

Abstract
Endogenous metabolites play essential roles in the regulation of cellular activity through their interactions with genes, transcripts and proteins. The potential for exploiting metabolomics combined with systems biology to enhance biotechnological applications and protein expression is a relatively unexplored area, although there are some notable recent examples. The bacterial generation of pertussis toxin protein for vaccine production was recently enhanced using genome-wide metabolic models iteratively coupled with metabolomics data; nucleobases and nucleosides were identified as substantial sinks of nitrogen. When the corresponding metabolites were used to compensate for this sink, a new understanding of the organism’s physiology and regulatory network allowed the efficiency of vaccine production to be increased 2.4-fold. Beyond commercial protein production, modulating protein levels to drive in vivo activity also has the potential for phenotypic modulation. For example the differentiation and maturation of oligodendrocyte precursor cells to a functional oligodendrocyte state, as well as the corresponding role of this process in the remyelination repair process that is required for disease remission in multiple sclerosis, is critically dependent on the production of myelin proteins including myelin basic protein (MBP). With the goal of identifying metabolites capable of enhancing oligodendrocyte formation and/or function, metabolomics analysis was performed on the differentiation process and the data was paired with previously developed systems level knowledge. As a result, taurine was identified and, when coupled with a known differentiation-inducing drug (e.g., miconazole), was found to amplify MBP expression by 2.5-fold and facilitate myelination. Overall, the traditional view of metabolites as being biological end products most suited for biomarker discovery is being upended as we discover new roles of these downstream products as upstream regulators; thus metabolite-induced protein expression is a relatively untapped area that can be explored in almost every aspect of biochemistry and bioengineering.
Systems Biology and Metabolomics
Protein expression is intimately linked with the genome and the genetic information transmitted downstream, however, under-appreciated is the role metabolomics and metabolites can play as a driving force in these upstream events. For example, the post-genome era is characterized by the rise of transcriptomics, proteomics and metabolomics, all designed to elucidate the composition and, ultimately, the function behind their information content. In this context, the central dogma of biology that has shaped several generations of life scientists [1] is that genome-encoded information is being transmitted downwards to the transcriptome and proteome leading to changes within the metabolic pool, the metabolome. Therefore, it is not surprising that the metabolome has been primarily linked with phenotypic changes while the transcriptome and proteome are seen as control centers. This downstream concept places metabolomics at the forefront of diagnostics and biomarker discovery, however minimizing metabolites’ and their more significant role as initiators of bioactivity. Integrating metabolomics within a systems biology framework facilitates the elucidation of metabolite’s upstream impact, and assists in the identification of biologically active metabolites that can promote protein expression and activity. While individual metabolites have been examined (e.g. transcription factors), systems biology has not yet been effectively combined with metabolomics to modulate systems wide activities.
An aim of systems biology is to model metabolic reactions and apply this information to control activity within a system. Traditionally these models have been generated using genomic data, with the recent integration of transcriptomic and proteomic information to improve their performance. However, while combining systems biology and metabolomics has been postulated [2] and to a limited extent applied (ref NBT in press) [3], relatively little effort has gone into its integration to help identify metabolites that can control activity, especially in the area of protein expression. We will focus on two recent reports that integrate metabolomics activity screening (MAS), systems, and experimental biology to identify metabolites that can control protein expression and production.
Nitrogen Metabolic Balance and Vaccine Production
A recent study by Branco dos Santos et al. [4] used a genome-wide metabolic model of the human pathogen Bordetella pertussis in order to increase pertussis vaccine production by more than a factor of two (Figure 2). Initially the systems biology based model of the pathogen revealed an apparent gap in the computer versus experimentally observed nitrogen metabolism. Constraint-based analysis techniques allowed a comprehensive accounting of mass flow in the organism, and a model for Bordetella pertussis was constructed and validated revealing a gap in the experimentally determined nitrogen output. Characterization of theoretical optimal flux distributions through the metabolic network was used to predict all possible nitrogen sinks compatible with the data; this list was subsequently used as a filter for untargeted metabolomics analysis of the growth media. Building an iterative cycle in which metabolomics was used to help decipher the mass balance of the simulations; the authors could improve/curate the original computer model. In this way, quite unexpectedly, nucleobases and nucleosides were identified as substantial novel sinks of nitrogen, at concentrations ranging up to the mM level.
Figure 2.
Schematic representation of the discussed examples: in the upper panel an increase of pertussis toxin production was produced using a combination of systems biology modelling and metabolomics. Lower panel presents how metabolomics combined with pathway analysis and an existing therapeutic helped discover taurine and its ability to impact myelin basic protein expression, an important protein in multiple sclerosis neuron regeneration [4, 5].
In addition to identifying the relevant nitrogen metabolite sinks; the models were also used to investigate sulphur balance. In the particular case of Pertussis Toxin (PT), it was known that sulphate inhibits PT production, and that sulphate accumulated in the traditional growth medium through excess methionine and glutathione (in the medium). The metabolic map provided an alternative pathway that was previously unknown for inorganic sulphur assimilation via thiosulphate; thus using thiosulphate as an alternative together with a careful balancing of amino acid requirements based on the metabolomics analysis of biomass composition; this led to a 2.4-fold increase in toxin production in a simpler medium. Thus, a careful metabolomics analysis of fermentations, combined with a systems biology approach, led to an out-of-the-box solution to improve productivity.
Taurine Amplified Myelin Basic Protein Expression
Recently the metabolomics was employed in combination with pathway analysis to identify metabolites that modulate the process of oligodendrocyte differentiation and/or maturation, an important regenerative process associated with demyelinating diseases [5]. Mass spectrometry-based metabolomics was used to investigate the mechanism, and whether endogenous metabolites could impact oligodendrocyte precursor cell (OPC) differentiation. Taurine and creatine pathways were found to be the most highly altered events associated with OPC differentiation. Taurine and hypotaurine increased by over 20 and 10-fold, respectively, with metabolites in the creatine pathway also upregulated by at least an order of magnitude. Quantitative analysis of associated metabolites was performed on both the upstream and downstream parts of the taurine and creatine pathways to examine the role of other metabolites associated with these pathways. When added exogenously at physiologically relevant concentrations, taurine was found to dramatically enhance drug-induced OPC differentiation and facilitate the in vitro myelination of co-cultured axons. Mechanistically, taurine-induced OPC differentiation- and myelination-enhancing activities appeared to be driven by taurine’s ability to increase serine levels, which is an initial building block required for the synthesis of the glycosphingolipid components of myelin. A key outcome in the taurine induced differentiation process was the increased generation of myelin basic protein (Figure 2) by 2.5 fold, a protein associated with the myelination of neurons.
Data Integration to identify metabolite bioactivity
The primary motivation for integrating metabolomics and systems biology has been in the generation of more comprehensive metabolic maps focusing primarily on altering metabolite levels, this coincides with recent reports indicating that our knowledge about metabolic biological activity is rapidly increasing [6]. However, the integration of such data has been slow moving, particularly considering that numerous metabolites exert important biological functions allowing them to shape the protein landscape, e.g. transcription factor control, GPCR activation, allosteric regulation, among others. We postulate that combining global metabolomics and systems biology data would provide information to manipulate protein expression and function through metabolic triggers, where metabolism could be harnessed to increase protein production or alter protein function.
While knowledge about receptors and signaling pathways related to specific metabolites is increasing, most modern metabolite databases, such as METLIN or HMDB, do not provide such information. A first step in the direction of providing metabolic activity has recently been described in the MAS approach (NBT in press) and incorporated into XCMS Online, where metabolite selection is made on multiple selection criteria including statistical significance, pathway analysis and multi-omics screening. However, as we are in the early stages a more sophisticated level of metabolite selection will likely evolve. Current system biology models concentrate on production of metabolites and flux with criteria that has yet to be fully understood including understanding how the proteins are made, their folding requirements and what the production inhibitory effects are. Ultimately, metabolomics guided systems biology based models will provide the necessary framework for the integration of all aforementioned approaches, in that it should allow predicting metabolic requirements for the production of key active metabolites. In turn, this will allow us to perturb metabolism and concurrently shape the proteomic landscape to our needs.
Metabolite-Induced Protein Expression (MIPE)
The examples described above underscore the utility of integrating metabolomics and systems biology to decipher and advance metabolite-induced protein expression (MIPE). Metabolites are a logical source to enhance and understand these biological systems especially as their bioactivities are becoming more apparent (NBT in press) as is the systems-wide role they are playing [2]. A significant challenge will be in deciphering which metabolites are best suited to alter the system, a challenge that will likely be solved by combining seemingly disparate information, including statistical analysis, pathway information, other omic technologies, and integrating this knowledge into existing databases. A task that is ripe for machine learning type approaches, once enough data is available to provide meaningful information. For example, combining genome wide metabolic models with experimental metabolomics data will likely lead to improved models and the identification of metabolites with unique activities. Most intriguing to us is that we are turning to these metabolic “end products” as the starting point to manipulate proteins (the presumed drivers of activity); maybe we should take a second look at who is behind the wheel.
Figure 1.
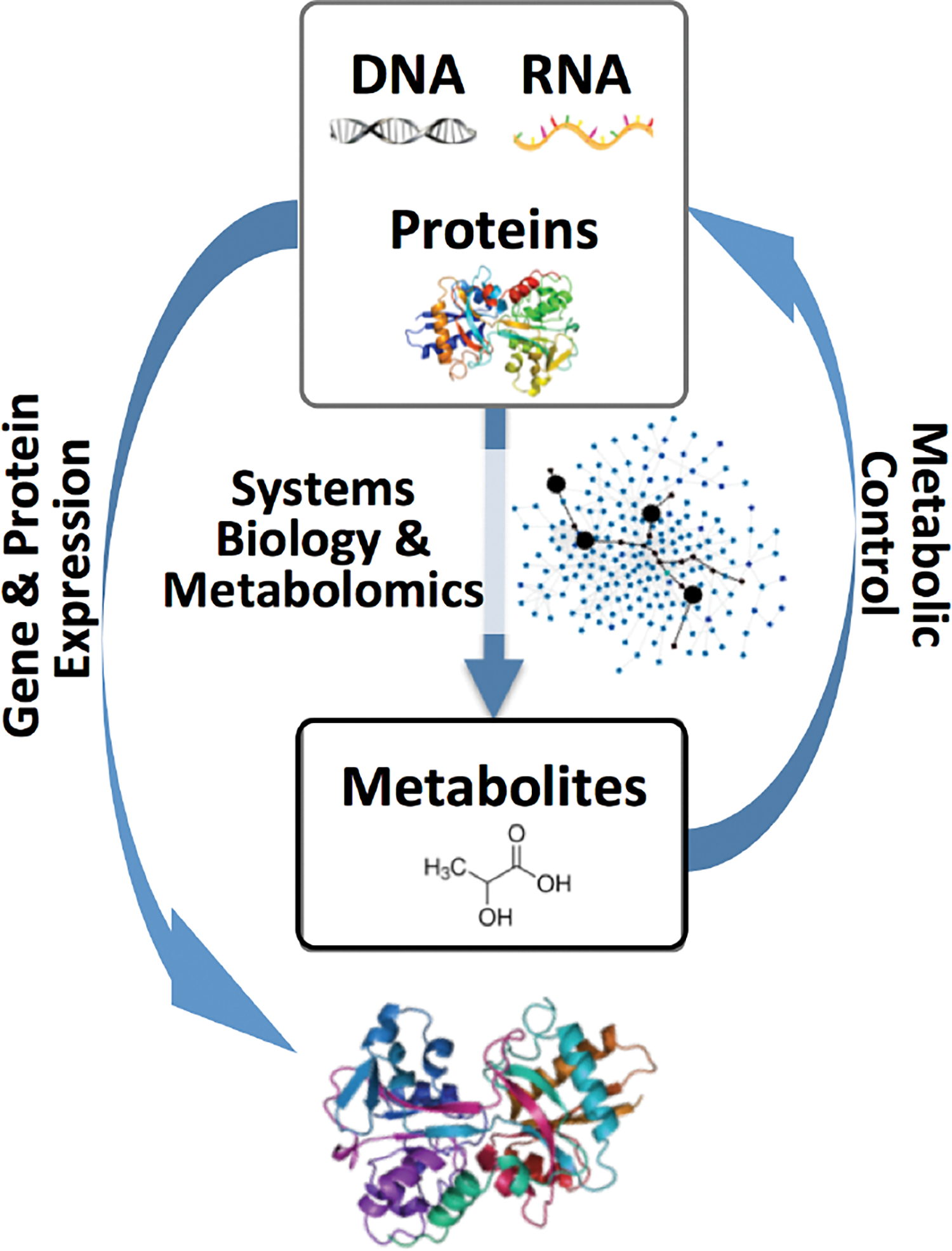
An overview of metabolite induced protein expression as guided by systems biology and metabolomics.
References
- [1].Crick F, Central Dogma of Molecular Biology, Nature, 227 (1970) 561. [DOI] [PubMed] [Google Scholar]
- [2].Huan T, Forsberg EM, Rinehart D, Johnson CH, Ivanisevic J, Benton HP, Fang M, Aisporna A, Hilmers B, Poole FL, Thorgersen MP, Adams MWW, Krantz G, Fields MW, Robbins PD, Niedernhofer LJ, Ideker T, Majumder EL, Wall JD, Rattray NJW, Goodacre R, Lairson LL, Siuzdak G, Systems biology guided by XCMS Online metabolomics, Nature Methods, 14 (2017) 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tomita M, Kami K, Systems Biology, Metabolomics, and Cancer Metabolism, Science, 336 (2012) 990–991. [DOI] [PubMed] [Google Scholar]
- [4].Branco dos Santos F, Olivier BG, Boele J, Smessaert V, De Rop P, Krumpochova P, Klau GW, Giera M, Dehottay P, Teusink B, Goffin P, Probing the Genome-Scale Metabolic Landscape of Bordetella pertussis, the Causative Agent of Whooping Cough, Applied and Environmental Microbiology, 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Beyer BA, Fang M, Sadrian B, Montenegro-Burke JR, Plaisted WC, Kok BPC, Saez E, Kondo T, Siuzdak G, Lairson LL, Metabolomics-based discovery of a metabolite that enhances oligodendrocyte maturation, Nature Chemical Biology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW, GPCR-Mediated Signaling of Metabolites, Cell Metabolism, 25 (2017) 777–796. [DOI] [PubMed] [Google Scholar]