

Abstract
We compared three cell isolation and two proteomic sample preparation methods for single-cell and near-single-cell analysis. Whole blood was used to quantify hemoglobin (Hb) and glycated-Hb (gly-Hb) in erythrocytes using targeted mass spectrometry and stable isotope-labeled standard peptides. Each method differed in cell isolation and sample preparation as follows: 1) FACS and automated preparation in one-pot for trace samples (autoPOTS); 2) limited dilution via microscopy and a novel rapid one-pot sample preparation method that circumvented the need for the solid-phase extraction, low-volume liquid handling instrumentation and humidified incubation chamber; and 3) CellenONE-based cell isolation and the same one-pot sample preparation method used for limited dilution. Only the CellenONE device routinely isolated single-cells from which Hb was measured to be 540–660 amol per red blood cell (RBC), which was comparable to the calculated SI reference range for mean corpuscular hemoglobin (390–540 amol/RBC). FACSAria sorter and limited dilution could routinely isolate single-digit cell numbers, to reliably quantify CMV-Hb heterogeneity. Finally, we observed that repeated measures, using 5–25 RBCs obtained from N = 10 blood donors, could be used as an alternative and more efficient strategy than single RBC analysis to measure protein heterogeneity, which revealed multimodal distribution, unique for each individual.
Keywords: Single-cell proteomics, quantitative, targeted, one-pot, hemoglobin, red blood cell, HbA1c, glycated hemoglobin, carboxymethyl hemoglobin
1. Introduction
Compared to standard bulk specimen analysis used routinely in proteomics, single-cell and/or near-single-cell analysis is a relatively challenging methodology requiring specialized instrumentation for cell isolation and sample preparation protocols. These optimized protocols aim to minimize adsorptive sample losses and allow the operation of liquid chromatography-mass spectrometry (LC-MS) instrumentation at their detection limits to achieve the needed sensitivity to detect and quantify low-abundance analytes. A general single-cell sample preparation method, namely nano-ProteOmic sample Preparation (nPOP), has been developed to deliver single-cells onto fluorocarbon-coated glass slides, with as low as 5 μm spatial separation.1 When combined with stable isotope labeling, nPOP can be used to increase sample throughput for global quantitative proteomic analyses, mitigating throughput limitations in single-cell assays. Other technological developments, methods refinement, and novel combinations of the two have been implemented to routinely identify ∼1500 proteins in a single-cell and more than 7000 proteins when using as little as 2 μg of protein input.2−19 Typically, cell isolation required dedicated instrumentation such as FACS, laser capture microdissection, specialized single-cell handling microscopy, microfluidics, or CellenONE, an instrument designed for single-cell isolation. Furthermore, low volume sample preparation required advanced liquid handling instrumentation and humidified chambers that can be mitigated using carrier peptide strategies that make use of isobaric tags, foregoing the need to adhere to low volume sample preparation. Lastly, aiming to improve proteome coverage has required nano-LC operation at lower than typical (vendor recommended) flow rates and the latest MS instrumentation offering the lowest detection limits. For these reasons, single-cell and near-single-cell proteomics are currently limited to laboratories that have access to this type of instrumentation. While CellenONE and FACS have been used for single-cell isolation, near-single-cell proteomics is demonstrated to be achievable for measuring cellular heterogeneity, without the use of specialized instrumentation.
We compared three cell isolation and two proteomic sample preparation methods for ease of use in single-cell and near-single-cell (nsc) analysis. We chose hemoglobin (Hb) and glycated-Hb (gly-Hb) as targets for evaluating the quantitation performance in erythrocytes isolated from whole blood. Measurement of the gly-Hb:Hb ratio at the single-cell level provides a frequency distribution curve of the analytes which has implication for improving diagnosis of diabetes nephropathy and blood doping in high performance sports. Peptide quantitation used standard tandem mass spectrometry and stable isotope-labeled standard peptide methods where the N-terminal proteolytic peptide and its N-terminal carboxymethyl valine (CMV) of Hb and gly-HB respectively were targeted. All the three methods used parallel reaction monitoring via nano-LC-MS/MS for data generation, a relatively common proteomic protocol for targeted analysis. Specifically, we compared the following three options for cell isolation and sample preparation: 1) FACS and automated preparation in one-pot for trace samples (autoPOTS);10 2) limiting dilution via microscopy and a novel rapid one-pot sample preparation method that circumvented the need for the solid-phase extraction, low-volume liquid handling instrumentation and humidified incubation chamber required with autoPOTS; and 3) CellenONE-based cell isolation and the same one-pot sample preparation method used for limiting dilution.
In addition to comparing three methods for conducting targeted single-cell proteomics, we also aimed to assess cell population heterogeneity, measured as the extent of hemoglobin carboxymethylation. To do so, a simplified one-pot protocol was implemented to perform quantitative single-cell proteomics, with the typical instrumentation accessible in a standard proteomics core LC-MS facility. Specifically, the present study used a simplified autoPOTS protocol that eliminated the need for specialized instrumentation that many laboratories do not possess. Finally, by targeting the N-terminal proteolytic peptide of hemoglobin beta subunit (HBB) and its advanced glycation end-products (AGEs) counterpart (CMV), the suitability for measuring RBC heterogeneity was determined.
The glycated hemoglobin (HbA1c) content in red blood cells (RBCs) is a function of average long-term blood glucose concentration and the life-span of RBCs, which is approximately 120 days.20 Formation of glycated hemoglobin in RBCs occurs through the Maillard reaction21 and advanced glycation end-products (AGEs), namely carboxymethyl valine (CMV) and carboxyethyl valine (CEV), on the hemoglobin β-chain.22 We note that these AGE modifications can occur on other amino acid residues and are not limited to the N-terminal valine on the β-chain. Besides CMV and CEV, HbA1c protein can contain a complex combination of AGEs on different amino acid residues.23−26
Bottom-up proteomics using high-resolution accurate mass spectrometry (HRAM)27−30 has shown CMV as the predominant modification on the N-terminal valine of the β-chain in HbA1c,31 which suggests CMV as a target to measure RBC age distribution using a single RBC or near-single RBCs (e.g., 5–25 RBCs) in a targeted bottom-up proteomics assay. It should be noted that CMV modifications have been previously suggested as a valuable marker for the diagnosis of diabetic nephropathy, but no correlation was found between CMV abundance and HbA1c32 likely due to the finding that oxidative stress accelerates CMV formation but not HbA1c formation.33 By targeting the N-terminal proteolytic peptide of hemoglobin β-subunit (HBB) and its AGE counterpart (CMV), the suitability of the assay for measuring RBC heterogeneity was assessed in this study.
2. Materials and Methods
2.1. Patient and Blood Specimen Collection
The certificate of ethical approval for harmonized minimal risk clinical study pertaining to this project/study is active and available via the Board of Record REB Number: BC20-0440 through the University of Victoria Human Research Ethics Board. Blood specimens were collected from N = 10 individuals at the Victoria Lipid Clinic Society (Victoria, BC, Canada) by a registered nurse via venipuncture into lithium heparin coated blood collection tubes. A total of ten specimens were collected over a 91-day time period. Blood specimens were stored at 4 °C until processed within 72 h of blood collection. These samples were subsequently processed using limiting dilution and modified rapid digestion Trypsin/Ly-C, described in sections 2.2 and 2.4, respectively.
For automated deposition of RBCs, using a FACSAria Fusion sorter (BD Biosciences, San Jose, USA), a 300 μL aliquot of blood was washed three times with 300 μL of 1× PBS and centrifugation at 500g for 10 min per cycle. The RBC sample was reconstituted to a concentration of ∼10 million cells/mL and filtered for FACS sorting as previously described.10
2.2. Limiting-Dilution and RBC Spotting
Here, 100 μL of whole blood was aliquoted into 1.5 mL microcentrifuge tubes and centrifuged at 300g for 5 min. The supernatant was pipetted out and discarded, and packed RBCs were resuspended in 100 μL of 6.67% human serum albumin (HSA) (Sigma-Aldrich, Canada, SKU: A9731), which was previously prepared by dissolving 66.7 mg of lyophilized powder in 1 mL of LC-MS grade water and filtered using a 0.22 μm filter. Limiting dilution (LD) was performed in a 2-fold dilution series in 6.67% HSA using 384-well deep well small volume microplate, polypropylene (Greiner Bio-One, Germany, REF: 784201). In detail, 10 μL of 6.67% HSA was first aliquoted per well, vertically from row A to K, the final dilution (in row K) being 2048-fold. Aiming to isolate 5–25 RBCs per spot, 0.25 μL from diluent wells (in the LD plate) was sampled using a 0.1–2.5 μL manual pipettor and delivered into a low volume microscopy compatible 384 well plate (high content imaging plate, with lid black with clear 127 μm bottom 384 well, tissue culture treated cyclic olefin copolymer, Corning, USA, REF: 4681), hereafter referred to as one-pot-384 (OP384). Sample spotting was performed by a (reverse) capillary action, rather than dispensing with the pipettor plunger, by contact of the pipette tip with the bottom of the well for 10 s. Care was taken not to apply a downward force on the pipettor as that could damage the 127 μm cyclic olefin copolymer film and thereby interfere with microscopy. Resting the weight of the pipettor while ensuring perpendicular contact with the surface was sufficient to achieve spotting/reverse capillary flow. The same pipette tip was used for spotting in the subsequent two wells, at which point the pipette tip was discarded and the procedure was repeated until the number of desired replicates (N = 10 per specimen) were spotted. In this way, the volume of sample per well was estimated at ∼80 nL (250 nL sample used to spot 3 wells, with residual volume in tip). Wells were examined using an inverted phase contrast microscope at 100× total magnification (Bausch and Lomb PhotoZoom 31-19-14 Inverted Phase Contrast Microscope) to view RBCs encapsulated in (dried) HSA. The plate was stored at 4 °C until sample preparation.
2.3. CellenONE Assisted RBC Spotting
This was a pilot experiment to determine the capability of the PRM assay (see section 2.5) for measuring peptide targets in single RBCs, as well as to assess the characteristics of the resultant response curve when the RBC input ranged from one to five cells. Spotting RBCs directly into OP384 was performed at the Cellenion laboratory (Cellenion SASU, Lyon, France) using a CellenONE X1 instrument software version 2.0.0.984. For these experiments, the concentration of HSA was lowered from 6.67% to 2.22% as the former reduced the stability of the droplet formation created by the proprietary dispensing technology. The following parameters were used to isolate and dispense RBCs directly into OP384: Nozzle voltage 96 V, pulse duration 49 μs, frequency 500 Hz, and LED delay 200 μs. For imaging, detection, and isolation, the background was set to ejection of 344 pix, sedimentation 180 pix. Detection parameters were lower gray 10 upper gray 255, minimum diameter 6.5 μm, max diameter 100 μm, and elongation 4. The isolation parameters were set to lower gray 10, upper gray 255, minimum diameter 7.5, max diameter 16.3, and elongation 1.65. Channel was transmission, selection mode was positive, and threshold 10–255. These settings resulted in an RBC deposit frequency of 0.12. In other words, for every 8.15 objects detected (non-RBC particles) 1 RBC was deposited.
RBCs were deposited in an OP384 plate as single RBCs, two RBCs, three RBCs, four RBCs, five RBCs, and as HSA without RBC. The OP384 plate containing the deposits was then shipped to the University of Victoria Genome BC Proteomics Centre for subsequent sample processing (described in section 2.4) and LC-MS analyses (described in section 2.5).
2.4. Modified Rapid Digestion Trypsin/Ly-C
The protocol used for sample preparation was based on previous studies2,9−11 but was modified to eliminate surfactant, alkylation agent, solid-phase extraction, and automated low-volume liquid handling instrumentation. Microliter volume, one-pot, organic solvent assisted, solid phase extraction-free sample preparation was performed as follows: Unless otherwise noted, all reagents were LC-MS grade. For wells containing RBC(s) encapsulated in HSA, 10 μL of 10% acetonitrile (MeCN) in water was added. The OP384 was covered with the accompanying lid and placed in a bath ultrasonic cleaner (Digital Ultrasonic Cleaner, PS-40A, Vevor, USA) operated at 240 W and 40 kHz with a bath water temperature of 70 °C for 15 min. The bath sonicator was covered with a lid to ensure 100% humidity and Styrofoam around the perimeter of the OP384 assisted level floatation in the bath (preventing flooding of the wells with bath water). After the 15 min incubation, the OP384 was placed in an ice bath to minimize sample evaporation. A modified rapid digestion protocol (Rapid Digestion Kit-Trypsin/Lys-C, CAT.#VA1061, Promega, USA) was used, substituting the rapid digestion buffer with 25 mM Tris pH 8, 2 mM Bond-Breaker TCEP solution (Thermo Scientific, USA, CAT. #77720). The Trypsin/Lys-C mix was diluted to 100 ng/μL in water and mixed with the substitute digestion buffer at a 1:15 (v/v). Then 16 μL of mixed digestion buffer was added per well (in the OP384). Iodoacetamide (IAA) was not included as an alkylating agent based on a previous study which demonstrated increased N-terminus peptide alkylation at temperatures exceeding 40 °C.34 While not performed in this study, alkylation with 40 mM iodoacetamide for 30 min at room temperature has been tested and may be included, after high temperature digestion to prevent interpeptide cysteine–cysteine bonds. High-temperature digestion was achieved by placing the OP384 in the bath sonicator as described above but without sonication for a 60 min incubation followed by 20 min of sonication at 240 W and 40 kHz. The OP384 was then placed in an ice bath and acidified with 1 μL of 10% formic acid and spiked with 2 μL of stable isotope-labeled peptide standards (SIL) at a concentration of 1 fmol/μL. As silicone mats are not commercially available for the OP384, aluminum foil was used instead and fixed in place using labeling tape at which point samples were considered prepared for nano-LC-MS. The volume of sample injected per LC-MS analysis was 15 μL which was approximated to be equivalent to ∼2.5 μg of protein, based on ∼80 nL of sample (containing 6.67% HSA) spotted per well.
2.5. Nano-LC-MS/MS at Site 1
Nano-LC-MS at site 1 (University of Victoria) encompassed PRM assay development, including LOD, repeatability, and specimen analysis. Nano-LC-MS was performed using an EASY-nLC 1000 liquid chromatography instrument (Thermo Scientific) and an Orbitrap Fusion ETD Tribrid mass spectrometer (Thermo Scientific) operated using Xcalibur software version 4.3.73.11 (Thermo Scientific).
The EASY-nLC 1000 was operated in the two-column mode using in-house packed (MAGIC C18AQ 100 Å 5 μm, Michrom Bioresources, USA) trap column (2 cm, 75 μm ID) upstream of an in-house packed (MAGIC C18AQ 100 Å 5 μm, Michrom Bioresources) analytical column (15 cm, 75 μm ID). While the 5 μm beads do not offer the same separation resolution as 3 and 2 μm beads, experience (in-house) has demonstrated them more robust and compatible with targeted quantification for trace materials, including single-cell input. It requires noting the MAGIC C18AQ 100 Å 5 μm from Michrom Bioresources may no longer be commercially available and the Princetonpher-100 C18 100-5U (Princeton Chromatography, USA) may serve as a potential substitute. Flow rate was maintained at 300 nL/min resulting in a back pressure range of ∼100–140 bar throughout the gradient, at ambient column temperature (22–27 °C). Mobile phase A and B consisted of 0.1% formic acid in water and 0.1% formic acid in 90% MeCN, respectively. The elution gradient was performed as follows: 0% B 0 min, 40% B 20 min, 80% B 2 min, 100% B 4 min, 0% B 6 min, total gradient time of 32 min. Precolumn and analytical columns were equilibrated with 4 μL each mobile phase A at variable flow rates determined by setting an instrument-controlled maximum pressure of 348 bar. Sample loading onto the trap column was performed at a flow rate of 4 μL/min with maximum pressure set at 348 bar. The total time for equilibration and sample loading was ∼15 min and the total time for analysis was 47 min per sample. A needle wash sequence consisting of five 25 μL volumes each of polar and organic solvents was programmed to be performed during the gradient to minimize cross-sample carryover between injections. Electrospray was achieved by connecting the analytical column with an emitter consisting of a 10 μm tip (PicoTip Emitter, New Objective, USA, FS360-20-10-N-20-C12). Note, since solid phase extraction is omitted a precolumn filter of 0.2 μm porosity fitted directly to the switching valve and upstream of the line-out may be considered to prevent particulates from entering the flow path which otherwise may increase system pressure and/or clog downstream components. Additionally, the well coordinates for injecting from an OP384 plate are not preprogrammed in the EASY-nLC 1000 software and require calibration using the instructions provided in the user guide (Thermo EASY-nLC 1000 User Guide (Touch-screen software version 3.0), 60053-97227 Revision C January 2012).
The nano-LC method for data dependent acquisition (DDA) and data independent acquisition (DIA) was identical to that described above for PRM analysis apart from the elution gradient which was as follows: 0% B 0 min, 25% B 100 min, 40% B 20 min, 90% B 15 min, 100% B 1 min, 0% B 4 min for a total gradient time of 140 min.
The Xcaliber acquisition settings for collecting MS spectra were based on previously developed methods for PRM (PRM_FusionLumos_SW3_1.pdf (washington.edu)), DDA, and DIA35 modes, and the details of the settings used in the present study are provided in Supplemental Figures S1, S2, and S3–S4, respectively.
2.6. Nano-LC-MS/MS at Site 2
Nano-LC-MS at site 2 (Brigham Young University) utilized PRM parameters developed at site 1 to reproduce LOD, perform LOQ, and measure endogenous targets in RBCs prepared by automated preparation in one-pot for trace samples (AutoPOTS).10 The LOD and LOQ were determined using a seven-point calibration curve consisting of the following peptide standard concentrations (in amol/μL): Blank 0, 0.975, 3.9, 15.6, 62.5, 250, and 1000; the SIL peptides were used as normalizers spiked-in at a constant concentration of 200 amol/μL. Triplicate injections were performed at each concentration to evaluate the intra-assay CV and to define the LOQ for use as Tier 2 assays for research application.36 A similar but not identical (to site 1) nano-LC method was used consisting of online SPE and a two column setup; precolumn (Jupiter 3.0, C18, 100 μm ID, 5 cm) (Phenomenex, Torrance, CA) and an analytical column (Dr. Maisch C18, 3.0 μm, 30 μm ID, 50 cm). The LC gradient was 2% to 40% B (20 min); 40% to 80% (2 min) and 80% to 99% (4 min) at a flow rate of 0.535 μL/min (1/9 split was applied before the analytical column). The LC and MS instruments at site 2 were an UltiMate 3000 RSLCnano with an UltiMate WPS-3000TPL/PL RSLCnano well Plate Autosampler (Thermo Fisher, Waltham, MA) and an Orbitrap Exploris 480 (Thermo Fisher) mass spectrometer with the Nanospray Flex ion source.
Endogenous peptide measurements in RBCs isolated using FACS were performed as previously described.10 One microliter of mobile phase A (water with 0.1% FA) was dispensed into each well prior to FACS sorting, and RBCs were deposited in four replicates at concentrations of 5, 50, and 250 RBCs per well into a Corning low volume 384-well plate (PN 3544). Two microliters of 0.5%N-dodecyl-β-d-maltoside (DDM) in 50 mM ammonium bicarbonate (ABC)/phosphate buffered saline (PBS) was added into each well for cell lysis and protein denaturation for 60 min at 70 °C. One microliter of enzyme solution with 2 ng of Lys-C in 50 mM ABC was added and incubated at 37 °C for 4 h. Followed by a 12 h 37 °C trypsin digestion step, with 1 μL of enzyme solution containing 2 ng of trypsin in 50 mM ABC. One microliter of 5% formic acid solution was used to quench the digestion, and 2 μL of 800 amol SIL peptide standards (VHLTPEEK(+8) and CM-VHLTPEEK (+8)) was spiked in. The 384 well plate was then sealed with a foil adhesive mat and stored at −20 °C. Nano-LC-MS was performed by injecting 7 μL of each sample using the same column and gradient setup described above for the LOD and LOQ experiments. Samples were injected in an ascending sequence from low to high concentrations. PRM was performed using the method developed at the University of Victoria but on an Orbitrap Exploris instrument (Thermo).
2.7. Peptide Standard Synthesis
Fmoc solid phase synthesis of the peptide VHLTPEEK, which can exist on the N-terminus of human hemoglobin beta subunit (HBB_HUMAN, UniProtKB - P68871) after N-terminal methionine excision, and of the advanced glycation product carboxymethyl valine (CM-VHLTPEEK), were performed as previously described,37,38 with modification. Natural (NAT) and stable isotope labeled (SIL) versions of the peptide were synthesized by incorporating 12C/14N amino acids for the NAT peptide and 13C/15N heavy isotope labeled l-lysine on the C-terminus of the SIL peptide. Peptides were synthesized in duplicate and prior to deprotection one pair of NAT and SIL peptide were subjected to on-resin N-terminal carboxymethylation. Carboxymethylation was achieved using glyoxylic acid and sodium cyanoborohydride treatment as previously described38 with modification. In a 5 mL glass vial, 21 mg of HCOCO2H·H2O (Sigma-Aldrich, Canada, G10601) and 47 mg of NaBH3CN (Sigma-Aldrich, Canada, 156159) were dissolved in 5 mL of dimethylformamide (DMF). From this solution 200 μL was added to peptides still protected and coupled to resin and incubated for 12 h at ambient temperature in a fume hood. The reaction solution was subsequently eluted, and 3 replicate washes were performed using 200 μL of DMF. The other pair of NAT and SIL peptides were treated with a similar protocol omitting the addition of HCOCO2H·H2O and NaBH3CN. Peptides were subsequently deprotected and decoupled from the solid phase resin using repeated incubations with a solution of trifluoroacetic acid, triisopropylsilane, and water (23.75:0.625:0.625 ratio, respectively). The eluents were precipitated with cold ether, evaporated, and subsequently freeze-dried. Peptides were resolubilized in 100 μL of 3% MeCN and subjected to matrix assisted laser desorption ionization time-of-flight MS analysis using an Ultraflex III-MALDI TOF/TOF mass spectrometer (Bruker Ltd., Milton, ON) for crude yield and purity assessment.
Peptides were purified by preparative HPLC using an Agilent 1290 Infinity II UHPLC equipped with an Eclipse Plus C18 RRHD 1.8 μm 2.1 × 150 mm analytical column (Agilent). Retention times for peptides were previously determined on the same LC and column using dynamic MRM on an Agilent 6495B Triple Quadrupole MS. Purity analysis for each fraction was performed using an Ultraflex III-MALDI TOF/TOF mass spectrometer (Bruker Ltd., Milton, ON), and only pure fractions were pooled for subsequent amino acid analysis. The purity of the pooled fractions was confirmed via UV-HPLC on an Agilent 1290 Infinity HPLC instrument.
Amino acid analysis was performed as previously described,39 with modification. Peptide standards were subjected to acid hydrolysis and subsequent derivatization via dansylation. Quantification via dynamic MRM was performed on an Agilent 1290 Infinity II UHPLC online with a 6495B triple quadrupole MS using a 13C or 2H labeled internal standard for each amino acid (Cambridge isotope Laboratories, Inc., MA, USA), except for the carboxymethyl valine, for which a standard was not available.
2.8. Carboxymethyl Valine Stability at High Temperature and Multiple Reaction Monitoring
The stabilities of the carboxymethyl modified peptide standard (CM-VHLTPEEK) and the unmodified standard (VHLTPEEK) were compared at 70 and 4 °C as follows. A working stock of SIL and NAT peptides was made to consist of ∼50 fmol/μL of each peptide in 3% MeCN and 3 uL was added in triplicate to 5 μL of 10% MeCN predispensed in 3 separate wells of a Greiner low volume 384 well microplate (8 μL final in each well). The exact triplicate composition and volume was prepared in 300 μL sample injection vials (PP screw vial, 12 × 32 mm, 9 mm thread, from Canadian Life Science, Edmonton, AB). The microplate was placed in a sonicator bath at 70 °C for 15 min while the vial samples were stored in a refrigerator (4 °C). After the incubation, the microplate was placed on an ice bath and allowed to cool and 16 μL of 25 mM Tris pH 8, 2 mM TCEP was added followed by 1 μL of enzyme (Promega rapid digest Trypsin/LysC). The microplate was incubated at 70 °C for 1 h followed by 20 min of sonication. The sample injection vials were incubated in the refrigerator (4 °C). One microliter of 10% formic acid was added to each well, and samples were analyzed via dynamic multiple reaction monitoring (MRM) using an Agilent 1290 Infinity II LC System and Agilent 6495B Triple Quadrupole MS instrument (Agilent) as previously described,40,41 with modification. Five microliters of sample was injected and separation was achieved using an Eclipse Plus C18 RRHD 1.8 μm 2.1 × 50 mm analytical column (Agilent) fitted with an upstream 1290 Infinity II in-line filter, 0.3 μm, 2 mm ID, SST (Agilent). The gradient was as follows: 2% (2 min); 2% to 25% (5 min) at a flow rate of 0.4 mL/min, 25% to 98% (2 min) at a flow rate of 0.6 mL/min and 98% to 80% (2 min) at a flow rate of 0.4 mL/min followed by a post time of 2 min of 2% B at a flow rate of 0.4 mL/min. The aqueous and organic mobile phase buffers consisted of 0.1% formic acid in water and 0.1% formic acid in MeCN, respectively. Targeted MS acquisitions were performed at unit resolution using 6 min detection windows, a 890 ms cycle time, resulting in a dwell time of ∼10 ms. Three time segments were used for dynamic MRM as follows: at a start time of 0 min, the valve was diverted to waste, at 2 min the valve was diverted to the MS, and at 7 min the valve was diverted to waste and the Delta EMV (+) was set to 500. The source settings were set as follows; gas temperature 150 °C, gas flow 15 L/min, nebulizer 30 psi, sheath gas temperature 250 °C, sheath gas flow 11 L/min, capillary 3500 and 3000 V (−ve) for positive and negative ion funneling, respectively, nozzle voltage was 300 and 1500 V for positive and negative ionization, respectively, iFunnel parameters for high pressure RF were 200 V for positive mode and 150 V for negative mode and those for low pressure RF were 110 and 60 V for positive and negative, respectively.
2.9. Analysis of Blood Specimens from Study Participants
Endogenous CM-VHLTPEEK and VHLTPEEK in blood samples were quantified via low cell input one-pot PRM and by standard bulk sample preparation and MRM. Detailed chronological specimen collection and processing for low cell input one-pot PRM is provided in Supplemental Document 1. After spotting, samples were processed as described in section 2.4 and analyzed as described in section 2.5. In addition to the specimens analyzed, a background control (HSA without RBC) was also subjected to 10 replicate spotting and processed in parallel as described in sections 2.4 and 2.5. Lastly, variability of the analytical workflow was assessed by preparing CM-VHLTPEEK and VHLTPEEK peptide standards in mobile phase A with identical SIL concentrations (to that used for specimens) and NAT concentrations at average endogenous (in RBCs) CM-VHLTPEEK and VHLTPEEK concentrations, respectively. Briefly, a master mix was made in a background of 0.1% FA in water. Peptide standards were spiked in at concentrations so that the following amounts of peptide standards would be injected on the column: 13 amol CMV-NAT, 12.7 fmol NAT, and 2 fmol each CMV-SIS and SIS. As with specimens and HSA control, 10 injections were analyzed as described in section 2.5.
Bulk sample preparation used 50 μL of gravity-packed RBCs dispensed into 1 mL of lysis/protein solubilization buffer consisting of 2% sodium deoxycholate and 50 mM ammonium bicarbonate in water. Cell disruption was achieved by incubating at 99 °C on a Thermo Mixer (Eppendorf, Hamburg, Germany) at 1000 rpm shaking for 15 min. Proteins were reduced and alkylated with 20 mM dithiothreitol and 40 mM IAA and 1:20 and 1:50 dilutions used for protein quantification using the Bradford assay. Trypsin digestion was achieved using a 1:1 trypsin to protein concentration ratio and incubatiion at 37 °C for 9 h with 750 rpm shaking on a Thermo Mixer (Eppendorf). Acidification to a final concentration of 1% formic acid was used to stop the digestion and precipitate the sodium deoxycholate. Samples were subjected to solid phase extraction, lyophilization, and subsequent solubilization in 0.1% formic acid to a protein concentration of 0.6 μg/μL. A 25 μL aliquot of the sample was mixed with 25 μL of SIL and CMV-SIL at 8000 fmol/μL and 80 fmol/μL, respectively. Using 5 μL, samples were analyzed via MRM as described in section 2.8. Endogenous peptides were quantified using external calibration curves. Nine-point calibration curves were generated in mobile phase A using SIL and CMV-SIL peptides as normalizers, at identical concentrations to those used for specimens. The NAT and CMV-NAT peptide concentrations reflected expected endogenous concentrations with ranges of 16 fmol/μL to 107 513 fmol/μL and 0.18 fmol/μL to 1200 fmol/μL, respectively.
2.10. Selectivity
Selectivity for measuring endogenous CM-VHLTPEEK was assessed as previously described.42 The calibration curves from the bulk analysis, described in section 2.9 revealed an LOQ of 75 fmol CM-VHLTPEEK injected on the column. Based on this LOQ, three-point calibration curves were generated in N = 6 bulk sample preparations, described in section 2.9, with the lowest point on the calibration curve being the endogenous signal. The middle point was spiked in at 25 × the LOQ (75 fmol × 25 = 1875 fmol NAT CM-VHLTPEEK injected on the column), and the highest point on the curve was 50 × the LOQ (75 fmol × 50 = 3750 fmol CM-VHLTPEEK on the column). The SIL CM-VHLTPEEK peptide was spiked in at 30 fmol/μL (600 fmol on column). Samples were prepared in duplicate, and each sample was injected in replicate for MRM analysis (described in section 2.8) with a wash injection included between each injection. Samples were prepared to a 60 μL final volume in each sample injection vial as follows: 55 μL trypsin digested sample, described in section 2.9, and 5 μL of peptide standard. The high concentration peptide standard mix contained 360 fmol/μL CMV-SIL and 2250 fmol/μL CMV-NAT. The medium concentration contained 360 fmol/μL CMV-SIL and 1126 fmol/μL CMV-NAT. The lowest concentration contained 360 fmol/uL CMV-SIL. Twenty microliter injections were used for MRM analysis, which was performed as described in section 2.8.
2.11. Data Analysis and Availability
Data sets for targeted experiments (PRM and MRM) are available through Panorama Public,43 which can be searched using the following link: https://panoramaweb.org/yMtekT.url. The associated raw data files can be obtained through ProteomeXchange using the ID: PXD030875. Data sets pertaining to DDA and DIA analyses have been deposited and are available from the Center for Computational Mass Spectrometry using the MassIVE identifiers MSV000088397 and MSV000088395, respectively.
Data acquired using DDA and DIA were analyzed using the MSFragger44 and DIA-Umpire,45 the default workflow was selected in FragPipe version 15.0 with MSFragger version 3.2 and Philosopher version 3.4.13 (build 1611589727). Detailed parameter settings and search log files are provided as text files in the Supplemental Documents 2, 3, 4, 5, and 6.
All targeted data sets were analyzed using Skyline.46 For associated figures generated in Graphpad Prism (GraphPad Prism version 8.2.1 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com), Skyline calculated peak areas were exported to Microsoft Excel (Microsoft Corporation, 2018. Microsoft Excel, available at: https://office.microsoft.com/excel.) to perform calculations that were not available directly through Skyline, such as percent difference of the slope of the line across response curves in replicate biological matrix, Violin plots for analyzing repeated measures in blood specimens and other similar data analyses.
For LOQ and repeatability determination, a coefficient of variation of less than 20% across triplicate injections was set as the cutoff and the LOD was based on the Skyline calculated value, which identifies LOD as the signal which is 3 standard deviations above the signal in blank measurements. This data analysis pertains to data generated as described in section 2.6.
Raw data files obtained from repeated measures of 5–25 RBCs in patient blood samples, control HSA samples, and peptide standards spiked-in mobile phase A were analyzed as described above. Normalized peak areas for endogenous CM-VHLTPEEK and VHLTPEEK were used to calculate % CM-VHLTPEEK values, which were then plotted in Graphpad Prism using Violin plots. For validation purposes, the distribution of the data set was also plotted (not presented) using the cumulative frequency distribution function in Graphpad Prism, selecting relative frequency, auto for center of first bin, and center of last bin and selecting “No bins. Tabulate exact cumulative frequency distribution”. For normality and lognormality tests, normal (Gaussian) distribution, log-normal distribution, and compute the relative likelihood of sampling from Gaussian (normal) vs a log-normal distribution (assuming no other possibilities) were selected. The method to test the distribution was set to Kolmogorov–Smirnov normality test with Dallal-Wilkinson-Lilliefor P value and create a QQ plot was selected. These data analyses pertain to data generated as described in section 2.9.
Data analysis for selectivity included using Skyline to integrate peak areas and subsequently export normalized peak areas as an excel file. To calculate the slope of the line for the three-point response curve for each of the six biological matrices independently, peak areas were pasted into GraphPad Prism and the group analysis function was used to perform a linear regression analysis. The calculated equations of the curves were used to average across all six slope values, which was then used to determine whether each of the individual slopes were within 10% of the average. In other words, a (greater than) 10% difference was used to identify interference.42
3. Results
3.1. RBC Isolation
Submicroliter volumes of RBCs were spotted in wells via limiting dilution but resulted in the sample drying within minutes of being spotted (data not shown). Saline, normally used to manipulate RBCs, resulted in salt crystallization and cell shrinkage/lysis, which interfered with downstream microscopy (data not shown). A compatible solution was experimentally identified by comparing isotonic solutions of glucose, fructose, sucrose, glycerol, and human serum albumin (HSA) at 6.67%, with the latter providing the optimal conditions for cell integrity and optical clarity for RBC identification via microscopy (Supplemental Figure S5).
The limiting dilution approach was routinely used for isolating less than 10 RBCs but became laborious for single RBC isolation. Automated single RBC isolation and deposition, compatible with downstream quantitative LC-MS, was first attempted with fluorescence-activated cell sorting (FACS). Experimentation revealed FACSAria used in this study was not suitable for reproducible delivery of single RBCs into individual wells of nanoPOTS or autoPOTS, the latter having the same dimensions of the OP384 (data not shown). The FACS model tested (FACSAria) had low efficiency for delivering cells to small well targets (data not shown). However, FACSAria was used to deliver higher numbers of RBCs into wells in the range of 5–250 RBCs per well (results of these experiments are presented in section 3.2). Automated single RBC isolation-deposition directly into OP384 was achieved using the CellenONE (Lyon, France) instrument (Supplemental Figure S6). The reproducibility of delivering a single RBC was first optimized and then demonstrated by isolation of 20 single RBCs in HSA and 5 controls lacking RBCs but containing HSA; example images are provided in Supplemental Figure S6. These experiments revealed reproducible and timely isolation-deposition of RBCs. However, optimal droplet formation required reducing the concentration of HSA to 2.22%, which rendered RBCs unviewable in OP384 wells under microscope, a noted limitation. Finally, given the high precision of CellenONE-based cell sorting, it was possible to eliminate the use of HSA in the buffer, as the verification with microscopy was not required.
3.2. One-Pot Rapid Digestion Evaluated with DIA and DDA Methods
Here our aim was to eliminate online solid phase extraction, which requires a modified 10-port switching valve, while adhering to one-pot sample preparation required to use a solvent composition that did not contain signal suppressing reagent(s) such as detergents. As an alternative, cell lysis was achieved in 10% MeCN with high-temperature sonication, which also solvated proteins via emulsification and denatured them for subsequent proteolytic digestion (high efficiency) with heat stable Trypsin and Lys-C. The reproducibility of sample preparation was assessed via nano-LC-MS using both DIA and DDA methods, and the results from the former are presented in Figure 1. The nano-LC pressure profiles during the gradient are provided in Supplemental Figure S7. Peptide peak areas were comparable between injections with a median CV of 28.5% without normalization and a median CV of 19.3% when peptide peak areas were normalized to the total ion chromatogram (TIC). Peptide peak areas displayed consistent relative peptide peak areas; the peptide with the highest peak area was the same across all samples.
Figure 1.

Consistency of peptide chromatographic peak areas, retention times, and identifications from nano-LC-MS analysis when samples were prepared with the detergent-free one-pot rapid digestion protocol. RBCs were spotted and counted in OP384 and processed using a detergent-free one-pot rapid digestion described herein. The input number of RBCs ranged from 0 to 8 RBCs with 0 RBCs serving as a background control. The X-axes denote the number of RBCs input. Panels A, B, and C depict peak areas and total ion chromatogram (TIC) normalized peak areas for trypsin and Lys-C derived HSA, HBB, and HBA peptides, respectively, and across all replicates. Panel D depicts peptide retention times for HSA (left), HBB (middle), and HBA (right) across all replicates; human serum albumin (HSA), hemoglobin beta subunit (HBB), hemoglobin alpha subunit (HBA).
However, the TICs showed inconsistency across samples, suggesting the workflow was not free of signal suppression, evident when the normalization to TIC improved the peptide peak area consistency; note the right panels in Figure 1A (HSA peptides), B (HBB peptides), and C (HBA peptides) and CV as the median of 19.3% across all hemoglobin and HSA peptides with normalization to TIC. The retention time of peptides was consistent across samples, depicted in Figure 1D for HSA, HBB, and HBA peptides, respectively. Lastly, HBB and HBA peptides were consistently detected in all samples with the exception of the HSA blank sample, as expected. The observed retention times in the HSA blank sample were from baseline noise selected for transitions in the absence of peptide signals. However, no linear relationship was observed for hemoglobin peptides with the increasing number of RBC input, suggesting either inaccuracy in the quantification of the number of RBC counts via microscopy and/or the peptide peak areas were below the limit of quantification when measured by DIA and/or DDA, possibly due to reduced signal-to-noise, which was adequate for detection but not quantification.
3.2.1. Endogenous Measure of Peptide Targets Using FACS and autoPOTS
Results pertaining to peptide standard synthesis and IDMS-PRM assay development including, LOD, LOQ, repeatability, and selectivity, are provided in Supplemental Figures S8–S13. Three approaches were used to determine the number of RBCs that would be required for repeatable measure of endogenous peptides (Figure 2 and Figure 3). In one approach FACS was used to isolate RBCs at three different absolute numbers; 5, 50, and 250 RBCs. RBCs were isolated in four replicates directly into autoPOTS and samples prepared as previously described10 with endogenous peptide targets measured via isotope dilution PRM (Figure 2A). A blank containing no RBCs served to assess background signal, which can be especially high for hemoglobin peptides since the high abundance of the protein renders many analytical instrumentations “contaminated” when used to measure blood specimens. Not surprisingly, background signal, measured as peak area ratio to labeled peptide standard counterparts spiked-in, was observed for VHLTPEEK and to a lesser extent for CM-VHLTPEEK when measuring samples prepared by the autoPOTS protocol, see Figure 2A PRM chromatograms labeled RBC = 0. However, endogenous peak area ratios were orders of magnitude higher for VHLTPEEK and significantly higher for CM-VHLTPEEK; see Figure 2A PRM chromatograms labeled RBC = 5, RBC = 50, and RBC = 250. Peak area ratios were then plotted against the number of RBC input and linear regression analysis revealed R-Squared values >0.95; see Figure 2A scatter plot. Using the equation of the line the endogenous concentrations were interpolated as 1144 amol VHLTPEEK and 7.53 amol of CM-VHLTPEEK per RBC, on average.
Figure 2.

Endogenous measure of peptide targets using near-single-cell and single-cell preparations. Panel A displays data generated at Site 2 using PRM to measure endogenous peptides prepared via the previously described autoPOTS workflow.10 RBCs were deposited directly into autoPOTS, in four replicates, aiming to deliver 0 (control background), 5, 50, and 250 RBCS which were subsequently processed for PRM using the autoPOTS workflow. The PRM chromatograms are representative of one of four replicate preparations and are labeled accordingly with the peptide targets. Data presented in panels B and C were generated at site 1 using PRM to measure endogenous peptide targets prepared with the one-pot rapid digestion protocol. Limiting dilution was used to isolate single RBCs in triplicate (RBC = 1 R1 through to RBC = 1 R3) along with triplicate control no RBC inputs (RBC = 0 R1 through to RBC = 0 R3). Single RBC isolation was confirmed via phase contrast microscopy at 100× magnification and can be identified in the images via the yellow arrow. Samples were prepared via one-pot rapid digestion and the resulting PRM chromatograms for both peptide targets are provided below the associated well (image) and labeled accordingly. Normalized peak area ratios (to CM-SIL and SIL spiked in at 2 fmol total) are displayed in panel C revealing significantly higher signals for peptide targets in wells containing single RBC compared to the no cell input wells.
Figure 3.

Endogenous analyte concentration in RBCs isolated via CellenONE and one-pot rapid digestion sample preparation. RBCs were isolated directly into OP384 wells and the resulting droplets viewed under 100× magnification using phase contrast microscopy and images are displayed in panel A. Panels B and C display PRM chromatograms for CM-VHLTPEEK and VHLTPEEK peptides with total endogenous signal and CM-SIL and SIL peptide chromatograms overlaid. The CM-SIL and SIL peptides were spiked in at 2 fmol total. Panel D displays the scatter plot of the endogenous peptide signal as peak area ratio normalized to counterpart CM-SIL and SIL peptides plotted against the number of input RBCs.
3.2.2. Endogenous Measure of Peptide Targets Using Limiting Dilution and One-Pot Rapid Digestion
The second approach used to measure endogenous peptides utilized limiting dilution to isolate RBCs into OP384 plates, after which samples were prepared for MS analysis using one-pot rapid digestion and analyzed via isotope dilution PRM (Figure 2B). As with the autoPOTS approach, control samples were included to measure background signal; see images and associated PRM chromatograms labeled RBC = 0 (Figure 2B). Background signal, measured as peak area ratio to spiked-in counterpart CM-SIL and SIL peptides, was observed for VHLTPEEK but not for CM-VHLTPEEK (Figure 2B panels labeled RBC = 0 R1, R2, and R3). For single RBCs isolated via limiting dilution, images are displayed in Figure 2B and labeled as RBC = 1 R1, R2, and R3 and can be seen as shown by the yellow arrow in each image. The associated PRM chromatograms for each image are displayed below the image as is the peak area ratios to heavy labels in bar graphs in Figure 2C. The VHLTPEEK signal was observed at more than an order of magnitude higher than the background, suggesting the one-pot rapid digestion protocol was suitable for endogenous measure of this peptide from a RBC. In contrast, the signal for CM-VHLTPEEK, while above the LOD and therefore higher than background signal, showed poor peak shape and inconsistent transition detection, indicating the measure of this peptide with acceptable repeatability would require more than one RBC. Lastly, single-point estimation of endogenous peptide concentrations (i.e., 2 fmol of heavy peptide standard spike-in) showed 1650 amol, 1840 amol, and 4260 amol of VHLTPEEK peptide across the three replicates, significantly higher than the interpolation value obtained via FACS-autoPOTS, suggesting inaccuracy in the limiting dilution approach for a single RBC in an OP384 well. This final point presents a notable limitation when using the limiting dilution approach in an attempt to isolate single RBCs.
3.2.3. Endogenous Measure of Peptide Targets Using CellenONE and One-Pot Rapid Digestion
The third approach tested for measuring endogenous peptides used the CellenONE instrument to isolate single RBCs directly into OP384 wells that were then subjected to one-pot rapid digestion sample preparation and subsequent isotope dilution PRM analysis (Figure 3). The highest concentration of carrier HSA that would not reduce the stability of the CellenONE droplet formation was empirically determined to be 2.22%. However, at this concentration, the RBCs were not identifiable in OP384 wells (Figure 3A) and RBC enumeration was reliant on the onboard imaging instrumentation of the CellenONE (Supplemental Figure S6). Alternatively or in parallel, subsequent interpolation of endogenous hemoglobin protein concentration could be used as a measure of RBC number since the mean corpuscular hemoglobin reference range has previously been established.47 Indeed, the latter approach revealed 1320 amol, 1080 amol, and 1310 amol of VHLTPEEK peptide per RBC equivalent to 660 amol, 540 amol, and 655 amol hemoglobin tetramer when single RBCs were measured (Figure 3), which was comparable to the MCH reference range of 390–540 amol/RBC.
While the endogenous VHLTPEEK signal was robust in single RBC input samples (Figure 3C,D), the same analysis showed below the LOD signal for CM-VHLTPEEK (Figure 3B,D). When the number of RBC deposited was >1, a signal was above the LOD (Figure 3C) but linear regression analysis of the scatter plot showed an R-Squared of 0.53 in the 2–5 RBC input range, suggesting 5 or >5 RBCs would be required for CM-VHLTPEEK measurement. This was corroborated by the R-Squared value of 0.95 when >5 RBCs were used (Figure 2A right-most panel). Lastly, while the CellenONE and one-pot rapid digestion protocol were applicable for single-cell peptide quantification by isotope dilution PRM, dispersion in the measurements (R-Squared values in Figure 3) suggested reliable RBC counting was achieved by measuring the total hemoglobin content rather than relying on optical counts.
3.3. One-Pot Isotope Dilution PRM to Assess RBC Heterogeneity
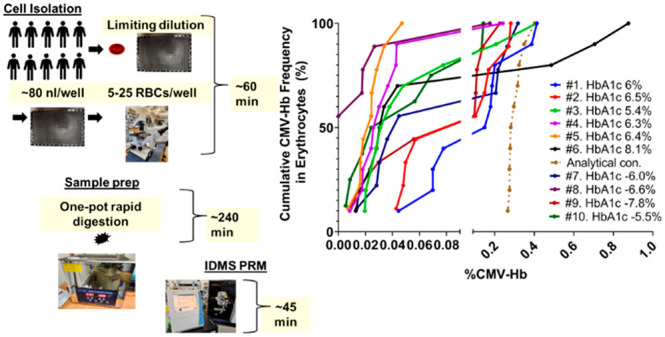
The results of the analytic LOD assessment, described in Supplemental Figure S11, and endogenous measurements, described in section 3.2, suggested the one-pot rapid digestion protocol would be suitable for endogenous measurement of CM-VHLTPEEK when the RBC input exceeded five cells. Since the assay could not be used to measure RBC heterogeneity, defined as the endogenous ratio of CM-VHLTPEEK to VHLTPEEK, using the single-cell approach, a small sample size approach was attempted via repeated sampling of specimens aiming to analyze 5–25 RBCs at each repeated measure. Initial endogenous measurements using the FACS-autoPOTS approach, described in section 3.2, suggested repeated measures would need to exceed N = 4. Therefore, it was decided to increase repeated measures to N = 10 per specimen to assess whether RBC heterogeneity could be evaluated. Therefore, a total of 50 to 250 RBCs were measured per individual and since absolute measurements were used to measure the ratio of CM-VHLTPEEK to VHLTPEEK the variability in the number of RBC input, which ranged from 5 to 25, was not considered as a variable in measurement.
Figure 4 depicts the results of repeated measures presented as a violin plot in the left panel. Details of the blood specimen collection and chronology are provided in Supplemental Document 1, and donor HbA1c values (range of 5.5% to 8.1%) were obtained from medical files (confidentially and known only by the clinician). Constricted by the time frame for sample collection, which occurred over the course of >90 days, samples were analyzed in two batches: 1) specimens 1–6 and 2) subsequently specimens 7–10. Data analysis, presented as violin plots, suggested increased variability associated with larger y-axis values. The coefficient of variation of repeated measures for each specimen was in the range of 47%–215% while repeated measures of peptide standards at concentrations within the range of endogenous values was 14%. The shape of violin plots was suggestive of multimodality, pronounced in specimens 1, 2, 6, and 7. Lastly, the lack of correlation when comparing bulk sample measurements to the mean %CM-VHLTPEEK in low input samples (Figure 4 right panel) suggested the sample size of 5–25 RBCs was small enough for measuring %CM-VHLTPEEK distribution.
Figure 4.

One-pot rapid digestion isotope dilution PRM to measure RBC heterogeneity. The heights of the violin plots depict the variance across repeated measures while the shapes are indicative of (multi)modality. The solid line in each plot represents medians and dashed lines represent quartiles. The corresponding HbA1c values, in order from individual 1 to 10, were: 6.0%, 6.5%, 5.4%, 6.3%, 6.4%, 8.1%, 6.0%, 6.6%, 7.8%, and 5.5%. The right panel depicts a scatter plot comparing %CM-VHLTPEEK measured via MRM in bulk sample preparations to the mean %CM-VHLTPEEK when measured in low input (5–25 RBCs) samples via PRM. Linear regression showed no correlation with an R-Squared value of 0.34.
4. Discussion
Isolation of RBCs for subsequent sample preparation was achieved with FACS, limiting dilution, and CellenONE, but only the latter showed capability for reproducible single RBC isolation. Definitive RBC counts in OP384 wells required the use of HSA that offered the unique advantage of flexible storage of isolated cells and could serve a practical advantage for shipping samples between laboratories or from collection of samples at one location with their subsequent analysis at a second location. However, the use of HSA presented disadvantages of potential interference during MS analysis and increased the dynamic range in protein concentration that could reduce sensitivity. In contrast, HSA served as a carrier minimizing absorptive losses, a significant factor leading to reduced sensitivity in single-cell and low cell number proteomics.
A one-pot rapid digestion protocol was also developed in this study and was used for processing single RBC’s and up to 25 RBCs per input in ∼1.5 h for subsequent LC-MS analysis. Consistent peak areas and shapes corresponding to endogenous trypsin/LysC digested peptides and compatibility with nano-LC, measured as consistent system back pressure, suggested this method was suitable for quantitative bottom-up proteomics. Furthermore, stability analysis showed the method did not compromise (breakdown) primary amine carboxymethylation and could therefore be used for measuring carboxymethylated peptides.
Synthesis of peptide standards to measure the N-terminal valine carboxymethylation of HBB is also reported here for the first time. Carboxymethylation was characterized as 58+ Dalton mass shift would be localized to the N-terminal valine based on the presence of representative fragment ions, including mass shifts for a, b, and c ions when monitored via MRM (used in measurements made by QQQ-MS) and PRM (used in measurements made by Q-orbitrap MS). Using these peptide standards, the absolute concentration of endogenous counterpart was interpolated in single RBCs at <10 amol, while that for the unmodified peptide was measured at a concentration comparable to the SI reference range for the hemoglobin tetramer. Both measurements are the first of their kind in single RBCs. The use of a peptide standard also identified an analyte of interest with a comparable precursor mass to CM-VHLTPEEK that was gated for fragmentation during both PRM and MRM and shared many of the product ions with the target of interest, including 58+ Dalton mass shifts in a, b, and c ions. This unknown analyte of interest may be a potential target for future study and may be misidentified as CM-VHLTPEEK in studies that do not utilize a peptide standard.
As discussed above, the isotope dilution PRM assay developed in this study could be used to measure endogenous targets in single RBCs where zeptomole sensitivity was achieved when using autoPOTS and low attomole sensitivity was achieved when using one-pot rapid digestion. While VHLTPEEK could be measured (robustly) in single RBCs, the one-pot rapid digestion protocol required >5 RBCs to measure CM-VHLTPEEK. However, when repeated sampling of specimens was performed aiming to analyze 5 to 25 RBC per measurement, the heterogeneous distribution of CM-VHLTPEEK (across an RBC population) was achievable, suggesting this approach as an alternative to single-cell measurements for assessment of RBC heterogeneity.
The limit of quantification reported in this study and assay throughput can likely be improved. Assay sensitivity was observed to be enhanced using a smaller ID analytical column (75 μm compared to 30 μm) and when combined with a 1/9 split before the analytical column, a 10-fold improvement in sensitivity was achieved, approaching zeptomole sensitivity. However, recent progress in capillary electrophoresis MS48 suggests both assay sensitivity and throughput can be enhanced for single-cell protein quantification.
5. Conclusions
We demonstrate a one-pot rapid digestion method for use with a simplified autoPOTS process that can be carried out with common laboratory instrumentation for bottom-up proteomics with quantitative sensitivity in the attomole range. This simplified autoPOTS process allowed reproducible quantification even in single cells when combined with isotope dilution PRM. Using this approach, a surrogate hemoglobin peptide was quantified in single RBCs revealing a single-cell concentration comparable to the SI reference range for mean corpuscular hemoglobin concentration. Lastly, this study showed empirical evidence that RBC heterogeneity, measured as the ratio of CM-VHLTPEEK to VHLTPEEK, could be assessed via repeated small sample size (5–25 RBCs) sampling of specimens as an alternative, more accessible strategy to single-cell analysis. A noted limitation of our study was that there may be a more suitable peptide target in RBCs for measuring cellular heterogeneity. This peptide target had nearly identical precursor mass as CM-VHLTPEEK and shared many product ions but eluted slightly earlier and at a significantly higher signal intensity. Lastly, we caution against generalization when assessing the performance discrepancy of the CellenONE instrument and FACSAria based on the observations in this study, where single erythrocyte isolation was only successful with the former. Other studies have demonstrated FACS functional for single-cell isolation, and these instruments will likely continue to serve as an option for single-cell sorting.
Acknowledgments
The authors acknowledge Vagisha Sharma, affiliated with the University of Washington, for assisting with data deposition to Panorama Public and ProteomeXchange. The authors also acknowledge the University of Victoria Human Research Ethics Board Michael Williams Building, R. B202 PO Box 1700 STN CSC Victoria, BC V8W 2Y2, Tel: 250-472-4545 for thorough review of the ethics application pertaining to this study. We are grateful for funding for technology development and platform support for The Pan-Canadian Proteomics Centre (PCPC), from Genome Canada, and Genome British Columbia through the Genomics Technology Platform (GTP) program for operations and technology development (264PRO). We are also grateful for support from Genome BC through the Sector Innovation Program (SIP7), and to The Partnership for Clean Competition (PCC).
Glossary
ABBREVIATIONS
- RBC
Red Blood Cell
- PRM
Parallel Reaction Monitoring
- LC
Liquid Chromatography
- MS
Mass Spectrometry
- SIL
Stable Isotope Label
- NAT
Natural Isotope Label
- CM
Carboxymethyl
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.2c00429.
Supplemental Document 2: MSFragger_log_file_for _DDA_search_log_2021–05–15_22–20–41 (TXT)
Supplemental Document 3: MSFragger_log_file_for _DIA_search_log_2021–05–15_22–20–41 (TXT)
Supplemental Document 4: MSFragger_log_file_for _DIA_speclibrary_log_2021–05–15_22–20–41(TXT)
Supplemental Document 5: MSFragger_parameters_for_searching_DDA_data (TXT)
Supplemental Document 6: MSFragger_parameters_for_searching_DIA_wide_window_data (TXT)
Supplemental Figures S1–S13 and Supplemental Document 1: Methods and results communicating; selective peptide standard N-terminal carboxymethylation, LOD and LOQ measurements, stability of CM-NAT and CM-SIL peptides in one-pot rapid digestion, selectivity and matrix effect. Detailed instrumentation parameters used for data acquisition, Figure S1, S2, S3 and S4. Single RBC isolation via limiting dilution Figure S5, CellenONE automated single RBC isolation Figure S6, nano-LC pressure profiles for samples prepared via one-pot rapid digestion, Figure S7, MALDI-TOF mass spectra of synthetic peptide standards Figure S8, MRM chromatograms fo synthetic peptide standards Figure S9, UV-HPLC chromatograms and MALDI-TOF mass spectra of purified peptide standards Figure S10, PRM assay chromatograms and LOD and LOQ measurements Figure S11, MRM chromatograms and peak areas of peptide standards subjected to the one-pot rapid digestion workflow Figure S12., assessment of selective measurement of endogenous CM-VHLTPEEK Figure S13. Details of the blood specimen collection, chronology, erythrocyte separation from whole blood and erythrocyte spotting in OP384 wells (PDF)
Author Contributions
The paper was drafted by Azad Eshghi and David Goodlett; all other authors provided suggestions, feedback, and edits. All authors have given approval to the final version of the paper.
The authors declare the following competing financial interest(s): Fabiana Izaguirre is an employee of Cellenion SASU.
Supplementary Material
References
- Leduc A.; Huffman R. G.; Cantlon J.; Khan S.; Slavov N. Exploring Functional Protein Covariation across Single Cells Using NPOP. Genome Biol. 2022, 23 (1), 261. 10.1186/s13059-022-02817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Clair G.; Chrisler W. B.; Shen Y.; Zhao R.; Shukla A. K.; Moore R. J.; Misra R. S.; Pryhuber G. S.; Smith R. D.; Ansong C.; Kelly R. T. Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew. Chem., Int. Ed. 2018, 57 (38), 12370–12374. 10.1002/anie.201802843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou M.; Clair G.; Tsai C. F.; Xu K.; Chrisler W. B.; Sontag R. L.; Zhao R.; Moore R. J.; Liu T.; Pasa-Tolic L.; Smith R. D.; Shi T.; Adkins J. N.; Qian W. J.; Kelly R. T.; Ansong C.; Zhu Y. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91 (20), 13119–13127. 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvillion S. P.; Zhu Y.; Nagy G.; Adkins J. N.; Ansong C.; Renslow R. S.; Piehowski P. D.; Ibrahim Y. M.; Kelly R. T.; Metz T. O. New Mass Spectrometry Technologies Contributing towards Comprehensive and High Throughput Omics Analyses of Single Cells. Analyst 2019, 144 (3), 794–807. 10.1039/C8AN01574K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellors J. S.; Jorabchi K.; Smith L. M.; Ramsey J. M. Integrated Microfluidic Device for Automated Single Cell Analysis Using Electrophoretic Separation and Electrospray Ionization Mass Spectrometry. Anal. Chem. 2010, 82 (3), 967–973. 10.1021/ac902218y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J.; Zheng X.; Xu P.; Xu J.; Cao C.; Fu Y.; Xu B.; Dai X.; Wang Y.; Liu H.; Yi Q.; Zhu Y.; Wang J.; Wang L.; Dong Z.; Huang L.; Huang Y.; Du W. Interfacial Nanoinjection-Based Nanoliter Single-Cell Analysis. Small 2020, 16, e1903739 10.1002/smll.201903739. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Scheibinger M.; Ellwanger D. C.; Krey J. F.; Choi D.; Kelly R. T.; Heller S.; Barr-Gillespie P. G.. Single-Cell Proteomics Reveals Changes in Expression during Hair-Cell Development. Elife 2019, 8. 10.7554/eLife.50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Xu H.; Wei X.; He H. Single-Cell Detection and Photostimulation on a Microfluidic Chip Aided with Gold Nanorods. Cytom. A 2020, 97, 39. 10.1002/cyto.a.23855. [DOI] [PubMed] [Google Scholar]
- Weke K.; Singh A.; Uwugiaren N.; Alfaro J. A.; Wang T.; Hupp T. R.; O’Neill J. R.; Vojtesek B.; Goodlett D. R.; Williams S. M.; Zhou M.; Kelly R. T.; Zhu Y.; Dapic I. MicroPOTS Analysis of Barrett’s Esophageal Cell Line Models Identifies Proteomic Changes after Physiologic and Radiation Stress. J. Proteome Res. 2021, 20 (5), 2195–2205. 10.1021/acs.jproteome.0c00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Acor H.; McCown M. A.; Nwosu A. J.; Boekweg H.; Axtell N. B.; Truong T.; Cong Y.; Payne S. H.; Kelly R. T. Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling. Anal. Chem. 2021, 93 (3), 1658–1666. 10.1021/acs.analchem.0c04240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Piehowski P. D.; Zhao R.; Chen J.; Shen Y.; Moore R. J.; Shukla A. K.; Petyuk V. A.; Campbell-Thompson M.; Mathews C. E.; Smith R. D.; Qian W. J.; Kelly R. T. Nanodroplet Processing Platform for Deep and Quantitative Proteome Profiling of 10–100 Mammalian Cells. Nat. Commun. 2018, 9 (1), 882. 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161. 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petelski A. A.; Emmott E.; Leduc A.; Huffman R. G.; Specht H.; Perlman D. H.; Slavov N. Multiplexed Single-Cell Proteomics Using SCoPE2. Nat. Protoc. 2021, 16 (12), 5398–5425. 10.1038/s41596-021-00616-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J.; Williams S. M.; Markillie L. M.; Feng S.; Tsai C.-F.; Aguilera-Vazquez V.; Sontag R. L.; Moore R. J.; Hu D.; Mehta H. S.; Cantlon-Bruce J.; Liu T.; Adkins J. N.; Smith R. D.; Clair G. C.; Pasa-Tolic L.; Zhu Y. High-Throughput and High-Efficiency Sample Preparation for Single-Cell Proteomics Using a Nested Nanowell Chip. Nat. Commun. 2021, 12 (1), 6246. 10.1038/s41467-021-26514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. A.; Rhoads A.; Cocco A. R.; Peckner R.; Haber A. L.; Schweitzer L. D.; Krug K.; Mani D. R.; Clauser K. R.; Rozenblatt-Rosen O.; Hacohen N.; Regev A.; Carr S. A. Streamlined Protocol for Deep Proteomic Profiling of FAC-Sorted Cells and Its Application to Freshly Isolated Murine Immune Cells*. Mol. Cell. Proteomics 2019, 18 (5), 995–1009. 10.1074/mcp.RA118.001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Palma S.; Stange D.; van de Wetering M.; Clevers H.; Heck A. J. R.; Mohammed S. Highly Sensitive Proteome Analysis of FACS-Sorted Adult Colon Stem Cells. J. Proteome Res. 2011, 10 (8), 3814–3819. 10.1021/pr200367p. [DOI] [PubMed] [Google Scholar]
- Hughes C. S.; Foehr S.; Garfield D. A.; Furlong E. E.; Steinmetz L. M.; Krijgsveld J. Ultrasensitive Proteome Analysis Using Paramagnetic Bead Technology. Mol. Syst. Biol. 2014, 10, 757. 10.15252/msb.20145625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf E. L.; Pellegrini D.; McDonnell L. A. Set of Novel Automated Quantitative Microproteomics Protocols for Small Sample Amounts and Its Application to Kidney Tissue Substructures. J. Proteome Res. 2016, 15 (12), 4722–4730. 10.1021/acs.jproteome.6b00889. [DOI] [PubMed] [Google Scholar]
- Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes upon Perturbation. Mol. Syst. Biol. 2022, 18 ( (3), ) e10798. 10.15252/msb.202110798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R. M.; Franco R. S.; Khera P. K.; Smith E. P.; Lindsell C. J.; Ciraolo P. J.; Palascak M. B.; Joiner C. H. Red Cell Life Span Heterogeneity in Hematologically Normal People Is Sufficient to Alter HbA1c. Blood 2008, 112 (10), 4284–4291. 10.1182/blood-2008-04-154112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard L. C. Action of Amino Acids on Sugars. Formation of Melanoidins in a Methodical Way. Compte-Rendu Acad. Sci. 1912, 154, 66–68. [Google Scholar]
- Wells-Knecht K. J.; Zyzak D. V.; Litchfield J. E.; Thorpe S. R.; Baynes J. W. Mechanism of Autoxidative Glycosylation: Identification of Glyoxal and Arabinose as Intermediates in the Autoxidative Modification of Proteins by Glucose. Biochemistry 1995, 34 (11), 3702–3709. 10.1021/bi00011a027. [DOI] [PubMed] [Google Scholar]
- Shapiro R.; McManus M. J.; Zalut C.; Bunn H. F. Sites of Nonenzymatic Glycosylation of Human Hemoglobin A. J. Biol. Chem. 1980, 255 (7), 3120–3127. 10.1016/S0021-9258(19)85860-X. [DOI] [PubMed] [Google Scholar]
- Muralidharan M.; Bhat V.; Mandal A. K. Structural Analysis of Glycated Human Hemoglobin Using Native Mass Spectrometry. FEBS J. 2020, 287 (6), 1247–1254. 10.1111/febs.15085. [DOI] [PubMed] [Google Scholar]
- Peterson K. P.; Pavlovich J. G.; Goldstein D.; Little R.; England J.; Peterson C. M. What Is Hemoglobin A1c? An Analysis of Glycated Hemoglobins by Electrospray Ionization Mass Spectrometry. Clin. Chem. 1998, 44 (9), 1951–1958. 10.1093/clinchem/44.9.1951. [DOI] [PubMed] [Google Scholar]
- Wang S.-H.; Wang T.-F.; Wu C.-H.; Chen S.-H. In-Depth Comparative Characterization of Hemoglobin Glycation in Normal and Diabetic Bloods by LC-MSMS. J. Am. Soc. Mass Spectrom. 2014, 25 (5), 758–766. 10.1007/s13361-014-0830-2. [DOI] [PubMed] [Google Scholar]
- Peterson A. C.; Russell J. D.; Bailey D. J.; Westphall M. S.; Coon J. J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteomics MCP 2012, 11 (11), 1475–1488. 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S.; Bourmaud A.; Kim S. Y.; Domon B. Technical Considerations for Large-Scale Parallel Reaction Monitoring Analysis. J. Proteomics 2014, 100, 147–159. 10.1016/j.jprot.2013.10.029. [DOI] [PubMed] [Google Scholar]
- Gallien S.; Duriez E.; Demeure K.; Domon B. Selectivity of LC-MS/MS Analysis: Implication for Proteomics Experiments. J. Proteomics 2013, 81, 148–158. 10.1016/j.jprot.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Heil L. R.; Remes P. M.; MacCoss M. J. Comparison of Unit Resolution Versus High-Resolution Accurate Mass for Parallel Reaction Monitoring. J. Proteome Res. 2021, 20 (9), 4435–4442. 10.1021/acs.jproteome.1c00377. [DOI] [PubMed] [Google Scholar]
- Jagadeeshaprasad M. G.; Batkulwar K. B.; Meshram N. N.; Tiwari S.; Korwar A. M.; Unnikrishnan A. G.; Kulkarni M. J. Targeted Quantification of N-1-(Carboxymethyl) Valine and N-1-(Carboxyethyl) Valine Peptides of β-Hemoglobin for Better Diagnostics in Diabetes. Clin Proteomics 2016, 13, 7. 10.1186/s12014-016-9108-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura T.; Nakano K.; Hashiguchi T.; Iwamoto H.; Miura K.; Yoshimura Y.; Hanyu N.; Hirata K.; Imakuma M.; Motomiya Y.; Maruyama I. Elevation of N-(Carboxymethyl)Valine Residue in Hemoglobin of Diabetic Patients. Its Role in the Development of Diabetic Nephropathy. Diabetes Care 2001, 24 (5), 891–896. 10.2337/diacare.24.5.891. [DOI] [PubMed] [Google Scholar]
- Shimada S.; Tanaka Y.; Ohmura C.; Tamura Y.; Shimizu T.; Uchino H.; Watada H.; Hirose T.; Nakaniwa T.; Miwa S.; Kawamori R. N-(Carboxymethyl)Valine Residues in Hemoglobin (CMV-Hb) Reflect Accumulation of Oxidative Stress in Diabetic Patients. Diabetes Res. Clin. Pract. 2005, 69 (3), 272–278. 10.1016/j.diabres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Suttapitugsakul S.; Xiao H.; Smeekens J.; Wu R. Evaluation and Optimization of Reduction and Alkylation Methods to Maximize Peptide Identification with MS-Based Proteomics. Mol. Biosyst 2017, 13 (12), 2574–2582. 10.1039/C7MB00393E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle B. C.; Swearingen K. E.; Barnes C. A.; Schmidt T.; Gessulat S.; Küster B.; Wilhelm M. Generating High Quality Libraries for DIA MS with Empirically Corrected Peptide Predictions. Nat. Commun. 2020, 11 (1), 1548. 10.1038/s41467-020-15346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr S. A.; Abbatiello S. E.; Ackermann B. L.; Borchers C.; Domon B.; Deutsch E. W.; Grant R. P.; Hoofnagle A. N.; Hüttenhain R.; Koomen J. M.; Liebler D. C.; Liu T.; MacLean B.; Mani D. R.; Mansfield E.; Neubert H.; Paulovich A. G.; Reiter L.; Vitek O.; Aebersold R.; Anderson L.; Bethem R.; Blonder J.; Boja E.; Botelho J.; Boyne M.; Bradshaw R. A.; Burlingame A. L.; Chan D.; Keshishian H.; Kuhn E.; Kinsinger C.; Lee J. S. H.; Lee S.-W.; Moritz R.; Oses-Prieto J.; Rifai N.; Ritchie J.; Rodriguez H.; Srinivas P. R.; Townsend R. R.; Van Eyk J.; Whiteley G.; Wiita A.; Weintraub S. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-Based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Proteomics MCP 2014, 13 (3), 907–917. 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzyk M. A.; Smith D.; Yang J.; Cross T. J.; Jackson A. M.; Hardie D. B.; Anderson N. L.; Borchers C. H. Multiple Reaction Monitoring-Based, Multiplexed, Absolute Quantitation of 45 Proteins in Human Plasma. Mol. Cell Proteomics 2009, 8 (8), 1860–1877. 10.1074/mcp.M800540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K.; Higashi T.; Sano H.; Jinnouchi Y.; Yoshida M.; Araki T.; Ueda S.; Horiuchi S. N (Epsilon)-(Carboxymethyl)Lysine Protein Adduct Is a Major Immunological Epitope in Proteins Modified with Advanced Glycation End Products of the Maillard Reaction. Biochemistry 1996, 35 (24), 8075–8083. 10.1021/bi9530550. [DOI] [PubMed] [Google Scholar]
- Han J.; Higgins R.; Lim M. D.; Lin K.; Yang J.; Borchers C. H. Short-Term Stabilities of 21 Amino Acids in Dried Blood Spots. Clin. Chem. 2018, 64 (2), 400–402. 10.1373/clinchem.2017.278457. [DOI] [PubMed] [Google Scholar]
- Eshghi A.; Pistawka A. J.; Liu J.; Chen M.; Sinclair N. J. T.; Hardie D. B.; Elliott M.; Chen L.; Newman R.; Mohammed Y.; Borchers C. H. Concentration Determination of > 200 Proteins in Dried Blood Spots for Biomarker Discovery and Validation. Mol. Cell Proteomics 2020, 19 (3), 540–553. 10.1074/mcp.TIR119.001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud S. A.; Sinclair N. J.; Pětrošová H.; Palmer A. L.; Pistawka A. J.; Zhang S.; Hardie D. B.; Mohammed Y.; Eshghi A.; Richard V. R.; Sickmann A.; Borchers C. H. Molecular Phenotyping of Laboratory Mouse Strains Using 500 Multiple Reaction Monitoring Mass Spectrometry Plasma Assays. Commun. Biol. 2018, 1 (1), 78. 10.1038/s42003-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker J. R.; Halusa G. N.; Hoofnagle A. N.; Sharma V.; MacLean B.; Yan P.; Wrobel J. A.; Kennedy J.; Mani D. R.; Zimmerman L. J.; Meyer M. R.; Mesri M.; Rodriguez H.; Clinical Proteomic Tumor Analysis C.; Paulovich A. G. CPTAC Assay Portal: A Repository of Targeted Proteomic Assays. Nat. Methods 2014, 11 (7), 703–704. 10.1038/nmeth.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Eckels J.; Schilling B.; Ludwig C.; Jaffe J. D.; MacCoss M. J.; MacLean B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteomics MCP 2018, 17 (6), 1239–1244. 10.1074/mcp.RA117.000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A. T.; Leprevost F. V.; Avtonomov D. M.; Mellacheruvu D.; Nesvizhskii A. I. MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat. Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou C.-C.; Avtonomov D.; Larsen B.; Tucholska M.; Choi H.; Gingras A.-C.; Nesvizhskii A. I. DIA-Umpire: Comprehensive Computational Framework for Data-Independent Acquisition Proteomics. Nat. Methods 2015, 12 (3), 258–264. 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinforma. Oxf. Engl. 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill V. L.; Simpson V. Z.; Higgins J. M.; Hu Z.; Stevens R. A.; Metcalf J. A.; Baseler M. Evaluation of the Performance of the Sysmex XT-2000i Hematology Analyzer With Whole Blood Specimens Stored at Room Temperature. Lab. Med. 2009, 40 (12), 709–718. 10.1309/T0FJYP2RBXEHX4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B.; Pade L. R.; Choi S. B.; Muñoz-LLancao P.; Manzini M. C.; Nemes P. Capillary Electrophoresis Mass Spectrometry for Scalable Single-Cell Proteomics. Front. Chem. 2022, 10, 863979. 10.3389/fchem.2022.863979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.