

Abstract
Although duplications have long been recognized as a fundamental process driving major evolutionary innovations, direct estimates of spontaneous chromosome duplication rates, leading to aneuploid karyotypes, are scarce. Here, from mutation accumulation (MA) experiments, we provide the first estimates of spontaneous chromosome duplication rates in six unicellular eukaryotic species, which range from 1 × 10−4 to 1 × 10−3 per genome per generation. Although this is ∼5 to ∼60 times less frequent than spontaneous point mutations per genome, chromosome duplication events can affect 1–7% of the total genome size. In duplicated chromosomes, mRNA levels reflected gene copy numbers, but the level of translation estimated by polysome profiling revealed that dosage compensation must be occurring. In particular, one duplicated chromosome showed a 2.1-fold increase of mRNA but translation rates were decreased to 0.7-fold. Altogether, our results support previous observations of chromosome-dependent dosage compensation effects, providing evidence that compensation occurs during translation. We hypothesize that an unknown posttranscriptional mechanism modulates the translation of hundreds of transcripts from genes located on duplicated regions in eukaryotes.
Keywords: chromosome duplication, mutation accumulation, aneuploidy, dosage compensation, translation efficiency, phytoplankton
Significance.
Aneuploid karyotypes cause genetic disease and decrease fitness due to gene dosage imbalance. The deleterious effects of aneuploidy can lead to the evolution of dosage compensation, a mechanism that restores ancestral ploidy and usually affects the transcription rate. Here, we estimated the rate of spontaneous aneuploidy in six eukaryotic phytoplankton species from mutation accumulation experiments and explored transcriptional and translational responses following whole chromosome duplications. We provide evidence for chromosome-specific posttranscriptional dosage compensation.
Introduction
Complete or partial chromosome duplications leading to aneuploid karyotypes are known to contribute to genetic diseases, such as trisomy 21 in humans or cancer (Rajagopalan and Lengauer 2004). Duplication of a single chromosome leads to an immediate imbalance in cellular metabolism because of the increased levels of transcripts and proteins encoded by the doubled chromosome. This is expected to have deleterious effects because it is costly and may disrupt the function of biochemical pathways and protein interactions (Dephoure et al. 2014; Veitia and Potier 2015). The deleterious effects of aneuploidy have been documented in many different biological model systems such as yeast (Torres et al. 2007), mouse, and human (Gearhart et al. 1987). In Caenorhabditis elegans, mutation accumulation (MA) experiments provided evidence of purifying selection against gene duplications, causing an excess of transcripts, as compared with gene duplications associated with invariant transcript levels (Konrad et al. 2018). Moreover, the consequences of aneuploidy on gene transcription are complex, and whereas gene transcription may increase with chromosome copy number, gene transcription changes may also spread outside the duplicated regions in Arabidopsis (Hou et al. 2018; Song et al. 2020), Drosophila (Devlin et al. 1988), and human cells (FitzPatrick et al. 2002).
In contrast, in some circumstances, aneuploidy may confer a selective advantage such as in resistance to drugs in Candida albicans (Selmecki et al. 2006) or Saccharomyces cerevisiae (Chen et al. 2012). Aneuploidy was also prevalent and tolerated across S. cerevisiae lineages (Scopel et al. 2021), where a fifth of the sequenced strains harbored atypical aneuploidy karyotypes (Peter et al. 2018), witnessing a lack of strong deleterious effects. Indeed, different dosage compensation mechanisms have evolved and may restore the ancestral gene dose leading to aneuploidy tolerance. Three possible targets of dosage compensation mechanisms for duplicated genes include the modification of the transcription rate (e.g. the amount of transcript products), the translation rate (e.g. the number of translated transcripts compared with the number of total produced transcripts), or the protein degradation rate (the number of degraded proteins compared with the total number of produced proteins). The most studied mechanisms are those involved in modifying the transcription rate during the evolution of heterogametic sex chromosomes, known both in plants (Muyle et al. 2012, 2017; Charlesworth 2019) and animals (Disteche 2012; Graves 2016). Different mechanisms evolved either by simulating the ancestral ploidy by doubling the transcription of the genes of the single-copy male X (Baker et al. 1994) or by equalizing the ploidy between the two sexes by halving the transcription by silencing of one X in female (Heard et al. 1997). Although dosage compensation is well studied in sex chromosome evolution, whether and how it evolves after a chromosome ploidy variation is unclear, particularly for autosomes (Kojima and Cimini 2019). Previous studies have reported inconsistent results; on the one hand, a significant increase of transcription for the duplicated genes has been observed in C. albicans (Selmecki et al. 2006), Drosophila (Loehlin and Carroll 2016), S. cerevisiae (Torres et al. 2007), and mammalian cells (Williams et al. 2008); on the other hand, decreased transcription of the duplicated genes has been reported in yeast and mammals, suggesting a compensation at the transcriptional level (Henrichsen et al. 2009; Qian et al. 2010). In the case of chromosome duplication, evidence for dosage compensation at the transcriptional level is scarce (Stenberg et al. 2009; Hose et al. 2020), and scaling of gene transcription with gene copy number seems more prevalent and has been reported in disomic yeasts (Kaya et al. 2020), C. albicans, and human cells (Kojima and Cimini 2019). Moreover, dosage compensation has been reported to occur at the posttranscriptional level, via the modification of translation efficiency, in Drosophila (Zhang and Presgraves 2017), or at the posttranslational level, via an increased degradation rate of proteins, for 20% of the proteome in yeast (Dephoure et al. 2014). Increased protein degradation is involved in the compensation mechanism in human Down's syndrome for proteins encoded on triplicated chromosome 21 (Liu et al. 2017).
Although the consequences of chromosome duplication on transcription rates—and to a lesser extent on translation rates and protein abundance—have been studied in many model organisms, our knowledge of chromosome duplication rates in eukaryotes is currently limited to S. cerevisiae (Lynch et al. 2008; Zhu et al. 2014; Liu and Zhang 2019) and humans (Nagaoka et al. 2012; Loane et al. 2013). Here, we investigate the spontaneous chromosome duplication rate in six unicellular species by analyzing sequence data from a MA experiment. The principle of MA experiments is to follow the descendants originated from a single cell under minimal selection, ensured by serial bottlenecks during dozens to thousands cell divisions (Halligan and Keightley 2009). The six species include five green algae and one diatom, all ecological relevant primary producers in the sunlit ocean (de Vargas et al. 2015), with a large phylogenetic spread encompassing 1.5 billion years of divergence (Yoon et al. 2004). We also recovered cryopreserved MA lines to investigate the consequence of chromosome duplication on the transcription and translation rates.
Results
Whole Chromosome Duplication Rate
In this study, we analyzed whole genome resequencing data from previous MA experiments (Krasovec et al. 2016, 2017, 2018a) in five haploid green algae species: Picochlorum costavermella RCC4223 (Krasovec et al. 2018b), Ostreococcus tauri RCC4221 (Blanc-Mathieu et al. 2014), Ostreococcus mediterraneus RCC2590 (Subirana et al. 2013), Bathycoccus prasinos RCC1105 (Moreau et al. 2012), and Micromonas commoda RCC299 (Worden et al. 2009) and one diploid diatom, Phaeodactylum tricornutum RCC2967 (Bowler et al. 2008; Giguere et al. 2022). We maintained 12–40 MA lines for a total of 1,595 to 17,250 generations depending of species (supplementary table S1, Supplementary Material online). Coverage was used as a proxy of copy number (fig. 1A and supplementary figs. S1 to S16, Supplementary Material online) and unveiled whole chromosome duplication events in four of the six species (table 1 and supplementary table S2, Supplementary Material online). Four whole chromosome duplications were observed in M. commoda (chromosomes C05, C12, C16, and C17), four in B. prasinos (chromosomes C04, C05, C06 ,and C19), one in O. mediterraneus (chromosome C14), and four in P. tricornutum (chromosomes C02, C14 and C23). The chromosome duplication events were mapped onto the genealogies of the MA lines to identify all independent chromosome duplication events (fig. 1B for B. prasinos). Genealogy analysis provided evidence that several chromosomes were duplicated twice over the course of the experiment: C19 in lines Bp25c and Bp28b, C17 in Mc08 and Mc09, and C23 in Pt11 and Pt10c. The probabilities of observing two independent whole chromosome duplications of the same chromosome in B. prasinos, M. commoda, and P. tricornutum are 0.46, 0.47, and 0.34, respectively (see Materials and Methods), and these probabilities are thus consistent with the null hypothesis of an equal probability of duplication across chromosomes. All independent duplication events inferred from coverage analysis and genealogies are summarized in table 1 and supplementary table S2, Supplementary Material online. Unexpectedly, the analyses revealed that the ancestral line (AL) of the MA experiment in B. prasinos carried two copies of chromosome C01. One copy was subsequently lost 11 times independently over 4,145 generations, corresponding to a spontaneous duplicated chromosome loss of 0.006 per duplicated chromosome per generation in B. prasinos.
Fig. 1.

(A) Normalized raw genomic coverage of the ancestral line (T0 of MA experiment), Bp20c, and Bp28 MA lines of B. prasinos. Vertical grey lines are chromosome separators. Read coverage highlighted in blue shows chromosomes in double copies. Raw coverage of all lines from all species are provided in supplementary figures S1 to S16, Supplementary Material online. (B) Pedigree of the MA lines from the B. prasinos experiment. Chromosome C01 is duplicated in the T0 line (named AL) of the experiment. This duplication is then lost several times, and five other duplications of chromosomes C04, C05, C06, and C19 occurred. In case of shared ancestry between lineages, the number of shared mutations and generations was counted only once, in order to include only independent generations and mutations.
Table 1.
Spontaneous whole chromosome duplication rate in six species
| Species | N lines | Gen | TotGen | N Chrom | N WCD | Duplicated Chrom | U WCD | U cell | U bs |
|---|---|---|---|---|---|---|---|---|---|
| Bathycoccus prasinos | 35 | 143 | 4,994 | 19 | 5 | C04, C05, C06, C19 | 0.000054 | 0.00103 | 0.0046 |
| Micromonas commoda | 37 | 112 | 4,145 | 17 | 5 | C05, C12, C16, C17 | 0.000071 | 0.00121 | 0.0171 |
| Ostreococcus tauri | 40 | 431 | 17,250 | 20 | 0 | — | <0.000003 | <0.00006 | 0.0054 |
| Ostreococcus mediterraneus | 37 | 235 | 8,710 | 19 | 1 | C14 | 0.000006 | 0.00011 | 0.0064 |
| Picochlorum costavermella | 12 | 133 | 1,596 | 10 | 0 | — | <0.00006 | <0.00063 | 0.0119 |
| Phaeodactylum tricornutum | 36 | 181 | 6,516 | 25 | 4 | C02, C14, C23 | 0.000025 | 0.00061 | 0.0132 |
N lines, number of MA lines; Gen, average number of generations per MA line; TotGen, total number of generations; NChrom, number of chromosomes in the ancestral karyotype; NWCD, number of independent whole chromosome duplications; UWCD, whole chromosome duplication rate per chromosome per cell division; UCell, whole chromosome duplication rate per cell division; and Ubs, base substitution mutation rate per cell division. The estimation of the upper limit of NWCD and Ucell in O. tauri and P. costavermella relies on the assumption of one duplication event.
Consequences of Chromosome Duplication on Transcription
To explore the consequences of whole or partial chromosome duplication on transcription, we compared transcription rates in a control strain (B. prasinos RCC4222) and one MA line of B. prasinos, which was revived after 4 years of cryopreservation. This cryopreserved culture originated from line Bp37, and the recovered culture is hereafter referred to as Bp37B. Whole genome coverage was used as a proxy for chromosome copy number in both lines. This confirmed the presence of two copies of chromosome C04 in Bp37B as in the original Bp37 (fig. 2A and supplementary fig. S6, Supplementary Material online) and also revealed an additional chromosome C01 in Bp37B (fig. 2A and supplementary fig. S17, Supplementary Material online). The heterogeneous coverage of chromosome C01 led us to divide it into two regions for subsequent analyses: region C01a (1.98 fold coverage) and C01b (1.35 fold coverage). The control line also contained duplicated regions (fig. 2A and supplementary fig. S17, Supplementary Material online), chromosome C10, and a region of chromosome C02 named C02b, whereas C02a was single copy. The boundaries of C01 and C02 subregions are provided in supplementary table S3, Supplementary Material online. We interpreted coverage values <2 (supplementary fig. S17, Supplementary Material online), for example, C01b in Bp37B, C10, and C02b as duplications that were carried by a subpopulation of cells. Cultures were grown up to ∼10 million cells per mL prior to extraction so that polymorphism is not unexpected.
Fig. 2.

(A) Genomic coverage of 1 kbp windows along the genome of B. prasinos Bp37B. (B) Distribution of gene transcription rates tr(i) between chromosomes. (C) Distribution of gene translation efficiencies te(i) between chromosomes. (B) and (C) Both the transcription and translation efficiencies ratios are significantly different between chromosomes (ANOVA, P value < 10−15). (D) Distribution of protein homeostasis (transcription rate × translation rate) between chromosomes. Y-axes are in log2. Blue, duplicated chromosome in Bp37B; yellow, duplicated chromosome in control RCC4222.
Comparative transcriptome analyses of Bp37B and the control line revealed that there were on average twice the number of transcripts for genes located on the duplicated chromosomes as compared with the control line—C04 (transcription rate tr(i) average = 2.13, median = 1.95, estimated from TPM, fig. 2B, raw data available in supplementary table S4, Supplementary Material online) and C01a (transcription rate tr(i) average = 2.29, median = 2.02). The transcription rate of genes on chromosomes C10 and C02b was also affected. Altogether, the transcript production was scaled up with the chromosome copy number in the Bp37B and control lines (Pearson correlation, rho = 0.87, P value < 0.001, fig. 3A).
Fig. 3.

Raw relative genomic coverage average (Bp37B/control) related to average transcription rate tr(i) (A), translation efficiency te(i) (B), and average protein synthesis (average transcription rate × average translation efficiency) per chromosome (C). Blue, chromosomes duplicated in Bp37B; yellow, chromosomes duplicated the control RCCC4222; black, nonduplicated chromosomes; grey, outlier chromosome C19. Data are provided in supplementary tables S6 and S7, Supplementary Material online.
Gene-by-Gene Variation of the Consequences of DNA Copy Number
Dosage invariant genes (Antonarakis et al. 2004; Lyle et al. 2004) are genes for which the transcription is not affected by copy number, and they may be involved in aneuploidy tolerance. We investigated differential gene transcription at the individual gene scale with DESeq2 (Love et al. 2014) to identify candidate invariant genes. We found that the transcription rate of 158 out of the 1,776 genes in two copies (9%) located on duplicated chromosomes or regions was not significantly higher than for genes in single copy (table 2). Two Gene Ontology (GO) categories (RNA processing, GO:0006396, and DNA metabolic process, GO:0006259) were overrepresented in this transcriptomic invariant gene set: first, genes involved in RNA processing (3.8 times more frequent, P value < 0.01) including members of heteromeric protein complexes such as subunits 7 and 2 of the U6 snRNA-associated Sm-like protein (supplementary table S5, Supplementary Material online, invariant 158 annotations) and, second, genes involved in DNA metabolic process (2.2 times more frequent in the invariant gene subset, P value <0.04) including the DNA Polymerase A and the DNA primase large subunit. Notable protein complex members of the invariant data set are histone 3 and the subunit E of the translation initiation factor 3. There are in total 12 genes annotated as subunits in the subset of invariant genes; this is significantly more than the frequency of protein coding genes annotated as subunits in the complete proteome (Fisher exact test, P value = 0.0006). This suggests that a higher proportion of genes coding for protein forming complexes have a gene-specific regulated transcription that is not affected by gene copy number.
Table 2.
Estimation of number of dosage insensitive genes (duplicated genes for which transcription is not changing)
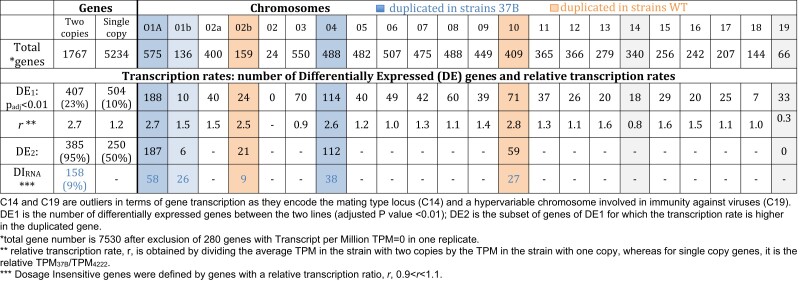
|
Consequences of Chromosome Duplication on Translation
The compelling evidence of overtranscription of the majority of duplicated genes prompted us to investigate whether posttranscriptional processes may temperate this excess of transcripts. This hypothesis was tested in the same B. prasinos line by sequencing mRNAs associated with ribosomes (polysomes) in order to compare the translation efficiency te(i) of genes located on duplicated and nonduplicated lines, by estimating the relative proportion of ribosome-bound mRNA in single versus duplicated genes. We found that the average translation efficiency of genes located on duplicated chromosome C04 was 0.71 (median = 0.54) as compared with the genes on this chromosome in the control line (fig. 2C). However, the translation efficiency of genes on region C01a was 0.93, whereas it was 1.54 for genes on region C01b; 1.04 and 0.76 for C02a and C02b, respectively; and 0.99 for chromosome C10. Translation rate modification is thus chromosome dependent (fig. 3): genes located on the C04 and C02b show a significant decrease in translation efficiency in the line with the duplicated regions, whereas genes located on duplicated region C01b show a significant increase in translation efficiency in the line with the duplicated region. Last, we estimated the expected protein production in Bp37B as compared with the control line by using the absolute translation rate for each gene: that is, the ratio of mRNA in polysomes in Bp37B as compared with the control (fig. 2D). This predicted that genes on C04 have a similar protein production rate in Bp37B and in the control line, despite a higher transcription rate as a consequence of the chromosome duplication, as well for the two parts of the chromosome C02 (fig. 2D). However, genes located on chromosome C01, despite different transcription and translation efficiency rates, produce twice the level of protein compared with the single-copy genes in the control line. Likewise, genes on chromosome C10 produce an excess of proteins compared with the single-copy genes in Bp37B. Altogether, we observed no dosage compensation at the transcription level for any of the duplicated regions, whereas dosage compensation occurs at the translational level in two out of five duplicated regions and is thus chromosome dependent (fig. 3). In addition, both transcription and translation efficiency averages are significantly different (ANOVA, P value < 10−15) among chromosomes or chromosomal regions.
Searching for Independent Molecular Signatures of Translation Modulation
The observed dosage compensation at the translational level on chromosome C04 suggests that posttranscriptional regulation occurs on transcripts from duplicated genes. As the length of the poly(A) tail is a key feature of many cytoplasmic mRNAs and is known to regulate translation (Subtelny et al. 2014; Eichhorn et al. 2016; Lim et al. 2016), we tested whether the translation of transcripts linked to duplicated chromosome C04 might be associated with a variation of poly(A) tail length. Using 3′RACE experiments, we measured the poly(A) tail length in Bp37B and RCC4222 (fig. 4 and supplementary fig. S18, Supplementary Material online) transcripts from three genes located on chromosome C04 (duplicated in line Bp37B: 04g00840, 04g01730, and 04g04360), one gene duplicated in the control line located on chromosome C01a (01g01300), and one control gene located on chromosome C08 (08g03470, single copy in both lines). Raw transcription TPM data of these five genes are provided in supplementary table S8, Supplementary Material online. We first ligated an RNA adapter (RA3) to the 3′ end of RNAs before reverse transcription. Then, the poly(A) tail was polymerase chain reaction (PCR) amplified with a gene-specific primer and the RA3 anchor primers. Finally, the PCR products were analyzed by gel electrophoresis. Short poly(A) tails tend to generate a band. However, in the case of long poly(A) tails, amplicons present diverse poly(A) tail lengths and create a smear on the gel (fig. 4A). Additionally, an internal control PCR was performed using a gene-specific primer in the 3′ untranslated region (UTR) to determine the size without a poly(A) tail (fig. 4B). For genes located on chromosome C04, a smear is observed in Bp37B and RCC4222 (fig. 4A). However, the smear range size decreases in Bp37B compared with RCC4222 for the three transcripts tested suggesting the poly(A) tail size is shorter in Bp37B. No difference is observed for the two other genes located on a nonduplicated chromosome (supplementary fig. S18, Supplementary Material online).
Fig. 4.

Transcripts from genes located on B. prasinos–duplicated chromosome C04 present reduced poly(A) tail lengths. Poly(A) tail measurement was performed using modified 3′RACE. PCR amplification was performed using primers flanking poly(A) tail (A) or primers anchored in 3′ UTR just before poly(A) tail as internal control (B). Experiments were performed using total RNA from RCC4222 line (control line) and BP37B line (with a duplicated chromosome C04). Illustrations representing PCR amplification are present on the right panel. A black line represents the messenger RNA, and the orange line represents the ligated adapter used for reverse transcription and PCR amplification. FW, Forward primer. RV, reverse primer.
Discussion
Rates of Whole Chromosome Duplication in Unicellular Eukaryotes
The rates of spontaneous whole chromosome duplication reported here in four out of six species vary between 6 × 10−4 and 1 × 10−3 events per haploid genome per generation and are thus one to two orders of magnitude lower than spontaneous point mutations per genome per generation. Theoretically, there are two possible mechanisms at the origin of chromosome duplication: (1) the supernumerary replication of one chromosome before cell division, leading to one cell with one chromosome and one cell with two chromosomes, or (2) the unequal segregation of chromosomes during cell division, leading to one cell with two copies and one cell without any copy. The latter scenario has been intensively experimentally studied in human cells (Ford and Correll 1992; Cimini et al. 2001) and seems all the more likely in the extremely small-sized Mamiellophyceae cells (1 µm cell diameter) as they have been reported to contain less kinetochore microtubules than chromosomes (Gan et al. 2011), which may induce a high error rate in the chromosome segregation process. However, the whole chromosome duplication rates reported here in the Mamiellophyceae are in line with those found in the diatom P. tricornutum and in yeast (9.7 × 10−5) (Zhu et al. 2014), yeast containing approximately as many kinetochore microtubules as chromosomes (Peterson and Ris 1976). Altogether, the spontaneous whole chromosome duplication rates reported previously in yeast (Zhu et al. 2014) and in the species from evolutionary distant eukaryotic lineages reported here (Yoon et al. 2004) suggest a high rate of spontaneous aneuploidy across unicellular eukaryotes. To investigate whether spontaneous whole chromosome duplication had been overlooked in other lineages, we screened publicly available resequencing data of MA lines from the freshwater green alga Chlamydomonas reinhardtii (Ness et al. 2015) and identified one event for chromosome 14 in the strain CC1373. Evidence of spontaneous whole chromosome duplication (four chromosomes in a single individual from a pedigree) has also been recently observed in the brown alga Ectocarpus (Krasovec et al. 2023).
Interestingly, we were able to estimate the rate of chromosome duplication loss during one experiment, and it is about three orders of magnitude higher than the spontaneous duplication rate. This is consistent with previous observations in S. cerevisiae and our observations that cells with duplicated chromosomes are ephemeral and unlikely to be maintained for a long time in batch culture. Indeed, the coverage of the large duplication events on C01b, C02b, C04, and C10 were 1.4, 1.6, 1.8, and 1.4 times the coverage of single-copy chromosomes respectively, suggesting that the proportion of cells carrying a supernumerary chromosome within the population is 0.4, 0.6, and 0.8. Consistent with this, we observed the loss of the duplication of chromosome C04 in one of the Bp37B lines over 4 months of sub-culturing.
As opposed to the aneuploidy generated during meiotic divisions, which has been intensively studied in mammalian cells (Nagaoka et al. 2012), the chromosomal duplication events reported here are generated during mitotic division. Since the molecular mechanisms involved in chromosomal segregation in mitosis, meiosis 1, and meiosis 2, are different, the associated aneuploidy rates are not expected to be equal. The aneuploidy rates in mitotically dividing human cells, such as HCT116 lines, have been estimated to be 7 × 10−2 (Thompson and Compton 2008), more than one order of magnitude higher than the highest estimation of 1.2 × 10−3 reported here in B. prasinos. The observed difference of the spontaneous point mutation rate per genome per generation is expected to vary between species as the consequence of different genome sizes, effective population sizes (Lynch et al. 2016), and the difference between the observed and expected GC content (Krasovec et al. 2017). However, the eight spontaneous aneuploidy rates currently available are not yet sufficient to investigate the origin of chromosome duplication rate variations, such as the effect of chromosome number or effective population size.
Fitness Effect of Whole Chromosome Duplication and Dosage Compensation
The high spontaneous rates of spontaneous chromosome duplication challenge the common idea that whole chromosome duplications are highly deleterious for individual cells, at least in mitotically dividing cells. Deleterious effects of chromosome duplication may be higher in meiosis than in mitosis as the molecular mechanisms involved in meiosis comprise additional checkpoints (Roeder and Bailis 2000). Using the number of cell divisions per days as a proxy of fitness during the MA experiment (Krasovec et al. 2016), only 1 out of the 15 MA lines with a whole chromosome duplication displayed a significant fitness decrease with generation time (Bp28b, Pearson correlation, P value = 0.016, ρ = −0.846, supplementary table S9, Supplementary Material online). Recent studies in yeast point toward slightly deleterious effects of aneuploidy, as well as a significant effect of the genetic background on aneuploidy tolerance (Scopel et al. 2021). Compensating point mutations have been previously linked to aneuploidy tolerance in yeast (Torres et al. 2010), notably in the gene encoding the deubiquitinating enzyme UBP6. Point mutations are unlikely to impact aneuploidy tolerance in the different MA lines reported here, because some lines with chromosome duplications do not carry any point mutation (Mc3, Bp28b, Bp26, and Bp25) or only carry synonymous mutations and mutations located in intergenic regions (i.e. Om3 or Mc28, supplementary table S10, Supplementary Material online). The deleterious effect of aneuploidy could be mitigated by a dosage compensation mechanism preserving relative protein homeostasis. In B. prasinos, despite the huge gene-to-gene variation in transcription and translation rates, transcription rates were overwhelmingly scaled up with chromosome copy number (fig. 3A), whereas translation rates differed significantly between chromosomes (fig. 3B). Nevertheless, genes on chromosome C19 displayed a unique pattern of DNA copy number-independent transcription and translation variation between the two lines. C19 is a small idiosyncratic chromosome found in all Mamiellophyceae species sequenced so far (Blanc-Mathieu et al. 2017; Yau et al. 2020) and is characterized by a lower GC content. Previous studies reported a strong variation in transcription rates of genes on this chromosome, which is associated with resistance to viruses (Yau et al. 2016).
In B. prasinos, genes located on duplicated regions fell into three different categories based on their translation efficiency. First, there were genes with no relative difference in translation rates, such as genes located on duplicated chromosome C10, which is duplicated in 40% percent of cells, and on region C01a, which is duplicated in 100% of cells. Proteins encoded by these genes were inferred to be overabundant in the cell (fig. 3C). Second, there were genes with a decreased translation rate on chromosome C04 and region C02b. More precisely, the estimated frequency of mRNA linked to ribosomes was inversely proportional to the excess of mRNA produced for duplicated genes on these two chromosomes, predicting approximately the same protein production rate as in cells without duplicated chromosomes (fig. 3C). Third, and very surprisingly, there were genes with higher translation rates associated with higher DNA copy numbers and higher transcription rates, such as genes located in the C01b region. As a consequence, the rate of protein synthesis on genes located on region C01b reached the same level of relative protein synthesis as genes located on region C01a, that is, twice the protein synthesis predicted in cells with a single copy of this chromosome. We speculate that the absence of dosage compensation on region C01a and the excess of translation on region C01b simulate the ancestral protein homeostasis in the ancestral line, which contained two complete chromosome C01.
Chromosome-wide regulation of gene translation is relatively unexplored and poorly understood. Indeed, whereas the position of a gene on a chromosome may allow its transcription rate to be predicted from epigenetic marks on the DNA, chromosome-wide epitranscriptomic mRNA modification mechanisms are yet to be discovered. Notwithstanding, chromosome-wide translational dosage compensation has been previously observed in Drosophila (Zhang and Presgraves 2016) and at the gene scale in several species (Zhang and Presgraves 2017; Chang and Liao 2020).
Is Poly(A) Tail Length a Potential Post-transcriptional Mechanism Involved in Dosage Compensation?
The role of poly(A) tail length in translation efficiency is starting to be better understood (Weill et al. 2012; Subtelny et al. 2014), and alternative polyadenylation is indeed implicated in several processes: transcription termination by RNAP II, mRNA stability, mRNA export, and translation efficiency (Zhang et al. 2010; Di Giammartino et al. 2011). In the cytoplasm, the poly(A) tail plays important roles in mRNA translation and stability and the modulation of its length has an important impact on translation efficiency (Subtelny et al. 2014; Eichhorn et al. 2016; Lim et al. 2016). For example, during oocyte maturation and early embryonic development, an increase in poly(A) tail length occurs for particular mRNAs resulting in an increase of translation (Subtelny et al. 2014; Eichhorn et al. 2016; Lim et al. 2016). This modulation of poly(A) tail length has also been found to activate some neuronal transcripts (Udagawa et al. 2012). Slobadin et al. (Slobodin et al. 2020) suggested that poly(A) tails can be modulated to balance mRNA levels and adjust translation efficiency since they observed that the CCR4–Not complex shortened the poly(A) tails, which reduced the stability of mRNAs. Here, our analysis of a subset of three genes suggests that, in a context of chromosome duplication, modulation of poly(A) tail length could be one of the posttranscriptional mechanisms involved in dosage compensation. However, the limited sample size of our observations does not allow us to draw any conclusion. Future genome-wide analyses will be necessary to validate this modulation and investigate the role of other mechanisms, such as adenosine methylation, in chromosome-wide translation compensation (Miao et al. 2022).
Transcription factories, nuclear structures where coexpressed or physically close genes are transcribed, have also been proposed to explain chromosome-dependent dosage compensation (Sutherland and Bickmore 2009). Our observation of a chromosome-scale similar translational behavior of transcripts could be due to the action of a single factory bound to that chromosome, whereas other chromosomes are under the control of their own transcription factories. The existence of such factories in unicellular eukaryotic species is unknown and needs further future investigations.
Conclusion
In conclusion, the high prevalence of whole chromosome duplication in five unicellular photosynthetic eukaryotes suggests that spontaneous whole chromosome duplication is pervasive in eukaryotes. In one species, B. prasinos, we provide evidence that a whole chromosome duplication event is associated with dosage compensation at the posttranscriptional level which might involve the adjustment of poly(A) tail length. These results suggest that a yet unknown posttranscriptional regulation mechanisms operate in dosage compensation of aneuploid karyotypes.
Materials and Methods
Sequencing Data from MA Experiments
MA experiments of six species—Picochlorum costavermella RCC4223 (Krasovec, et al. 2018b), O. tauri RCC4221 (Blanc-Mathieu et al. 2014), O. mediterraneus RCC2590 (Subirana et al. 2013; Yau et al. 2020), B. prasinos RCC1105 (synonym to RCC4222) (Moreau et al. 2012), M. commoda RCC299 (Worden et al. 2009), and P. tricornutum RCC2967 (Bowler et al. 2008; Giguere et al. 2022)—were conducted with a flow cytometry protocol described previously for phytoplankton species in liquid medium (Krasovec et al. 2016). Briefly, a MA experiment consists in monitoring MA lines that have evolved from a same cell (the AL) during hundreds of generations. Relaxed selection pressure on spontaneous mutations is ensured by maintaining all MA lines at very low effective population sizes (6 < Ne < 8.5) throughout the experiment (Krasovec et al. 2016). As a consequence, comparison of the complete genome sequence of the ancestral and MA lines enables the direct estimation of spontaneous mutation rates, excluding lethal mutations. Here, MA lines came from a single cell obtained by dilution serving as T0 culture (named the AL) and were maintained in 24-well plates (with one well per MA line) in L1 medium at 20 °C with a 16 h:8 h dark:light life cycle. One-cell bottlenecks were performed by dilution every 14 days to keep a low effective population size to limit selection. Effective population size was calculated by the harmonic mean of cell number over the 14-day period (Krasovec et al. 2016). The number of cell divisions per day between bottlenecks was calculated from the total number of cells obtained by flow cytometry for each MA line (Krasovec et al. 2016), knowing that inoculation is done with one cell. DNA of initial line (AL) and final time of MA lines were extracted with chloroform protocol and sequencing done with Illumina HiSeq or MiSeq by GATC Biotech (Germany). To detect duplications, raw reads were mapped against the reference genomes for each strain with bwa mem v0.7.12 (Li and Durbin 2010). Next, bam files were treated with samtools v1.3.1 (Li et al. 2009) and bedtools v 2-2.18.0 (Quinlan and Hall 2010) to extract the coverage.
Probability Calculations
To test whether our observations were compatible with the null hypothesis of “equal duplication rates between chromosomes” in each species, we calculated the probability of observing two independent duplications in the same chromosome for a given duplication events. The probability of drawing one chromosome twice out of k whole-chromosome duplication (WCD) events chosen from n chromosomes, P(k, n), is equal to the ratio of the number of combinations of (k−1) out of n without replacement multiplied by k−1, which corresponds to the number of possible k−1 different chromosomes with one in two copies, out of n, divided by the number of combinations of k out of n with replacement (k−1)*C(k−1, n)/(n + k−1), which corresponds to the total number of possible subsamples of k chromosomes out of n.
All statistical analyses were performed with R (R Core Team 2022).
Dosage Compensation Analysis
This study was started 3 years after the end of the MA experiments, for which some MA lines had been cryopreserved which allowed us to restart a culture for one B. prasinos MA line with a duplicated chromosome C04 (Bp37) to investigate the effect of the duplication of transcription and translation rates. The restarted culture from the cryopreserved Bp37 was renamed Bp37B. As a control, we used the reference culture of the strain B. prasinos RCC4222, that is, the derived from the RCC1105 used for the MA experiment. All cultures (Bp37B and RCC4222) were maintained under a 12:12 h light:dark regime under 50 μmol photon/m2 s−1 white light at 20 °C. The karyotype of the defrozen cultures Bp37B and the control RCC4222 were checked by DNAseq resequencing at the Bioenvironment platform (UPVD, Perpignan). The karyotype of Bp37B contained on additional chromosome copy number as compared with Bp37, and the karyotype of RCC4222 contained one additional copy number and one less chromosome copy number as compared with the ancestral line (supplementary fig. S17, Supplementary Material online). The relative transcription rate was corrected by the number of DNA copy in each line for further analysis below.
For transcription analyses, total RNA was extracted using the Direct-zol RNA MiniPrep Kit (Zymo Research, California, USA) from pooling flasks of cultures (100 mL cultures with 200 million cells per mL) taken 6 h before and 1 h before the light on, in triplicates for the control RCC4222 and duplicates for Bp37B. Next, polysome extraction was performed for Ribo-Seq as described previously (Carpentier et al. 2020) with few modifications. Briefly, 600 mL of B. prasinos culture were centrifuged at 8,000 × g for 20 min. After centrifugation, pellets were resuspended in 2.4 mL of polysome extraction buffer. After 10 min of incubation on ice and centrifugation, 2 mL of supernatant was loaded on a 9 mL 15–60% sucrose gradient and centrifuged for 3 h at 38,000 rpm with rotor SW41 Ti. Fractions corresponding to polysomes were pooled, and polysomal RNA was extracted as previously described (Carpentier et al. 2020). RNA library preparation was performed on total or polysomal RNA using a NEBNext Poly(A) mRNA Magnetic Isolation Module and a NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs) according to the manufacturer's instructions with 1 μg of RNA as a starting point. Libraries were multiplexed and sequenced on a NextSeq 550. Raw reads were mapped against the reference transcriptome of B. prasinos with RSEM with standard parameters (Li and Dewey 2011). We obtained the TPM average of total RNA and polysome-linked RNA that we compared between B. prasinos RCC4222 and the MA line Bp37B. TPM stands for transcripts per kilobase million, and the sum of all TPM values is the same in all samples, such that a TPM value represents a relative transcription level that is comparable between samples. At the gene scale, we first used the total RNA TPM values to estimate the transcription difference ratio, rRNA for each gene i, between two copies and single-copy genes as:
These relative transcription rates rRNA(i) were normalized by the transcription rate median of genes located on nonduplicated chromosomes in Bp37B (C03, C05, C06, C07, C08, C09, C011, C012, C013, C014, C015, C016, C017, and C018), in order to estimate the transcription rate tr(i) of duplicated chromosomes related to nonduplicated chromosomes. Chromosome C19 was not considered because it is an idiosyncratic chromosome (see Discussion).
Second, we used the total RNA and polysome-linked RNA TPM values for each gene i to calculate the translation rates of each gene, r(i), as:
The translation efficiency between a gene i in the two lines in single versus two copies, te(i), was estimated as:
The te(i) ratio was then normalized by the median translation efficiency of all genes on nonduplicated chromosomes in strain BP37B (5,267 genes).
To estimate the dosage compensation at the scale of the chromosomes, the values were normalized by the average TPM of all single-copy chromosomes prior to the calculation of the ratios (supplementary table S7, Supplementary Material online).
Poly(A) tail analysis was performed as previously described with slight modifications (Sement and Gagliardi 2014). PCR products were resolved on a 2.5% agarose gel. Primers used in this study are available in supplementary table S11, Supplementary Material online.
Differential Gene Transcription Analyses
The statistical significance of the genes differential transcription levels was further estimated with DESeq2 (Love et al. 2014). Transcriptional invariant genes were defined as genes present on duplicated regions and for which the relative transcription rate rRNA(i) was comprised between 0.9 and 1.1. Genes with no transcription were removed from this analysis. The overrepresentation of a certain GO term in the transcriptional invariant gene set was compared with the genome-wide GO term background frequency using the GO enrichment analysis with default values implemented in pico-PLAZA workbench (Vandepoele et al. 2013).
Supplementary Material
Acknowledgements
We are grateful to Claire Hemon and Elodie Desgranges for technical assistance with the MA experiments. We acknowledge the GenoToul Bioinformatics platform (Toulouse, France) for bioinformatics analysis support and cluster availability, the BIOPIC platform for support with the cytometry, and the sequencing facility of the Université de Perpignan Via Domitia BioEnvironnement platform. This work was funded by ANRJCJC-SVSE6-2013-0005 and ANR PHYTOMICS (ANR-21-CE02-0026). This study is set within the framework of the “Laboratoires d’Excellences (LABEX)” TULIP (ANR-10-LABX-41) and of the “École Universitaire de Recherche (EUR)” TULIP-GS (ANR-18-EURE-0019).
Contributor Information
Marc Krasovec, CNRS, Biologie Intégrative des Organismes Marins, UMR7232, Sorbonne Universités, Banyuls/Mer, France.
Rémy Merret, Laboratoire Génome et Développement des Plantes, UMR5096, Centre National de la Recherche Scientifique, Perpignan, France; Laboratoire Génome et Développement des Plantes, UMR5096, Université de Perpignan Via Domitia, Perpignan, France.
Frédéric Sanchez, CNRS, Biologie Intégrative des Organismes Marins, UMR7232, Sorbonne Universités, Banyuls/Mer, France.
Sophie Sanchez-Brosseau, CNRS, Biologie Intégrative des Organismes Marins, UMR7232, Sorbonne Universités, Banyuls/Mer, France.
Gwenaël Piganeau, CNRS, Biologie Intégrative des Organismes Marins, UMR7232, Sorbonne Universités, Banyuls/Mer, France.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Author Contributions
M.K., S.-S.B., and G.P. performed the MA experiments. M.K. performed the bioinformatics analysis. F.S. performed the cell cultures, RNA extractions, PCR, and gel migration experiments. R.M. designed and performed the polysome analyses, poly(A) tail tests, and RNAseq preparation. G.P. performed statistical analyses and coordinated the project. M.K. drafted the first version of the manuscript, and all authors participated to writing the final version.
Data Availability
Genomic raw reads are available under the bioprojects PRJNA531882 (Ostreoccocus tauri, O. mediterraneus, M. commoda, and B. prasinos), PRJNA453760 and PRJNA389600 (P. costavermella), and PRJNA478011 (P. tricornutum). Transcriptomic raw reads of the B. prasinos MA lines 37 (Bp37B) and control line RCC4222 are available under the bioproject PRJNA715163. A summary is provided in supplementary table S12, Supplementary Material online.
Literature Cited
- Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. 2004. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet. 5:725–738. [DOI] [PubMed] [Google Scholar]
- Baker BS, Gorman M, Marín I. 1994. Dosage compensation in Drosophila. Annu Rev Genet. 28:491–521. [DOI] [PubMed] [Google Scholar]
- Blanc-Mathieu R, et al. 2014. An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genomics. 15:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc-Mathieu R, et al. 2017. Population genomics of picophytoplankton unveils novel chromosome hypervariability. Sci Adv. 3:e1700239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler C, et al. 2008. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456:239–244. [DOI] [PubMed] [Google Scholar]
- Carpentier M-C, et al. 2020. Monitoring of XRN4 targets reveals the importance of cotranslational decay during Arabidopsis development. Plant Physiol. 184:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AY-F, Liao B-Y. 2020. Reduced translational efficiency of eukaryotic genes after duplication events. Mol Biol Evol. 37:1452–1461. [DOI] [PubMed] [Google Scholar]
- Charlesworth D. 2019. Young sex chromosomes in plants and animals. New Phytol. 224:1095–1107. [DOI] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, Li R. 2012. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, et al. 2001. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 153:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, et al. 2014. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3:e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vargas C, et al. 2015. Eukaryotic plankton diversity in the sunlit ocean. Science 348:1261605. [DOI] [PubMed] [Google Scholar]
- Devlin RH, Holm DG, Grigliatti TA. 1988. The influence of whole-arm trisomy on gene expression in Drosophila. Genetics 118:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, Manley JL. 2011. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 43:853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM. 2012. Dosage compensation of the sex chromosomes. Annu Rev Genet. 46:537–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn SW, et al. 2016. mRNA poly(A)-tail changes specified by deadenylation broadly reshape translation in Drosophila oocytes and early embryos. eLife 5:e16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzPatrick DR, et al. 2002. Transcriptome analysis of human autosomal trisomy. Hum Mol Genet. 11:3249–3256. [DOI] [PubMed] [Google Scholar]
- Ford JH, Correll AT. 1992. Chromosome errors at mitotic anaphase. Genome 35:702–705. [DOI] [PubMed] [Google Scholar]
- Gan L, Ladinsky MS, Jensen GJ. 2011. Organization of the smallest eukaryotic spindle. Curr Biol. 21:1578–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearhart JD, Oster-Granite ML, Reeves RH, Coyle JT. 1987. Developmental consequences of autosomal aneuploidy in mammals. Dev Genet. 8:249–265. [DOI] [PubMed] [Google Scholar]
- Giguere DJ, et al. 2022. Telomere-to-telomere genome assembly of Phaeodactylum tricornutum. PeerJ 10:e13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JAM. 2016. Evolution of vertebrate sex chromosomes and dosage compensation. Nat Rev Genet. 17:33–46. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 40:151–172. [Google Scholar]
- Heard E, Clerc P, Avner P. 1997. X-chromosome inactivation in mammals. Annu Rev Genet. 31:571–610. [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, et al. 2009. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 41:424–429. [DOI] [PubMed] [Google Scholar]
- Hose J, et al. 2020. The genetic basis of aneuploidy tolerance in wild yeast. eLife 9:e52063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, et al. 2018. Global impacts of chromosomal imbalance on gene expression in Arabidopsis and other taxa. Proc Natl Acad Sci. 115:E11321–E11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya A, et al. 2020. Molecular signatures of aneuploidy-driven adaptive evolution. Nat Commun. 11:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Cimini D. 2019. Aneuploidy and gene expression: is there dosage compensation? Epigenomics 11:1827–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, et al. 2018. Mutational and transcriptional landscape of spontaneous gene duplications and deletions in Caenorhabditis elegans. Proc Natl Acad Sci. 115:7386–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, et al. 2016. Fitness effects of spontaneous mutations in picoeukaryotic marine green algae. G3 Genes Genomes Genet. 6:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, et al. 2018b. Genome analyses of the microalga Picochlorum provide insights into the evolution of thermotolerance in the green lineage. Genome Biol Evol. 10:2347–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, Eyre-Walker A, Sanchez-Ferandin S, Piganeau G. 2017. Spontaneous mutation rate in the smallest photosynthetic eukaryotes. Mol Biol Evol. 34:1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, Hoshino M, Zheng M, Lipinska AP, Coelho SM. 2023. Low spontaneous mutation rate in complex multicellular eukaryotes with a haploid-diploid life cycle. Mol Biol Evol. :msad105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, Sanchez-Brosseau S, Grimsley N, Piganeau G. 2018a. Spontaneous mutation rate as a source of diversity for improving desirable traits in cultured microalgae. Algal Res. 35:85–90. [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinforma Oxf Engl. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lee M, Son A, Chang H, Kim VN. 2016. mTAIL-seq reveals dynamic poly(A) tail regulation in oocyte-to-embryo development. Genes Dev [Internet]. Available from:http://genesdev.cshlp.org/content/early/2016/07/14/gad.284802.116.
- Liu Y, et al. 2017. Systematic proteome and proteostasis profiling in human trisomy 21 fibroblast cells. Nat Commun. 8:1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang J. 2019. Yeast spontaneous mutation rate and spectrum vary with environment. Curr Biol CB. 29:1584–1591.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane M, et al. 2013. Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: impact of maternal age and prenatal screening. Eur J Hum Genet. 21:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehlin DW, Carroll SB. 2016. Expression of tandem gene duplicates is often greater than twofold. Proc Natl Acad Sci. 113:5988–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE. 2004. Gene expression from the aneuploid chromosome in a trisomy mouse model of down syndrome. Genome Res. 14:1268–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci. 105:9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, et al. 2016. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 17:704–714. [DOI] [PubMed] [Google Scholar]
- Miao Z, Zhang T, Xie B, Qi Y, Ma C. 2022. Evolutionary implications of the RNA N6-methyladenosine methylome in plants. Mol Biol Evol. 39:msab299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau H, et al. 2012. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 13:R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyle A, et al. 2012. Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLOS Biol. 10:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyle A, Shearn R, Marais GA. 2017. The evolution of sex chromosomes and dosage compensation in plants. Genome Biol Evol. 9:627–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka SI, Hassold TJ, Hunt PA. 2012. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet. 13:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res. 25:1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, et al. 2018. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556:339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JB, Ris H. 1976. Electron-microscopic study of the spindle and chromosome movement in the yeast Saccharomyces cerevisiae. J Cell Sci. 22:219–242. [DOI] [PubMed] [Google Scholar]
- Qian W, Liao B-Y, Chang AY-F, Zhang J. 2010. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 26:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan H, Lengauer C. 2004. Aneuploidy and cancer. Nature 432:338–341. [DOI] [PubMed] [Google Scholar]
- R Core Team . 2022. R: a language and environment for statistical computing. Vienna: (Austria: ): R Foundation for Statistical Computing. https://www.R-project.org/. [Google Scholar]
- Roeder GS, Bailis JM. 2000. The pachytene checkpoint. Trends Genet. 16:395–403. [DOI] [PubMed] [Google Scholar]
- Scopel EFC, Hose J, Bensasson D, Gasch AP. 2021. Genetic variation in aneuploidy prevalence and tolerance across Saccharomyces cerevisiae lineages. Genetics 217:iyab015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313:367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sement FM, Gagliardi D. 2014. Detection of uridylated mRNAs. Methods Mol Biol. 1125:43–51. [DOI] [PubMed] [Google Scholar]
- Slobodin B, et al. 2020. Transcription dynamics regulate poly(A) tails and expression of the RNA degradation machinery to balance mRNA levels. Mol Cell. 78:434–444.e5. [DOI] [PubMed] [Google Scholar]
- Song MJ, Potter BI, Doyle JJ, Coate JE. 2020. Gene balance predicts transcriptional responses immediately following ploidy change in Arabidopsis thaliana. Plant Cell. 32:1434–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg P, et al. 2009. Buffering of segmental and chromosomal aneuploidies in Drosophila melanogaster. PLOS Genet. 5:e1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subirana L, et al. 2013. Morphology, genome plasticity, and phylogeny in the genus Ostreococcus reveal a cryptic species, O. mediterraneus sp. nov. (Mamiellales, Mamiellophyceae). Protist 164:643–659. [DOI] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, Bartel DP. 2014. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland H, Bickmore WA. 2009. Transcription factories: gene expression in unions? Nat Rev Genet. 10:457–466. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. 2008. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 180:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, et al. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317:916–924. [DOI] [PubMed] [Google Scholar]
- Torres EM, et al. 2010. Identification of aneuploidy-tolerating mutations. Cell 143:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa T, et al. 2012. Bidirectional control of mRNA translation and synaptic plasticity by the cytoplasmic polyadenylation complex. Mol Cell. 47:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K, et al. 2013. pico-PLAZA, a genome database of microbial photosynthetic eukaryotes. Environ Microbiol. 15:2147–2153. [DOI] [PubMed] [Google Scholar]
- Veitia RA, Potier MC. 2015. Gene dosage imbalances: action, reaction, and models. Trends Biochem Sci. 40:309–317. [DOI] [PubMed] [Google Scholar]
- Weill L, Belloc E, Bava F-A, Méndez R. 2012. Translational control by changes in poly(A) tail length: recycling mRNAs. Nat Struct Mol Biol. 19:577–585. [DOI] [PubMed] [Google Scholar]
- Williams BR, et al. 2008. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322:703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden AZ, et al. 2009. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324:268–272. [DOI] [PubMed] [Google Scholar]
- Yau S, et al. 2016. A viral immunity chromosome in the marine picoeukaryote, Ostreococcus tauri. PLoS Pathog. 12:e1005965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau S, et al. 2020. Virus-host coexistence in phytoplankton through the genomic lens. Sci Adv. 6:eaay2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, Hackett JD, Ciniglia C, Pinto G, Bhattacharya D. 2004. A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol. 21:809–818. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Presgraves DC. 2016. Drosophila X-linked genes have lower translation rates than autosomal genes. Mol Biol Evol. 33:413–428. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Presgraves DC. 2017. Translational compensation of gene copy number alterations by aneuploidy in Drosophila melanogaster. Nucleic Acids Res. 45:2986–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Virtanen A, Kleiman FE. 2010. To polyadenylate or to deadenylate: that is the question. Cell Cycle Georget Tex. 9:4437–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 111:E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic raw reads are available under the bioprojects PRJNA531882 (Ostreoccocus tauri, O. mediterraneus, M. commoda, and B. prasinos), PRJNA453760 and PRJNA389600 (P. costavermella), and PRJNA478011 (P. tricornutum). Transcriptomic raw reads of the B. prasinos MA lines 37 (Bp37B) and control line RCC4222 are available under the bioproject PRJNA715163. A summary is provided in supplementary table S12, Supplementary Material online.