholar]
- 14.U.S. Congress . 1994. Social Security Act. 42 U.S.C. § 1396(b)https://www.ssa.gov/OP_Home/ssact/title19/1903.htm [Google Scholar]
- 15.Chakradhar S. STAT News; 2019. Maisie’s Army’: How a Grassroots Group Is Mobilizing to Help Toddlers Access a Lifesaving Drug.https://www.statnews.com/2019/08/20/maisies-army-zolgensma-access-spinal-muscular-atrophy/ [Google Scholar]
- 16.Ballreich J., Ezebilo I., Khalifa B.A., Choe J., Anderson G. Coverage of genetic therapies for spinal muscular atrophy across fee-for-service Medicaid programs. J. Manag. Care Spec. Pharm. 2022;28:39–47. doi: 10.18553/jmcp.2022.28.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MIT NEWDIGS FoCUS Project . New Drug Development Paradigm Initiative; 2019. Tracking Medicaid Coverage of Durable Cell and Gene Therapies.https://newdigs.tuftsmedicalcenter.org/tracking-medicaid-coverage-of-durable-cell-and-gene-therapies/ [Google Scholar]
- 18.Berry D., Wellman J., Allen J., Mayer C. Assessing the state of Medicaid coverage for gene and cell therapies. Mol. Ther. 2022;30:2879–2880. doi: 10.1016/j.ymthe.2022.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sachs R.E., Donohue J.M., Dusetzina S.B. Confronting state Medicaid drug spending pressures. JAMA. 2020;324:1831. doi: 10.1001/jama.2020.19325. [DOI] [PubMed] [Google Scholar]
- 20.U.S. Congress . 1994. Social Security Act. 42 U.S.C. § 1302(438)https://www.govinfo.gov/content/pkg/CFR-2010-title42-vol4/pdf/CFR-2010-title42-vol4-part438.pdf [Google Scholar]
- 21.FDA . 2019. Highlights of Prescribing Information - ZOLGENSMA.https://www.fda.gov/media/126109/download [Google Scholar]
- 22.Medicaid and CHIP Payment and Access Commission . 2018. Elements of the Medicaid Appeals Process under Fee for Service, by State.https://www.macpac.gov/publication/elements-of-the-medicaid-appeals-process-under-fee-for-service-by-state/ [Google Scholar]
- 23.Juliusson G., Hagberg O., Lazarevic V.L., Lehmann S., Höglund M. Impact of treatment delay in acute myeloid leukemia revisited. Blood Adv. 2021;5:787–790. doi: 10.1182/bloodadvances.2020003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray S.J. Timing of gene therapy interventions: the earlier, the better. Mol. Ther. 2016;24:1017–1018. doi: 10.1038/mt.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Institute for Clinical and Economic Review . 2022. Betibeglogene Autotimer for Beta Thalassemia: Effectiveness and Value [Evidence Report]https://icer.org/wp-content/uploads/2021/11/ICER_Beta-Thalassemia_Evidence-Report_060222-1.pdf [Google Scholar]
- 26.Gifford K., Winter A., Wiant L., Dolan R., Tian M., Garfield R. 2020. How State Medicaid Programs Are Managing Prescription Drug Costs.https://files.kff.org/attachment/How-State-Medicaid-Programs-are-Managing-Prescription-Drug-Costs.pdf [Google Scholar]
- 27.Center for Medicaid and CHIP Services . 2022. Beneficiary Protections and Medicaid Drug Coverage - under Value Based Purchasing (VBP) and Other Innovative Payment Arrangements [Informational Bulletin]https://www.medicaid.gov/federal-policy-guidance/downloads/cib07212022.pdf [Google Scholar]
- 28.Center for Medicare & Medicaid Services Hospital Price Transparency. https://www.cms.gov/hospital-price-transparency
- 29.Department of Health and Human Services Information Blocking. https://www.healthit.gov/topic/information-blocking
- 30.Centers for Medicare & Medicaid Services . 2020. Medicaid Program: Establishing Minimum Standards in Medicaid State Drug Utilization Review (DUR) and Supporting Value-Based Purchasing (VBP) for Drugs Covered in Medicaid, Revising Medicaid Drug Rebate and Third Party Liability (TPL) Requirements.https://www.govinfo.gov/content/pkg/FR-2020-12-31/pdf/2020-28567.pdf [Google Scholar]
- 31.The White House . 2022. Executive Order on Lowering Prescription Drug Prices for Americans.https://www.whitehouse.gov/briefing-room/presidential-actions/2022/10/14/executive-order-on-lowering-prescription-drug-costs-for-americans [Google Scholar]
- 32.U.S. Department of Health and Human Services . 2023. A Report in Response to the Executive Oder on Lowering Prescription Drug Costs for Americans.https://innovation.cms.gov/data-and-reports/2023/eo-rx-drug-cost-response-report [Google Scholar]
- 33.Medicaid and CHIP Payment and Access Commission . 2021. Report to Congress on Medicaid and CHIP.https://www.macpac.gov/wp-content/uploads/2021/06/June-2021-Report-to-Congress-on-Medicaid-and-CHIP.pdf [Google Scholar]
- 34.Centers for Medicare and Medicaid Services . 2020. State Medicaid and CHIP Telehealth Toolkit: Policy Considerations for States Expanding Use of Telehealth.https://www.medicaid.gov/medicaid/benefits/downloads/medicaid-chip-telehealth-toolkit.pdf [Google Scholar]
- 35.FDA . 2017. Highlights of Prescribing Information - KYMRIAH.https://www.fda.gov/media/107296/download [Google Scholar]
- 36.FDA . 2017. Highlights of Prescribing Information - YESCARTA.https://www.fda.gov/media/108377/download [Google Scholar]
- 37.Gustafson K., Jackson J., Olsen M., Moorman A. Avalere Health; 2020. CMS Finalizes New Reimbursement Model for CAR T Treatments.https://avalere.com/insights/cms-proposes-significant-changes-to-reimbursement-mechanisms-for-car-t [Google Scholar]
- 38.Maud S., Laetsch J., Buechner S., Rivers M., Boyer H., Bittencourt P., Bader M., Verneris H., Stefanski G., Myers M., et al. Tisagenlecleucel in children and young adults with B-cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:443–447. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.FDA . 2017. Highlights of Prescribing Information - LUXTURNA.https://www.fda.gov/media/109906/download [Google Scholar]