

Abstract
Tuberous sclerosis complex (TSC) is a neurogenetic disorder leading to epilepsy, developmental delay, and neurobehavioral dysfunction. The syndrome is caused by pathogenic variants in TSC1 (coding for hamartin) or TSC2 (coding for tuberin). Recently, we reported a progressive frontotemporal dementia-like clinical syndrome in a patient with a mutation in TSC1, but the neuropathological changes seen in adults with TSC with or without dementia have yet to be systematically explored. Here, we examined neuropathological findings in adults with TSC (n = 11) aged 30 to 58 years and compared them to age-matched patients with epilepsy unrelated to TSC (n = 9) and non-neurological controls (n = 10). In 3 of 11 subjects with TSC, we observed a neurofibrillary tangle-predominant “TSC tauopathy” not seen in epilepsy or non-neurological controls. This tauopathy was observed in the absence of pathological amyloid beta, TDP-43, or alpha-synuclein deposition. The neurofibrillary tangles in TSC tauopathy showed a unique pattern of post-translational modifications, with apparent differences between TSC1 and TSC2 mutation carriers. Tau acetylation (K274, K343) was prominent in both TSC1 and TSC2, whereas tau phosphorylation at a common phospho-epitope (S202) was observed only in TSC2. TSC tauopathy was observed in selected neocortical, limbic, subcortical, and brainstem sites and showed a 3-repeat greater than 4-repeat tau isoform pattern in both TSC1 and TSC2 mutation carriers, but no tangles were immunolabeled with MC1 or p62 antibodies. The findings suggest that individuals with TSC are at risk for a unique tauopathy in mid-life and that tauopathy pathogenesis may involve TSC1, TSC2, and related molecular pathways.
Keywords: Tuberous Sclerosis Complex (TSC), tau, acetylation, tauopathy, TSC1, TSC2, neurofibrillary tangle
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominantly inherited, childhood-onset disorder in which affected individuals may develop tumors of the brain, kidney, heart, and other organs [1, 8, 10, 15]. TSC is also associated with diverse neurological manifestations, including developmental delay, neurobehavioral dysfunction (along the autism spectrum), and epilepsy [5], but many with TSC escape these symptoms and lead normal adult lives [12]. TSC is caused by a pathogenic variant in either TSC1 (coding for hamartin) located on chromosome 9q34 [23] or TSC2 (coding for tuberin) located on chromosome 16p13 [7]. These proteins form a poly-protein complex [17, 25] that inhibits the mammalian target of rapamycin complex 1 (mTORC1) pathway, which controls a host of cellular processes, including translation, autophagy, and cell growth [8].
Previous studies have reported that cortical tubers of infants with TSC-related intractable epilepsy exhibit phospho-tau immunoreactivity, in dysmorphic neurons (without frank neurofibrillary tangles) and glial cells, as well as in cellular processes in the neuropil [21]. These observations prompted the suggestion that TSC represents an “infantile tauopathy”. The link between TSC and tau in adults, however, is only now beginning to emerge. Recently, we reported a TSC1 mutation carrier who presented with a progressive neurodegenerative syndrome that met clinical research criteria for behavioral variant frontotemporal dementia (bvFTD) and showed prominent right anterior temporal lobe atrophy [16]. Frontal lobe tissue surgically resected for treatment of TSC-related epilepsy from the patient’s living sibling showed elevated tau phosphorylation without evidence of tau aggregation or tangle formation [16]. A tuberous sclerosis-associated neuropsychiatric disorder (TAND) has also been described, characterized in adults by changes in behavior, language, and executive functioning [6]. We studied a cohort of adults with mild TSC to determine whether they exhibited features consistent with TAND or FTD [12]. This TSC cohort exhibited a range of cognitive phenotypes, including mild behavioral and cognitive deficits that are seen, more prominently, in patients with FTD. Adults with TSC further showed mildly elevated CSF phosphorylated tau (pTau-181) levels and focal uptake on 18F-flortaucipir positron emission tomographic (FTP-PET) imaging, often in the vicinity of radiographically identified tubers. In recent model-based studies, TSC1 haploinsufficiency induced tau acetylation (at K343 and 6 other sites) without affecting tau phosphorylation [2]. Despite this momentum, little is known about the prevalence or characteristics of a tauopathy in adults with TSC.
To gain insight regarding the relationship between TSC and tau in adults, we screened post-mortem brain tissue, using tau immunohistochemistry, in a cohort of individuals with TSC and compared the findings to those from age-matched patients with epilepsy (unrelated to TSC) and healthy controls. We identified a novel human tauopathy, which we term “TSC tauopathy”, characterized by neurofibrillary tangles in the cerebral cortex, limbic system, subcortical regions, and brainstem, with prominent tau acetylation and 3-repeat (3R) > 4-repeat (4R) tau isoform-containing inclusions.
Materials and Methods
Subjects and specimens
We searched the NIH NeuroBioBank for adults with a clinical diagnosis of TSC. Subjects with fixed and frozen tissue available for our regions-of-interest were prioritized. Selection was not based on TSC1/TSC2 genotyping, which was not available from the source. This strategy yielded a TSC group (n = 11), which we matched, as closely as possible for age and sex, to groups composed of patients with epilepsy unrelated to TSC (n = 9) and controls without neurological disease (n = 10). Subject characteristics are detailed in Table 1. Clinical information about the subjects with TSC was limited, but available information suggests that most patients showed recurrent seizures and varying degrees of developmental disability. None of the cases was labeled or described as having a neurodegenerative syndrome. The purpose of the non-TSC epilepsy group was to ensure that changes observed in TSC were not merely the result of recurrent seizures, but quantitative seizure frequency information was not available. Limited data were available regarding seizure etiology in this group. Four had a known etiology, including prior intracranial hemorrhage (EP 7), aneurysmal rupture/clipping (EP 6), and neurodevelopmental abnormalities (EP 3, EP 9). For the remaining five participants, available data were sparse/uninformative (EP 1, EP 2, EP 4) or absent (EP 3, EP 8). Seizure etiology is relevant because prior reports suggest that CSF total tau and phospho-tau levels are elevated only in patients with epilepsy due to a known etiology [17], although how these changes relate to tau aggregation remains unknown. In an initial screening phase, formalin-fixed tissue blocks for the TSC and epilepsy groups and 5 μm-thick formalin-fixed paraffin embedded (FFPE) tissue sections for the healthy control group were obtained. These materials represented three brain regions-of-interest, anterior cingulate cortex, inferior frontal gyrus, and entorhinal/hippocampal complex, chosen because they are frequently affected in a range of tau-based disorders. In addition, frozen tissue blocks from these regions were obtained from 10/11 TSC, 8/9 epilepsy, and 10/10 control subjects. Formalin-fixed tissue blocks were processed and embedded in paraffin, cut into 5 μm-thick sections, and mounted on glass slides. Sections underwent an initial round of immunostaining, and 3 patients with TSC (TS 6, TS 7, and TS 10) showing significant tauopathy were identified. These patients underwent additional mapping and characterization studies. For regional mapping, we obtained 22 formalin-fixed tissue blocks representing 43 areas of interest, including cortical, subcortical, limbic, and brainstem regions. These blocks were also processed and embedded in paraffin, cut into 5 μm-thick sections, and mounted on glass slides.
Table 1.
Subject clinical and demographic characteristics
| Case# | Age (years) | Sex (M:F) | PMI (hours) |
Clinical diagnosis | NIH Biobank (source) | |
|---|---|---|---|---|---|---|
| TSC | TS 1 (TSC2) | 30 | M | 3.0 | Tuberous Sclerosis | Maryland |
| TS 2 (TSC2) | 34 | F | 5.0 | Tuberous Sclerosis | Maryland | |
| TS 3 (N/A) | 39 | F | 16.0 | Tuberous Sclerosis | Maryland | |
| TS 4 (TSC2) | 40 | F | 18.0 | Tuberous Sclerosis | Maryland | |
| TS 5 (TSC2) | 42 | F | 15.0 | Tuberous Sclerosis | Maryland | |
| TS 6 (TSC1) | 46 | M | 1.0 | Tuberous Sclerosis | Maryland | |
| TS 7 (TSC2) | 47 | F | 3.0 | Tuberous Sclerosis | Maryland | |
| TS 8 (TSC2) | 55 | M | 23.0 | Tuberous Sclerosis | Maryland | |
| TS 9 (TSC2) | 56 | F | 14.0 | Tuberous Sclerosis | Maryland | |
| TS 10 (TSC2) | 58 | M | 17.0 | Tuberous Sclerosis | Maryland | |
| TS 11 (TSC2) | 58 | F | 20.0 | Tuberous Sclerosis | Maryland | |
| 45.9 (SD) | 4:7 | 12.2 (SD) | ||||
| Epilepsy (non-TSC) | EP 1 | 26 | F | 19.0 | Epilepsy | Maryland |
| EP 2 | 32 | F | 28.0 | Epilepsy | Maryland | |
| EP 3 | 39 | M | 15.0 | Epilepsy; Intellectual Disability NOS | Maryland | |
| EP 4 | 42 | F | 29.0 | Epilepsy | Maryland | |
| EP 5 | 42 | F | 21.0 | Epilepsy | Maryland | |
| EP 6 | 43 | M | 22.0 | Epilepsy | Maryland | |
| EP 7 | 46 | M | 14.0 | Epilepsy; Cerebrovascular Accident | Maryland | |
| EP 8 | 51 | F | 32.3 | Epilepsy | Harvard | |
| EP 9 | 55 | F | 18.0 | Epilepsy; ASD | Maryland | |
| 41.8 (SD) | 3:6 | 22.0 (SD) | ||||
| Control | Control 1 | 24 | F | 17.9 | Alcohol abuse, BPD 1, cocaine abuse | Miami |
| Control 2 | 34 | M | 26.7 | Control | Mt. Sinai | |
| Control 3 | 41 | F | 10.7 | Control | Mt. Sinai | |
| Control 4 | 42 | M | 6.0 | Control | Mt. Sinai | |
| Control 5 | 43 | F | 21.5 | MDD | Miami | |
| Control 6 | 45 | F | 24.5 | BPD | Mt. Sinai | |
| Control 7 | 46 | M | 14.8 | Control | Mt. Sinai | |
| Control 8 | 49 | M | 12.8 | Control | Mt. Sinai | |
| Control 9 | 56 | M | 18.8 | MDD | Mt. Sinai | |
| Control 10 | 58 | M | 16.8 | Control | Mt. Sinai | |
| 43.8 (SD) | 6:4 | 17.0 (SD) |
ASD, autism spectrum disorder; BPD, borderline personality disorder; MDD, major depressive disorder; NOS, not otherwise specified; TSC, Tuberous Sclerosis Complex; F, female; M, male; PMI, postmortem interval from death to autopsy; SD, standard deviation.
Having conducted a broad regional survey in the three of eleven TSC subjects showing prominent tau-related changes, we returned to the remaining eight TSC subjects (TS 1–5, 8, 9, and 11) to re-screen four susceptible brain regions identified through the mapping survey: superior frontal gyrus, ventral striatum, amygdala, and temporal pole. We performed p-tau (CP13, S202) and ac-tau (MAB359, K274) immunohistochemistry on these regions.
Genetic screening
DNA was isolated from TSC patients’ frozen tissue using the NucleoSpin DNA Lipid Tissue kit (Macherey-Nagel, REF #740471.50). Briefly, ~40 mg of tissue was resected from the inferior frontal gyrus of each TSC patient except TS 3, for whom frozen tissue was not available. Tissue was mechanically disrupted by agitation using NucleoSpin® Beads and 40 μL of lysis buffer (LT buffer) supplemented with 10 μL of Liquid Proteinase K. After lysis, cleared liquid supernatant was transferred onto the NucleoSpin® DNA Lipid Tissue Column and centrifuged for 30 s at 11,000 x g to bind the DNA to the columns. Column membranes were washed and then DNA eluted from the column, using 100 μL of elution buffer (Buffer BE). DNA samples were screened at the University of California, Los Angeles (UCLA) using targeted sequencing of a panel of genes previously implicated in neurodegenerative disorders, including TSC1 and TSC2, as previously described [12, 16, 18]. Coding and exon-intron boundary regions of the TSC1 and TSC2 genes were screened for (likely) pathogenic variants, as previously described [19]. Pathogenic variants were confirmed by Sanger sequencing.
Immunohistochemical staining
Brain tissue sections were deparaffinized in xylene for 30 minutes, washed in 100% and 95% ethanol, and then treated with 3% H2O2 in 10% methanol for 30 minutes to quench endogenous peroxidase activity. Sections were pre-treated with 88% formic acid for 5 min (for amyloid beta, MAB359, RD3, RD4 and acK343) or with 2 mg/ml of proteinase K (for alpha-synuclein) for ten minutes. All sections then underwent antigen retrieval by autoclaving at 121°C for 5 minutes in 0.01M citrate-buffer pH 6 (for amyloid beta, TDP-43, alpha-synuclein, CP13, MAB359, RD3, RD4, PHF1, and acK343) or 0.01M Tris-buffer pH 9 (for MC1 and p62). After cooling for 15 minutes, sections were washed with PBS-T (0.2% Tween 20 in 0.01 M PBS, pH 7.4) for 15 minutes, blocked for 30 minutes in blocking solution (10% horse serum for mouse primaries or 10% goat serum for rabbit primaries in PBS-T) and then incubated in primary antibodies against amyloid beta (anti-mouse, 1:500, Millipore, MAB5206), TDP-43 (anti-rabbit, 1:4K, Proteintech, 10782–2), alpha-synuclein (anti-mouse, 1:5K, LB509, gift from John Trojanowski and Virginia Lee, University of Pennsylvania, USA), CP13 (anti-mouse, 1:250, targeting pSer202, gift from Peter Davies, Feinstein Institute for Medical Research, USA), MAB359 (anti-rabbit, 1:250, targeting K274, gift from Li Gan, Weill Cornell Medicine, USA), PHF1 (anti-mouse, 1:3K, targeting pSer396–404, gift from Peter Davies), acK343 (anti-mouse, targeting K343, 1:250, gift from Aimee Kao, University California of San Francisco [2]), RD3 (anti-mouse, 1:2K, Millipore, 05–803), RD4 (anti-mouse, 1:2K, Millipore, 05–804), MC1 (anti-mouse, 1:500, tau conformational changes, gift from Peter Davies), or p62 (anti-mouse, 1:500, BD, 610832) at room temperature overnight. After two changes of PBS-T, the sections were incubated in biotinylated secondary antibody (1:200, horse anti-mouse, BA-2000 Vector Laboratories or 1:200, goat anti-rabbit IgG or BA-1000, Vector Laboratories) for 40 minutes at room temperature, then washed and incubated with avidin-biotin-peroxidase complexes (1:100, ABC, PK-6100, Vector Laboratories) for 30 minutes at room temperature. After washing, immunostaining was visualized with 0.05% chromogen 3,3’-diaminobenzidine (DAB)/0.01% H2O2 solution and counterstained with hematoxylin, dehydrated, and coverslipped in Permaslip (Alban Scientific). All immunohistochemical runs included positive controls to exclude technical factors. Digital images were captured using a high-resolution digital camera (Nikon Digital Sight DS-Fi1) mounted on a Nikon Eclipse 80i microscope using NIS-Elements (Version 3.22, Nikon) imaging software.
Semi-quantitative rating of immunohistochemical findings
Stained sections were evaluated using a Nikon bright field microscope using a 10x objective. The densities of tau-positive neurofibrillary tangles (NFTs), visualized with a variety of immunostains, were semi-quantitatively designated as (−) = absent, (+) = 1–5 tangles, (++) = 5–15 tangles, or (+++) = more than 15 tangles across the entire region-of-interest on a single section. Other potential (non-tau) pathological protein deposits were rated similarly.
NFT regional mapping
Regional distribution of phosphorylated tau (CP13, S202) and acetylated tau (MAB359, K274) immunoreactivity was assessed in cortical, subcortical, limbic, and brain stem regions in TS 6, 7, and 10 using the semi-quantitative rating system for neurofibrillary tangles. Scores were converted into integers (0–3), and available data were averaged across subjects for each region. These averages were used to create heat map representations of NFT density on a series of schematic brain slab illustrations.
Western blotting
Postmortem frozen inferior frontal gyrus tissue from two non-neurological controls, two non-TSC epilepsy controls, and TS 6, 7, and 10 was subjected to biochemical analysis. Total protein was extracted from the inferior frontal cortex by adding ice cold Pierce radio-immunoprecipitation assay (RIPA) buffer [25 mM tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS; Thermo Fisher Scientific, #89900] containing protease (MilliporeSigma, #4693124001), phosphatase (MilliporeSigma, #4906837001), and deacetylase inhibitors 3μM trichostatin A (Cayman Chemical Company, #89730) and 10 mM niacinamide (MilliporeSigma, #N5535) and homogenizing using Kimble Pellet Pestle Cordless Motor for 1 min/sample. Samples were then sonicated on high power, 30 sec on, 15 sec off for 15 cycles followed by centrifugation at 15,000 rpm for 20 minutes at 4°C. Soluble proteins isolated were from supernatant. To concentrate tau protein, 100 μL of the resultant supernatant was heated at 95°C for 30 minutes. After the heat shock, samples were centrifuged again at 15,000 rpm at 4°C for 20 minutes. The supernatant corresponding to the heat stable fraction, which mostly contains tau protein, was collected. Protein concentration was determined by bicinchoninic acid (BCA) assay (Thermo). Equivalent amounts of protein were separated on 4–12% SDS-PAGE (Thermo), then transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked at room temperature with Odyssey Blocking Buffer (LI-COR 927–40100) and incubated at 4°C overnight with primary antibodies and 1 hour at room temperature with appropriate fluorescent secondary antibodies (1:5K) (LI-COR). Blots of heat stable fraction containing concentrated tau were normalized to total protein stained using Licor Revert 700 Total Protein (926–11010; 926–11016). Immunoreactive bands were visualized using a LI-COR Odyssey CLx image scanner. The following primary antibodies were used: CP13, PHF1 and MAB359 (as described above), and GAPDH (anti-mouse, 1:500, Abcam, ab8245).
Results
Subject characteristics
The TSC group (4 males, 7 females) ranged in age from 30 to 58 years, with a median of 46 years (Table 1). Genetic screening revealed 9 TSC2 carriers and one TSC1 mutation carrier; one subject (TS 3) had an unknown genetic status due to lack of available frozen tissue. The specific TSC1 and TSC2 mutations found are detailed in Supplementary Table 1, online resource. As dictated by the study design, the three groups did not differ in age (F = 0.4, p = 0.6) or sex distribution (X2 = 1.7, p = 0.4).
Adult TSC is associated with a neurofibrillary “TSC tauopathy” involving cortical, subcortical, and limbic regions
To screen for pathological tau-related changes in all subjects, we performed phospho-tau (p-tau; CP13, S202) and acetyl-tau (ac-tau; MAB359, K274) immunohistochemistry on sections taken from the anterior cingulate gyrus, inferior frontal gyrus, and entorhinal/hippocampal complex. We chose CP13 because it is a widely used p-tau antibody, highly sensitive to early tau aggregation across a range neurodegenerative tauopathies. We chose MAB359 because of our suspicion, based on cell-based models [2], that ac-tau may prove more sensitive than p-tau for TSC-related changes and because this antibody stains tangles and other tau inclusions across a range of tauopathies [9]. Medial temporal neurofibrillary tauopathy is a common finding in middle-aged adults, and screening revealed low rates of Braak 1–2 NFT stages in all three groups (Table 2), consistent with literature-based norms [3]. In addition, screening revealed p-tau and ac-tau patterns inconsistent with age-related neurofibrillary change in 3 of 11 TSC subjects (TS 6, 7, and 10; see Table 2). These three subjects were among the older members of the TSC cohort. In TS 7 and 10, both TSC2 mutation carriers, p-tau and, especially, ac-tau staining revealed an NFT-predominant tauopathy (Figure 1). A morphologically indistinguishable tauopathy was seen in TS 6, a TSC1 mutation carrier, except that this subject’s NFTs were stained only for ac-tau (K274) and not for p-tau (S202) in this initial screen. In these three TSC subjects, the tau immunostaining was almost completely restricted to neurons, with NFTs located primarily in Layers 3 and 5, accompanied by a notable paucity of neuropil threads. Diffuse/granular or “pre-tangle” inclusion material was rare. In contrast to age-related neurofibrillary change, which begins in entorhinal cortex before spreading to CA1/subiculum, in the three TSC tauopathy cases the inferior frontal gyrus was the most prominently and consistently affected region among those initially assessed. In only one section did we identify astroglial tau inclusions, which were observed in the depth of a cortical sulcus; otherwise, there was no evidence of a perivascular tau pattern. Morphologically, the neuronal tau inclusions were highly fibrillar (Figure 1, insets). In all three groups, amyloid beta deposition (diffuse or neuritic plaques or vascular amyloid), TDP-43 inclusions, and Lewy body disease were absent in all three screening brain regions (Table 2).
Table 2.
Neuropathological evaluation and semi-quantitative assessment of tau pathology
| Case # | Estimated Braak stage | amyloid beta | TDP-43 | alpha-synuclein | CP13 (p-tau, S202) | MAB359 (ac-tau, K274) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | IFG | ERC/Hip | ACC | IFG | ERC/Hip | ACC | IFG | ERC/Hip | ACC | IFG | ERC/Hip | ACC | IFG | ERC/Hip | |||
| TSC | TS 1 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| TS 2 | 0 | - | - | - | - | - | - | - | - | - | - | ++ | - | - | - | - | |
| TS 3 | 0 | - | N/A | - | - | N/A | - | - | N/A | - | - | N/A | - | - | N/A | - | |
| TS 4 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| TS 5 | 0 | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | |
| TS 6 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | + | +++ | +++ | |
| TS 7 | 0 | - | - | - | - | - | - | - | - | - | + | ++ | - | + | ++ | - | |
| TS 8 | 0 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | |
| TS 9 | 2 | - | - | - | - | - | - | - | - | - | - | + | + | - | + | + | |
| TS 10 | 2* | - | - | - | - | - | - | - | - | - | + | +++ | +++ | ++ | +++ | +++ | |
| TS 11 | 1 | - | - | - | - | - | - | - | - | - | - | - | + | - | - | + | |
| Epilepsy (non-TSC) | EP 1 | 0 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - |
| EP 2 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| EP 3 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| EP 4 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| EP 5 | 0 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | |
| EP 6 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| EP 7 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| EP 8 | 1 | - | - | - | - | - | - | - | - | - | - | - | ++ | - | - | ++ | |
| EP 9 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Control | Control 1 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Control 2 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Control 3 | 0 | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | |
| Control 4 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Control 5 | 0 | - | - | - | - | - | - | - | - | - | + | - | - | + | - | - | |
| Control 6 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Control 7 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Control 8 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Control 9 | 1 | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | |
| Control 10 | 2 | - | - | - | - | - | - | - | - | - | - | + | ++ | - | - | - | |
Semi-quantitative ratings of immunoreactivity were observed using a 10 objective across the entire region on a single section and designated as - (absent), + (1–5 tangles), ++ (5–15 tangles), or +++ (> 15 tangles). ACC, anterior cingulate cortex; ERC, entorhinal cortex; Hip, Hippocampus; IFG, inferior frontal gyrus; N/A, not assessed.
Braak stage assessment is tentative given the well-developed TSC tauopathy in this case; the NFTs seen throughout the limbic system and basal temporal areas were not accompanied by prominent neuropil threads, as would be expected in AD-type neurofibrillary tauopathy; therefore, the true Braak stage may be lower.
Fig. 1. TSC tauopathy is a neurofibrillary tauopathy with prominent tau acetylation.
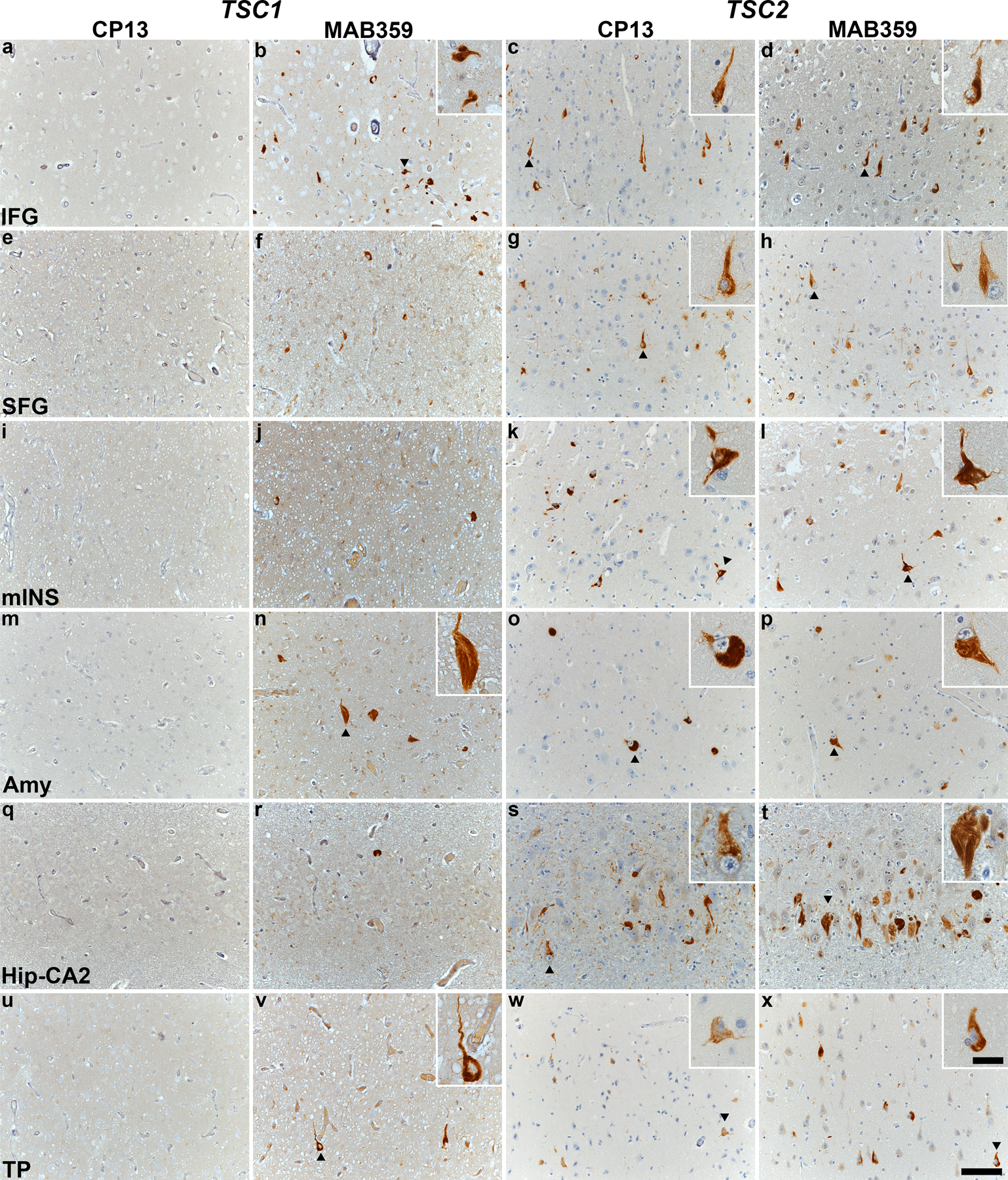
Immunohistochemical staining with CP13 and MAB359 antibodies in TSC subjects carrying pathogenic variants in TSC1 (TS 6, a-b, e-f, i-j, m-n, q-r, u-v) and TSC2 (TS 10, c-d, g-h, k-l, o-p, s-t, w-x). Representative images of immunostaining for CP13 and MAB359 in inferior frontal gyrus (IFG, a-d), superior frontal gyrus (SFG, e-h), middle insula (mINS, i-l), amygdala (Amy, m-p), hippocampal CA2 (Hip-CA2, q-t) and temporal pole (TP, u-x) are shown, highlighting neurofibrillary tangles (arrowheads) in the insets. All sections were counterstained with hematoxylin. Scale bars in panel x are 100 µm and 25 µm (inset) and apply to all images.
Having identified a novel “TSC tauopathy” in ~30% of our small TSC cohort, we expanded the study of these three subjects to map the pattern of tau lesions, using p-tau (S202) and ac-tau (K274) immunostaining, across the brain. These studies confirmed that TS 6 (a TSC1 mutation carrier) showed acetylated tau-positive NFTs in the absence of p-tau (S202) immunoreactivity (Supplementary Table 2, online resource, and Figure 1). The regional distribution of NFTs, however, was similar in all three subjects, involving cortical, subcortical, limbic, and selected brainstem regions (Supplementary Table 2, online resource, and Figure 2). The regions with the most consistent NFT burden included the inferior frontal gyrus, temporal pole, superior frontal gyrus, amygdala, and ventral striatum. Although the middle insula was inconsistently affected across cases, in TS 10 this region was the most severely affected region in the secondary screen.
Fig. 2. TSC tauopathy most prominently involves neocortical and paralimbic fronto-temporal-insular regions, ventral striatum, and amygdala.
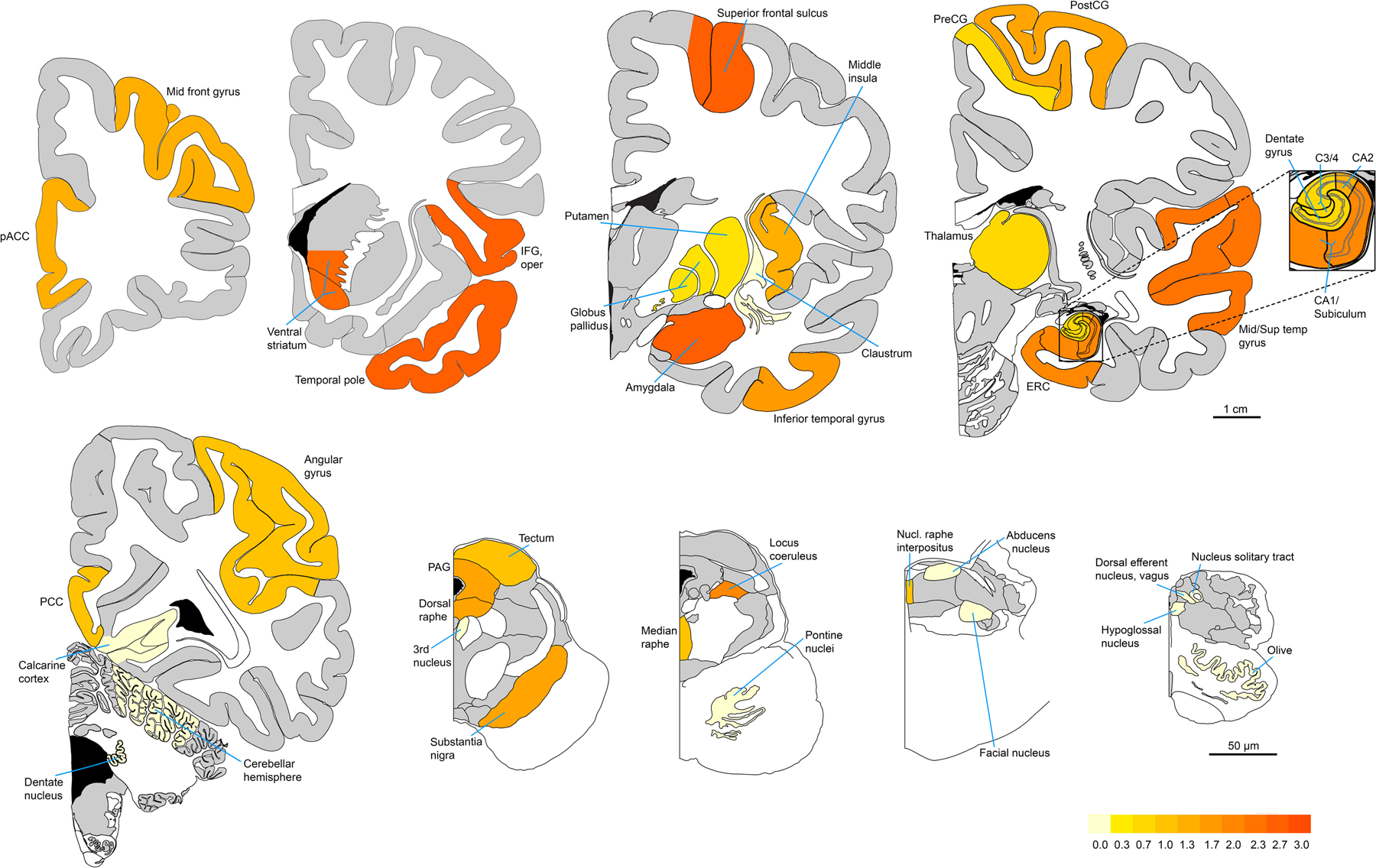
Heat map represents MAB359 (ac-tau, K274) immunoreactive NFT density averaged, region-wise, across the three subjects (TS 6, 7, and 10) with TSC tauopathy. The pattern emphasizes cortical, subcortical, and limbic regions and does not conform to the Braak NFT staging system. Figures are adapted mainly from sections 9, 18, 32, 54, and 82 of the Allen Human Brain Reference Atlas (atlas.brain-map.org), and from plates 10, 22, 30, and 34 of Olszewski and Baxter’s Cytoarchitecture of the Human Brainstem.
Mapping the most susceptible regions in the three subjects with TSC tauopathy (TS 6, TS 7, and TS 10) enabled us to re-screen the remaining eight TSC subjects based on those regions. We observed neurofibrillary tangles only in older TSC subjects (TS 8, TS 9, and TS 11, ages 55– 58), and most of these tangles were accompanied by neuropil threads and concentrated in the limbic regions, a pattern more consistent with age-related neurofibrillary tauopathy. These results are presented in Table 3. On this basis, the re-screening procedure identified no new subjects with TSC tauopathy within the TSC group.
Table 3.
Rescreening results: NFT burden in TSC tauopathy-vulnerable areas
| Case # | Estimated Braak stage | CP13 (p-tau, S202) | MAB359 (ac-tau, K274) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VS | Amy | SFG | TP | VS | Amy | SFG | TP | |||
| TSC | TS 1 (TSC2) | 0 | - | - | + | - | - | - | - | - |
| TS 2 (TSC2) | 0 | - | - | - | - | - | - | - | - | |
| TS 3 (N/A) | 0 | - | - | - | - | - | - | - | - | |
| TS 4 (TSC2) | 0 | N/A | - | - | - | N/A | - | - | - | |
| TS 5 (TSC2) | 0 | - | - | - | - | - | - | - | - | |
| TS 6 (TSC1)* | 0 | N/A | - | - | - | N/A | +++ | ++ | +++ | |
| TS 7 (TSC2)* | 0 | +++ | ++ | +++ | + | +++ | ++ | +++ | ++ | |
| TS 8 (TSC2) | 0 | + | ++ | + | + | - | - | - | - | |
| TS 9 (TSC2) | 2 | + | +++ | - | +++ | + | ++ | - | ++ | |
| TS 10 (TSC2)* | 4* | ++ | +++ | +++ | ++ | ++ | +++ | +++ | +++ | |
| TS 11 (TSC2) | 1 | + | ++ | + | - | - | + | - | + | |
Data for the TSC tauopathy subjects reprinted from Supplementary Table 2, online resource, for ease of comparison across the TSC group.
Amy, amygdala; SFG, superior frontal gyrus; TP, temporal pole; VS, ventral striatum; N/A, not assessed.
TSC tauopathy has distinctive characteristics that include 3R tau predominance, more acetylated than phosphorylated tau, and absent MC1 and p62 staining
Next, we sought to provide a more in-depth immunohistochemical characterization of the neurofibrillary lesions found in TSC tauopathy by evaluating the relative 3R and 4R tau isoform involvement, additional disease-relevant p-tau and ac-tau epitopes, and other key properties related to tau conformation and labeling. These studies were limited to the initial screening regions due to limited section availability. Remarkably, NFTs in TS 6, 7 and 10 all showed a predominance of 3R tau (RD3) immunostaining, with only 3R tau staining observed in TS 6 (except in medial temporal regions where typical age-related neurofibrillary tauopathy, with more prominent neuropil threads, appeared to comingle). Antibodies to tau phosphorylated at S396–404 (PHF1) and acetylated at K343 (acK343) again revealed a predominance of tau acetylation, although TS 6 did show PHF1 immunoreactivity despite the absence of p-tau staining at S202 (CP13). Finally, and to our surprise, we observed that MC1 and p62 staining was absent in regions containing many tau-positive NFTs. These findings are summarized in Supplementary Table 3, online resource, and images are shown in Figure 3.
Fig. 3. TSC tauopathy shows a unique pattern of immunohistochemical staining characteristics.
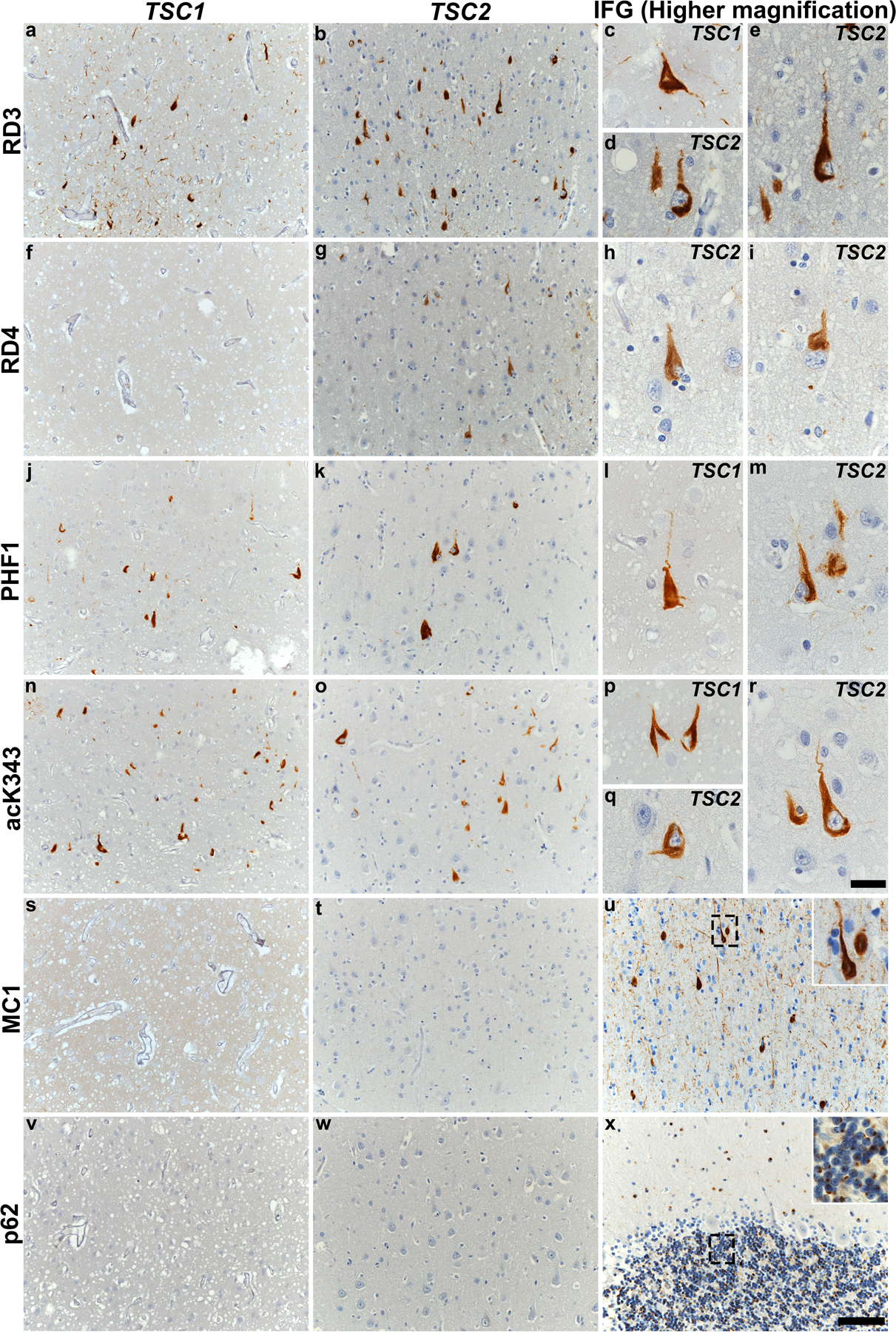
Immunohistochemical staining performed on inferior frontal gyrus with various tau antibodies and p62 is shown in subjects with TSC tauopathy (TS 6 and TS 10). Representative images highlight immunostaining for 3R tau (RD3, a-e), 4R tau (RD4, f-i), p-tau S396–404 (PHF1, j-m), ac-tau K343 (acK343, n-r), conformational tau (MC1, s-u) and p62 (v-x). Neither TSC1 nor TSC2 subjects showed MC1 (s and t) or p62 (v and w) immunoreactivity. Positive controls are shown for MC1 (u, inferior temporal gyrus in a subject with AD) and p62 (x, cerebellum in a subject with C9orf72-FTD, to capture p62-positive dipeptide repeat protein inclusions). Boxes in panels u and x are magnified in insets. All sections were counterstained with hematoxylin. Scale bar: 100 µm in panel x and applies to all other images, 25 µm in panel r and all other insets (u, x).
Immunoblotting with a subset of the tau antibodies used for immunohistochemistry generally corroborated the key immunohistochemical findings (Figure 4), including the consistency of tau acetylation across TSC tauopathy cases and the relative prominence of tau acetylation vs. phosphorylation, especially in the TSC1 mutation carrier.
Fig. 4. TSC1 and TSC2 variants show differing tau phosphorylation and acetylation in TSC.
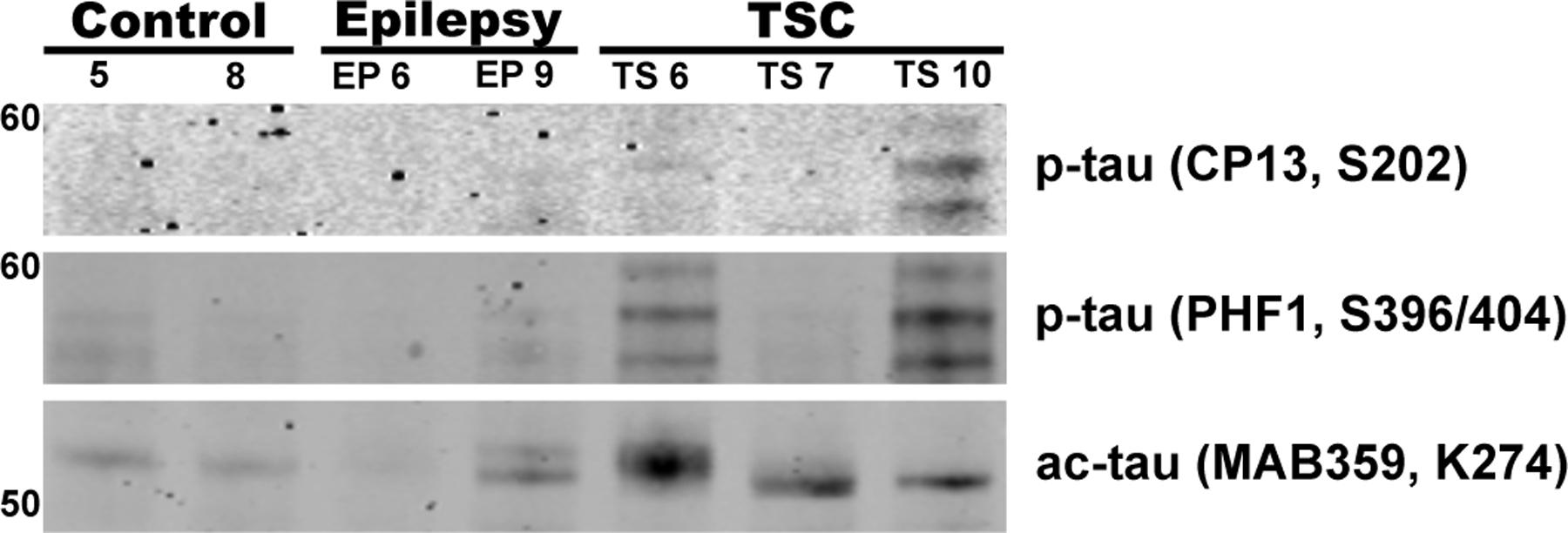
Immunoblots performed on frozen tissue homogenates from inferior frontal gyrus tissue of control, non-TSC epilepsy (EP 6, EP 9), and TSC subjects with TSC1 (TS 6) or TSC2 mutations (TS 7, TS 10). Antibodies used include CP13, PHF1, and MAB359 and were normalized to total protein stain. Ac-tau and, to a lesser extent, p-tau, was more abundant in subjects with TSC tauopathy compared with other groups.
Discussion
Abnormal tau has been found in autopsy specimens from children with TSC [21] and in mechanistic experiments in cultured cells and model organisms [2], but the presence and characteristics of a tauopathy in adults with TSC has not been systematically explored. Here, by screening a cohort of 11 individuals with TSC aged 30–58 years, we identified a novel aging-associated tauopathy, for which we offer the term “TSC tauopathy”. This NFT-predominant tauopathy appears to be most prominent in frontal, insular, and temporal cortex, as well as ventral striatum and amygdala and occurs in the absence of amyloid-beta deposition or other pathological protein aggregates. Remarkably, TSC tauopathy was characterized by more acetylated than phosphorylated tau, by more 3R than 4R tau, and by the absence of MC1 or p62 staining.
TSC tauopathy appears to differ from all other well-characterized tauopathies. The compact NFTs observed are morphologically most like those seen in primary age-related tauopathy (PART) [4], Alzheimer’s disease (AD) [22], and chronic traumatic encephalopathy (CTE) [13, 14], but the topographical distribution and immunostaining characteristics differ from these more common tauopathies in several respects. Unlike PART and AD, which follow typical Braak stage topography, TSC tauopathy appears unlikely to begin in the medial temporal lobe, as it is most prominent in neocortical and paralimbic fronto-temporal-insular regions, amygdala, and ventral striatum. Medial temporal and brainstem neurofibrillary changes were observed in our older TSC subjects, as expected, but at a rate comparable to that observed in the epilepsy and non-neurological control groups and age-based norms [3]. Like TSC tauopathy, CTE features early NFTs in cortex, striatum, and amygdala, and individuals with head trauma due to repeated seizures or autism-related self-injurious behaviors may be at risk for CTE; indeed, we detected a single patch of CTE-like tauopathy in one section from one TSC subject. In contrast to CTE, however, TSC tauopathy shows no perivascular patterning, no predilection for sulcal depths, and little (or no) astroglial tauopathy. Moreover, the TSC-linked tauopathy was not observed in our epilepsy control group, which was included to control for head trauma risk and other epilepsy-related factors. Finally, PART, AD, and CTE all show a balanced 3R/4R tauopathy, prominent neuropil threads, and ready detection with CP13, MC1, and p62 antibodies [14, 20, 24]; all of these features were absent in TSC tauopathy. The regional distribution, 3R and acetylated tau predominance, and inconspicuous or absent staining with CP13, MC1, and p62 combine to set TSC tauopathy apart. A recent study [11] including TSC subjects examined the relationship between p-tau and AV1451 (flortaucipir) staining, but this study did not provide sufficient characterization of the tau lesions in TSC to differentiate them from other tauopathies. TSC tauopathy also differs from other TSC-related tau abnormalities previously reported in the TSC literature by our group [12, 16] and others [6, 11]. Generally, prior reports depict tau-related changes as elevated p-tau staining in the absence of frank neurofibrillary tangles. It is possible that prior reports, including ours, may have reflected the consequences of seizures, neurosurgery, or both, which motivated us to design a study that included non-TSC epilepsy subjects to facilitate direct comparisons. One limitation of this study is the lack of detailed clinical information regarding the etiology, localization, and frequency of seizures in both groups, but the specificity of TSC tauopathy to the TSC group remains an important component of this study.
Although TSC tauopathy appears to show overlapping characteristics in TSC1 and TSC2 mutation carriers, our preliminary findings raise the possibility of important, if subtle differences. The single TSC1 carrier (TS 6) showed prominent tau acetylation despite a dearth of tau phosphorylation, especially when assessed at S202. In keeping with this observation, our recent study [2] suggested that TSC1 haploinsufficiency led to specific tau acetylation (at K343 and other sites) and tau accumulation, mediated by dysregulation of p300 acetyltransferase and SIRT1 deacetylase. Additional neuropathological studies in TSC1 carriers are needed to determine whether the lack of p-tau (S202) staining is a consistent feature.
TSC tauopathy represents a primary tauopathy, and TSC has been linked to progressive neurodegeneration and FTD-like symptoms [12, 16]. In this light, should TSC tauopathy be classified as a subtype of frontotemporal lobar degeneration with tau-immunoreactive inclusions (FTLD-tau)? Although this designation may become appropriate, we consider it premature based on available data. Based on its relatively low NFT burden compared to other neurodegenerative tauopathies, the tauopathy characterized in this study appears most likely to represent an incipient, preclinical stage. Accordingly, reclassifying TSC tauopathy as a subtype of FTLD-tau should require, in our opinion, the description of a patient or patients with a progressive FTD-like neurodegenerative syndrome and more fully developed TSC tauopathy-like features at autopsy. In that context, the most appropriate designation, in keeping with existing FTLD nosology, would be “FTLD-tau with TSC1 (or TSC2) mutation”, which could be shortened to FTLD-tau/TSC1 (or TSC2). In this way, TSC tauopathy may serve as a placeholder until such time that a more definitive designation can be made.
The next steps in this work are to expand the pool of adult TSC subjects to screen for TSC tauopathy and to explore and confirm potential differences between TSC1 and TSC2 carriers. Converging experimental and neuropathological studies will be needed to determine the mechanisms that lead to the unique characteristics of this tauopathy. The lessons learned could have important implications for tauopathy pathogenesis more broadly, and TSC1/2-related signaling represents a compelling new potential avenue for tauopathy treatment.
Supplementary Material
Acknowledgments
We thank the NIH NeuroBioBank for providing the human brain tissue. This work was supported by the Rainwater Charitable Foundation, the Bluefield Project to Cure FTD, and NIH grants P30AG062422, P01AG019724, and U01AG057195.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Al-Saleem T, Wessner LL, Scheithauer BW, Patterson K, Roach ES, Dreyer SJ, Fujikawa K, Bjornsson J, Bernstein J, Henske EP (1998) Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 83: 2208–2216 [PubMed] [Google Scholar]
- 2.Alquezar C, Schoch KM, Geier EG, Ramos EM, Scrivo A, Li KH, Argouarch AR, Mlynarski EE, Dombroski B, DeTure M et al. (2021) TSC1 loss increases risk for tauopathy by inducing tau acetylation and preventing tau clearance via chaperone-mediated autophagy. Sci Adv 7: eabg3897 Doi 10.1126/sciadv.abg3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: 960–969 Doi 10.1097/NEN.0b013e318232a379 [DOI] [PubMed] [Google Scholar]
- 4.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH et al. (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128: 755–766 Doi 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curatolo P, Moavero R, de Vries PJ (2015) Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol 14: 733–745 Doi 10.1016/S1474-4422(15)00069-1 [DOI] [PubMed] [Google Scholar]
- 6.de Vries PJ, Whittemore VH, Leclezio L, Byars AW, Dunn D, Ess KC, Hook D, King BH, Sahin M, Jansen A (2015) Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol 52: 25–35 Doi 10.1016/j.pediatrneurol.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.European Chromosome 16 Tuberous Sclerosis C (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75: 1305–1315 Doi 10.1016/0092-8674(93)90618-z [DOI] [PubMed] [Google Scholar]
- 8.Feliciano DM (2020) The Neurodevelopmental Pathogenesis of Tuberous Sclerosis Complex (TSC). Front Neuroanat 14: 39 Doi 10.3389/fnana.2020.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grinberg LT, Wang X, Wang C, Sohn PD, Theofilas P, Sidhu M, Arevalo JB, Heinsen H, Huang EJ, Rosen H et al. (2013) Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathologica: Doi 10.1007/s00401-013-1080-2 [DOI] [PMC free article] [PubMed]
- 10.Kerfoot C, Wienecke R, Menchine M, Emelin J, Maize JC Jr., Welsh CT, Norman MG, DeClue JE, Vinters HV (1996) Localization of tuberous sclerosis 2 mRNA and its protein product tuberin in normal human brain and in cerebral lesions of patients with tuberous sclerosis. Brain Pathol 6: 367–375 Doi 10.1111/j.1750-3639.1996.tb00866.x [DOI] [PubMed] [Google Scholar]
- 11.Liu AJ, Lusk JB, Ervin J, Burke J, O’Brien R, Wang SJ (2022) Tuberous sclerosis complex is a novel, amyloid-independent tauopathy associated with elevated phosphorylated 3R/4R tau aggregation. Acta neuropathologica communications 10: 27 Doi 10.1186/s40478-022-01330-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu AJ, Staffaroni AM, Rojas-Martinez JC, Olney NT, Alquezar-Burillo C, Ljubenkov PA, La Joie R, Fong JC, Taylor J, Karydas A et al. (2020) Association of Cognitive and Behavioral Features Between Adults With Tuberous Sclerosis and Frontotemporal Dementia. JAMA Neurol 77: 358–366 Doi 10.1001/jamaneurol.2019.4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W et al. (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131: 75–86 Doi 10.1007/s00401-015-1515-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM et al. (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136: 43–64 Doi 10.1093/brain/aws307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizuguchi M, Ikeda K, Takashima S (2000) Simultaneous loss of hamartin and tuberin from the cerebrum, kidney and heart with tuberous sclerosis. Acta Neuropathol 99: 503–510 Doi 10.1007/s004010051152 [DOI] [PubMed] [Google Scholar]
- 16.Olney NT, Alquezar C, Ramos EM, Nana AL, Fong JC, Karydas AM, Taylor JB, Stephens ML, Argouarch AR, Van Berlo VA et al. (2017) Linking tuberous sclerosis complex, excessive mTOR signaling, and age-related neurodegeneration: a new association between TSC1 mutation and frontotemporal dementia. Acta Neuropathol 134: 813–816 Doi 10.1007/s00401-017-1764-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmio J, Suhonen J, Keranen T, Hulkkonen J, Peltola J, Pirttila (2009) Cerebrospinal fluid tau as a marker of neuronal damage after epileptic seizure. Seizure 18: 474–477 doi: 10.1016/j.seizure.2009.04.006 [DOI] [PubMed] [Google Scholar]
- 18.Ramlaul K, Fu W, Li H, de Martin Garrido N, He L, Trivedi M, Cui W, Aylett CHS, Wu G (2021) Architecture of the Tuberous Sclerosis Protein Complex. J Mol Biol 433: 166743 Doi 10.1016/j.jmb.2020.166743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramos EM, Dokuru DR, Van Berlo V, Wojta K, Wang Q, Huang AY, Miller ZA, Karydas AM, Bigio EH, Rogalski E et al. (2019) Genetic screen in a large series of patients with primary progressive aphasia. Alzheimers Dement 15: 553–560 Doi 10.1016/j.jalz.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424 Doi 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salminen A, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H, Alafuzoff I (2012) Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog Neurobiol 96: 87–95 Doi 10.1016/j.pneurobio.2011.11.005 [DOI] [PubMed] [Google Scholar]
- 22.Sarnat HB, Flores-Sarnat L (2015) Infantile tauopathies: Hemimegalencephaly; tuberous sclerosis complex; focal cortical dysplasia 2; ganglioglioma. Brain Dev 37: 553–562 Doi 10.1016/j.braindev.2014.08.010 [DOI] [PubMed] [Google Scholar]
- 23.Togo T, Akiyama H, Iseki E, Uchikado H, Kondo H, Ikeda K, Tsuchiya K, de Silva R, Lees A, Kosaka K (2004) Immunohistochemical study of tau accumulation in early stages of Alzheimer-type neurofibrillary lesions. Acta Neuropathol 107: 504–508 Doi 10.1007/s00401-004-0842-2 [DOI] [PubMed] [Google Scholar]
- 24.van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J et al. (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277: 805–808 Doi 10.1126/science.277.5327.805 [DOI] [PubMed] [Google Scholar]
- 25.Weaver CL, Espinoza M, Kress Y, Davies P (2000) Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging 21: 719–727 Doi 10.1016/s0197-4580(00)00157-3 [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Yu Z, Chen X, Li J, Li N, Cheng J, Gao N, Yuan HX, Ye D, Guan KL et al. (2021) Structural insights into TSC complex assembly and GAP activity on Rheb. Nat Commun 12: 339 Doi 10.1038/s41467-020-20522-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.