

Abstract
Bicyclo[3.2.1] lactones are chemical scaffolds found in numerous bioactive natural products. Herein, we detail the development of a novel palladium(II)-catalyzed tandem intramolecular β-C(sp3)–H olefination and lactonization reaction that rapidly transforms linear carboxylic acid possessing a tethered olefin into the bicyclo[3.2.1] lactone motif. This transformation features a broad substrate scope, shows excellent functional group compatibility, and can be extended to the preparation of the related seven-membered bicyclo[4.2.1] lactones. Additionally, we demonstrate the synthetic potential of this annulation by constructing the 6,6,5-tricyclic lactone core structure of the meroterpenoid cochlactone A. We anticipate that this compelling reaction may provide a novel synthetic disconnection that can be broadly applied towards the preparation of a variety of bioactive natural products.
Graphical Abstract
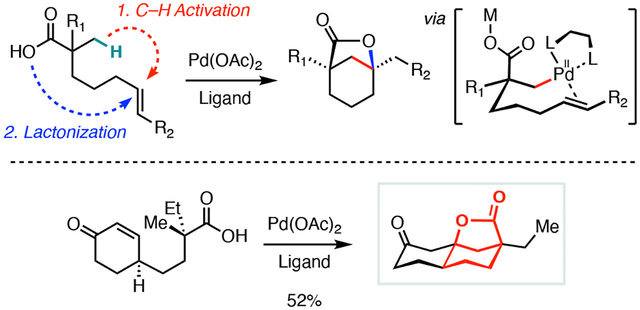
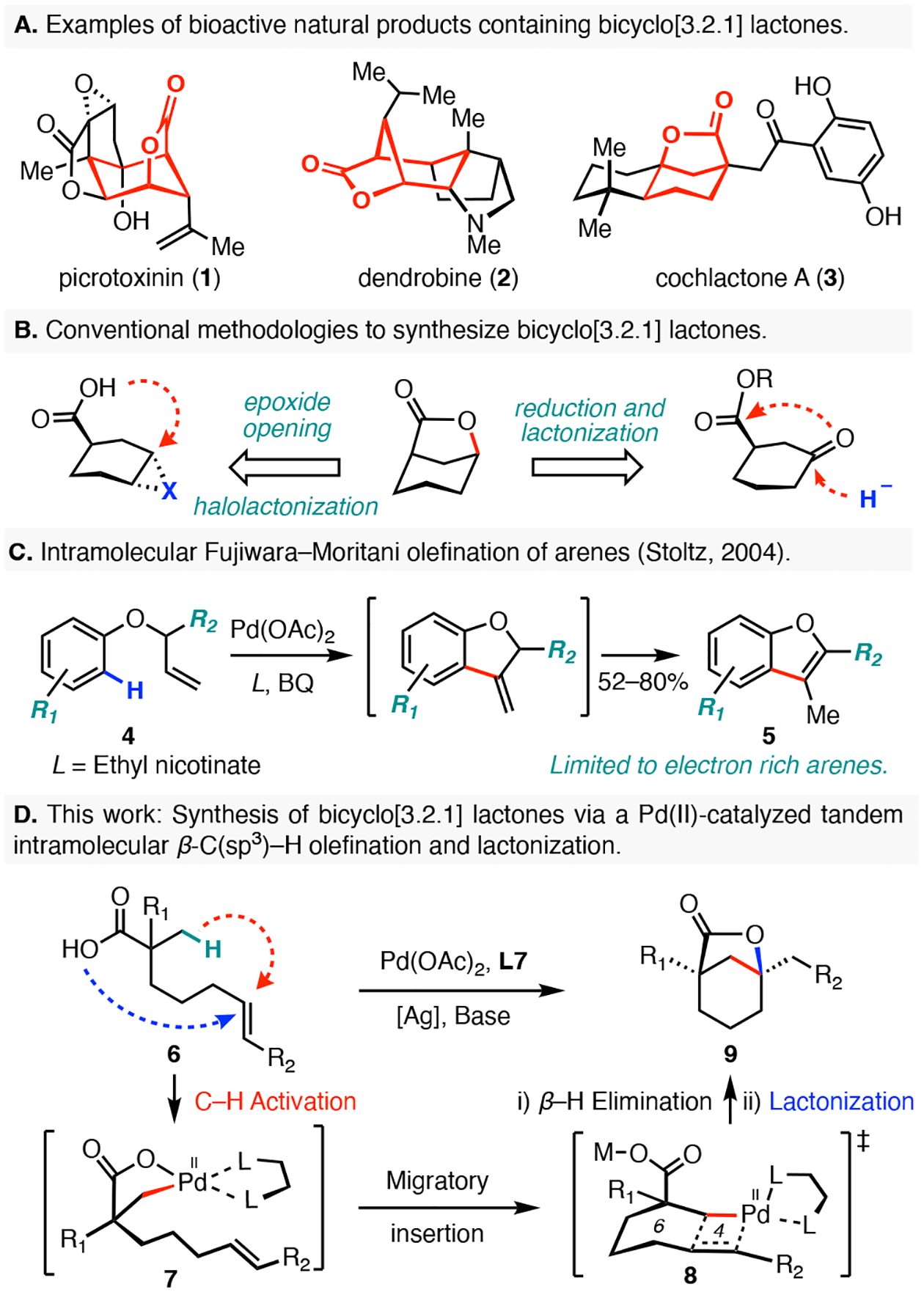
Bicyclo[3.2.1] lactones represent a structural motif present in numerous bioactive natural products, such as picrotoxinin (1), dendrobine (2), and cochlactone A (3, Figure 1A).1 Conventionally, this structure is accessed by synthesis of a functionalized six-membered ring with a pendant carboxylic acid (Figure 1B). From elaborated cyclohexenes, halolactonization or an acid/base-mediated opening of an epoxide ring can be used to form the lactone; however, both of these approaches introduce a potentially undesirable heteroatom that may need to be removed.2 An alternative preparation of bicyclo[3.2.1] lactones from cyclohexanones employs a two-step protocol comprising of a an initial carbonyl reduction followed by a hydroxyl-directed lactonization reaction.3 Successful implementations of this protocol can be limited by stereoselectivity issues in the initial reduction step. Given the recent developments in carboxylic-acid directed C–H functionalization reactions,4 we reasoned that it might be possible to synthesize this intriguing lactone scaffold de novo via a palladium(II)-catalyzed tandem intramolecular β-C(sp3)–H olefination and lactonization sequence of linear carboxylic acid precursors possessing a tethered olefin (Figure 1D). Such approach would significantly streamline synthetic access to these bridged scaffolds and would also provide a novel disconnection that was previously inaccessible via conventional methodologies.
Figure 1.
Pd(II)-catalyzed synthesis of bicyclo[3.2.1] lactones.
To date, intramolecular C–H olefination strategies have only been successfully coupled with C(sp2)–H activation reactions. For instance, palladium mediated oxidative couplings between indoles and olefins have been reported as key steps in syntheses of several natural products.5 In 2004, Stoltz and co-workers disclosed the catalytic intramolecular Fujiwara–Moritani/oxidative Heck cyclization of arenes (Figure 1C).6 Although this impressive transformation provided access to functionalized benzofurans and dihydrobenzofurans, the non-directed electrophilic palladation mechanism was limited to highly electronrich arenes. Additionally, Wang and co-workers disclosed a directed macrocyclization protocol for the synthesis of stapled peptides via the intramolecular olefination of phenylalanines,7 arylsulfonamides,8 and arylacetamide.9 While intermolecular β-C(sp3)–H olefination reactions have been achieved with numerous directing groups, including perfluorinated N-arylamide,10 8-aminoquinoline,11 pyridine,12 free carboxylic acid,4e and native amides,13 the development of the corresponding intramolecular C(sp3)–H olefination reaction remains a significant challenge.
As part of our research program aimed at developing new C–H functionalization reactions that rapidly increase molecular complexity, we herein report a palladium(II)-catalyzed synthesis of bicyclo[3.2.1] lactones from linear carboxylic acids (Figure 1D). At the outset of our work, we hypothesized that this transformation may proceed by an initial carboxylic-acid directed β-C(sp3)–H activation followed by intramolecular olefin migratory insertion, β–H elimination of the resulting Pd(II)alkyl complex, and lactonization via a conjugate addition of the carboxylic acid. However, we recognized that several challenges might make this transformation difficult to realize. First, competitive chelation of the pendant double bond to the palladium center was recognized as a potential impediment to C–H activation. We surmised that the use of an electron-deficient alkene might solve this problem, but this raises the potential for a competitive intramolecular 7-exo-trig oxy-Michael addition prior to the C–H activation event. Second, the introduction of the olefin moiety into the backbone of the linear substrate 6 introduces a reactive allylic C–H bond that can compete with the desired β-C–H bond activation.14 Third, the putative carbon–carbon bond forming step would involve intramolecular migratory insertion of the alkylpalladium species 7 via the 6–4 bicyclic palladacycle 8, which has been previously described only for a few examples of highly activated benzyl halide mediated C(sp3)–C(sp2) Heck reactions.15
We began our investigation by preparing the linear cyclization substrate 10a, which was easily accessed in four steps from the commercial isobutyric acid (see the Supporting Information). Our group and others have showed that mono-N-protected amino acids (MPAAs) are excellent ligands for enabling a range of C(sp3)–H activation reactions of free carboxylic acids.16 Guided by these findings, our initial reaction conditions examined a series of commercially available MPAAs. We were pleased to find that the phenylalanine derivative (L1) provided the desired bicyclo[3.2.1] lactone product 11a in a modest 43% yield without any detectable side products other than the unreacted 10a (Scheme 1). Several additional ligands were examined, including the pyridine-based L2 and L3 and the monoprotected aminoethyl amine derivative L4, but these did not improve the reaction yield (39–42%). Additionally, we found that the use of the bidentate acyl-protected amino oxazoline L5 or the monodentate pyridone L6 resulted in a decreased yield (25% and 20%, respectively). Next, we turned our attention to thioether containing ligands that can act as soft σ-donors capable of stabilizing the palladium catalyst through their strong coordination.4e,17 When the N-acyl aminoethyl phenyl thioether (L7) was employed, we observed a significant increase in the yield of the product. Following additional modifications to the reaction conditions such as varying the base, bystanding oxidant, and solvent we were able to obtain the bicyclo[3.2.1] lactone product 11a in 87% yield (see the Supporting Information).18
Scheme 1.
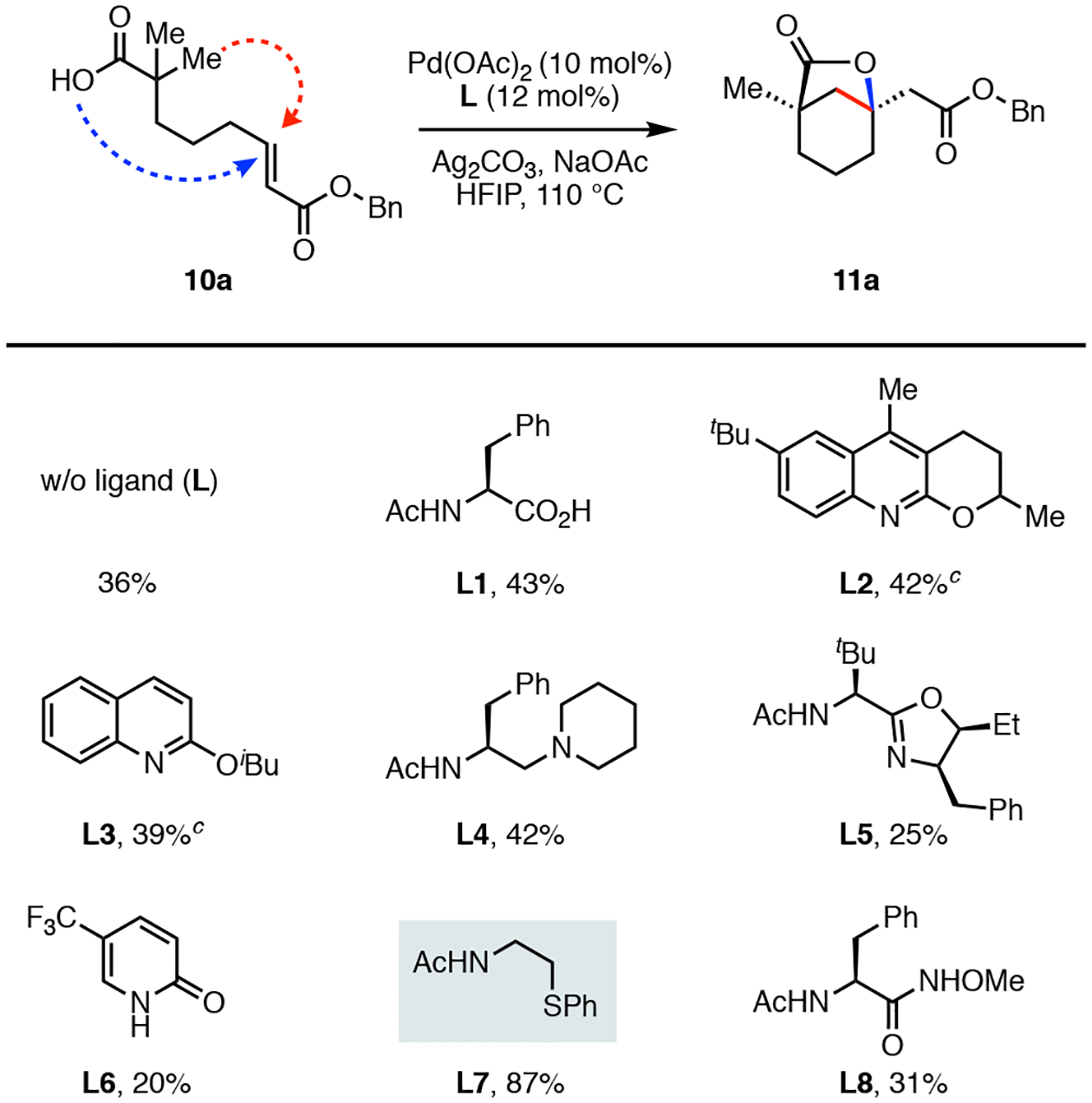
Ligand Investigation for the Tandem Intramolecular β-C(sp3)–H Olefination and Lactonization.a,b
aConditions: 10a (0.1 mmol), Pd(OAc)2 (10 mol%), Ligand (L) (12 mol%), Ag2CO3 (2.0 equiv), NaOAc (1.0 equiv), HFIP (1.0 mL), 110 °C, 15h. bYields were determined by 1H NMR analysis using CH2Br2 as an internal standard. c Ligand (L) (20 mol%).
With the optimal reaction conditions in hand, we next investigated the substrate scope with respect to the olefin moieties (Scheme 2). Using olefin cross metathesis with the appropriate acrylate, it was possible to prepare a range of annulation precursors bearing a variety of different functional groups. We found that simple alkyl- (11b–11e) and the methoxyethyl ether acrylates 11f were tolerated in excellent yields ranging from 73% to 82%. Other electron-withdrawing substituents such, as the methyl ketone (11g), cyanide (11h), and phosphonate (11i) also provided the bicyclo[3.2.1] lactones in high yields (82–88%). While the tertiary amides (11j–11l) were compatible with the intramolecular β-C(sp3)–H olefination and lactonization reaction, products were formed in slightly lower yields (53–64%). We attribute this to the reduced electron withdrawing nature of the unsaturated amides compared to the corresponding unsaturated esters. Lastly, the sulfonyl phenyl group was also tolerated and provided the crystalline 11m product in 78% yields. In the absence of a terminal electron withdrawing group we did not observe formation of the bicyclo[3.2.1] lactone but instead obtained product derived from allylic C–H functionalization (see the Supporting Information).19
Scheme 2.
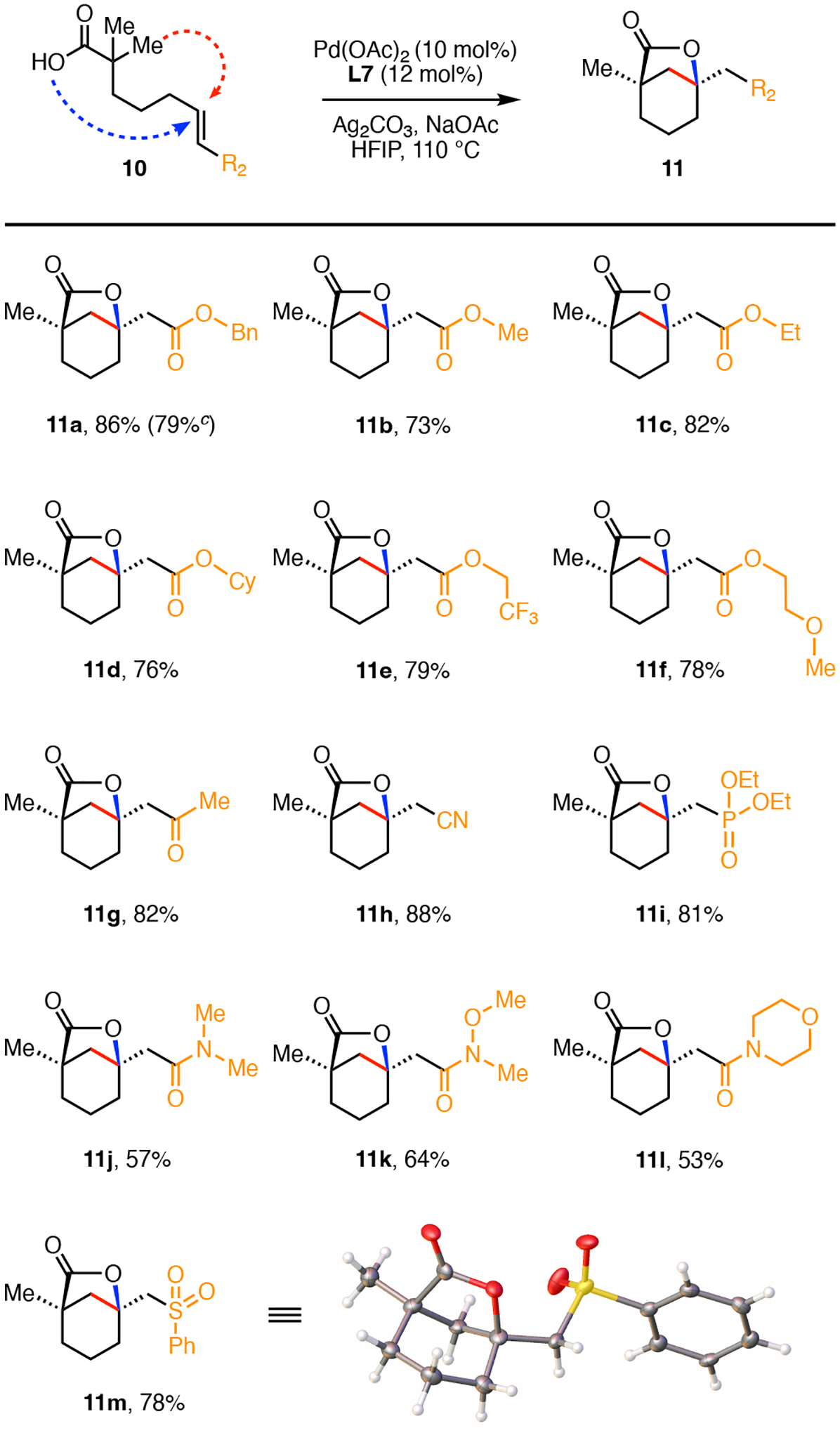
Substrate Scope of the Alkene Residue.a,b
aConditions: 10 (0.1 mmol), Pd(OAc)2 (10 mol%), Ligand (L7) (12 mol%), Ag2CO3 (2.0 equiv), NaOAc (1.0 equiv), HFIP (1.0 mL), 110 °C, 15h. bIsolated Yields. cIsolated yield on 1.0 mmol scale.
A wide variety of substituents at the β-position of the carboxylic acid residue were found to be compatible with reactivity occurring exclusively at the methyl C−H bond (Scheme 3). For example, the alkyl bearing substrates (13a–13d) provided the desired lactone products in 61–74% isolated yields. The alkyl chloride 12e was transformed to the lactone 13e in 60% yield. This substituent provides a useful synthetic handle for further product elaboration. The α-hydrogen substrate 12f that lacks the favorable Thorpe−Ingold effect coupled with the typically interfering acidic C−H bond displayed a significantly reduced reactivity. However, we found that increasing the palladium and ligand loadings by twofold resulted in the formation of the α-hydrogen bicyclo[3.2.1] lactone 13f in 27% yield. Substrates containing the chelating oxygen heteroatoms such as the methoxy-12g and the benzyloxy-12h substituents showed good reactivity and provided the desired products in 73% and 65% yields.
Scheme 3.
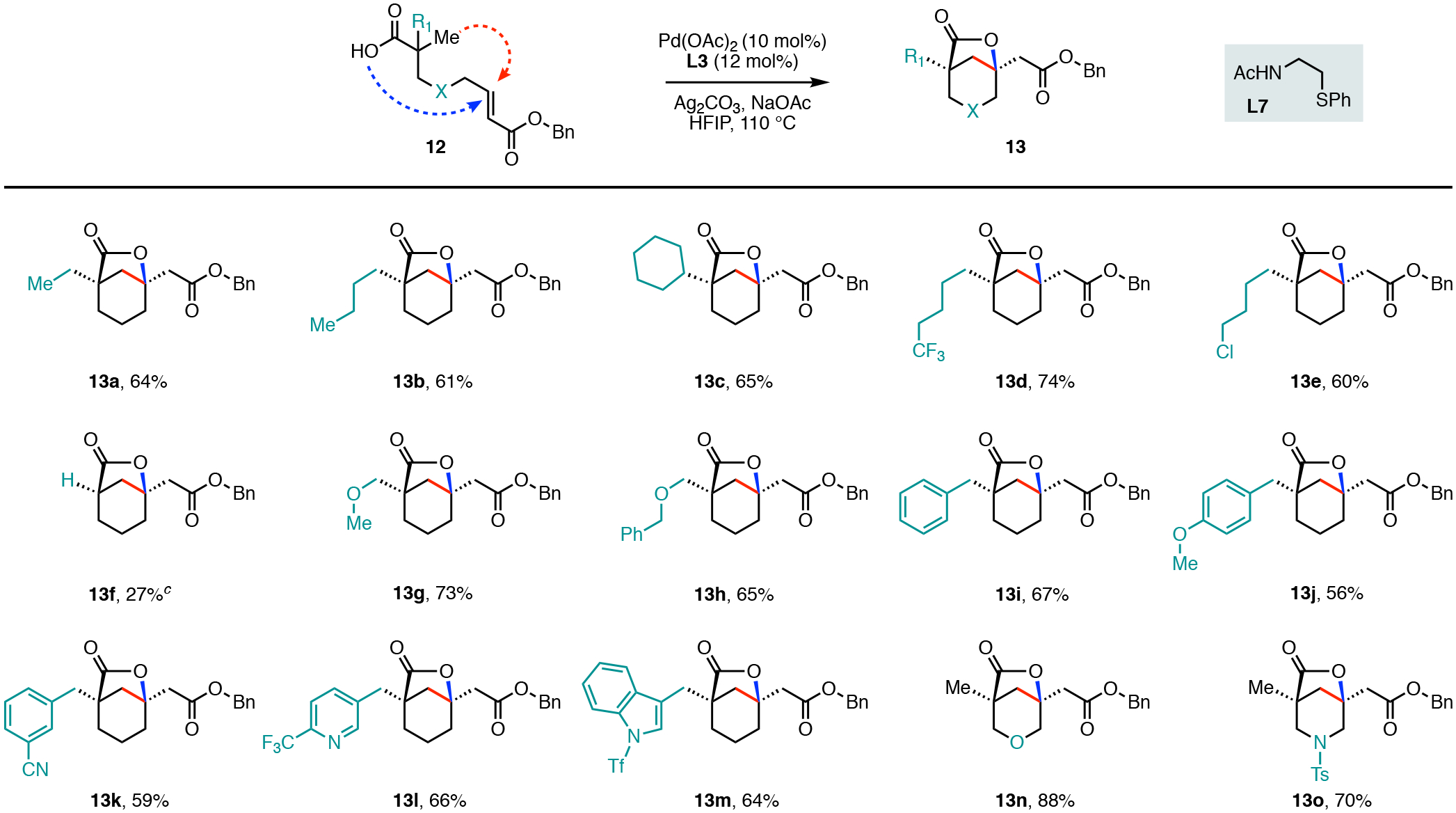
Carboxylic Acids Substrate Scope.a,b
aConditions: 12 (0.1 mmol), Pd(OAc)2 (10 mol%), Ligand (L7) (12 mol%), Ag2CO3 (2.0 equiv), NaOAc (1.0 equiv), HFIP (1.0 mL), 110 °C, 15h. bIsolated yield. cPd(OAc)2 (20 mol%), Ligand (L7) (24 mol%).
Our reaction protocol was also compatible with the incorporation of electron-neutral (13i), electron-donating (13j), and electron-withdrawing (13k) aryl substituents and furnished the lactone products in overall good yields (56–67%, Scheme 3). This transformation also tolerated the 2-(trifluoromethyl)pyridine and tosyl protected indole heterocycles and provided the annulated lactone products 13l (66%) and 13m (64%). Lastly, we evaluated the influence of oxygen and nitrogen substituents in the linear backbone and prepared the corresponding precursors 12n and 12o. Both of these substrates were well tolerated and generated the tetrahydropyran and piperidine bicyclo[3.2.1] lactones in 88% and 70%, respectively.
We also sought to investigate what role, if any, the geometry of alkene plays in the tandem intramolecular β-C(sp3)–H olefination and lactonization reaction (Scheme 4). The preparation of all of the substrates shown in Schemes 2 and 3 employed olefin cross metathesis and consequently furnished the depicted E-alkene configuration for all substrates. To arrive at the opposite Z-alkene isomer, we utilized the Z-selective Still–Gennari20 olefination reaction to generate the linear precursor (Z)-10c. Exposure of the (Z)-10c to our standard reaction conditions provided the bicyclo[3.2.1] lactone 11c in 84% yield. This result highlighted that both olefin isomers are transformed to product in comparable reaction yield, and that this transformation can be used in a setting where it may not be possible to exclusively prepare a single olefin isomer.
Scheme 4.
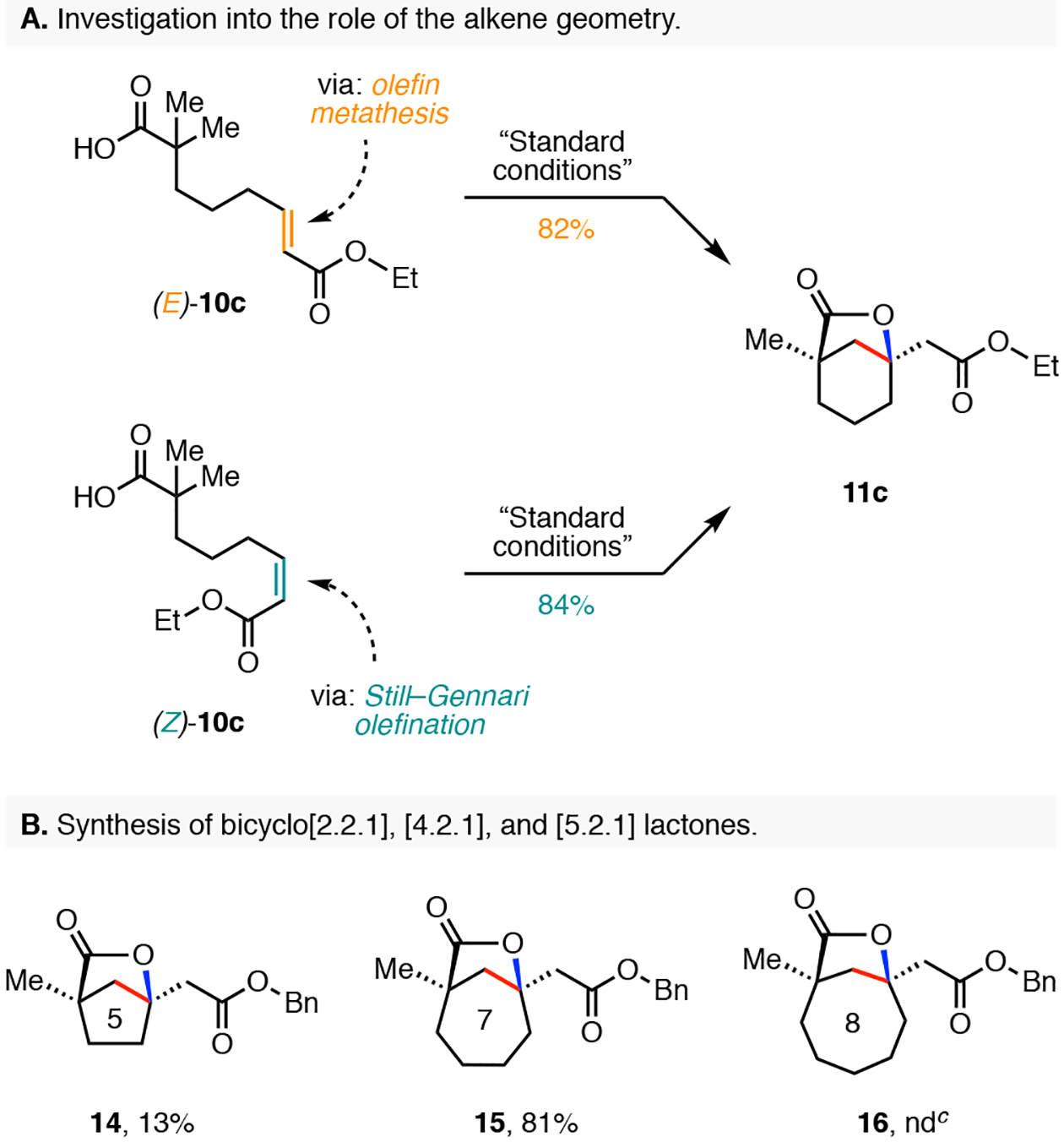
Alkene Geometry and Synthetic Application.a,b
aConditions: Carboxylic acid (0.1 mmol), Pd(OAc)2 (10 mol%), Ligand (L7) (12 mol%), Ag2CO3 (2.0 equiv), NaOAc (1.0 equiv), HFIP (1.0 mL), 110 °C, 15h. bIsolated yield. cNone detected.
It was of interest to determine if this methodology is amenable to preparation of bicyclic lactones with varying numbers of carbon atoms in the central ring system (Scheme 4B). When this strategy was applied towards the synthesis of bicyclo[2.2.1] lactone, we observed formation of the product 14 in 13% yield. We attribute the reduction in product yield to the considerable increase in steric strain about the alkylpalladium species during the intramolecular migratory insertion step. Pleasingly, we found that the addition of one extra carbon into the backbone of the annulation precursor was tolerated and the reaction provided the seven-membered bicyclo[4.2.1] lactone 15 in 81% yield. A limitation of this methodology was met when we attempted to prepare the eight-membered bicyclo[5.2.1] lactone 16. Exposure of the corresponding starting material to our reaction conditions resulted in a recovery of the starting material and no detectable levels of the lactone 16 were observed.
To showcase the potential synthetic utility of this reaction, we applied this method to the synthesis of the 6,6,5-tricyclic lactone 24 that constitutes the core of the meroterpenoid cochlactone A (3, Scheme 5). Cochlactone A (3) was isolated as a racemate from the fungus Ganoderma cochlear in 2018 and showed promising anti-inflammatory activity (nitric oxide production inhibitory assay: IC50 = 5.9 ± 0.1 μM).1d Our short approach began with a sequential alkylation of 17 (lithium diisopropylamide, methyl iodide followed by lithium diisopropylamide, ethyl iodide) to provide the α-quaternary ester 18 (88% over two steps). Subsequent oxidative cleavage (ozone, triphenyl phosphine) of the terminal alkene provided the aldehyde 19 (94%). Robinson annulation between methyl vinyl ketone and the aldehyde 19 was catalyzed by (diethylamino)trimethylsilane to first generate the Michael adduct 20, which was subsequently treated with potassium hydroxide and tetrabutylammonium hydroxide to provide the cyclohexanone 21 (81% yield, 1:1 diastereomers at C5).21 Removal of the trimethylsilyl ethyl ester (refluxing trifluoroacetic acid) generated the cyclization precursor 23 (49%) along with the C5 diastereomer 22 (49%). The pivotal tandem intramolecular β-C(sp3)–H olefination and lactonization reaction of 23 using standard conditions with two equivalents of lithium carbonate provided the 6,6,5-tricyclic lactone core 24 in 52% yield. Our effort towards this interesting tricyclic scaffold underscores the ability of this newly developed transformation to provide access to synthetically useful building blocks and, importantly, suggests that it may find use in complex natural product syntheses.
Scheme 5.
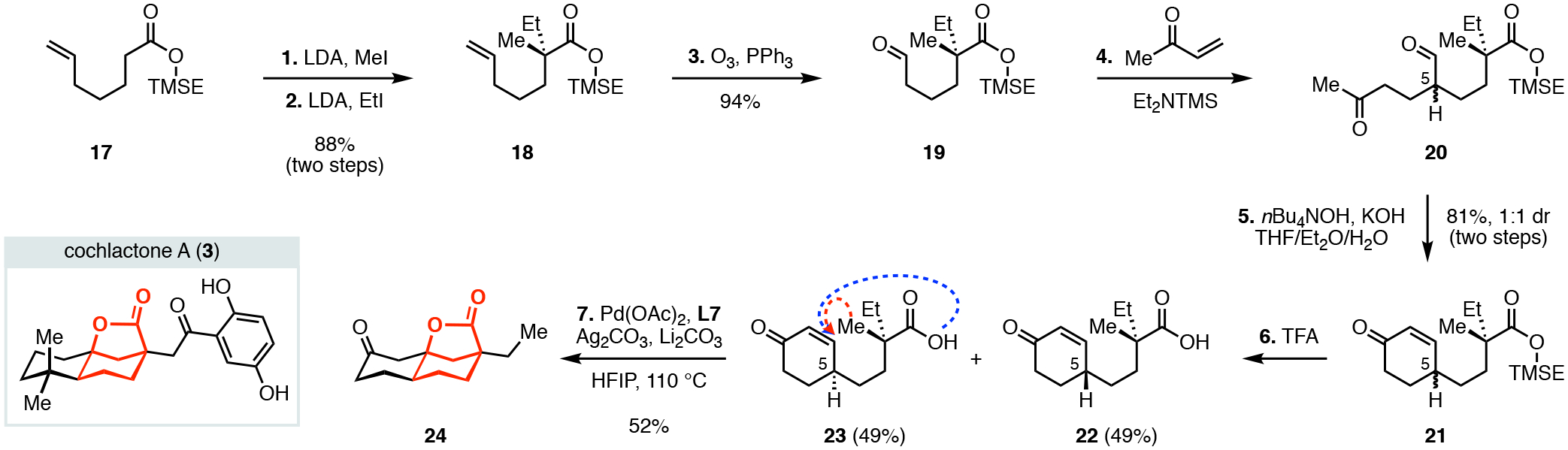
Synthesis of the 6,6,5-Tricyclic Lactone Core Structure of Cochlactone A (3).a,b
aReagents and conditions: (1) LDA (1.2 equiv), MeI (1.5 equiv), THF, −78→0 °C; (2) LDA (1.2 equiv), EtI (1.5 equiv), THF, −78→0 °C; (3) O3, PPh3 (1.5 equiv), DCM−MeOH (10:1 v/v) −78→0 °C; (4) MVK (1.5 equiv), Et2NTMS (0.2 equiv), CH3CN, reflux; (5) KOH (0.9 equiv), nBu4NOH (0.25 equiv, 40% aq), Et2O−THF (3:1 v/v) 50 °C; (6) TFA (6.5 equiv), DCM, 40 °C; (7) Pd(OAc)2 (20 mol%), Ligand (L7) (24 mol%), Ag2CO3 (2.0 equiv), Li2CO3 (2.0 equiv), HFIP, 110 °C, 15h. bTMSE = 2-(trimethylsilyl)ethyl
In conclusion, we have developed a palladium(II)-catalyzed tandem intramolecular β-C(sp3)–H olefination and lactonization reaction that rapidly transforms a linear free acid with a tethered olefin into the bicyclo[3.2.1] lactone scaffold. This transformation is amenable to broad structural variability and is compatible with a range of common functional groups. Co-ordinating heteroatoms such as oxygen and nitrogen on the backbone of the annulation substrates are also tolerated and provide a concise synthetic entry to novel tetrahydropyran and piperidine bicyclo[3.2.1] lactones. This reaction can be extended to the preparation of the related seven-membered bicyclo[4.2.1] lactones and proceeds with comparable efficiency using (Z)- and (E)-alkene isomers. We demonstrate the synthetic utility of this reaction by constructing the 6,6,5-tricyclic lactone core structure of the meroterpenoid cochlactone A (3) in seven steps. We anticipate that this newly developed synthetic strategy may find use in the preparation of other natural products and can also be used in medicinal chemistry to generate a library of potentially bioactive compounds.
Supplementary Material
ACKNOWLEDGMENT
Financial support from The Scripps Research Institute and the NIH (NIGMS R01 GM084019) is gratefully acknowledged. We thank Dr. Jason Chen, Brittany Sanchez, and Quynh Nguyen Wong of the TSRI Automated Synthesis Facility for assistance with HRMS and Prep SFC. We also acknowledge M. Gembicky (UCSD) for X-ray analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full experimental details, product characterization, and X-ray crystallographic data (PDF)
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Suzuki H; Keimatsu I; Ito K, Structure determination of (−)-dendrobine. J. Pharm. Soc. Jpn 1934, 54, 802. [Google Scholar]; (b) Porter LA, Picrotoxinin and Related Substances. Chem. Rev 1967, 67, 441–464. [DOI] [PubMed] [Google Scholar]; (c) Jin Z.-l.; Gao N; Xu W; Xu P; Li S; Zheng Y.-y.; Xue M, Receptor and transporter binding and activity profiles of albiflorin extracted from Radix paeoniae Alba. Sci. Rep 2016, 6, 33793. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Peng X-R; Lu S-Y; Shao L-D; Zhou L; Qiu M-H, Structural Elucidation and Biomimetic Synthesis of (±)-Cochlactone A with Anti-Inflammatory Activity. J. Org. Chem 2018, 83, 5516–5522. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dowle MD; Davies DI, Synthesis and synthetic utility of halolactones. Chem. Soc. Rev 1979, 8, 171–197. [Google Scholar]; (b) Aitken RA; Gopal J; Hirst JA, Catalytic asymmetric synthesis of highly functionalised compounds with six contiguous stereocentres. J. Chem. Soc., Chem. Commun 1988, 632–634. [Google Scholar]; (c) Chini M; Crotti P; Flippin LA; Macchia F; Pineschi M, Regiochemical control of the ringopening of 1,2-epoxides by means of chelating processes. 2. Synthesis and reactions of the cis- and trans-oxides of 4-[(benzyloxy)methyl]cyclohexene, 3-cyclohexenemethanol, and methyl 3-cyclohexenecarboxylate. J. Org. Chem 1992, 57, 1405–1412. [Google Scholar]; (d) Trost B; Krische MJ, Palladium-Catalyzed Enyne Cycloisomerization Reaction in an Asymmetric Approach to the Picrotoxane Sesquiterpenes. 2. Second-Generation Total Syntheses of Corianin, Picrotoxinin, Picrotin, and Methyl Picrotoxate. J. Am. Chem. Soc 1999, 121, 6131–6141. [Google Scholar]
- 3.(a) Kende AS; Bentley TJ; Mader RA; Ridge D, Simple total synthesis of (+)-dendrobine. J. Am. Chem. Soc 1974, 96, 4332–4334. [DOI] [PubMed] [Google Scholar]; (b) Beale MH, Conjugate addition of lithium methylcuprates to a gibberell-1(10)en-2-one; preparation of 10-epi-gibberellin A53. J. Chem. Soc., Perkin Trans 1 1985, 1147–1150. [Google Scholar]; (c) Kaneko T; Wong H, Total synthesis of (±) podophyllotoxin. Tetrahedron Lett 1987, 28, 517–520. [Google Scholar]; (d) Poirel C; Renard P-Y; Lallemand J-Y, Stereoselective access to polyfunctionalized decalins. Tetrahedron Lett 1994, 35, 6485–6488. [Google Scholar]; (e) Takahashi Y; Yoshimura F; Tanino K; Miyashita M, Total Synthesis of Zoanthenol. Angew. Chem. Int. Ed 2009, 48, 8905–8908. [DOI] [PubMed] [Google Scholar]
- 4.(a) Giri R; Maugel N; Li J-J; Wang D-H; Breazzano SP; Saunders LB; Yu J-Q, Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C−H Bonds in Simple Carboxylic Acids. J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]; (b) Zhu Y; Chen X; Yuan C; Li G; Zhang J; Zhao Y, Pd-catalysed ligand-enabled carboxylate-directed highly regioselective arylation of aliphatic acids. Nat. Commun 2017, 8, 14904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh KK; van Gemmeren M, Pd-Catalyzed β-C(sp3)−H Arylation of Propionic Acid and Related Aliphatic Acids. Chem. – Eur. J 2017, 23, 17697–17700. [DOI] [PubMed] [Google Scholar]; (d) Shen P-X; Hu L; Shao Q; Hong K; Yu J-Q, Pd(II)-Catalyzed Enantioselective C(sp3)–H Arylation of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhuang Z; Yu C-B; Chen G; Wu Q-F; Hsiao Y; Joe CL; Qiao JX; Poss MA; Yu J-Q, Ligand-Enabled β-C(sp3)–H Olefination of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hu L; Shen P-X; Shao Q; Hong K; Qiao JX; Yu J-Q, PdII-Catalyzed Enantioselective C(sp3)−H Activation/Cross-Coupling Reactions of Free Carboxylic Acids. Angew. Chem. Int. Ed 2019, 58, 2134–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ghosh KK; Uttry A; Koldemir A; Ong M; van Gemmeren M, Direct β-C(sp3)–H Acetoxylation of Aliphatic Carboxylic Acids. Org. Lett 2019, 21, 7154–7157. [DOI] [PubMed] [Google Scholar]; (h) Zhuang Z; Yu J-Q, Lactonization as a general route to β-C(sp3)–H functionalization. Nature 2020, 577, 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zhuang Z; Herron AN; Fan Z; Yu J-Q, Ligand-Enabled Monoselective β-C(sp3)–H Acyloxylation of Free Carboxylic Acids Using a Practical Oxidant. J. Am. Chem. Soc 2020, 142, 6769–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ghiringhelli F; Uttry A; Ghosh KK; van Gemmeren M, Direct β- and γ-C(sp3)−H Alkynylation of Free Carboxylic Acids**. Angew. Chem. Int. Ed 2020, 59, 23127–23131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wang Z; Hu L; Chekshin N; Zhuang Z; Qian S; Qiao Jennifer X; Yu J-Q, Ligand-controlled divergent dehydrogenative reactions of carboxylic acids via C–H activation. Science 2021, 374, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Zhuang Z; Herron AN; Yu J-Q, Synthesis of Cyclic Anhydrides via Ligand-Enabled C–H Carbonylation of Simple Aliphatic Acids. Angew. Chem. Int. Ed 2021, 60, 16382–16387. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Lucas EL; Lam NYS; Zhuang Z; Chan HSS; Strassfeld DA; Yu J-Q, Palladium-Catalyzed Enantioselective β-C(sp3)–H Activation Reactions of Aliphatic Acids: A Retrosynthetic Surrogate for Enolate Alkylation and Conjugate Addition. Acc. Chem. Res 2022, 55, 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Trost BM; Godleski SA; Genet JP, A total synthesis of racemic and optically active ibogamine. Utilization and mechanism of a new silver ion assisted palladium catalyzed cyclization. J. Am. Chem. Soc 1978, 100, 3930–3931. [Google Scholar]; (b) Baran PS; Corey EJ, A Short Synthetic Route to (+)-Austamide, (+)-Deoxyisoaustamide, and (+)-Hydratoaustamide from a Common Precursor by a Novel Palladium-Mediated Indole → Dihydroindoloazocine Cyclization. J. Am. Chem. Soc 2002, 124, 7904–7905. [DOI] [PubMed] [Google Scholar]; (c) Baran PS; Guerrero CA; Corey EJ, Short, Enantioselective Total Synthesis of Okaramine N. J. Am. Chem. Soc 2003, 125, 5628–5629. [DOI] [PubMed] [Google Scholar]; (d) Ferreira EM; Stoltz BM, Catalytic C−H Bond Functionalization with Palladium(II): Aerobic Oxidative Annulations of Indoles. J. Am. Chem. Soc 2003, 125, 9578–9579. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H; Ferreira EM; Stoltz BM, Direct Oxidative Heck Cyclizations: Intramolecular Fujiwara–Moritani Arylations for the Synthesis of Functionalized Benzofurans and Dihydrobenzofurans. Angew. Chem. Int. Ed 2004, 43, 6144–6148. [DOI] [PubMed] [Google Scholar]
- 7.Bai Z; Cai C; Yu Z; Wang H, Backbone-Enabled Directional Peptide Macrocyclization through Late-Stage Palladium-Catalyzed δ-C(sp2)−H Olefination. Angew. Chem. Int. Ed 2018, 57, 13912–13916. [DOI] [PubMed] [Google Scholar]
- 8.Tang J; Chen H; He Y; Sheng W; Bai Q; Wang H, Peptideguided functionalization and macrocyclization of bioactive peptidosulfonamides by Pd(II)-catalyzed late-stage C–H activation. Nat. Commun 2018, 9, 3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan J; Wu J; Liu S; Yao H; Wang H, Macrocyclization of peptidoarylacetamides with self-assembly properties through late-stage palladium-catalyzed C(sp2)–H olefination. Sci. Adv 2019, 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Wasa M; Engle KM; Yu J-Q, Pd(II)-Catalyzed Olefination of sp3 C−H Bonds. J. Am. Chem. Soc 2010, 132, 3680–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J; Li S; Deng Y; Fu H; Laforteza Brian N; Spangler Jillian E; Homs A; Yu J-Q, Ligand-Controlled C(sp3)–H Arylation and Olefination in Synthesis of Unnatural Chiral α–Amino Acids. Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B; Lu C; Zhang S-Y; He G; Nack WA; Chen G, Palladium-Catalyzed Stereoretentive Olefination of Unactivated C(sp3)–H Bonds with Vinyl Iodides at Room Temperature: Synthesis of β-Vinyl α-Amino Acids. Org. Lett 2014, 16, 6260–6263. [DOI] [PubMed] [Google Scholar]
- 12.Stowers KJ; Fortner KC; Sanford MS, Aerobic Pd-Catalyzed sp3 C−H Olefination: A Route to Both N-Heterocyclic Scaffolds and Alkenes. J. Am. Chem. Soc 2011, 133, 6541–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park H; Li Y; Yu J-Q, Utilizing Carbonyl Coordination of Native Amides for Palladium-Catalyzed C(sp3)−H Olefination. Angew. Chem. Int. Ed 2019, 58, 11424–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Fraunhoffer KJ; Prabagaran N; Sirois LE; White MC, Macrolactonization via Hydrocarbon Oxidation. J. Am. Chem. Soc 2006, 128, 9032–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pàmies O; Margalef J; Cañellas S; James J; Judge E; Guiry PJ; Moberg C; Bäckvall J-E; Pfaltz A; Pericàs MA; Diéguez M, Recent Advances in Enantioselective Pd-Catalyzed Allylic Substitution: From Design to Applications. Chem. Rev 2021, 121, 4373–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Wu GZ; Lamaty F; Negishi E, Metal-promoted cyclization. 26. Palladium-catalyzed cyclization of benzyl halides and related electrophiles containing alkenes and alkynes as a novel route to carbocycles. J. Org. Chem 1989, 54, 2507–2508. [Google Scholar]; (b) Wu G; Shimoyama I; Negishi E, Palladium-catalyzed carbonylative cyclization of o-allylbenzyl halides to produce benzo-annulated enol lactones and/or bicyclo[3.3.0]hept-3-en-6-ones. An efficient route to U-68,215. J. Org. Chem 1991, 56, 6506–6507. [Google Scholar]; (c) Liu Z; Shi C; Chen Y, Synthesis of 3-Alkyl-1H-quinolin-2-ones via Palladium-Catalyzed Intramolecular Cyclization of Benzyl Halides and α,β-Unsaturated Amides. Synlett 2008, 2008, 1734–1736. [Google Scholar]; (d) Gharpure SJ; Shelke YG; Reddy SRB, Synthesis of isochromene derivatives using an intramolecular benzylic C(sp3)–C(sp2) bond forming Heck reaction on vinylogous carbonates. RSC Advances 2014, 4, 46962–46965. [Google Scholar]; (e) Kurandina D; Chuentragool P; Gevorgyan V, Transition-Metal-Catalyzed Alkyl Heck-Type Reactions. Synthesis 2019, 51, 985–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Engle KM, The mechanism of palladium(II)-mediated C–H cleavage with mono-N-protected amino acid (MPAA) ligands: origins of rate acceleration. Pure Appl. Chem 2016, 88, 119–138. [Google Scholar]; (b) Shao Q; Wu K; Zhuang Z; Qian S; Yu J-Q, From Pd(OAc)2 to Chiral Catalysts: The Discovery and Development of Bifunctional Mono-N-Protected Amino Acid Ligands for Diverse C–H Functionalization Reactions. Acc. Chem. Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Naksomboon K; Valderas C; Gómez-Martínez M; Álvarez-Casao Y; Fernández-Ibáñez MÁ, S,O-Ligand-Promoted Palladium-Catalyzed C–H Functionalization Reactions of Nondirected Arenes. ACS Catal 2017, 7, 6342–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jerhaoui S; Djukic J-P; Wencel-Delord J; Colobert F, Asymmetric, Nearly Barrierless C(sp3)–H Activation Promoted by Easily-Accessible N-Protected Aminosulfoxides as New Chiral Ligands. ACS Catal 2019, 9, 2532–2542. [Google Scholar]; (c) Sukowski V; Jia W-L; van Diest R; van Borselen M; Fernández-Ibáñez MÁ, S,O-Ligand-Promoted Pd-Catalyzed C−H Olefination of Anisole Derivatives. Eur. J. Org. Chem 2021, 2021, 4132–4135. [Google Scholar]
- 18. We examined the enantioselectivity of the chiral thioether ligands L9, L10, and the N-Ac-Phe (L1) but in all instances the ee was <10% (see the Supporting Information).
- 19. We also explored the viability of a trisubstituted olefin substrate S21 in our transformation. Exposure of S21 to our standard reaction conditions did not provide any of the corresponding bicyclo[3.2.1] lactone product (see the Supporting Information).
- 20.Still WC; Gennari C, Direct synthesis of Z-unsaturated esters. A useful modification of the Horner-Emmons olefination. Tetrahedron Lett 1983, 24, 4405–4408. [Google Scholar]
- 21.Chen K; Ishihara Y; Galán MM; Baran PS, Total synthesis of eudesmane terpenes: cyclase phase. Tetrahedron 2010, 66, 4738–4744. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.