

Abstract
Acute lymphoblastic leukaemia (ALL) is the most common cancer of childhood. Here, we map emerging evidence suggesting that children with ALL at the time of diagnosis may have a delayed maturation of the gut microbiome compared with healthy children. This finding may be associated with early-life epidemiological factors previously identified as risk indicators for childhood ALL, including caesarean section birth, diminished breast feeding and paucity of social contacts. The consistently observed deficiency in short-chain fatty-acid-producing bacterial taxa in children with ALL has the potential to promote dysregulated immune responses and to, ultimately, increase the risk of transformation of preleukaemic clones in response to common infectious triggers. These data endorse the concept that a microbiome deficit in early life may contribute to the development of the major subtypes of childhood ALL and encourage the notion of risk-reducing microbiome-targeted intervention in the future.
Subject terms: Acute lymphocytic leukaemia, Microbiome, Risk factors, Paediatric cancer
In this Perspective article, Mel Greaves and co-workers outline emerging evidence that suggests that children with newly diagnosed acute lymphoblastic leukaemia may have a delayed maturation of the gut microbiome compared with healthy children, a deficit that might be associated with early-life epidemiological factors and could contribute to the risk of transformation of preleukaemic clones in response to common infectious triggers.
Introduction
Acute lymphoblastic leukaemia (ALL) accounts for one-third of paediatric cancer cases in developed societies1. Although treatment of paediatric ALL is highly successful with cure rates of around 90%2, the longer-term complications of therapy and impact on quality of life during treatment are substantial. Two-thirds of childhood ALL survivors face severe morbidity for decades after their disease is eradicated, as well as 20 times higher mortality compared with their healthy, age-matched counterparts3. This largely unaccounted for burden of ALL justifies the key importance of ongoing research into its aetiology and pursuit of a longer-term goal of primary prevention4,5.
Research over recent decades has unravelled the natural history and clonal evolution of the main genetic subtypes of B cell precursor-ALL (BCP-ALL), which account for the majority of childhood ALL cases and have a peak incidence around the ages of 2–6 years. These observations have endorsed a two-stage model for this cancer originally termed the ‘delayed infection’ hypothesis6,7, which has features in common with the so-called hygiene hypothesis proposed for allergies and type 1 diabetes8. The first stage is manifested by genomic lesions arising in progenitor cells in utero leading to the development of clinically covert preleukaemic clones with only modest, proliferative advantage9. Backtracking of BCP-ALL cases with neonatal Guthrie cards and umbilical cord blood samples as well as comparative genomics in monozygotic twins with ALL have confirmed the prenatal origin of the predominant initiating chromosomal aberrations ETS translocation variant 6 (ETV6)::runt-related transcription factor 1 (RUNX1)10 and high hyperdiploidy11,12. However, as shown in mouse models13 and human umbilical cord blood samples14, these initiating events are not sufficient for leukaemic transformation15,16. ETV6::RUNX1 fusions (in-frame and in lymphocytes) are present in 1–5% of healthy newborn babies, but the overwhelming majority (~99%) will not develop leukaemia indicating low penetrance of the disease and the need for additional, postnatal mutational events14,17.
The ‘delayed infection’ hypothesis predicted that persistent preleukaemic clones acquire the essential, postnatal, secondary mutations as a result of a dysregulated immune response to common infections or chronic inflammation7. The nature and diversity of these ‘triggering’ infections for ALL remain uncertain, although respiratory viruses are implicated by epidemiological studies18,19. Experimental modelling data have highlighted possible mechanisms via which inflammatory cytokines might both expand preleukaemic clones and trigger the commonly observed secondary genetic changes20. The highly recurrent secondary genetic changes are primarily copy-number alterations (deletions) in genes elicited by off-target immunoglobulin heavy chain V(D)J recombination-activating protein (RAG) activity20. In this context, activation-induced cytidine deaminase (AID), which can be expressed in BCPs following repetitive strong inflammatory signals, has been shown to cooperate with RAG to result in genomic instability that can drive the evolution of pre-leukaemic clones13.
But, critically, the delayed infection model also predicted that the infection-driven dysregulated immune response triggering these crucial second hits was contingent upon a deficit of microbial exposure in infancy and a consequent failure of adequate immune network priming or maturation. Epidemiological evidence supports that contention via surrogate measures7. The risk of BCP-ALL is increased by caesarean section (C-section) birth21,22, brief or absent breastfeeding23,24 and paucity of social contacts during infancy25–27. We note that these social risk factors are shared with type 1 diabetes and allergies, raising the possibility of a common underlying immune priming deficit22. More recently, such early-life exposures were shown to have a profound impact on the acquisition and robustness of the neonatal and infant gut microbiome28–31, which, in turn, is recognized as fundamental to the maturation of the naive immune network of infants32. This led to the suggestion that the key risk factor of microbial underexposure (or ‘delay’) in BCP-ALL resides in the pivotal role of the microbiome and the prediction of a deficient gut microbiome in patients who develop BCP-ALL4,7.
Recent longitudinal studies have now revealed that a delayed maturation of the gut microbiome by the age of 12 months is associated with an increased risk of asthma diagnosis by 5 years of age33–35. Although the emerging role of the gut microbiome in the pathogenesis of childhood ALL has been discussed in recent review articles5,7,36–41, the rarity of the disease has thus far precluded any prospective longitudinal studies. Here, we comprehensively analyse the findings of existing case–control gut microbiome studies of childhood ALL at the time of diagnosis in the context of newly discovered maturation patterns of the gut microbiome. We discuss how early-life exposures associated with an increased risk of childhood ALL can induce gut microbiome instability and perturb its maturation, which in turn can jeopardize the integrity of the immune network. Finally, we propose methods to further delineate the role of the gut microbiome in BCP-ALL pathogenesis in future clinical trials and mouse models in the era of rapidly evolving gut microbiome research.
Conservation of gut microbiome maturation
Longitudinal studies support the existence of distinct phases of gut microbiome development during early childhood30,42,43 (Fig. 1a). The neonatal gut microbiome, which is primarily shaped by the maternal gut microbiome44, is characterized by a high relative abundance of the phyla Proteobacteria (for example, Enterobacteriaceae) and Actinobacteria (mainly Bifidobacterium spp.)28,30, with the latter playing a catalytic role in the foraging of glycans from human breast milk45. Cessation of breastfeeding, introduction of a solid diet and increased social exposure are associated with maturation of the gut microbiome towards an adult-like state46. This is characterized by a rapid expansion of Firmicutes, which become the dominant phylum beyond the first year of life (>50% relative abundance) and the primary source of short-chain fatty acids (SCFAs, especially butyrate)47,48 (Fig. 1b). More recently, the evolutionary-conserved development of the gut microbiome in healthy children was further elucidated through the identification of multiple successive trajectories involving specific bacterial genera49. Such strong changes in the early-life gut microbiome community are reflected by a rapid increase in species richness and evenness within individual samples (α-diversity), followed by gross stabilization by the age of 5 years30,42,43. The gut microbiome of children also exhibits progressively fewer differences in composition compared with adults (β-diversity) over time, correlating with increased exposure to a shared environment49,50.
Fig. 1. Early development of the gut microbiome in healthy children.

The development of the gut microbiome during early childhood consists of four different stages (acquisition, developmental, transitional and stable) that are characterized by stereotypic changes in diversity and composition30. a, During the first 3 years of life, the gut microbiome exhibits increasing richness and evenness within individual samples (α-diversity)50,56 and decreasing compositional differences compared with adults (β-diversity)61, followed by gross stabilization. Different early-life exposures can either advance or delay the maturation trajectories of the gut microbiome. b, Recent longitudinal studies of healthy children have also revealed evolutionary-conserved changes in the relative abundance of major bacterial phyla during the first 3 years of life30,50. Depicted trends in different gut microbiome metrics are estimates on the basis of the results of three recently published studies (Supplementary Table 1).
Early-life exposures that disrupt stereotypic waves of gut colonization and induce gut microbiome instability are associated with an altered composition of the gut microbiome during infancy, while also affecting the dynamic succession and functional capacity of taxa associated with later stages of development51–54 (Fig. 1). The mode of birth is the predominant factor shaping gut microbiota composition during the neonatal period28. The primary source of the neonatal microbiome is the maternal gut, as maternal skin and vaginal microbiota colonize the newborn baby only transiently44. At birth, neonates are colonized by strains of Bacteroides matching those of the maternal gut, irrespective of the mode of delivery55. However, children born by C-section demonstrate reduced colonization stability of Bacteroides by day 14 (ref. 55) and reduced abundance throughout infancy28,30,56,57 and up to 5 years of life49. The low-Bacteroides profile of neonates delivered by C-section is accompanied by an increased relative abundance of opportunistic pathogens (for example, Enterococcus spp. and Klebsiella spp.), which is also observed in a small proportion of vaginally delivered neonates with a similar low-Bacteroides profile28. Furthermore, children born by C-section exhibit an overall reduced gut microbiome stability29 and reduced stool levels of SCFAs (especially acetate)58 during the first months of life as well as a delayed maturation of the gut microbiome by the second year of life46.
Breastfeeding becomes the predominant factor shaping gut microbiota composition after the neonatal period and until weaning30. Lack of exclusive breastfeeding and early cessation is associated with reduced relative abundance of Bifidobacterium spp.59 and lower stool levels of acetate60. Cessation of breastfeeding initiates the transition towards an adult-like gut microbiome composition, which is characterized by expansion of Roseburia spp. and Anaerostipes spp., restriction of Lactobacillus spp. and a functional shift towards an increased capacity to degrade complex polysaccharides61. Although breast milk may temporarily suppress maturation of the gut microbiome during infancy, children who are predominantly breastfed develop a more mature gut microbiome by the second year of life46.
Intrapartum antibiotics have been associated with gut microbiome instability and account for substantial variation in the composition of the gut microbiome even among vaginally delivered neonates during the first week of life28,46. Exposure is associated with reduced relative abundance of Bacteroides and reduced levels of the SCFA propionate at birth62, as well as increased relative abundance of potentially pathogenic Proteobacteria63. Although antibiotic-induced gut microbiome instability in children and adults appears to grossly resolve within a month of exposure64, the interaction with other host and environmental factors and the impact of the timing of exposure and repeated administration may potentiate the magnitude and duration of their effects on the gut microbiome community64,65. Postnatal antibiotic exposure has been shown to delay the compositional and functional maturation of the gut microbiome, which is manifested through a reduced relative abundance of Lachnospiraceae (for example, Dialister spp. and Lachnospira spp.) and Ruminococcaceae and increased relative abundance of Veillonella46.
Gut microbiome maturation can be further influenced by the immediate social environment, geography and diet. A lack of older siblings is associated with reduced gut microbiome α-diversity and reduced relative abundance of Faecalibacterium30,66, as well as delayed maturation of the gut microbiome by the age of 12 months33. Similarly, delayed entry into daycare is associated with reduced relative abundance of Lachnospiraceae, Ruminococcaceae and Prevotella spp. and delayed maturation of the gut microbiome31. Furthermore, the gut microbiome composition of children living in urban areas in Africa is more similar to European children living in urban areas compared with children living in African villages67. School-age children living in urban areas show reduced relative abundance of bacteria (for example, Prevotella spp.) capable of fermenting complex carbohydrates for the production of SCFAs and reduced stool levels of all major SCFAs67. These observations have been linked to a shift in dietary habits towards reduced fibre consumption, increased food variety and increased calorie intake67.
The aforementioned findings provide evidence for the potential impact of early-life exposures with established epidemiological links to BCP-ALL on the compositional and functional maturation of the gut microbiome. The common denominator among these adverse exposures appears to be the delayed expansion of key, evolutionary-conserved bacterial taxa46 during critical periods of immune system development, which can have a catalytic role in the development of aberrant immune responses68. Collectively, these observations emphasize the need to meticulously characterize potential differences in the gut microbiome of children with leukaemia and healthy children.
Gut dysbiosis in children with ALL
We searched the MEDLINE and Embase databases from inception until 20 October 2022 to identify existing clinical studies examining the diversity and composition of the gut microbiome of children with ALL. We only included studies in which sample collection occurred before the administration of any systemic chemotherapy, given that chemotherapy can have a substantial impact on the diversity and composition of the gut microbiome69. None of the identified studies differentiated between different subtypes of childhood ALL. The search strategy and a summary of the results of the database search is provided in Supplementary Table 2. Differences in the diversity and composition (relative abundance of taxa) of the gut microbiome between healthy children and children with ALL, as well as levels of statistical significance, were directly extracted from the published results of selected studies. Only for the study of Liu et al.70, which reported differences in relative abundance at the level of bacterial species, we used publicly available individual participant data (taxon-relative abundance) to conduct a linear discriminant analysis of effect size (LEfSE) at other taxonomic levels (Supplementary methods).
Gut microbiome diversity at the time of diagnosis
We identified six case–control studies investigating gut microbiome α-diversity at the time of childhood ALL diagnosis (Table 1). The Shannon diversity index was reported to be lower in children and adolescents with ALL compared with healthy siblings or unrelated children, reaching statistical significance (P < 0.05) in four studies70–74 (Fig. 2, top panel). Other measures of α-diversity, such as the inverse Simpson index and Chao1, showed a similar pattern (Fig. 2, top panel). Matching of cases and controls by age and antibiotic exposure varied across studies. In the study of Bai et al.74, antibiotic exposure was associated with a diminished difference in α-diversity between ALL cases and controls, when compared with participants without antibiotic exposure in the past 90 days before sample collection. The reduction in Shannon diversity index between children with ALL and healthy children was not statistically significant in the study of Liu et al.70, in which participants were free of antibiotics for 90 days before specimen collection. However, the mean age of participants was greater compared with other cohorts, raising the possibility that previous differences in α-diversity during the first 5 years of life were no longer detectable70.
Table 1.
Case–control studies investigating differences in gut microbiome diversity and composition between healthy children and children with acute lymphoblastic leukaemia at the time of diagnosis
| Study | Country | Age of casesa | Cases (N) | Ab (time)b % of n | Type of control | Age of controla | Control (N) | Ab (time)b % of n | Seq | HVR | Reported levels of taxonomy | Method of analysis of relative abundance | Reporting threshold for relative abundance | FDR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| De Pietri et al., 2020 (ref. 71) | USA | 3.7 | 51 | NR | HS | 6.8 | 18 | No (30 days) | 16s rRNA | V3–V4 | NR | NR | NR | NR |
| Gao et al., 2020 (ref. 72) | China | 6.3 | 18 | No (14 days) | UHC | 5.9 | 18 | No (14 days) | 16s rRNA | V3–V4 | Genus | WMW | FC > 1.5 | <0.05 |
| Chua et al., 2020 (ref. 75) | Malaysia | 2–6 | 7 | Yes (30 days), 100% | UHC | 2–6 | 7 | No (30 days) | 16s rRNA | V4 | Phylum, Genus | DESeq2 | FC > 4 | <0.1 BHP |
| Rajagopala et al., 2020 (ref. 73) | USA | 5.0 | 29 | Yes (60 days), 61% | HS | 6.0 | 23 | No (60 days) | 16s rRNA | V4 | Phylum, Genus | DESeq2 | FC > 1.5 | <0.05 BHP |
| Bai et al., 2017c (ref. 74) | China | 5.5 | 20 | Yes (30 days), 100% | UHC | 4.3 | 16 | Yes (30 days), 100% | 16s rRNA | V3–V4 | Phylum, Class, Order, Family, Genus | LEfSE | LDA > 2 | <0.05 |
| 5.6 | 10 | No (90 days) | UHC | 4.5 | 17 | No (90 days) | ||||||||
| Liu et al., 2020 (ref. 70) | China | 7.2 | 58 | No (90 days) | UHC | 7.6 | 23 | No (90 days) | 16s rDNA micro -array | V1–V9 | Phylumd, Classd, Orderd, Familyd, Genusd, Species | LEfSE | LDA > 2 | <0.05 |
Ab, antibiotics; BHP, Benjamini–Hochberg procedure; DESeq2, differential gene expression analysis based on the negative binomial distribution; FC, fold change; FDR, false discovery rate; HS, healthy sibling; HVR, hypervariable region (the region of the 16s rRNA gene targeted for sequencing); LDA, linear discriminant analysis score; LEfSE, linear discriminant analysis (LDA) of effect size; N, sample number; NR, not reported; Seq, sequencing platform; UHC, unrelated healthy child; WMW, Wilcoxon–Mann–Whitney test. aAge reported as arithmetic mean or range (in years). bTime interval within which patients were exposed to antibiotics before sample collection or antibiotic-free time interval before sample collection. cThe study included two independent cohorts of antibiotic-exposed and antibiotic-unexposed participants. dFor this taxonomic level, we used participant-level data provided by the study to determine, through linear discriminant analysis of effect size, differences in relative abundances between study groups.
Fig. 2. Case–control studies investigating differences in gut microbiome diversity between children with newly diagnosed acute lymphoblastic leukaemia and healthy children.
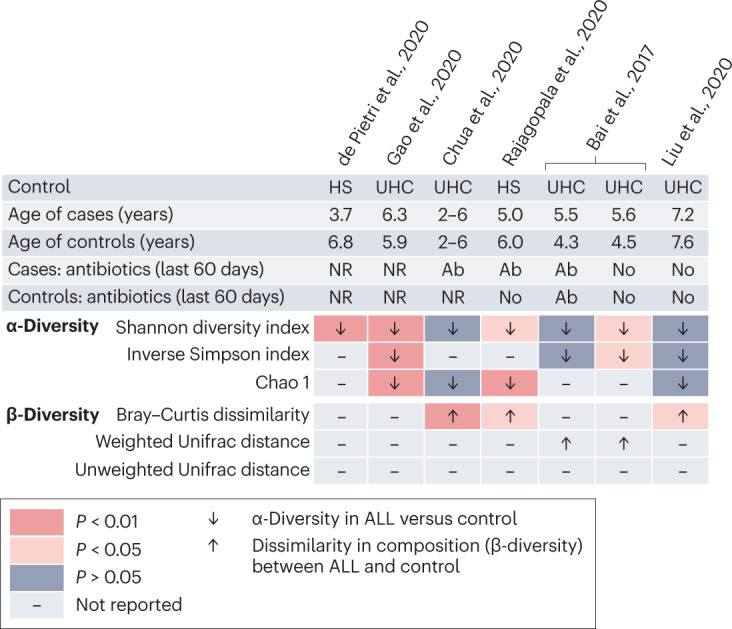
We systematically searched the MEDLINE and Embase databases from inception until 20 October 2022 to identify existing clinical studies examining the diversity and composition of the gut microbiome of children with newly diagnosed acute lymphoblastic leukaemia (ALL). The search strategy and the criteria for study selection are provided in Supplementary Table 2 and in Supplementary methods. Differences in α-diversity (top panels) and β-diversity (bottom panels) between children with ALL and healthy children at the time of diagnosis (de Pietri et al. 2020 (ref. 71), Gao et al. 2020 (ref. 56), Chua et al. 2020 (ref. 75), Rajagopala et al. 2020 (ref. 73), Bai et al. 2017 (ref. 74) and Liu et al. 2020 (ref. 70)). Ab, antibiotics; HS, healthy sibling; NR, not reported; UHC, unrelated healthy children.
Three studies investigating differences in β-diversity of the gut microbiome between healthy children and children with ALL at the time of diagnosis reported a statistically significant Bray–Curtis dissimilarity between the two groups70,73,75 (Fig. 2, bottom panel). A consistent difference in gut microbiome β-diversity between children with ALL and healthy children, irrespective of previous antibiotic exposure, was also reported by Bai et al.74 on the basis of the analysis of the weighted Unifrac distance. The reduced α-diversity and the persistence of significant differences in gut microbiome composition (β-diversity) observed in children with ALL compared with healthy children are in line with a stunted progression along the developmental and transitional phases of gut microbiome maturation.
Gut microbiome composition at the time of diagnosis
We identified four case–control studies of childhood ALL examining differences in gut microbiome composition at the level of phylum. All four studies revealed a significant reduction in the relative abundance of Firmicutes in children with ALL at the time of diagnosis compared with controls (Fig. 3). In three studies, a corresponding increase in the relative abundance of the phylum Bacteroidetes was reported in children with ALL compared with healthy children (Fig. 3). The latter observation was not replicated in our re-analysis of participant-level data from the study of Liu et al.70, in which participants were older and free of antibiotics for 90 days before specimen collection. Differences at the levels of class, order and family are presented in Supplementary Fig. 2.
Fig. 3. Case–control studies investigating differences in gut microbiome composition at the level of phylum and genus between healthy children and children with newly diagnosed acute lymphoblastic leukaemia.

We systematically searched the MEDLINE and Embase databases from inception until 20 October 2022 to identify existing clinical studies examining the diversity and composition of the gut microbiome of children with newly diagnosed acute lymphoblastic leukaemia (ALL). The search strategy and the criteria for study selection are provided in Supplementary Table 2 and in Supplementary methods. Only for the study of Liu et al. (2020), we used publicly available participant-level data to analyse differences in relative abundance between study groups at the level of phylum and genus using linear discriminant analysis of effect size (LEfSE) as described in Supplementary methods. For all other studies, data were directly extracted as reported in individual publications (Gao et al. 2020 (ref. 56), Chua et al. 2020 (ref. 75), Rajagopala et al. 2020 (ref. 73), Bai et al. 2017 (ref. 74) and Liu et al. 2020 (ref. 70)). a, Differences in the relative abundance of different bacterial phyla between children with ALL at the time of diagnosis and healthy children. b, Selected bacterial genera showing consistent differences in their relative abundance in children with ALL compared with healthy children. Only genera with consistent differences between study groups across three or more independent studies are presented. The complete list of genera identified by all studies (including known developmental trajectories) is provided in Supplementary Fig. 2.c, Physiological changes in the relative abundance of the five identified genera in healthy children during early childhood as reported by Roswall et al.61. Ab, antibiotics; FC, fold change; HS, healthy sibling; LDA, linear discriminant analysis score; NR, not reported; UHC, unrelated healthy children.
Five identified studies reported differences in gut microbiome composition at the level of genus between children with newly diagnosed ALL and healthy controls (Supplementary Fig. 2, which shows all identified taxa). Despite differences in antibiotic exposure, age matching of participants, geographical location, the sequencing platform used, the method of composition analysis and reporting thresholds, five genera showed consistent differences in relative abundance between children with ALL and healthy children across three or more studies (Fig. 3b). Roseburia, Dialister, Prevotella, Faecalibacterium and Anaerostipes showed reduced relative abundance in children with ALL at the time of diagnosis compared with healthy children. Two of the identified studies used machine learning to show that differences in the relative abundance at the level of genus can effectively discriminate between newly diagnosed children with ALL and healthy children, as evident by an area under the receiver operating characteristic curve (AUC) greater than 0.8 (refs. 70,73).
Implications for the two-hit model of ALL
Within the limitations imposed by differences in study methodology and the populations involved, the results of the identified studies suggest that the gut microbiome of children with ALL at the time of diagnosis exhibits reduced α-diversity and significantly different β-diversity compared with healthy children. They also show a decrease in the relative abundance of Firmicutes, the phylum with the most substantive expansion during the developmental phase of gut microbiome maturation (3–14 months). Furthermore, children with ALL at the time of diagnosis have reduced relative abundance of multiple genera that belong to older developmental trajectories; that is, genera which in healthy children exhibit low (<1%) relative abundance at birth and rapid expansion after weaning61 (Fig. 3c). Although these studies are somewhat preliminary with modest numbers and heterogenous design (including differences in sequencing methods, reporting thresholds and adjustment for confounders such as antibiotics), their results are in line with a suboptimal enrichment and delayed development of the gut microbiome postnatally in children with ALL and support the contention that immaturity of the gut microbiome is a key risk variable in the pathogenesis of the disease (Fig. 3). We also note that many of the affected taxa are well-known producers of SCFAs, which have a pivotal role in the regulation of gut immunity and the maintenance of an intact immune barrier76–78. More specifically, Liu et al.70 reported that newly diagnosed children with ALL exhibit reduced relative abundance of both butyrate-producing species (Roseburia faecis, R. intestinalis, R. inulinivorans, Anaerostipes hardus, Faecalibacterium prausnitzii, Eubacterium ramulus) and acetate-producing species (Prevotella maculosa, P. aurantiaca, Bacteroides uniformis, B. ovatus) (Supplementary Fig. 3).
The cross-sectional nature of the studies analysed, as well as their variable control for important confounders of gut microbiome composition, poses particular challenges to data interpretation and the inference of a causal relationship between gut microbiome maturation and progression of covert preleukaemic clones to overt leukaemia based solely on these preliminary data. For example, the chronological age of participants is a major determinant of gut microbiome maturation and could account for significant discrepancies in the relative abundance of different bacterial taxa between study groups49. Similarly, recent antibiotic exposure (depending on the class of the antibacterial agent, duration of exposure and its precise timing relative to specimen collection) can cause transient disturbances in the composition of the gut microbiome69,74,79 and has the potential to obfuscate study group differences in the relative abundance of a subset of implicated taxa. However, given that antibiotic-induced dysbiosis and reduction in SCFA-producing taxa in mice and humans are usually reversible within a few weeks of exposure64,65, antibiotic exposure is unlikely to account for the consistent differences observed across all included studies. This notion is also supported by the results of two of the analysed case–control studies that revealed a consistent decrease in the relative abundance of Firmicutes, as well as major SCFA-producing genera in children with newly diagnosed ALL who were not exposed to antibiotics for at least 90 days before sample collection70,74.
The consistent reduction in the relative abundance of specific bacterial taxa, and especially those belonging to older developmental trajectories across multiple studies despite variable matching of study groups (including different ethnicities, geographical locations and exposure to antibiotics) and analytic methods, supports a pervasive lag in the gut microbiome maturation of children with ALL compared with healthy children, which is likely to be long-standing and to originate from adverse exposures that occurred during the first year of life. Interestingly, longitudinal case–control studies in asthma have recently highlighted that delayed maturation of the gut microbiome (characterized by reduced relative abundance of the genera Roseburia, Dialister, Prevotella, Faecalibacterium and Blautia and increased abundance of Enterococcus), as well as reduced stool levels of butyrate by the age of 12 months, is associated with aberrant immune responses and an increased risk of asthma diagnosis by the age of 5 years33–35. These observations suggest that gut microbiome immaturity during critical periods of immune priming increases the propensity for dysregulated immune responses80, which in turn can pave the way for second chromosomal hits in preleukaemic clones upon exposure to common childhood infectious triggers7 (Fig. 4).
Fig. 4. The two-hit model of childhood B cell precursor-acute lymphoblastic leukaemia.

Proposed role of the gut microbiome in the two-hit model of childhood B cell precursor-acute lymphoblastic leukaemia (BCP-ALL) based on the results of currently available preliminary studies analysed in the present work. Chromosomal first hits are necessary events for the development of BCP-ALL, but are not sufficient to drive leukaemogenesis. The synergistic effect of adverse early-life exposures, such as caesarean section, reduced or absent breastfeeding, reduced dietary fibrea, antibioticsa, lack of older siblings and delayed entry into daycare, may lead to gut microbiome immaturity during a critical time in the development of the immune system. A deficiency in short-chain fatty acid (SCFA)-producing bacterial taxa can compromise gut microbiome-mediated immune priming leading to suppression of regulatory T (Treg) cells and promotion of T helper 17 (TH17)-dominated immune responses. SCFA deficiency can also compromise the integrity of the gut epithelial barrier and facilitate the systemic translocation of opportunistic pathogens, as well as increase susceptibility to viral infections. The resulting dysregulated pro-inflammatory immune responses against common infectious triggers can ultimately lead to an increased risk of leukaemic transformation in a small proportion (about 1%) of children with in utero-acquired preleukaemic clones. ETV6::RUNX1, ETS translocation variant 6::runt-related transcription factor 1. aThese variables have not been systematically evaluated as risk factors for ALL.
Adverse early-life exposures can compromise gut microbiome-mediated immune priming
The gut microbiome is a powerful mediator of the impact of early-life exposures on the development of the immune system, which also follows a stereotypical pattern of development during the first few months of life32. Νeonates normally possess a high relative frequency of myeloid-derived suppressor cells (MDSCs), CD4+ forkhead box P3 (FOXP3)+ regulatory T (Treg) cells and regulatory B cells with a polyreactive immunoglobulin repertoire, as well as high IL-10 and IL-27 levels32,81. This initial period of immune tolerance facilitates gut colonization with organisms that, in turn, are indispensable for subsequent development of the immune system and the maintenance of an intact gut barrier80. Microbial signals at the level of the intestinal mucosa, such as microorganism-associated molecular patterns (MAMPs)68 and microorganism-derived metabolites (for example, SCFAs)82, are thus utilized for mediating immune training83,84. Principal aspects of this gut microbiome–immune system interplay include the induction of Treg cells that orchestrate the complex immune network, as well as modulation of various aspects of B cell development78,85–87.
Commensal gut microbiota have been shown to tightly regulate RAG-dependent editing in pro-B cells located in the lamina propria and to diversify the early-B cell receptor repertoire88. Mice grown in germ-free facilities display smaller Peyer’s patches, reduced numbers of intestinal IgA-expressing B cells89, decreased numbers of Treg cells90 and increased levels of IgE, which can be normalized by colonization with commensal bacteria during the first 4 weeks of life91. Similarly, perturbation of the gut microbiome by antibiotics can precipitate exaggerated T helper 17 (TH17) immune responses to inhaled allergens in mice when exposure occurs during the first few postnatal days, but not when exposure happens in adult life68. One of the best-studied examples of microbial-mediated immune training involves polysaccharide A, a Toll-like receptor (TLR) 2 ligand, of Bacteroides fragilis, which induces Treg cells and suppresses pro-inflammatory TH17 cell responses, thus promoting gut immune tolerance and host–microbial symbiosis92. Other Bacteroides species (for example, B. ovatus and B. uniformis), whose successful and timely colonization is determined by the mode of delivery28, can have a profound impact on stool IgA levels during infancy93 and thus shape the stability of other immunomodulatory bacterial taxa that colonize the infant gut at later stages of gut microbiome maturation94. Elective C-section, an established risk factor for BCP-ALL21,22,95, induces persisting compositional and functional instability in the gut microbiome (especially in Bacteroides spp.28,55) that has been shown to suppress Treg cell differentiation and compromise gut immune tolerance, leading to the promotion of pro-inflammatory responses96,97. In this context, we note that children with ALL at the time of diagnosis were reported to have reduced relative abundance of B. vulgatus, B. ovatus and B. uniformis70; that is, immunomodulatory bacterial species whose transmission from mother to child is disrupted by C-section28.
Reduced or absent breastfeeding is another established risk factor for childhood ALL23,24 with pronounced effects on the development of the gut microbiome–immune system axis. Maternal IgA was recently shown to set the initial homeostatic level of Treg cells in the colon of offspring98. Maternal IgA transferred through breastfeeding stabilizes the infant gut microbiome and maintains gut immune tolerance until the establishment of endogenous secretory IgA production94,99, which begins after the first 30 days of life32. In parallel, human milk oligosaccharides (HMOs) facilitate the expansion of Bifidobacterium species, which have an instrumental role in the development of T cell-dependent IgA responses99 and have been linked to higher numbers of memory B cells during infancy100. Exclusive formula feeding is associated with lower numbers of Treg cells and increased production of pro-inflammatory cytokines101. A low relative abundance of Bifidobacteria and depletion of HMO utilization genes in the gut microbiome of human infants was also recently shown to be associated with systemic inflammation and polarization of naive CD4+ T cells towards TH17 cells, which could be reversed upon supplementation with Bifidobacterium infantis102. Geographical variations in the prevalence of specific Bifidobacterium spp. with different capacity to utilize HMOs have been associated with important differences in the overall composition of the gut microbiome community in the first year of life and may contribute to inefficient early immune priming and dysregulated immune responses in later life (for example, as encountered in allergy and autoimmunity)103.
Diet plays a major part in determining the composition and function of the developing gut microbiome and in shaping its interactions with the host104. SCFAs have emerged as being instrumental for gut immune homeostasis82. SCFAs, particularly butyrate, appear to be critical for the induction of IL-10-producing Treg cells in the gut through inhibition of histone deacetylases (HDACs), which leads to the activation of the FOXP3 promoter on naive CD4+ T cells105–108. Butyrate has also been shown to reduce the expression of co-stimulatory molecules on dendritic cells in response to MAMPs (for example, lipopolysaccharide (LPS))109, as well as to promote the differentiation of naive B cells into regulatory B cells that are capable of suppressing inflammatory responses110. Fibre-derived butyrate and propionate production by gut microbiota is capable of directly suppressing AID through inhibition of HDACs and ameliorating B cell-mediated immunopathology in mouse models111.
SCFA deficiency has been increasingly linked to impaired gut immune tolerance76,112,113. Reduced dietary fibre consumption observed in urban societies has been associated with reduced stool levels of all major SCFAs67, as well as aberrant activation of pro-inflammatory pathways114. In mice, a low-fibre diet during pregnancy and lactation leads to reduced SCFA levels and impaired thymic Treg cell differentiation in the offspring115. Reduced postnatal fibre intake by the offspring has also been associated with reduced SCFA levels (especially butyrate) and induction of pro-inflammatory pathways67. Although robust epidemiological evidence to support a role for reduced dietary fibre intake in the pathogenesis of childhood ALL is still lacking, reduced maternal fibre consumption during pregnancy has been linked to increased incidence of childhood ALL116. Interestingly, SCFA supplementation has also been shown to increase leptin sensitivity in mice fed a Western diet117. This may constitute an additional pathway through which diet and the gut microbiome can modulate the risk of childhood ALL, given that fasting may inhibit the transformation of preleukaemic clones through enhanced leptin receptor signalling in mouse models118.
Early-life family structure and social exposure have been associated with differential risk of developing infection-induced cancer119. The lack of older siblings and delayed entry into daycare have been associated with both delayed maturation of the gut microbiome31,33 and increased risk of childhood BCP-ALL25–27,120–122. Infants with older siblings exhibit increased gut microbiome α-diversity, faster gut microbiome maturation and earlier colonization with Faecalibacterium species58, which are known to increase the Treg/TH17 cell ratio via inhibition of HDACs and to ameliorate gut inflammation123,124. Living in a larger household and attending daycare has also been associated with an increased number of naive Treg cells and reduced incidence of allergies by 12 months of age125. Attending daycare facilities in which children are oriented to have a closer relationship to nature (for example, through extended contact with soil) can increase gut microbiome α-diversity, augment the number of Treg cells and suppress TH17 immune responses within 30 days of attendance126.
In summary, social risk factors associated with increased risk of childhood ALL can compromise gut microbiome-mediated early immune priming and disrupt gut immune tolerance during critical periods of immune system development. These observations are in line with the finding that children who develop ALL have reduced levels of IL-10 and enhanced pro-inflammatory signatures at birth127, which in turn have been linked to impaired B cell development and increased B cell DNA damage in mouse models of childhood ALL128.
The gut microbiome and susceptibility to common infections
Emerging evidence suggests that the gut bacterial microbiome can also affect host susceptibility to viral infections, as well as clinical outcomes129. The gut microbiota has been shown to enhance systemic antiviral immunity by regulating tonic interferon type I responses at distal sites through membrane vesicles containing bacterial DNA130. In parallel, gut microbiome-derived SCFAs also contribute to the priming of antiviral immunity131. Increased abundance of SCFA-producing bacteria appears to offer protection against many common respiratory viruses including rhinovirus, respiratory syncytial virus, adenovirus, influenza132 and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)133,134. Furthermore, the gut microbiome has emerged as an important determinant of the efficacy of oral and parenteral antiviral vaccines135,136. Gut microbiome-derived SCFAs can facilitate antiviral immunity by regulating interferon responses and antiviral effector cells, including circulating monocytes and CD8+ T cells137.
In summary, the importance of gut SCFA-producing bacterial taxa in the pathogenesis of childhood ALL is likely to be twofold. First, a delayed enrichment of these taxa during the first year of life, as a result of the synergistic effect of adverse life exposures, can compromise early immune training and shift the TH17/Treg cell balance towards dysregulated, pro-inflammatory responses. Second, a persisting deficiency in SCFA-producing bacterial taxa over time can additionally compromise the integrity of the gut epithelial barrier and increase host susceptibility to opportunistic pathogens, as well as commonly encountered pathogens of childhood (for example, respiratory viruses). This notion is supported by studies of mouse models of acute leukaemia in which antibiotic-induced disruption of the gut microbiome accelerates disease development, potentially through translocation of pro-inflammatory bacterial products (for example, LPS) via a leaky gut epithelial barrier that has been deprived of SCFAs138,139. Previous experimental studies have shown that preleukaemic clones residing in the bone marrow are resistant to suppression by pro-inflammatory cytokines (IL-6, IL-1β and tumour necrosis factor (TNF))140 or apoptosis by transforming growth factor β (TGFβ) secreted by bone marrow mesenchymal stromal cells (BMMSCs)141, leading to their survival advantage over normal BCPs. At the same time, the genome of preleukaemic clones has been shown to be increasingly vulnerable to repeated inflammatory stimuli and pro-inflammatory cytokines, which can trigger the acquisition of second chromosomal hits13. Evidence suggests that the gut microbiome may directly modulate the ability of BMMSCs to induce the maturation or apoptosis of haematopoietic cell lines, given that BMMSCs from germ-free mice have been shown to secrete increased pro-inflammatory cytokines (for example, IL-23)142.
Future perspectives
The low incidence of ALL compared with other childhood diseases associated with altered gut microbiome diversity and composition, such as allergy, makes the design of prospective longitudinal studies particularly challenging. A more feasible approach with potential for clinical translation is the implementation of large, cross-sectional multicentre studies encompassing a range of geographical areas, age groups, early-life social exposures, antibiotic treatments and disease cytogenetics to establish ALL-associated gut microbiome signatures. Controlling for these variables will be of paramount importance for confirming the ALL-associated gut microbiome maturation delay that has been observed in the presented preliminary studies. Given the complexity of gut microbiome communities, the multiplex co-abundance patterns and the different dispersal properties of individual bacterial taxa across human communities52, the use of machine learning is going to be indispensable for identifying the dynamic imprint of early-life exposures on gut microbiome maturation143,144. Provision of publicly available datasets accompanied by thorough patient metadata and utilization of standardized reporting tools, such as the Strengthening The Organization and Reporting of Microbiome Studies (STORMS) checklist145, can facilitate comprehensive meta-analyses, as well as meaningful comparisons with other diseases of childhood, including allergies and auto-immune conditions.
A necessary extension of the studies reviewed earlier will be to analyse the gut microbiome of newly diagnosed children with BCP-ALL in comparison with other types of childhood acute leukaemia (for example, T cell-ALL and acute myeloid leukaemia) to confirm the anticipated selective impact that gut microbiome dysbiosis may have on causation of subtypes of leukaemia. If this is confirmed, it will further endorse the causal link. In parallel with these patient-based studies, mouse modelling of infection-driven ALL138 may both confirm an association between microbiome status and risk of leukaemia and provide a test bed for prevention trials with faecal microbiome or specific bacterial transplants.
Conclusion
Herein, we summarized current progress in the emerging research field exploring the role of the gut microbiome in the pathogenesis of childhood ALL. These preliminary results are consistent with a delayed maturation of the gut microbiome in children with ALL, as detected at the time of diagnosis. This raises the possibility that adverse early-life exposures associated with BCP-ALL perturb the development of the gut microbiome–immune system axis away from evolutionary-conserved maturation trajectories. We propose that the resulting deficiency of specific SCFA-producing taxa at the early stages of gut microbiome development compromises immune network stabilization, increasing the risk that later infectious exposures prompt chronic inflammation and trigger ALL. The latter will occur infrequently and only in children who carry silent pre-malignant clones generated before birth. Pending confirmation in larger studies with harmonized study design and reporting, we anticipate that the gut microbiome maturity status will likely be established as a decisive factor in the pathogenesis of the major genetic subtypes of childhood BCP-ALL and offer opportunities for primary prevention4,5.
Finally, we note the parallels between the social risk factors and likely pivotal role of the early-life gut microbiome–immune system axis in ALL and other diseases that are increasingly more prevalent in young members of modern societies including allergies, type 1 diabetes and possibly other auto-immune diseases, such as multiple sclerosis4. These diseases have distinctive immunopathologies and background genetic risk variables, but they may share a common, immune priming deficiency that is microbiome dependent4,146. This possibility requires further exploration but raises the prospect of a common prophylactic intervention strategy that could be risk-reducing for a rare disease such as ALL as well as more common debilitating illnesses of childhood. This speculative and ambitious vision is encouraged by recent clinical studies in which Bifidobacterium and Lactobacillus species have demonstrable risk-reducing efficacy in infants with sepsis147, a pre-term birth148 and allergies149. Yet the future investigation of disease prevention via microbiome modification or boosting in infancy might benefit from more interaction between scientists and clinicians working on these different illnesses of childhood.
Supplementary information
Acknowledgements
The authors acknowledge support from the Cancer Research UK (CRM 171X), The Children’s Cancer and Leukaemia Group (CCLGA2019.02), The Royal Marsden Cancer Charity, the Wood family-in memory of Artemis and The Institute for Cancer Research, London.
Glossary
- α-Diversity
A measure of the number of different taxa (richness) and/or the degree of evenness in their relative abundance within a single sample.
- β-Diversity
A measure of the degree of similarity or distance between the composition of the microbial communities of two samples.
- Activation-induced cytidine deaminase
(AID). An enzyme required for somatic hypermutation and class-switch recombination of immunoglobulin genes during B cell maturation and immune response.
- Area under the receiver operating characteristic curve
(AUC). An aggregate measure of the performance of a predictive model across all possible classification thresholds.
- Bray–Curtis dissimilarity
A measure of β-diversity that quantifies the degree of dissimilarity in the composition of the microbial communities of two samples.
- Chao1
A measure of α-diversity that takes into account the number of different taxa (richness) within a sample.
- Human milk oligosaccharides
(HMOs). Human milk oligosaccharides are unconjugated complex glycans that have a central role in the development of the gut microbiome–immune system axis.
- High hyperdiploidy
A genetic aberration characterized by chromosomal gains (>51 chromosomes) that is commonly found in preleukaemic clones of childhood B cell precursor ALL.
- Inverse Simpson index
A measure of α-diversity that takes into account both richness and evenness within a sample, giving more weight to common taxa.
- Leptin
A hormone produced by adipose tissue that has a central role in the regulation of energy balance and has widespread effects in multiple organ systems, including haematopoietic cells.
- Linear discriminant analysis of effect size
(LEfSe). Determines the taxa most likely to explain differences between study groups. It uses standard statistical tests to detect taxa with significant difference in relative abundance between the groups, as well as additional tests to assess the biological significance and relevance of these taxa.
- Microorganism-associated molecular patterns
(MAMPs). Molecular structures conserved among classes of microorganisms that can be recognized by pattern recognition receptors to elicit immune responses.
- Neonatal Guthrie cards
Samples of dried blood routinely collected after birth via heel prick for the purpose of universal screening for genetic conditions.
- Peyer’s patches
Gut-associated lymphoid tissue found in the small intestine that forms the interface of the gut microbiome-mediated immune system priming.
- Shannon diversity index
A measure of α-diversity that takes into account both richness and evenness of taxa within a sample, giving more weight to rare taxa.
- Short-chain fatty acids
(SCFAs). Metabolites produced by gut commensals through the fermentation of non-digestible fibre.
- Weighted Unifrac distance
A measure of β-diversity that incorporates abundance information and places more weight to common species. By contrast, the unweighted Unifrac distance is a measure of β-diversity that takes into account the presence and absence of taxa.
Author contributions
The authors contributed equally to all aspects of the article.
Peer review
Peer review information
Nature Reviews Cancer thanks Martin Blaser, Stephen Sallan and Josef Vormoor for their contribution to the peer review of this work.
Data availability
The primary data that support the findings presented in this Perspective article, including the results of our re-analysis of participant-level data of the study of Liu et al.71, are available as Supplementary figures.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41568-023-00584-4.
References
- 1.Public Health England. Children, teenagers and young adults UK cancer statistics report 2021 1–30 (2021).
- 2.Pui CH, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin. Hematol. 2013;50:185–196. doi: 10.1053/j.seminhematol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kane E, et al. Excess morbidity and mortality among survivors of childhood acute lymphoblastic leukaemia: 25 years of follow-up from the United Kingdom Childhood Cancer Study (UKCCS) population-based matched cohort. BMJ Open. 2022;12:e056216. doi: 10.1136/bmjopen-2021-056216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greaves M, Cazzaniga V, Ford A. Can we prevent childhood leukaemia? Leukemia. 2021;35:1258–1264. doi: 10.1038/s41375-021-01211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hauer J, Fischer U, Borkhardt A. Toward prevention of childhood ALL by early-life immune training. Blood. 2021;138:1412–1428. doi: 10.1182/blood.2020009895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia. 1988;2:120–125. [PubMed] [Google Scholar]
- 7.Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer. 2018;18:471–484. doi: 10.1038/s41568-018-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bach J. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 9.Ford AM, Colman S, Greaves M. Covert pre-leukaemic clones in healthy co-twins of patients with childhood acute lymphoblastic leukaemia. Leukemia. 2023;37:47–52. doi: 10.1038/s41375-022-01756-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiemels JL, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354:1499–1503. doi: 10.1016/S0140-6736(99)09403-9. [DOI] [PubMed] [Google Scholar]
- 11.Maia AT, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17:2202–2206. doi: 10.1038/sj.leu.2403101. [DOI] [PubMed] [Google Scholar]
- 12.Taub JW, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood. 2002;99:2992–2996. doi: 10.1182/blood.V99.8.2992. [DOI] [PubMed] [Google Scholar]
- 13.Swaminathan S, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat. Immunol. 2015;16:766–774. doi: 10.1038/ni.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori H, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl Acad. Sci. USA. 2002;99:8242–8247. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuzuki S, Seto M, Greaves M, Enver T. Modeling first-hit functions of the t(12;21) TEL-AML1 translocation in mice. Proc. Natl Acad. Sci. USA. 2004;22:8443–8448. doi: 10.1073/pnas.0402063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fidanza M, et al. Inhibition of precursor B cell malignancy progression by toll-like receptor ligand-induced immune responses. Leukemia. 2016;30:2116–2119. doi: 10.1038/leu.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schäfer D, et al. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood. 2018;131:821–826. doi: 10.1182/blood-2017-09-808402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francis SS, Selvin S, Yang W, Buffler PA, Wiemels JL. Unusual space–time patterning of the Fallon, Nevada leukemia cluster: evidence of an infectious etiology. Chem. Biol. Interact. 2012;196:102–109. doi: 10.1016/j.cbi.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cazzaniga G, et al. Possible role of pandemic AH1N1 swine flu virus in a childhood leukemia cluster. Leukemia. 2017;31:1819–1821. doi: 10.1038/leu.2017.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papaemmanuil E, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcotte EL, et al. Caesarean delivery and risk of childhood leukaemia: a pooled analysis from the Childhood Leukemia International Consortium (CLIC) Lancet Haematol. 2016;3:e176–e185. doi: 10.1016/S2352-3026(16)00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sevelsted A, Stokholm J, Bønnelykke K, Bisgaard H. Cesarean section chronic immune disorders. Pediatrics. 2015;135:e92–e98. doi: 10.1542/peds.2014-0596. [DOI] [PubMed] [Google Scholar]
- 23.Amitay EL, Keinan-Boker L. Breastfeeding and childhood leukemia incidence: a meta-analysis and systematic review. JAMA Pediatr. 2015;169:e151025. doi: 10.1001/jamapediatrics.2015.1025. [DOI] [PubMed] [Google Scholar]
- 24.Su Q, et al. Breastfeeding and the risk of childhood cancer: a systematic review and dose-response meta-analysis. BMC Med. 2021;19:90. doi: 10.1186/s12916-021-01950-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudant J, et al. Childhood acute lymphoblastic leukemia and indicators of early immune stimulation: a Childhood Leukemia International Consortium study. Am. J. Epidemiol. 2015;181:549–562. doi: 10.1093/aje/kwu298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urayama KY, Buffler PA, Gallagher ER, Ayoob JM, Ma X. A meta-analysis of the association between day-care attendance and childhood acute lymphoblastic leukaemia. Int. J. Epidemiol. 2010;39:718–732. doi: 10.1093/ije/dyp378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamper-Jørgensen M, et al. Childcare in the first 2 years of life reduces the risk of childhood acute lymphoblastic leukemia. Leukemia. 2008;22:189–193. doi: 10.1038/sj.leu.2404884. [DOI] [PubMed] [Google Scholar]
- 28.Shao Y, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature. 2019;574:117–121. doi: 10.1038/s41586-019-1560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reyman M, et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat. Commun. 2019;10:4997. doi: 10.1038/s41467-019-13014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart CJ, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2018;562:583–588. doi: 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amir A, Erez-granat O, Braun T, Sosnovski K, Hadar R. Gut microbiome development in early childhood is affected by day care attendance. NPJ Biofilms Microbiomes. 2022;8:2. doi: 10.1038/s41522-021-00265-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olin A, et al. Stereotypic immune system development in newborn children. Cell. 2018;174:1277–1292.e14. doi: 10.1016/j.cell.2018.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stokholm J, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat. Commun. 2018;9:141. doi: 10.1038/s41467-017-02573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Depner M, et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat. Med. 2020;26:1766–1775. doi: 10.1038/s41591-020-1095-x. [DOI] [PubMed] [Google Scholar]
- 35.Stokholm J, et al. Delivery mode and gut microbial changes correlate with an increased risk of childhood asthma. Sci. Transl. Med. 2020;12:eaax9929. doi: 10.1126/scitranslmed.aax9929. [DOI] [PubMed] [Google Scholar]
- 36.Wen Y, Jin R, Chen H. Interactions between gut microbiota and acute childhood leukemia. Front. Microbiol. 2019;10:1300. doi: 10.3389/fmicb.2019.01300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cobaleda C, Vicente-Duenas C, Sanchez-Garcia I. An immune window of opportunity to prevent childhood B cell leukemia. Trends Immunol. 2021;42:371–374. doi: 10.1016/j.it.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Ma T, Chen Y, Li L-J, Zhang L-S. Opportunities and challenges for gut microbiota in acute leukemia. Front. Oncol. 2021;11:692951. doi: 10.3389/fonc.2021.692951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uribe-Herranz M, Klein-González N, Rodríguez-Lobato LG, Juan M, de Larrea CF. Gut microbiota influence in hematological malignancies: from genesis to cure. Int. J. Mol. Sci. 2021;22:1026. doi: 10.3390/ijms22031026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oldenburg M, Rüchel N, Janssen S, Borkhardt A, Gössling KL. The microbiome in childhood acute lymphoblastic leukemia. Cancers. 2021;13:4947. doi: 10.3390/cancers13194947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masetti R, et al. Gut microbiome in pediatric acute leukemia: from predisposition to cure. Blood Adv. 2021;5:4619–4629. doi: 10.1182/bloodadvances.2021005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arrieta M, et al. The intestinal microbiome in early life: health and disease. Front. Immunol. 2014;5:427. doi: 10.3389/fimmu.2014.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derrien M, Alvarez AS, de Vos WM. The gut microbiota in the first decade of life. Trends Microbiol. 2019;27:997–1010. doi: 10.1016/j.tim.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Ferretti P, et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe. 2018;24:133–145.e5. doi: 10.1016/j.chom.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirmiz N, Robinson RC, Shah IM, Barile D, Mills DA. Milk glycans and their interaction with the infant-gut microbiota. Annu. Rev. Food Sci. Technol. 2018;9:429–450. doi: 10.1146/annurev-food-030216-030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bokulich N.A., et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016;8:343ra82. doi: 10.1126/scitranslmed.aad7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuyama M, et al. Breastfeeding: a key modulator of gut microbiota characteristics in late infancy. J. Dev. Orig. Health Dis. 2019;10:206–213. doi: 10.1017/S2040174418000624. [DOI] [PubMed] [Google Scholar]
- 48.Tsukuda N, et al. Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life. ISME J. 2021;15:2574–2590. doi: 10.1038/s41396-021-00937-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roswall J, et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe. 2021;29:765–776.e3. doi: 10.1016/j.chom.2021.02.021. [DOI] [PubMed] [Google Scholar]
- 50.Odamaki T, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;16:90. doi: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao L, Wang J, Zheng J, Li X, Zhao F. Deterministic transition of enterotypes shapes the infant gut microbiome at an early age. Genome Biol. 2021;22:243. doi: 10.1186/s13059-021-02463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hildebrand F, et al. Dispersal strategies shape persistence and evolution of human gut bacteria. Cell Host Microbe. 2021;29:1167–1176.e9. doi: 10.1016/j.chom.2021.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beller L, et al. Successional stages in infant gut microbiota maturation. mBio. 2021;12:e01857-21. doi: 10.1128/mbio.01857-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cox LM, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–721. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitchell CM, et al. Delivery mode affects stability of early infant gut microbiota. Cell Rep. Med. 2020;1:100156. doi: 10.1016/j.xcrm.2020.100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niu J, et al. Evolution of the gut microbiome in early childhood: a cross-sectional study of Chinese children. Front. Microbiol. 2020;11:439. doi: 10.3389/fmicb.2020.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Azad MB, et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. Br. J. Obstetr. Gynecol. 2016;123:983–993. doi: 10.1111/1471-0528.13601. [DOI] [PubMed] [Google Scholar]
- 58.Martin R, et al. Early-life events, including mode of delivery and type of feeding, siblings and gender, shape the developing gut microbiota. PLoS ONE. 2016;11:e0158498. doi: 10.1371/journal.pone.0158498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho NT, et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat. Commun. 2018;9:4169. doi: 10.1038/s41467-018-06473-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bridgman SL, et al. Fecal short-chain fatty acid variations by breastfeeding status in infants at 4 months: differences in relative versus absolute concentrations. Front. Nutr. 2017;4:00011. doi: 10.3389/fnut.2017.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bäckhed F, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Nogacka A, et al. Impact of intrapartum antimicrobial prophylaxis upon the intestinal microbiota and the prevalence of antibiotic resistance genes in vaginally delivered full-term neonates. Microbiome. 2017;5:93. doi: 10.1186/s40168-017-0313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prescott S, et al. Impact of intrapartum antibiotic prophylaxis on offspring microbiota. Front. Pediatr. 2021;9:754013. doi: 10.3389/fped.2021.754013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yassour M, et al. Natural history of the infant gut microbiome and impact of antibiotic treatments on strain-level diversity and stability. Sci. Transl. Med. 2016;8:1173–1178. doi: 10.1126/scitranslmed.aad0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McDonnell L, et al. Association between antibiotics and gut microbiome dysbiosis in children: systematic review and meta-analysis. Gut Microbes. 2021;13:1870402. doi: 10.1080/19490976.2020.1870402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laursen MF, et al. Having older siblings is associated with gut microbiota development during early childhood. BMC Microbiol. 2015;15:154. doi: 10.1186/s12866-015-0477-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Filippo C, et al. Diet, environments, and gut microbiota. A preliminary investigation in children living in rural and urban Burkina Faso and Italy. Front. Microbiol. 2017;8:1979. doi: 10.3389/fmicb.2017.01979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Borbet TC, et al. Influence of the early-life gut microbiota on the immune responses to an inhaled allergen. Mucosal Immunol. 2022;15:1000–1011. doi: 10.1038/s41385-022-00544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hakim H, et al. Gut microbiome composition predicts infection risk during chemotherapy in children with acute lymphoblastic leukemia. Clin. Infect. Dis. 2018;67:541–548. doi: 10.1093/cid/ciy153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu X, et al. Pediatric acute lymphoblastic leukemia patients exhibit distinctive alterations in the gut microbiota. Front. Cell Infect. Microbiol. 2020;10:558799. doi: 10.3389/fcimb.2020.558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Pietri S, et al. Gastrointestinal toxicity during induction treatment for childhood acute lymphoblastic leukemia: the impact of the gut microbiota. Int. J. Cancer. 2020;147:1953–1962. doi: 10.1002/ijc.32942. [DOI] [PubMed] [Google Scholar]
- 72.Gao X, et al. A new insight into acute lymphoblastic leukemia in children: influences of changed intestinal microfloras. BMC Pediatr. 2020;20:290. doi: 10.1186/s12887-020-02192-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajagopala SV, et al. Persistent gut microbial dysbiosis in children with acute lymphoblastic leukemia (ALL) during chemotherapy. Microb. Ecol. 2020;79:1034–1043. doi: 10.1007/s00248-019-01448-x. [DOI] [PubMed] [Google Scholar]
- 74.Bai L, Zhou P, Li D, Ju X. Changes in the gastrointestinal microbiota of children with acute lymphoblastic leukaemia and its association with antibiotics in the short term. J. Med. Microbiol. 2017;66:1297–1307. doi: 10.1099/jmm.0.000568. [DOI] [PubMed] [Google Scholar]
- 75.Chua LL, et al. Temporal changes in gut microbiota profile in children with acute lymphoblastic leukemia prior to commencement-, during-, and post-cessation of chemotherapy. BMC Cancer. 2020;20:151. doi: 10.1186/s12885-020-6654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 77.Kim M, Kim CH. Regulation of humoral immunity by gut microbial products. Gut Microbes. 2017;8:392–399. doi: 10.1080/19490976.2017.1299311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu B, Wang L, Chu Y. Gut microbiota shape B cell in health and disease settings. J. Leukoc. Biol. 2021;110:271–281. doi: 10.1002/JLB.1MR0321-660R. [DOI] [PubMed] [Google Scholar]
- 79.Dunn KA, et al. Antibiotic and antifungal use in pediatric leukemia and lymphoma patients are associated with increasing opportunistic pathogens and decreasing bacteria responsible for activities that enhance colonic defense. Front. Cell. Infect. Microbiol. 2022;12:924707. doi: 10.3389/fcimb.2022.924707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gensollen T, Iyer SS, Kasper DL, Blumberg RS, Medical H. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–544. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat. Rev. Immunol. 2017;17:495–507. doi: 10.1038/nri.2017.54. [DOI] [PubMed] [Google Scholar]
- 82.Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016;16:341–352. doi: 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Planer JD, et al. Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature. 2016;534:263–266. doi: 10.1038/nature17940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turroni F, et al. The infant gut microbiome as a microbial organ influencing host well-being. Ital. J. Pediatr. 2020;46:16. doi: 10.1186/s13052-020-0781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H, et al. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature. 2020;584:274–278. doi: 10.1038/s41586-020-2564-6. [DOI] [PubMed] [Google Scholar]
- 86.New JS, et al. Neonatal exposure to commensal-bacteria-derived antigens directs polysaccharide-specific B-1 B cell repertoire development. Immunity. 2020;53:172–186.e6. doi: 10.1016/j.immuni.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen H, et al. BCR selection and affinity maturation in Peyer’s patch germinal centres. Nature. 2020;582:421–425. doi: 10.1038/s41586-020-2262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wesemann DR, et al. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature. 2013;501:112–115. doi: 10.1038/nature12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hapfelmeier S, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–1709. doi: 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sefik E, et al. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. 2015;349:993–997. doi: 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cahenzli J, Köller Y, Wyss M, Geuking MB, McCoy KD. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe. 2013;14:559–570. doi: 10.1016/j.chom.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Round JL, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang C, et al. Fecal IgA levels are determined by strain-level differences in bacteroides ovatus and are modifiable by gut microbiota manipulation. Cell Host Microbe. 2020;27:467–475.e6. doi: 10.1016/j.chom.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pabst O, Cerovic V, Hornef M. Secretory IgA in the coordination of establishment and maintenance of the microbiota. Trends Immunol. 2016;37:287–296. doi: 10.1016/j.it.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 95.Marcotte E, et al. Cesarean delivery and risk of childhood leukemia: findings from the Childhood Leukemia International Consortium (CLIC) Cancer Res. 2015;75:LB-194. doi: 10.1158/1538-7445.AM2015-LB-194. [DOI] [Google Scholar]
- 96.Zachariassen LF, et al. Cesarean section induces microbiota-regulated immune disturbances in C57BL/6 mice. J. Immunol. 2019;202:142–150. doi: 10.4049/jimmunol.1800666. [DOI] [PubMed] [Google Scholar]
- 97.Busi SB, et al. Persistence of birth mode-dependent effects on gut microbiome composition, immune system stimulation and antimicrobial resistance during the first year of life. ISME Commun. 2021;1:8. doi: 10.1038/s43705-021-00003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramanan D, et al. An immunologic mode of multigenerational transmission governs a gut Treg setpoint. Cell. 2020;181:1276–1290.e13. doi: 10.1016/j.cell.2020.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van den Elsen LWJ, Garssen J, Burcelin R, Verhasselt V. Shaping the gut microbiota by breastfeeding: the gateway to allergy prevention? Front. Pediatr. 2019;7:47. doi: 10.3389/fped.2019.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lundell A-C, et al. Infant B cell memory differentiation and early gut bacterial colonization. J. Immunol. 2012;188:4315–4322. doi: 10.4049/jimmunol.1103223. [DOI] [PubMed] [Google Scholar]
- 101.Wood H, et al. Breastfeeding promotes early neonatal regulatory T-cell expansion and immune tolerance of non-inherited maternal antigens. Allergy. 2021;76:2447–2460. doi: 10.1111/all.14736. [DOI] [PubMed] [Google Scholar]
- 102.Henrick BM, et al. Bifidobacteria-mediated immune system imprinting early in life. Cell. 2021;184:3884–3898.e11. doi: 10.1016/j.cell.2021.05.030. [DOI] [PubMed] [Google Scholar]
- 103.Vatanen T, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165:842–853. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019;16:35–56. doi: 10.1038/s41575-018-0061-2. [DOI] [PubMed] [Google Scholar]
- 105.Arpaia N, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate conditions human dendritic cells to prime type 1 regulatory T cells via both histone deacetylase inhibition and G protein-coupled receptor 109A signaling. Front. Immunol. 2017;8:1429. doi: 10.3389/fimmu.2017.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Furusawa Y, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 108.Smith PM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cait A, et al. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 2018;11:785–795. doi: 10.1038/mi.2017.75. [DOI] [PubMed] [Google Scholar]
- 110.Rosser EC, et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1β and interleukin-6 production. Nat. Med. 2014;20:1334–1339. doi: 10.1038/nm.3680. [DOI] [PubMed] [Google Scholar]
- 111.Sanchez HN, et al. B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat. Commun. 2020;11:60. doi: 10.1038/s41467-019-13603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Makki K, Deehan EC, Walter J, Bäckhed F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe. 2018;23:705–715. doi: 10.1016/j.chom.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 113.Corte V, et al. Microbiota derived short chain fatty acids, propionate and butyrate, contribute to modulate the inflammatory response in chronic kidney disease. Nephrol. Dial. Transplant. 2020;35:gfaa140.MO046. doi: 10.1093/ndt/gfaa140.MO046. [DOI] [Google Scholar]
- 114.Stražar M, et al. The influence of the gut microbiome on BCG-induced trained immunity. Genome Biol. 2021;22:275. doi: 10.1186/s13059-021-02482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nakajima A, et al. Maternal high fiber diet during pregnancy and lactation influences regulatory T cell differentiation in offspring in mice. J. Immunol. 2017;199:3516–3524. doi: 10.4049/jimmunol.1700248. [DOI] [PubMed] [Google Scholar]
- 116.Kwan ML, et al. Maternal diet and risk of childhood acute lymphoblastic leukemia. Public Health Rep. 2009;124:503–514. doi: 10.1177/003335490912400407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heiss CN, et al. The gut microbiota regulates hypothalamic inflammation and leptin sensitivity in Western diet-fed mice via a GLP-1R-dependent mechanism. Cell Rep. 2021;35:109163. doi: 10.1016/j.celrep.2021.109163. [DOI] [PubMed] [Google Scholar]
- 118.Lu Z, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat. Med. 2017;23:79–90. doi: 10.1038/nm.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blaser, M., Nomura, A., Lee, J., Stemmerman, G. & Perez-Perez GI. Early-life family structure and microbially induced cancer risk. PLoS ONE4, e100 (2007). [DOI] [PMC free article] [PubMed]
- 120.Wellbrock M, et al. 28-year incidence and time trends of childhood leukaemia in former East Germany compared to West Germany after German reunification: a study from the German Childhood Cancer Registry. Cancer Epidemiol. 2021;73:101968. doi: 10.1016/j.canep.2021.101968. [DOI] [PubMed] [Google Scholar]
- 121.Steliarova-Foucher E, et al. Changing geographical patterns and trends in cancer incidence in children and adolescents in Europe, 1991–2010 (automated childhood cancer information system): a population-based study. Lancet Oncol. 2018;19:1159–1169. doi: 10.1016/S1470-2045(18)30423-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Linet MS, et al. International long-term trends and recent patterns in the incidence of leukemias and lymphomas among children and adolescents ages 0–19 years. Int. J. Cancer. 2016;138:1862–1874. doi: 10.1002/ijc.29924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou L, et al. Faecalibacterium prausnitzii produces butyrate to maintain Th17/Treg balance and to ameliorate colorectal colitis by inhibiting histone deacetylase 1. Inflamm. Bowel Dis. 2018;24:1926–1940. doi: 10.1093/ibd/izy182. [DOI] [PubMed] [Google Scholar]
- 124.Zhang M, et al. Faecalibacterium prausnitzii produces butyrate to decrease c-Myc-related metabolism and Th17 differentiation by inhibiting histone deacetylase 3. Int. Immunol. 2019;31:499–514. doi: 10.1093/intimm/dxz022. [DOI] [PubMed] [Google Scholar]
- 125.Ponsonby A, et al. Household size, T regulatory cell development, and early allergic disease: a birth cohort study. Pediatr. Allergy Immunol. 2022;33:e13810. doi: 10.1111/pai.13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roslund MI, et al. Biodiversity intervention enhances immune regulation and health-associated commensal microbiota among daycare children. Sci. Adv. 2020;6:eaba2578. doi: 10.1126/sciadv.aba2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang JS, et al. Profound deficit of IL10 at birth in children who develop childhood acute lymphoblastic leukemia. Cancer Epidemiol. Biomark. Prev. 2011;20:1736–1740. doi: 10.1158/1055-9965.EPI-11-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fitch B, et al. Decreased IL-10 accelerates B-cell leukemia/lymphoma in a mouse model of pediatric lymphoid leukemia. Blood Adv. 2021;6:854–865. doi: 10.1182/bloodadvances.2021005522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Harper A, et al. Viral infections, the microbiome, and probiotics. Front. Cell Infect. Microbiol. 2021;10:596166. doi: 10.3389/fcimb.2020.596166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Erttmann SF, et al. The gut microbiota prime systemic antiviral immunity via the cGAS-STING-IFN-I axis. Immunity. 2022;55:847–861.e10. doi: 10.1016/j.immuni.2022.04.006. [DOI] [PubMed] [Google Scholar]
- 131.Wirusanti NI, Baldridge MT, Harris VC. Microbiota regulation of viral infections through interferon signaling. Trends Microbiol. 2022;30:778–792. doi: 10.1016/j.tim.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Haak BW, et al. Impact of gut colonization with butyrate-producing microbiota on respiratory viral infection following allo-HCT. Blood. 2018;131:2978–2986. doi: 10.1182/blood-2018-01-828996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brown JA, et al. Gut microbiota-derived metabolites confer protection against SARS-CoV-2 infection. Gut Microbes. 2022;14:2105609. doi: 10.1080/19490976.2022.2105609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Albrich WC, et al. A high-risk gut microbiota configuration associates with fatal hyperinflammatory immune and metabolic responses to SARS-CoV-2. Gut Microbes. 2022;14:2073131. doi: 10.1080/19490976.2022.2073131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huda MN, et al. Stool microbiota and vaccine responses of infants. Pediatrics. 2014;134:3937. doi: 10.1542/peds.2013-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.de Jong SE, Olin A, Pulendran B. The impact of the microbiome on immunity to vaccination in humans. Cell Host Microbe. 2020;28:169–179. doi: 10.1016/j.chom.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Trompette A, et al. Dietary fiber confers protection against flu by shaping Ly6c− patrolling monocyte hematopoiesis and CD8+ T cell metabolism. Immunity. 2018;48:992–1005.e8. doi: 10.1016/j.immuni.2018.04.022. [DOI] [PubMed] [Google Scholar]
- 138.Vicente-Dueñas C, et al. An intact gut microbiome protects genetically predisposed mice against leukemia. Blood. 2020;136:2003–2017. doi: 10.1182/blood.2019004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang R, et al. Gut microbiota regulates acute myeloid leukaemia via alteration of intestinal barrier function mediated by butyrate. Nat. Commun. 2022;13:2522. doi: 10.1038/s41467-022-30240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beneforti L, et al. Pro-inflammatory cytokines favor the emergence of ETV6-RUNX1-positive pre-leukemic cells in a model of mesenchymal niche. Br. J. Haematol. 2020;190:262–273. doi: 10.1111/bjh.16523. [DOI] [PubMed] [Google Scholar]
- 141.Dander E, Palmi C, D’amico G, Cazzaniga G. The bone marrow niche in B-cell acute lymphoblastic leukemia: the role of microenvironment from pre-leukemia to overt leukemia. Int. J. Mol. Sci. 2021;22:4426. doi: 10.3390/ijms22094426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xiao E, et al. Microbiota regulates bone marrow mesenchymal stem cell lineage differentiation and immunomodulation. Stem Cell Res. Ther. 2017;8:213. doi: 10.1186/s13287-017-0670-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Marcos-Zambrano LJ, et al. Applications of machine learning in human microbiome studies: a review on feature selection, biomarker identification, disease prediction and treatment. Front. Microbiol. 2021;12:634511. doi: 10.3389/fmicb.2021.634511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McCoubrey LE, Elbadawi M, Orlu M, Gaisford S, Basit AW. Harnessing machine learning for development of microbiome therapeutics. Gut Microbes. 2021;13:1872323. doi: 10.1080/19490976.2021.1872323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mirzayi C, et al. Reporting guidelines for human microbiome research: the STORMS checklist. Nat. Med. 2021;27:1885–1892. doi: 10.1038/s41591-021-01552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Blaser MJ. The theory of disappearing microbiota and the epidemics of chronic diseases. Nat. Rev. Immunol. 2017;17:461–463. doi: 10.1038/nri.2017.77. [DOI] [PubMed] [Google Scholar]
- 147.Panigrahi P, et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature. 2017;548:407–412. doi: 10.1038/nature23480. [DOI] [PubMed] [Google Scholar]
- 148.Korpela K, et al. Probiotic supplementation restores normal microbiota composition and function in antibiotic-treated and in caesarean-born infants. Microbiome. 2018;6:182. doi: 10.1186/s40168-018-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Durack J, et al. Delayed gut microbiota development in high-risk for asthma infants is temporarily modifiable by Lactobacillus supplementation. Nat. Commun. 2018;9:707. doi: 10.1038/s41467-018-03157-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The primary data that support the findings presented in this Perspective article, including the results of our re-analysis of participant-level data of the study of Liu et al.71, are available as Supplementary figures.