

Abstract

Here, a facile and selective synthesis method for cationic azatriphenylene derivatives was established by electrochemical intramolecular cyclization, where atom-economical C–H pyridination without a transition-metal catalyst or an oxidant is a key step. The proposed protocol is a practical strategy for the late-stage introduction of cationic nitrogen (N+) into π-electron systems and broadens the scope of molecular design of N+-doped polycyclic aromatic hydrocarbons.
Polycyclic aromatic hydrocarbons (PAHs) are a class of compounds consisting of two or more fused benzene rings; they are actively studied as a model of two-dimensional (2D) graphene in a wide range of fields, including the pharmaceutical, environmental science, electronics, and materials chemistry fields.1 Among PAHs, some disk-shaped molecules, including triphenylenes and hexabenzocoronenes, are known to exhibit liquid-crystalline properties arising from their highly stacked nature.2 The bottom-up synthesis of such graphene-like molecules provides access to a group of functional materials such as buckybowls (e.g., corannulenes and sumanenes)3 and helical PAHs (e.g., helicenes).4 In addition, doping of PAHs with heteroatoms (e.g., chalcogen, nitrogen, phosphine, and boron) has been attracting attention because it can drastically alter their electronic structures.5 In heteroatom doping of PAHs, introducing heteroatoms in a desired form at a desired position in the parent skeleton is important; therefore, precise organic synthesis is required to realize an efficient bottom-up approach for preparing heteroatom-doped PAHs.
Among the heteroatom-doped PAHs, cationic nitrogen (N+)-doped PAHs are known to exhibit functionalities (e.g., stable electrochemical properties, peculiar aggregation behavior,6 and fluorescent properties) that have led to their application in organic photocatalysts,7 ion-conductive materials,8 and bioimaging.9 Three main synthesis methods for N+-doped PAHs have been reported, as summarized in Figure 1a. Among them, photocyclization of polyaryl-substituted pyridinium salts is the most simple but powerful way to prepare N+-doped PAHs (Figure 1a(i)).6 Such Scholl-type reactions proceed by photoirradiation in the presence of an oxidant; however, N-arylpyridinium salts with limited synthesis methods are required as precursors. A number of methods based on ring-closing metathesis reactions of N-vinylpyridinium salts (Figure 1a(ii))10 and [4 + 2] annulation of 2-phenylpyridines or N-arylpyridinium salts with alkynes (Figure 1a(iii))11 have been proposed. The latter approach can be used to prepare N+-doped PAHs with imidazole, pyrazole, and triazole units as well as pyridinium units. However, all of these synthesis methods have shortcomings, such as difficulty controlling selectivity and the need to use transition-metal catalysts and chemical oxidants that have adverse environmental impacts. Therefore, a simpler and more environmentally benign synthesis method for N+-doped PAHs is desired.
Figure 1.

Concept of the present study.
In this context, we focused on the nucleophilic aromatic substitution (SNAr) reaction as a key step in the synthesis of cationic azatriphenylene derivatives. In our previous study, the SNAr reaction was used for the intramolecular cyclization of phenylpyridine derivatives possessing an electron-deficient perfluoroaryl moiety to give the corresponding cationic azatriphenylene derivatives in high yields under moderate conditions without the use of a transition-metal catalyst (Figure 1b).12 However, one of the challenges in the synthesis of N+-doped PAHs via the SNAr reaction is poor functional-group applicability. The development of a new synthesis method is required to overcome this problem and further expand the range of molecular design and accompanying properties.
In the present study, an electro-oxidative C–H activation was used for intramolecular cyclization to afford the corresponding cationic azatriphenylene derivatives (Figure 1c). The key reaction is the anodic pyridination of aromatic compounds reported by Yoshida et al.13 The anodic oxidation of an arene generates its radical cation, followed by nucleophilic reaction of pyridines. Total two-electron oxidation and deprotonation gives arylpyridinium derivatives via C–H activation. Therefore, our proposed strategy is not only an efficient and atom-economical method to access N+-doped PAHs without any transition-metal catalysts or oxidants but also a promising protocol with a wide scope of aromatic rings in both the precursors and products.
2-Phenylpyridine derivative 1a bearing a methoxyphenyl group was prepared by the Suzuki–Miyaura coupling reaction of 2-(2-bromophenyl)pyridine with 3-methoxyphenylboronic acid. In the cyclic voltammogram of 1a (Figure S1), an irreversible oxidation wave was observed at a peak potential of 1.5 V (vs saturated calomel electrode (SCE)), suggesting that the electron-rich methoxyphenyl moiety of 1a preferentially underwent one-electron oxidation, followed by chemical reaction. The highest occupied molecular orbital (HOMO) of 1a, as calculated by density functional theory (DFT), was localized at the methoxyphenyl moiety (Figure S1), supporting the above estimation of the discharging part for oxidation. Accordingly, the preparative-scale anodic oxidation of 1a was carried out under constant-current conditions in a divided cell equipped with a carbon felt anode and a Pt plate cathode (1 × 1 cm) under the reported standard conditions for anodic pyridination of arenes (Figure 2a).13 The amount of charge (2.6 F/mol) was determined by monitoring the consumption of 1a by thin-layer chromatography (TLC). The intramolecular pyridination proceeded to yield cationic azatriphenylene derivative 2a in 84% NMR yield. The counterion of 2a (BF4–) was derived from the supporting electrolyte. It was possible to purify the product from the supporting electrolyte (Bu4NBF4) by washing with cold MeOH, but the isolated yield was considerably reduced (60% isolated yield). The structure of 2a was determined by NMR, high-resolution mass spectrometry, elemental analysis, and X-ray analysis.
Figure 2.

(a) Anodic pyridination of 1a–1c, including yields of cyclized products 2a–2c. (b) The plausible reaction mechanism for the intramolecular pyridination of 1a–1c, with diagrams of the β-LUMO of 1a–1c in their one-electron-oxidized state.
Other regioisomers of 1a were subjected to the anodic pyridination under the optimized conditions shown in Figure 2a. The intramolecular anodic pyridination of 1b resulted in poor NMR yield (40%) and isolated yield (14%) of 2b because of a side reaction that afforded a complex mixture. In the case of 1c, the corresponding cationic azatriphenylene derivatives (2c) was not detected at all; instead, only a complex mixture was obtained.
A plausible reaction mechanism of the intramolecular cyclization of 1 is shown in Figure 2b. As an anodic reaction, one-electron oxidation of 1 generates the radical cation form with its charge localized at the methoxyphenyl moiety, where nucleophilic attack by a pyridine moiety occurs. Further one-electron oxidation and deprotonation (aromatization) results in the formation of 2. In the cathodic chamber, protons are reduced to generate H2 bubbles. Such an intramolecular substitution reaction is easily achieved by the electrochemical umpolung of the methoxyphenyl moiety. The difference in reactivity of the intramolecular anodic pyridination of 1a–1c can be explained on the basis of DFT calculations. Yoshida and co-workers reported on the selectivity in the intermolecular anodic pyridination of arenes, where the position of the large β-LUMO coefficient in the one-electron-oxidized state of arenes corresponds to the position of the nucleophilic attack by pyridine.13 On the basis of this knowledge, we conducted DFT calculations of the β-LUMO of 1a–1c in their one-electron-oxidized state. As shown in Figures 2b and S2, large coefficients are observed for the carbons of 1a and 1b, where subsequent pyridination occurs to form the corresponding products 2a and 2b, respectively. However, no coefficient is observed for the carbon of the 1c radical cation, where expected intramolecular pyridination should occur. This result strongly supports the experimental fact that 2c was not obtained.
We next investigated the scope of the intramolecular pyridination (Scheme 1). For 1d–1g bearing two methoxy groups or an ethylenedioxy group, 1d, 1e, and 1g provided the corresponding cationic azatriphenylene derivatives in good NMR yields (80% for 2d, 78% for 2e, and 83% for 2g); by contrast, 2f was not obtained at all despite complete consumption of starting material 1f. In this case, spiro compound 2f′ was obtained via pyridination at the ipso position of the dimethoxyphenyl moiety (63% NMR yield). A large β-LUMO coefficient is observed for the carbon at the ipso position of 1f; therefore, spirocyclic formation and demethylation occurred to give 2f′ (Figures S2 and S3). Other phenylpyridine derivatives bearing a tolyl (1h), phenyl (2i), or naphthyl (2j) group were also successfully prepared but afforded the products in low to moderate NMR yields (36% for 2h, 51% for 2i, and 73% for 2j). Less-activated arenes are known to not be suitable for this type of anodic coupling reaction, although Waldvogel et al. overcame this problem by using a boron-doped diamond (BDD) electrode.14 We could obtain the desired N+-doped PAHs from precursors lacking methoxy groups because of the advantage of intramolecular cyclization. The regioselectivity for the reaction was also consistent with the DFT simulation results for the β-LUMO coefficients (Figure S2). In particular, the structure of 2j was also determined by single-crystal X-ray diffraction analysis (vide infra).
Scheme 1. Scope of the Intramolecular Anodic Pyridination to Provide Cationic Azatriphenylene Derivatives.

NMR yield. Isolated yields are shown in parentheses. Detected.
Not detected.
0.3 M Et4NBF4 was used.
We next investigated the optical and electrochemical properties of the cationic azatriphenylene products. As representative examples, the data for 2a, 2d, 2i, and 2j are summarized in Figure 3. All other data are summarized in the Supporting Information (Figures S4 and Table S1).
Figure 3.
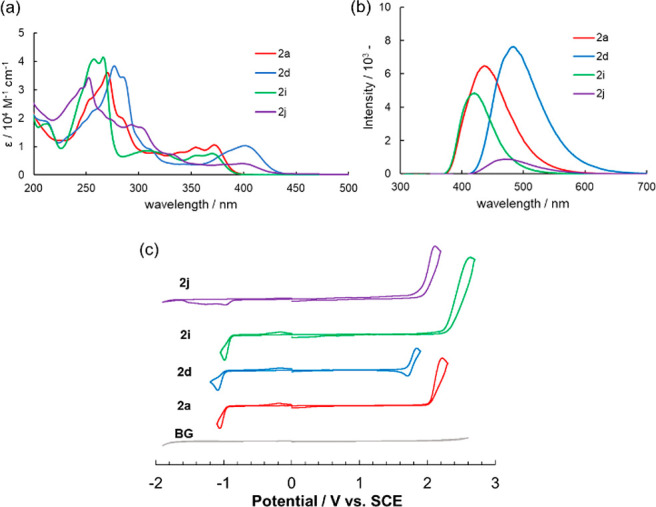
(a) UV–vis absorption spectra and (b) fluorescence spectra for 2a, 2d, 2i, and 2j in MeCN solutions. (c) Cyclic voltammograms of 2a, 2d, 2i, and 2j (3 mM) in 0.1 M Bu4NPF6/MeCN using a Pt working electrode (φ = 3 mm) at a scan rate of 100 mV/s.
UV–vis absorption spectra of the cationic azatriphenylene derivatives in acetonitrile (MeCN) solutions show an absorption maximum at 250–300 nm and a weaker absorption band at 300–450 nm (Figure 3a), which are attributed to the π–π* transition and the intramolecular charge transfer (ICT) transition, respectively, as supported by time-dependent DFT (TD-DFT) calculations (Figures S6–S13, Table S2). The pyridinium moiety, where the LUMO is mainly located, behaves as an electron-accepting unit, and the aryl groups attached to the nitrogen atom, where the HOMO is mainly located, behave as an electron-donating unit. The simulated transition for the weak absorption bands was assignable to the HOMO–LUMO transition. Fluorescence spectra of the cationic azatriphenylene products in MeCN solutions show a unimodal emission peak (Figure 3b). The fluorescence quantum efficiencies ΦFL of 2a (0.57) and 2d (0.62), which bear methoxy groups, are substantially higher than those of 2i (0.27) and 2j (0.11). The emission wavelengths compared with that of 2i were red-shifted by the introduction of methoxy groups (2a and 2d) or the expansion of the π-system (2j).
Cyclic voltammetry (CV) measurements were performed for the cationic azatriphenylene derivatives in MeCN solutions (Figure 3c). The cyclic voltammogram of 2a shows an irreversible oxidation current (E1/2ox = 2.11 V vs SCE) and an irreversible reduction current (E1/2red = −1.00 V vs SCE). The oxidation potential of 2a was markedly shifted to a positive region from that of precursor 1a (Figure S1) because the methoxyphenyl moiety was incorporated into the cationic azatriphenylene skeleton. Because of the electron-deficient nature of the cationic azatriphenylene derivative, the reduction potential was relatively positive, whereas the reduction wave did not appear in the CV of the precursor (Figure S1). The irreversible electron-transfer behavior indicates that neither the oxidized nor the reduced state of 2a was sufficiently stable for it to undergo subsequent chemical reactions. However, the cyclic voltammogram of 2d, which possesses two electron-donating methoxy groups, shows a reversible oxidation response at a slightly negative potential compared with that of 2a, indicating that the oxidized state was sufficiently stable to prevent possible side reactions such as dimerization. Among the cationic azatriphenylene products, 2i, which lacks methoxy groups, showed a higher oxidation potential and a large HOMO–LUMO energy gap. The cyclic voltammogram of π-extended 2j shows interesting irreversible multiple-reduction behavior, presumably due to the competitive reduction of a reducible pyridinium moiety and naphthalene moiety. As summarized in Figure S5, the HOMO–LUMO gaps of 2a–2j are mainly influenced by the HOMO level (i.e., the structure of the substituted benzene moiety). Because compound 2i is an N+-doped analogue of triphenylene, we comprehensively compared its physical properties with those of triphenylene (see the Supporting Information, Figure S14 and Table S3).
Finally, single-crystal X-ray diffraction analysis of the cationic azatriphenylene derivatives was performed (Figure 4). The crystal structure of 2a is highly planar; its torsion angle around the N atom is 4.2° (Figure 4a). In our previous study, the torsion angle of the fluorinated cationic azatriphenylene derivative was 20°. The high planarity of 2a resulted in well-stacked pairs with a large contact area. However, 2j, which has a naphthalene moiety, has a [4]-helicene structure with a torsion angle of 27.8° (Figure 4b) because of steric repulsion between the hydrogen atoms of the pyridinium and naphthalene moieties. The columnar stacking structure of 2j was composed of a racemic mixture (i.e., alternating P-helicene and M-helicene). Because of the highly twisted structure of 2j, the contact area in the stack was smaller than that of 2a. The proposed simple cyclization method can be a straightforward way to provide helical N+-doped PAHs with chiroptoelectronic properties.15
Figure 4.
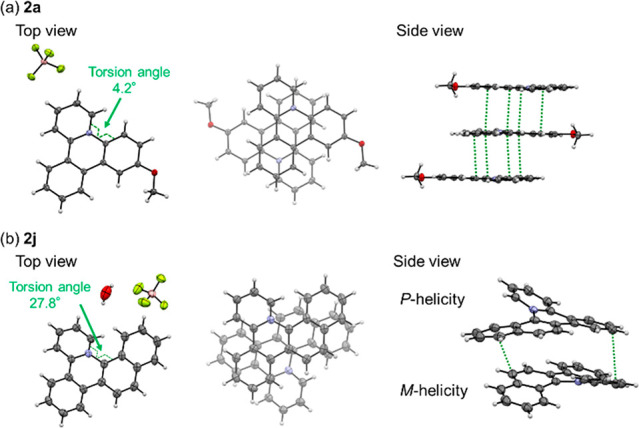
Crystal structures of (a) 2a and (b) 2j. Short atomic contacts of C···C are indicated by green dotted lines.
In conclusion, we have demonstrated a facile intramolecular cyclization reaction based on anodic pyridination to afford various cationic azatriphenylene derivatives. The reaction mechanism, particularly the reaction selectivity, was elucidated with the aid of DFT calculations. The cationic azatriphenylene products were found to have ICT interactions because of the donor–acceptor structure composed of an arene moiety and a pyridinium moiety. The intramolecular anodic pyridination method is useful for introducing a nitrogen cation into PAHs to perturb the π-electron system. The development of additional π-expanded N+-doped PAHs that include helicene architectures is currently underway in our lab.
Acknowledgments
This research was supported by Support for Tokyo Tech Advanced Researchers [STAR] Grant funded by the Tokyo Institute of Technology Fund (Tokyo Tech Fund, for S.I.) and JST SPRING grant (No. JPMJSP2106, for Y.O.). Y.O. acknowledges a support from the Kato Foundation for Promotion of Science (KS-3501). Y.N. is grateful for financial support from JST-ERATO (No. JPMJER1903) and JSPS-WPI. We thank Dr. Yoshihisa Sei (single-crystal X-ray analysis) and Dr. Masato Koizumi (HR-MS) at the Materials Analysis Division, Open Facility Center, Tokyo Institute of Technology.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.3c01341.
Detailed experimental procedures, characterization data, DFT studies, single-crystal X-ray data, and copies of NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Wu J.; Pisula W.; Müllen K. Graphenes as Potential Material for Electronics. Chem. Rev. 2007, 107, 718–747. 10.1021/cr068010r. [DOI] [PubMed] [Google Scholar]; b Narita A.; Wang X. Y.; Feng X.; Müllen K. New Advances in Nanographene Chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. 10.1039/C5CS00183H. [DOI] [PubMed] [Google Scholar]
- Wöhrle T.; Wurzbach I.; Kirres J.; Kostidou A.; Kapernaum N.; Litterscheidt J.; Haenle J. C.; Staffeld P.; Baro A.; Giesselmann F.; Laschat S. Discotic Liquid Crystals. Chem. Rev. 2016, 116, 1139–1241. 10.1021/acs.chemrev.5b00190. [DOI] [PubMed] [Google Scholar]
- a Mehta G.; Rao H.S. P. Synthetic Studies Directed Towards Bucky-Balls and Bucky-Bowls. Tetrahedron 1998, 54, 13325–13370. 10.1016/S0040-4020(98)00661-9. [DOI] [Google Scholar]; b Sakurai H.; Daiko T.; Hirao T. A Synthesis of Sumanene, a Fullerene Fragment. Science. 2003, 301, 1878. 10.1126/science.1088290. [DOI] [PubMed] [Google Scholar]; c Muzammil E. M.; Halilovic D.; Stuparu M. C. Synthesis of Corannulene-Based Nanographenes. Commun. Chem. 2019, 2, 58. 10.1038/s42004-019-0160-1. [DOI] [Google Scholar]
- Shen Y.; Chen C. F. Helicenes: Synthesis and Applications. Chem. Rev. 2012, 112, 1463–1535. 10.1021/cr200087r. [DOI] [PubMed] [Google Scholar]
- a Stępień M.; Gońka E.; Żyła M.; Sprutta N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. 10.1021/acs.chemrev.6b00076. [DOI] [PubMed] [Google Scholar]; b Liu J.; Feng X. Bottom-Up Synthesis of Nitrogen-Doped Polycyclic Aromatic Hydrocarbons. Synlett 2020, 31, 211–222. 10.1055/s-0039-1690767. [DOI] [Google Scholar]; c Borissov A.; Maurya Y. K.; Moshniaha L.; Wong W.-S.; Żyła-Karwowska M.; Stępień M. Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds. Chem. Rev. 2022, 122, 565–788. 10.1021/acs.chemrev.1c00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu D.; Zhi L.; Bodwell G. J.; Cui G.; Tsao N.; Müllen K. Self-Assembly of Positively Charged Discotic PAHs: From Nanofibers to Nanotubes. Angew. Chem., Int. Ed. 2007, 46, 5417–5420. 10.1002/anie.200700515. [DOI] [PubMed] [Google Scholar]; b Wu D.; Feng X.; Takase M.; Haberecht M. C.; Müllen K. Synthesis and Self-Assembly of Dibenzo[jk,mn]naphtho[2,1,8-fgh]thebenidinium Derivates. Tetrahedron 2008, 64, 11379–11386. 10.1016/j.tet.2008.08.063. [DOI] [Google Scholar]; c Wu D.; Pisula W.; Enkelmann V.; Feng X.; Müllen K. Controllable Columnar Organization of Positively Charged Polycyclic Aromatic Hydrocarbons by Choice of Counterions. J. Am. Chem. Soc. 2009, 131, 9620–9621. 10.1021/ja902420u. [DOI] [PubMed] [Google Scholar]; d Yang C.; Wu D.; Zhao W.; Ye W.; Xu Z.; Zhang F.; Feng X. Anion-Induced Self-Assembly of Positively Charged Polycyclic Aromatic Hydrocarbons towards Nanostructures with Controllable Two-Dimensional Morphologies. CrystEngComm 2016, 18, 877–880. 10.1039/C5CE02171E. [DOI] [Google Scholar]; e Aracena A.; Rezende M. C.; Encinas M. V.; Vergara C.; Vásquez S. O. Aggregation Phenomena in Photobicyclised Pyridinium Salts. New J. Chem. 2017, 41, 14589–14594. 10.1039/C7NJ02788E. [DOI] [Google Scholar]
- a Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. Electron-Transfer State of 9-Mesityl-10-methylacridinium Ion with a Much Longer Lifetime and Higher Energy Than That of the Natural Photosynthetic Reaction Center. J. Am. Chem. Soc. 2004, 126, 1600–1601. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]; b Nicewicz D. A.; Nguyen T. M. Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014, 4, 355–360. 10.1021/cs400956a. [DOI] [Google Scholar]
- Wu D.; Liu R.; Pisula W.; Feng X.; Müllen K. Two-Dimensional Nanostructures from Positively Charged Polycyclic Aromatic Hydrocarbons. Angew. Chem., Int. Ed. 2011, 50, 2791–2794. 10.1002/anie.201004245. [DOI] [PubMed] [Google Scholar]
- a Ghosh S.; Pal S.; Rajamanickam S.; Shome R.; Mohanta P. R.; Ghosh S. S.; Patel B. K. Access to Multifunctional AEEgens via Ru(II)-Catalyzed Quinoxaline-Directed Oxidative Annulation. ACS Omega 2019, 4, 5565–5577. 10.1021/acsomega.9b00274. [DOI] [Google Scholar]; b Ma W.; Zhang L.; Shi Y.; Ran Y.; Liu Y.; You J. Molecular Engineering to Access Fluorescent Trackers of Organelles by Cyclization: Chemical Environment of Nitrogen Atom-Modulated Targets. Adv. Funct. Mater. 2020, 30, 2004511–2004519. 10.1002/adfm.202004511. [DOI] [Google Scholar]; c Mule R. D.; Roy R.; Mandal K.; Chopra D.; Dutta T.; Sancheti S. P.; Shinde P. S.; Banerjee S.; Lal Koner A.; Bhowal R.; Senthilkumar B.; Patil N. T. Interplay of Anion-Π+ and Π+-Π+ Interactions in Novel Pyrido[2,1-a]Isoquinolinium-Based AIEgens - Substituent- and Counterion-Dependent Fluorescence Modulation and Applications in Live Cell Mitochondrial Imaging. Chem. Eur. J. 2022, 28, e202200632. 10.1002/chem.202200632. [DOI] [PubMed] [Google Scholar]
- a Núñez A.; Cuadro A. M.; Alvarez-Builla J.; Vaquero J. J. A New Approach to Polycyclic Azonia Cations by Ring-Closing Metathesis. Org. Lett. 2007, 9, 2977–2980. 10.1021/ol070773t. [DOI] [PubMed] [Google Scholar]; b Nuñez A.; Abarca B.; Cuadro A. M.; Alvarez-Builla J.; Vaquero J. J. Ring-Closing Metathesis Approach to Heteroaromatic Cations: Synthesis of Benzo[a]Quinolizinium Salts. Eur. J. Org. Chem. 2011, 2011, 1280–1290. 10.1002/ejoc.201001373. [DOI] [Google Scholar]
- a Li F.; Cho J.; Tan S.; Kim S. Synthesis of Quinolizinium-Type Heteroaromatics via a Carbene Intermediate. Org. Lett. 2018, 20, 824–827. 10.1021/acs.orglett.7b03964. [DOI] [PubMed] [Google Scholar]; b Toriumi N.; Asano N.; Miyamoto K.; Muranaka A.; Uchiyama M. N -Alkynylpyridinium Salts: Highly Electrophilic Alkyne-Pyridine Conjugates as Precursors of Cationic Nitrogen-Embedded Polycyclic Aromatic Hydrocarbons. J. Am. Chem. Soc. 2018, 140, 3858–3862. 10.1021/jacs.8b00356. [DOI] [PubMed] [Google Scholar]; c Shaikh A. C.; Banerjee S.; Mule R. D.; Bera S.; Patil N. T. External Oxidant-Dependent Reactivity Switch in Copper-Mediated Intramolecular Carboamination of Alkynes: Access to a Different Class of Fluorescent Ionic Nitrogen-Doped Polycyclic Aromatic Hydrocarbons. J. Org. Chem. 2019, 84, 4120–4130. 10.1021/acs.joc.9b00120. [DOI] [PubMed] [Google Scholar]; d Karak P.; Rana S. S.; Choudhury J. Cationic π-Extended Heteroaromaticsviaa Catalytic C-H Activation Annulative Alkyne-Insertion Sequence. Chem. Commun. 2021, 58, 133–154. 10.1039/D1CC05590A. [DOI] [PubMed] [Google Scholar]
- a Asanuma Y.; Eguchi H.; Nishiyama H.; Tomita I.; Inagi S. Synthesis of Ring-Fused Pyridinium Salts by Intramolecular Nucleophilic Aromatic Substitution Reaction and Their Optoelectronic Properties. Org. Lett. 2017, 19, 1824–1827. 10.1021/acs.orglett.7b00590. [DOI] [PubMed] [Google Scholar]; b Shida N.; Nishimi H.; Asanuma Y.; Tomita I.; Inagi S. Synthesis of a Conjugated Polymer with Ring-Fused Pyridinium Units via a Postpolymerization Intramolecular Cyclization Reaction. Polym. J. 2020, 52, 1401–1406. 10.1038/s41428-020-0388-8. [DOI] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. I. Electrochemical C-H Amination: Synthesis of Aromatic Primary Amines via N-Arylpyridinium Ions. J. Am. Chem. Soc. 2013, 135, 5000–5003. 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- a Herold S.; Möhle S.; Zirbes M.; Richter F.; Nefzger H.; Waldvogel S. R. Electrochemical Amination of Less-Activated Alkylated Arenes Using Boron-Doped Diamond Anodes. Eur. J. Org. Chem. 2016, 2016, 1274–1278. 10.1002/ejoc.201600048. [DOI] [Google Scholar]; b Möhle S.; Herold S.; Richter F.; Nefzger H.; Waldvogel S. R. Twofold Electrochemical Amination of Naphthalene and Related Arenes. ChemElectroChem. 2017, 4, 2196–2210. 10.1002/celc.201700476. [DOI] [Google Scholar]
- Xu K.; Fu Y.; Zhou Y.; Hennersdorf F.; Machata P.; Vincon I.; Weigand J. J.; Popov A. A.; Berger R.; Feng X. Cationic Nitrogen-Doped Helical Nanographenes. Angew. Chem., Int. Ed. 2017, 56, 15876–15881. 10.1002/anie.201707714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.