

Abstract
Background
Berberine effectively alleviates non-alcoholic fatty liver disease (NAFLD). Nevertheless, the mechanism is incompletely comprehended. It has been reported that SIRT1 mediates lipid metabolism in liver and berberine promotes the expression of SIRT1 in hepatocytes. We hypothesized that SIRT1 mediated the effect of berberine on NAFLD.
Methods
The effects of berberine on NAFLD were evaluated in C57BL/6J mice fed a high-fat diet (HFD) and in mouse primary hepatocytes and cell lines exposed to palmitate. The change of fatty acid oxidation (FAO) and the activity of CPT1A were observed in HepG2 cells. Quantitative real-time polymerase chain reaction and Western blot were employed to observe the expression of SIRT1 and lipid metabolism-related molecules. The interaction between SIRT1 and CPT1A was investigated by using co-immunoprecipitation assay in HEK293T cells.
Results
Berberine treatment attenuated hepatic steatosis, reduced triglyceride (190.1 ± 11.2 μmol/g liver vs 113.6 ± 7.6 μmol/g liver, P < 0.001) and cholesterol (11.3 ± 2.5 μmol/g liver vs 6.3 ± 0.4 μmol/g liver, P < 0.001) concentration in the liver, and improved lipid and glucose metabolism disorders compared with the HFD group. The expression of SIRT1 was reduced in the liver of NAFLD patients and mouse models. Berberine increased the expression of SIRT1 and promoted the protein level of CPT1A and its activity in HepG2 cells. SIRT1 overexpression mimicked the effect of berberine on reducing triglyceride levels in HepG2 cells, whereas SIRT1 knock-down attenuated the effect of berberine. Mechanistically, berberine increased the expression of SIRT1. SIRT1 deacetylated CPT1A at the Lys675 site, which suppressed its ubiquitin-dependent degradation, thereby promoting FAO and alleviating non-alcoholic liver steatosis.
Conclusions
Berberine promoted SIRT1 deacetylation of CPT1A at the Lys675 site, which reduced the ubiquitin-dependent degradation of CPT1A and ameliorated non-alcoholic liver steatosis.
Keywords: berberine, non-alcoholic fatty liver disease, SIRT1, carnitine palmitoyltransferase 1A, acetylation
Graphical Abstract
Graphical Abstract.

The mechanism of BBR in ameliorating non-alcoholic hepatic steatosis. BBR increased the expression of SIRT1. SIRT1 deacetylated CPT1A at the Lys675 site, which suppressed the ubiquitination degradation of CPT1A. The increased CPT1A promoted fatty acid oxidation, thus alleviating non-alcoholic hepatic steatosis. BBR, berberine; HFD, high-fat diet; Ac, acetylation; Ub, ubiquitin; FAO, fatty acid oxidation.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a prevalent global health problem that is associated with abnormal triglyceride (TG) accumulation in hepatocytes, insulin resistance, and abnormal glucose metabolism [1]. The spectrum of NAFLD includes simple steatosis, non-alcoholic steatohepatitis (NASH), the latter of which can progress to cirrhosis and even hepatocellular carcinoma. Following recent research, the global prevalence of NAFLD was ∼29.8% [2, 3]. NAFLD has become a considerable health and economic burden. Currently, no specific drug has been approved for the treatment of NAFLD by the Food and Drug Administration.
Berberine (BBR) is an isoquinoline extracted from natural herbs, such as Coptis chinensis, Rhizoma coptidis, and Hydrastis canadensis, and is typically used to treat diarrhea and gastrointestinal infectious diseases in Traditional Chinese Medicine [4]. Recent studies showed that BBR significantly reduced liver steatosis, improved insulin resistance, alleviated glucose and lipid metabolism disorders, and exhibited therapeutic effects on NAFLD in humans and mice. The therapeutic effects of BBR are partly due to its ability to induce autophagy and fibroblast growth factor 21, activate the AMPK pathway, and suppress lipid synthesis [5–7]. Nonetheless, the specific mechanism of BBR in the treatment of NAFLD remains unclear.
Sirtuins are a group of conserved nicotinamide adenine dinucleotide (NAD+)-dependent histone and/or protein deacetylases. Mammals have seven types of sirtuins (SIRT1 to SIRT7) that are located in various regions, including the nucleus (SIRT1, 2, 6, and 7), cytoplasm (SIRT1, 2), and mitochondria (SIRT1, 3, 4, and 5) [8–10]. As a metabolic regulator, SIRT1 extensively partakes in the cell energy metabolism process [11]. The deacetylase activity of SIRT1 is influenced by NAD+. The increasing NAD+ boosts SIRT1 activity, whereas nicotinamide suppresses it [12]. Many studies have confirmed that SIRT1 improves NAFLD through various mechanisms, such as suppressing lipid synthesis, promoting fatty acid oxidation (FAO), and reducing oxidative stress [13, 14]. Moreover, many studies demonstrated that BBR increased the expression of SIRT1 in hepatocytes [15, 16]. Therefore, we speculate that SIRT1 may participate in the beneficial effects of BBR on NAFLD.
This research aimed to explore whether BBR improves NAFLD through SIRT1 and its downstream molecules. The present study revealed that the expression of SIRT1 was reduced in NAFLD patients, high-fat diet (HFD)-fed mice, and hepatocytes exposed to palmitate (PA). BBR increased the expression of SIRT1, which stabilized CPT1A by promoting the deacetylation of CPT1A at the Lys675 site and hence promoted FAO, which ultimately improved non-alcoholic hepatic steatosis. Targeting the SIRT1–CPT1A pathway might be a promising strategy for NAFLD treatment.
Materials and methods
Reagent preparation
BBR (#B3251; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in dimethyl sulfoxide and heated in a 65°C water bath until it was completely dissolved and filtered using a 0.2-μm filter to prepare a 80-mM storage solution. Sodium palmitate (#P9767; Sigma-Aldrich) was dissolved in deionized water at 75°C in a water bath and prepared into a 40-mM solution. Then, 40% de-fatty acid bovine serum albumin was prepared using deionized water and dissolved in a 50°C water bath. Following complete dissolution, the two solutions were mixed in equal volumes and then filtered using a 0.2-μm filter to prepare a 20-mM PA solution.
Animal treatment
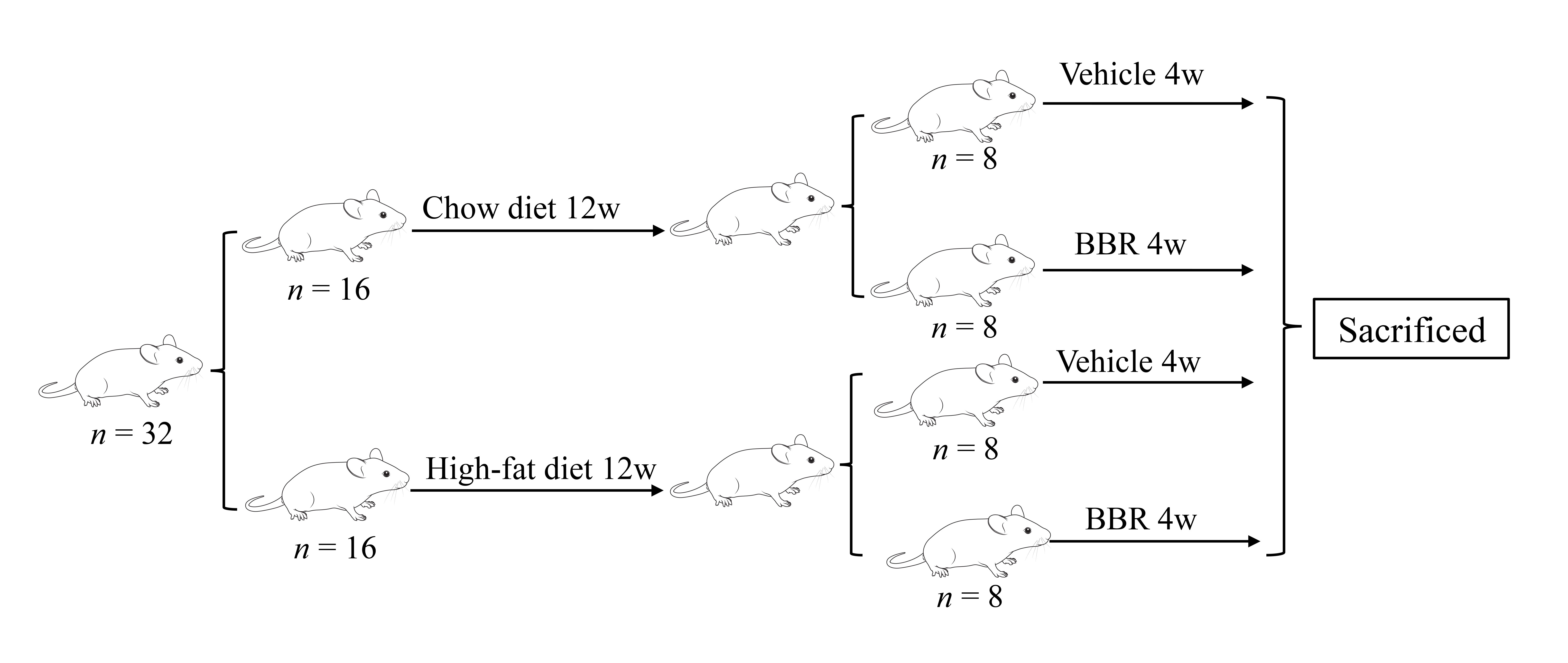
The study was performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal experiment protocol was approved by the Animal Ethics Committee of the Experimental Animal Center of Jinan University (IACUC-20210112–03). We purchased 7-week-old C57BL/6J male mice from Beijing Weitong Lihua Experimental Animal Center. All animals were kept under standard conditions of constant temperature and humidity and a 12-h light/12-h dark light cycle. All animals had free access to food and water. Following 1 week of adaptive feeding, the mice were randomly classified into two initial groups and fed with a chow diet (4% fat, #M10001; MolDiets, Guangzhou, China) or HFD (60% fat, #D12492; Research Diets, New Brunswick, NJ, USA). Following continuous administration of the chow diet or HFD for 12 weeks, each group was randomly classified into the BBR subgroup (n = 8, BBR 200 mg/kg‧d) or vehicle subgroup (n = 8, equal volume of 0.5% methylcellulose). During the feeding of a chow diet or HFD, BBR treatments were administered by gavage for 4 weeks, once a day, and the body weight was monitored weekly. The mice were anesthetized with pentobarbital sodium (40 mg/kg), sacrificed at the end of the experiment, and the liver and serum were collected (Supplementary Figure 1).
Intraperitoneal glucose tolerance test
At the end of the third week of BBR treatment, an intraperitoneal glucose tolerance test (IPGTT) was performed to evaluate glucose tolerance. The mice were fasted overnight (10–14 h) and 2 g/kg of glucose was injected intraperitoneally. Blood samples were obtained from the tail vein. Blood glucose was observed at each time point of 0 (before glucose injection), 15, 30, 60, and 120 min after the glucose injection by using test stripes and a glucose meter (Accu-Chek; Roche, Mannheim, Germany).
Intraperitoneal insulin tolerance test
The intraperitoneal insulin resistance test (IPITT) was conducted 2 days after the IPGTT to analyse insulin sensitivity. Mice were fasted for 4–6 h and 0.8 U/kg of insulin (Novolin R; Novo Nordisk, Bagsvaerd, Denmark) was injected intraperitoneally. Blood samples were obtained from the tail vein and blood glucose was measured at 0 (before insulin injection), 15, 30, 60, and 120 min after insulin injection utilizing a glucose meter (Accu-Chek; Roche).
Serum biochemical test
The blood was obtained and placed in a clean 1.5-mL EP tube, enabled to stand for 1 h at room temperature, and centrifuged at 2,000 r/min for 10 min to collect the mouse serum. The TG, total cholesterol (TC), high density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) contents in the serum were detected using a biochemical analyser (Hitachi, Yokohama, Japan).
Histopathology analysis
Mouse liver tissues were fixed in 10% neutral formaldehyde, embedded in paraffin, processed into 4-µm-thick paraffin sections, and placed on glass slides for hematoxylin and eosin (H&E) staining. Another portion of liver tissues were frozen sectioned, placed on a glass slide, and stained with oil red O (#D027; Jiancheng Biotech, Nanjing, China) to evaluate the degree of hepatic steatosis following the manufacturer’s instructions.
For cell oil red O staining, cells were washed using phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 30 min, and subsequently stained with oil red O in line with the instructions of the oil red O staining kit (#D027; Jiancheng Biotech).
Cell culture and treatment
The Human HepG2 cell line, mouse AML12 cell line, and human HEK293T cell line were procured from American Type Culture Collection. HepG2 cells and HEK293T cells were cultured in DMEM media (#10569044; Gibco, Waltham, MA, USA) containing 10% fetal bovine serum (#16140071; Gibco) at 37°C with 5% CO2. AML12 cells were cultured in DMEM/F12 media (#11320033; Gibco) containing 10% fetal bovine serum, 1 × ITS supplement (#PB180429; Procell, Wuhan, China), 40 ng/mL dexamethasone at 37°C with 5% CO2. For in vitro experiments, HepG2 cells were treated with or without 20 μM of BBR in DMEM media containing 250 μM of PA for 24 h. AML12 cells were treated with or without 10 μM of BBR in DMEM/F12 media containing 200 μM of PA for 24 h. Mouse primary hepatocytes were extracted by using a two-step perfusion method using type IV collagenase [17]. Mouse primary hepatocytes were cultured in DMEM/F12 media containing 200 μM of PA and treated with or without 10 μM of BBR for 24 h. All control cells were supplemented with the same volume of dimethyl sulfoxide or bovine serum albumin to eliminate the influence of the solvent on the experiment. The BBR and PA concentrations in the cell experiments were chosen following CCK8 experiments and pertinent literature [7].
CCK8 assay
A CCK8 assay kit (#BS350B; Biosharp, Hefei, China) was employed to identify cell viability. HepG2 cells, AML12 cells, and mouse primary hepatocytes were seeded at a density of 5 × 103 cells/well in 96-well plates in 100 μL of medium and cultured overnight. The cells were then treated with different concentrations of PA or BBR for 24 h. Next, 10 μL of CCK8 solution was added to each well incubated for 2 h at 37°C. The absorbance values were quantified at 450 nm utilizing a microplate reader (BioTek, Minneapolis, MN, USA).
Lentivirus and plasmid transfection
Human SIRT1 overexpression and knock-down lentivirus (Genechem, Shanghai, China), human CPT1A overexpression, and knockout lentivirus (Genechem) were diluted in PBS and added to the media for 24 h based on the multiplicity of infection. Human wild-type and mutant plasmids of SIRT1 and CPT1A (Tsingke, Beijing, China) were diluted in jetPRIME reagent (#114–15; Polyplus, Illkirch, France) and added to the media for 24 h following the manufacturer’s instructions.
Triglyceride measurement
For liver TG measurements, an appropriate amount of tissue block was rinsed using pre-cooled PBS, blot dried with filter paper, put in a homogenizer, and pre-cooled isopropanol added in a ratio of weight (g):volume (mL) = 1:9. The tissue was homogenized and centrifuged at 4°C, 10,000 g, for 10 min. The supernatant was utilized to observe the TG level using a TG detection kit (#E-BC-K261-M; Elabscience, Wuhan, China) following the instructions.
For the cellular TG measurements, HepG2, AML12 cells, and mouse primary hepatocytes were collected and split into two equal parts: one part for measuring protein concentration and the other part for quantifying intracellular TG concentration; the protein concentration was utilized to standardize the TG level.
FAO assay
FAO was detected using an FAO assay kit (#BR00001; Assay Genie, Dublin, Ireland). Briefly, HepG2 cells were lysed on ice for 5 min, then centrifuged at 14,000 r/min for 5 min; the lysate protein concentration was identified by using the BCA technique. Afterward, 20 µL of each sample was added to a 96-well plate in duplicate, 50 µL of control solution was swiftly added to one set of wells, and 50 µL of reaction solution was added to the other set of wells, and incubated in a 37°C incubator for 30 min. The absorbance values were measured at 492 nm using a microplate reader (BioTek). The FAO activity was calculated following the manufacturer’s instructions.
NAD+ and NADH assay
Intracellular NAD+ and NADH levels were detected in HepG2 cells by using a NAD+/NADH assay kit (#S0175; Beyotime, Haimen, China) following the manufacturer’s instructions. Protein was extracted from HepG2 cells and measured by using the BCA method; the protein concentration was utilized to standardize the NAD+ and NADH levels.
CPT1A activity assay
A CPT1A activity detection kit (#GMS50118; Genmed, Arlington, MA, USA) was used to reveal CPT1A enzymatic activity following the manufacturer’s instructions in HepG2 cells. Mitochondrial protein was extracted from HepG2 cells and measured by using the BCA method; the mitochondrial protein concentration was utilized to standardize the CPT1A activity.
Quantitative real-time PCR
Total RNA from liver tissue and cultured cells was extracted utilizing the Trizol reagent (#9108; Takara, Kyoto, Japan) and total RNA was reversely transcribed into cDNA using the reverse transcription reagent kit with genomic DNA eraser (#FSQ-201; Toyobo, Osaka, Japan) based on the manufacturer’s instructions. Quantitative real-time PCR was conducted using a Bio-Rad CFX system (Bio-Rad, Hercules, CA, USA) using cDNA transcribed from total RNA with gene-specific primers and SYBR Green Premix (#QPK-201; Toyobo) following the manufacturer’s instructions. The sequences of the primers used in this research are outlined in Table 1.
Table 1.
Sequence of primers for quantitative real-time PCR
| Gene Species Forward primer Reverse prime |
|---|
|
Western blot analysis and co-immunoprecipitation
Western blot was conducted using primary antibodies against SIRT1 (1:1,000, #8469; Cell Signaling Technology, Danvers, MA, USA), FASN (1:1,000, #3180; Cell Signaling Technology), ACC (1:1,000, #3676; Cell Signaling Technology), p-ACC (1:1,000, #3661; Cell Signaling Technology), SCD1 (1:1,000, #2438; Cell Signaling Technology), CPT1A (1:1,000, #15184–1-AP; Proteintech, Rosemont, IL, USA), SREBP1 (1:1,000, #14088–1-AP; Proteintech), CD36 (1:1,000, #18836–1-AP; Proteintech), HA (1:1,000, #201113; ZENBIO, Chengdu, China), FLAG (1:1,000, #390002; ZENBIO), and β-actin (1:2,000, #4970; Cell Signaling Technology). The protein bands were detected using the ECL Western blot substrate (#E412-01; Vazyme, Nanjing, China) in line with the manufacturer’s instructions and quantified using ImageJ (https://imagej.nih.gov/ij/).
A co-immunoprecipitation (co-IP) experiment was performed using antibodies against SIRT1 (1:50, #8469; Cell Signaling Technology), CPT1A (1:50, #15184–1-AP; Proteintech), acetylated lysine (1:1,000, #9441; Cell Signaling Technology), ubiquitin (1:200, #sc-8017; Santacruz, Dallas, Texas, USA), anti-HA magnetic beads (1:50, # HY-K0201; MedChemExpress, NJ, USA), and anti-Flag magnetic beads (1:50, #HY-K0207; MedChemExpress). For the co-IP experiment, an appropriate volume of antibody was added to the cell lysate and incubated for 4 h at 4°C to immunoprecipitate the target protein. Next, protein A/G magnetic beads (#HY-K0202; MedChemExpress) were added, followed by incubation overnight at 4°C. The next day, the magnetic beads were washed four times using PBST (1 × PBS + 0.5% Tween-20), and the immune complexes were dissolved in diluted 1 × protein SDS loading buffer and heated at 95°C for 5 min and then obtained by using the magnetic stand. The immune complexes were separated utilizing SDS–PAGE, which was followed by immunoblotting with primary antibodies against CPT1A, SIRT1, HA, Flag, ubiquitin, and acetylated lysine.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (version 8.0); all data are presented as the mean ± standard deviation (SD). For two groups, an unpaired two-tailed t-test was conducted for intergroup comparisons. For more than two groups, one-way analysis of variance and the post hoc multiple comparisons (least significant difference (LSD)) method) were used for intergroup comparisons. P-values of <0.05 was considered to be statistically significant.
Results
BBR improves lipid and glucose metabolism disorders induced by HFD in mice
From the third week of HFD feeding, the body weight of mice in the HFD + Vehicle group demonstrated a significant growth compared with mice in the chow diet group and it lasted until the end of the experiment. Moreover, compared with the HFD + Vehicle group, BBR treatment significantly lowered the body weight of mice (Figure 1A). Furthermore, the results of IPGTTs and IPITTs showed that HFD feeding impaired glucose tolerance and insulin sensitivity in mice, whereas BBR treatment improved those (Figure 1B and C). Additionally, HFD elevated the fasting blood glucose (5.1 ± 0.75 vs 6.4 ± 0.71 mmol/L, P = 0.004) in mice and BBR treatment reduced the abnormal fasting blood glucose (6.4 ± 0.71 vs 5.3 ± 0.74 mmol/L, P = 0.01) (Figure 1D). To investigate the effect of BBR on dyslipidemia induced by HFD, the blood lipid profile was measured. Compared with the chow diet group, HFD increased serum TG (0.59 ± 0.09 vs 0.98 ± 0.24 mmol/L, P = 0.001), TC (4.64 ± 0.29 vs 5.53 ± 0.79 mmol/L, P = 0.01), LDL-C (0.39 ± 0.04 vs 0.53 ± 0.12 mmol/L, P = 0.01), and HDL-C (1.89 ± 0.18 vs 2.21 ± 0.16 mmol/L, P = 0.002) levels. BBR treatment significantly reduced the serum TG (0.98 ± 0.24 vs 0.67 ± 0.09 mmol/L, P = 0.004), TC (5.53 ± 0.79 vs 3.85 ± 0.56 mmol/L, P < 0.001), and LDL-C (0.53 ± 0.12 vs 0.29 ± 0.03 mmol/L, P < 0.001) levels, but did not significantly affect HDL-C levels (Figure 1E–H). The results showed that BBR improved the glucose and lipid metabolism disorders induced by HFD in mice.
Figure 1.

BBR alleviates systematic lipid and glucose disorders in HFD-fed mice. C57BL/6J mice fed a chow diet or an HFD for 12 weeks were randomly classified into either the vehicle or the BBR treatment subgroup (administered by gavage for 4 weeks). (A) Body weight change of mice during the experiment. (B) IPGTT results following BBR treatment. (C) IPITT findings after BBR treatment. (D) Fasting blood glucose levels in serum. (E) TG, (F) TC, (G) LDL-C, and (H) HDL-C levels in serum. The data are expressed as means ± SD; n = 8; *P < 0.05 (HFD + Vehicle vs Chow + Vehicle group); #P < 0.05 (HFD + Vehicle vs HFD + BBR group); &P < 0.05 (Chow+ BBR vs Chow + Vehicle group). BBR, berberine; HFD, high-fat diet; IPGTT, intraperitoneal glucose tolerance test; IPITT, intraperitoneal insulin resistance test; TG, triglyceride; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; HDL-C, high density lipoprotein cholesterol.
BBR improves hepatic steatosis induced by HFD in mice
To detail the effect of BBR on liver lipid accumulation in HFD-fed mice, livers of mice were weighed and the liver sections were stained with H&E and oil red O. The contents of TG and TC in liver tissues were quantified by using a lipid detection kit. The results showed that HFD significantly induced liver TG (66.6 ± 13.8 vs 190.1 ± 11.2 μmol/g liver, P < 0.001) and TC (8.2 ± 2.0 vs 11.3 ± 2.5 μmol/g liver, P = 0.01) deposition. BBR treatment attenuated the contents of TG (190.1 ± 11.2 vs 113.6 ± 7.6 μmol/g liver, P < 0.001) and TC (11.3 ± 2.5 vs 6.3 ± 0.4 μmol/g liver, P < 0.001) in liver tissues induced by HFD (Figure 2A–C). Moreover, HFD increased the liver weight (1.0 ± 0.1 vs 2.4 ± 0.3 g, P < 0.001) and liver-to-body ratio (3.5 ± 0.4% vs 4.6 ± 0.9%, P = 0.01). BBR treatment attenuated liver weight (2.4 ± 0.3 vs 1.2 ± 0.2 g, P < 0.001) and liver-to-body ratio (4.6 ± 0.9% vs 3.1 ± 0.4%, P = 0.001) compared with the HFD + Vehicle group (Figure 2D and E). Additionally, HFD increased the ALT (46.7 ± 13.1 vs 298.9 ± 91.7 U/L, P < 0.001) and AST (243.1 ± 37.4 vs 450.1 ± 68.3 U/L, P < 0.001) levels. BBR treatment decreased the ALT (298.9 ± 91.7 vs 102.5 ± 24.6 U/L, P < 0.001) and AST (450.1 ± 68.3 vs 221.2 ± 31.1 U/L, P < 0.001) levels compared with the HFD + Vehicle group (Figure 2F and G). These findings indicated that BBR attenuated hepatic steatosis and decreased the elevated ALT and AST levels induced by HFD in mice.
Figure 2.

BBR improves hepatic steatosis induced by HFD in mice. (A) Representative images of liver pathology, H&E, and oil red O staining; images were obtained at original magnification, scale bars = 50 μm. (B) Liver TG concentration. (C) Liver TC concentration. (D) Liver weight. (E) The liver weight to body weight ratio. (F) and (G) Serum ALT and AST levels. The data are expressed as means ± SD; n = 8; *P < 0.05 (compared with the Chow + Vehicle group); #P < 0.05 (compared with the HFD + Vehicle group). BBR, berberine; HFD, high-fat diet; H&E, hematoxylin and eosin; TG, triglyceride; TC, total cholesterol; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
BBR attenuates hepatic TG accumulation induced by PA in vitro
HepG2 cells, AML12 cells, and mouse primary hepatocytes were treated with PA for 24 h in the presence or absence of BBR so as to investigate whether BBR reduces lipid accumulation in hepatocytes in vitro. The lipid content was then observed by using oil red O staining and a TG detection kit. Following the CCK8 experiment and related literature, PA and BBR concentrations of HepG2 cells (250 μM, 20 μM), AML12 cells (200 μM, 10 μM), and mouse primary hepatocytes (200 μM, 10 μM) were identified (Figure 3A–F) [7]. The results of oil red O staining showed that PA treatment increased the lipid contents of HepG2 cells, AML12 cells, and primary hepatocytes, and BBR treatment reduced the lipid deposition (Figure 3G–I). Subsequently, the TG detection kit was employed to detail the TG contents of HepG2 cells, AML12 cells, and mouse primary hepatocytes. The results implied that PA induced TG deposition in HepG2 cells (71.3 ± 7.9 vs 138.4 ± 20.4 μmol/mg protein, P = 0.006), AML12 cells (51.1 ± 6.8 vs 94.1 ± 5.1 μmol/mg protein, P < 0.001), and primary hepatocytes (49.4 ± 10.1 vs 206.2 ± 36.1 μmol/mg protein, P = 0.002). BBR reduced the intracellular TG concentration in HepG2 cells (138.4 ± 20.4 vs 81.4 ± 7.5 μmol/mg protein, P = 0.01), AML12 cells (94.1 ± 5.1 vs 58.2 ± 7.1 μmol/mg protein, P = 0.002), and primary hepatocytes (206.2 ± 36.1 vs 68.8 ± 28.1 μmol/mg protein, P = 0.006) induced by PA (Figure 3J–L). These findings indicated that BBR significantly declined TG deposition in hepatocytes induced by PA in vitro.
Figure 3.

BBR attenuates hepatic TG accumulation induced by PA in vitro. (A) and (B) The CCK8 results of HepG2 cells treated with different concentrations of PA or BBR for 24 h. (C) and (D) The CCK8 results of AML12 cells. (E) and (F) The CCK8 findings of mouse primary hepatocytes. (G)–(I) Representative photomicrographs of (G) HepG2 cells, (H) AML12 cells, and (I) primary hepatocytes for oil red O staining (×200), scale bars = 50 μm. (J)–(L) Quantitative analysis of TG in (J) HepG2 cells, (K) AML12 cells, and (L) primary hepatocytes. All results are representative of three independent experiments; data are expressed as means ± SD; *P < 0.05 (compared with the control group); #P < 0.05 (compared with the PA group). BBR, berberine; TG, triglyceride; PA, palmitate; CCK8, cell counting kit 8.
BBR increases the protein levels of SIRT1 and CPT1A in vivo and in vitro
To determine whether SIRT1 is involved in NAFLD pathogenesis, we analysed the published microarray data generated from the liver tissue of control and NAFLD patients (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48452) in the Gene Expression Omnibus database. Data from this database demonstrated that the hepatic SIRT1 mRNA level was decreased in the hepatic steatosis group and NASH group compared with that in normal subjects (Figure 4A). Then, we explored the protein levels of SIRT1 and molecules associated with lipid metabolism such as lipid uptake (CD36), synthesis (SCD1, SREBP1, FASN, ACC, p-ACC), and β-oxidation (CPT1A) by Western blot. The results demonstrated that the HFD challenge had different effects on the protein levels of SIRT1 and CPT1A. HFD reduced the protein level of SIRT1, but increased CPT1A. BBR treatment elevated the protein levels of SIRT1 and CPT1A in both the chow diet and HFD groups (Figure 4B–D). The protein levels of SIRT1 and CPT1A showed a similar trend in HepG2 cells, AML12 cells, and primary hepatocytes as shown by Western blot analysis (Figure 4E–M). We then detected the mRNA levels of SIRT1 and CPT1A in mouse liver tissues and HepG2 cells. The findings demonstrated that HFD or PA treatment decreased the mRNA level of SIRT1 but increased CPT1A. BBR significantly increased the transcription of SIRT1 but had no significant effect on the transcription of CPT1A (Figure 4N–Q). These results indicated that SIRT1 and CPT1A participated in the therapeutic effects of BBR on NAFLD, in which BBR boosted the expression of SIRT1 at the transcriptional level but influenced the protein level of CPT1A at the post-transcriptional level.
Figure 4.

BBR increases the protein levels of SIRT1 and CPT1A in vivo and in vitro. (A) For the expression of SIRT1 in the control group, liver steatosis group, and NASH group, the data were acquired from the GEO database GSE48452. (B) C57BL/6J mice were fed a chow diet or an HFD for 12 weeks and then treated with vehicle or BBR. The protein levels of SIRT1 and proteins related to lipid metabolism, such as lipid uptake (CD36), synthesis (SCD1, SREBP1, FASN, ACC, p-ACC), and β-oxidation (CPT1A), were assayed by using Western blot. (C) and (D) Quantitative analysis of (C) CPT1A and (D) SIRT1 in different groups. (E)–(G) The levels of proteins associated with lipid metabolism in (E) HepG2 cells, (F) AML12 cells, and (G) primary hepatocytes. (H)–(J) Quantitative analysis of CPT1A in (H) HepG2 cells, (I) AML12 cells, and (J) primary hepatocytes. (K)–(M) Quantitative analysis of SIRT1 in (K) HepG2 cells, (L) AML12 cells, and (M) primary hepatocytes. (N) and (O) Representative mRNA levels of (N) SIRT1 and (O) CPT1A in livers of chow diet or HFD-fed mice with or without BBR treatment. (P) and (Q) Representative mRNA levels of (P) SIRT1 and (Q) CPT1A in HepG2 cells. All results are representative of three independent experiments; data are expressed as means ± SD; ns, not significant; *P < 0.05 (compared with the chow diet group or control group); #P < 0.05 (compared with the HFD group or PA group). BBR, berberine; NASH, non-alcoholic steatohepatitis; GEO, Gene Expression Omnibus; HFD, high-fat diet.
BBR increases the NAD+ level and the activity of CPT1A in HepG2 cells
We detected the NAD+ level and the NAD+/NADH ratio in HepG2 cells because SIRT1 is NAD+-dependent. Results demonstrated that PA decreased the intracellular NAD+ level (5.7 ± 0.3 vs 3.7 ± 0.5 pmol/μg protein, P = 0.005) and NAD+/NADH ratio (1.7 ± 0.1 vs 1.1 ± 0.1, P = 0.003), whereas BBR reversed the PA-reduced NAD+ level (3.7 ± 0.5 vs 6.2 ± 0.9 pmol/μg protein, P = 0.01) and the NAD+/NADH ratio (1.1 ± 0.1 vs 2.1 ± 0.2, P = 0.002) in HepG2 cells (Figure 5A–C). We then detected the activity of CPT1A in HepG2 cells. Results demonstrated that PA caused a decrease in CPT1A activity (13.1 ± 1.5 vs 8.2 ± 0.5 nmol/min per milligram protein, P = 0.006), while BBR treatment significantly enhanced the activity of CPT1A (8.2 ± 0.5 vs 15.9 ± 1.7 nmol/min per milligram protein, P = 0.001) (Figure 5D). These results implied that BBR reversed the PA-reduced NAD+ level in HepG2 cells, which enhanced the activation of SIRT1. Furthermore, in HepG2 cells, PA increased the protein level of CPT1A but inhibited its activity. In contrast, BBR not only increased the protein level of CPT1A but also enhanced its activity.
Figure 5.

BBR increases the NAD+ level and the activity of CPT1A in HepG2 cells. (A)–(C) Intracellular levels of the (A) NAD+, (B) NADH, and (C) NAD+/NADH ratio in HepG2 cells treated with PA and BBR as indicated. (D) The activity of CPT1A in HepG2 cells treated with PA and BBR as indicated. All findings are representative of three independent experiments; data are presented as means ± SD; *P < 0.05 (compared with the control group); #P < 0.05 (compared with the PA group). BBR, berberine; NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide; PA, palmitate.
SIRT1 mediates the effect of BBR on decreasing TG accumulation and increasing FAO through CPT1A in hepatocytes
To investigate the effect of CPT1A on TG deposition in hepatocytes, overexpression and knockout of CPT1A by lentivirus transfection in HepG2 cells were performed, then cells were treated with PA for 24 h, and the TG content was measured by using a TG detection kit. In HepG2 cells, CPT1A overexpression attenuated PA-induced TG accumulation and CPT1A knockout aggravated TG deposition (Figure 6A and B). To explore the effect of SIRT1 on the TG content, the overexpression and knock-down of SIRT1 were performed in HepG2 cells. Following 24 h of PA treatment, the TG content was measured. The results disclosed that SIRT1 overexpression attenuated TG deposition, whereas SIRT1 knock-down had the opposite result (Figure 6C). To investigate whether SIRT1 regulates the expression of CPT1A, we knocked down or overexpressed SIRT1 in HepG2 cells, and then observed the mRNA and protein levels of CPT1A. The overexpression of SIRT1 in HepG2 cells increased the protein level of CPT1A, whereas SIRT1 knock-down attenuated the protein level of CPT1A, but there was no difference at the transcription level (Figure 6D and E). Next, we detected the expression levels of CPT1A in control or shSIRT1-transfected HepG2 cells with or without BBR treatment. Western blot results revealed that BBR increased CPT1A expression in control cells. Nevertheless, following SIRT1 knock-down, BBR could not effectively increase CPT1A expression (Figure 6F). These results showed that CPT1A and SIRT1 regulated TG deposition, and SIRT1 mediated the increased expression of CPT1A induced by BBR at the post-transcriptional level. Furthermore, we transfected CPT1A in control and SIRT1 knock-down HepG2 cells exposed to PA, with or without BBR treatment, and then detected the intracellular TG content. The results exposed that BBR considerably reduced TG accumulation induced by PA. Nevertheless, SIRT1 knock-down attenuated the effect of BBR on reducing TG. Re-expression of CPT1A in shSIRT1 HepG2 cells restored the effect of BBR on reducing TG (Figure 6G). To investigate the effect of CPT1A on FAO, we quantified FAO in PA (250 μM)-treated CPT1A knockout HepG2 cells and the knockout cells transfected with restored CPT1A wild-type synonymous mutant plasmid. The findings implied that CPT1A knockout significantly attenuated FAO in HepG2 cells and the restoration of FAO was observed when a synonymous mutant of CPT1A was reintroduced in the knockout cells (Figure 6H). Next, we measured FAO in HepG2 cells treated with 250 μM of PA and different concentrations of BBR (0, 10, 20 μM). The results demonstrated BBR promoted FAO in a dose-dependent manner (Figure 6I). Together, these results indicated that SIRT1 mediated the effect of BBR on decreasing TG accumulation by increasing the expression of CPT1A, which promoted FAO in hepatocytes.
Figure 6.

SIRT1 mediates the effect of BBR on decreasing TG accumulation and increasing fatty acid oxidation via CPT1A in hepatocytes. (A) TG concentrations of CPT1A overexpressing HepG2 cells treated with PA (250 μM). (B) TG concentrations of CPT1A knockout HepG2 cells treated with PA. (C) Quantitative analysis of TG concentrations in SIRT1 knock-down and overexpression HepG2 cells induced by PA. (D) The expression levels of CPT1A in SIRT1 knock-down and overexpression HepG2 cells were identified by using Western blot analysis. (E) The mRNA levels of CPT1A in shSIRT1- or LvSIRT1-transfected HepG2 cells. (F) Representative images of CPT1A and SIRT1 in shNC or shSIRT1 HepG2 cells treated with or without BBR. (G) Quantitative analysis of the TG contents and representative images of CPT1A and SIRT1 expression levels in PA-treated shNC or shSIRT1 HepG2 cells with or without BBR or CPT1A plasmids treatment. (H) Fatty acid oxidation was computed in PA (250 μM)-treated CPT1A knockout cells and the knockout cells transfected with restored CPT1A wild-type synonymous mutant plasmid. (I) Fatty acid oxidation assay of HepG2 cells treated with PA (250 μM) and different concentrations of BBR (0, 10, 20 μM). Results are representative of three independent experiments; data are provided as means ± SD; *P < 0.05 (compared with the control group); #P < 0.05 (compared with the CPT1A knockout group). BBR, berberine; TG, triglyceride; PA, palmitate; NC, normal control; sh, short hairpin; Lv, lentivirus; KO, knockout; FAO, fatty acid oxidation.
SIRT1 interacts with CPT1A and suppresses its degradation
SIRT1 regulated the expression of CPT1A at the post-transcriptional level as shown in Figure 6. To further investigate how SIRT1 regulates the expression of CPT1A at the post-transcriptional level, we immunoprecipitated SIRT1 and CPT1A in HepG2 cells and detected the direct binding between SIRT1 and CPT1A by Western blot. The results showed that SIRT1 interacted with CPT1A (Figure 7A). Moreover, co-IP with total proteins from HEK293T implied a similar result (Figure 7B). Hence, we proposed that SIRT1 interacts with CPT1A and influences its protein stability. To investigate this hypothesis, the proteasome inhibitor MG132 (10 μM) was treated in HepG2 cells with or without SIRT1 knock-down for 24 h. The protein level of CPT1A was quantified and the results showed that the expression of CPT1A significantly decreased following SIRT1 knock-down, and the protein level of CPT1A was significantly increased after treatment with MG132 (Figure 7C). The half-life of CPT1A in control or shSIRT1 HepG2 cells was measured after treatment with 100 μg/μL of cycloheximide (CHX) so as to analyse the effect of SIRT1 on CPT1A. SIRT1 knock-down reduced the half-life time of the CPT1A (Figure 7D and E). These results indicated that SIRT1 interacted with CPT1A and influenced its protein stability.
Figure 7.

SIRT1 interacts with CPT1A and suppresses its degradation. (A) HepG2 cell lysates were used for co-IP experiments with anti-SIRT1 or anti-CPT1A antibodies, respectively, followed by Western blot using indicated antibodies. (B) co-IP experiments of HEK293T cells co-transfected with HA-CPT1A and Flag-SIRT1, utilizing anti-HA antibody for IP and anti-Flag antibody for immunoblotting or anti-Flag antibody for IP and anti-HA antibody for immunoblotting. (C) CPT1A expression levels in shNC- or shSIRT1-transfected HepG2 cells treated with protease inhibitor MG132 (10 μM) for 24 h. (D) Representative images of CPT1A expression levels in shNC- or shSIRT1-transfected HepG2 cells treated with CHX (100 μg/μL) for indicated times. (E) CPT1A half-life time in shNC- or shSIRT1-transfected HepG2 cells after treatment with CHX for indicated times. All results are representative of three independent experiments. Values are presented as means ± SD; *P < 0.05 (compared with the control group). co-IP, co-immunoprecipitation; HEK, human embryonic kidney; IP, immunoprecipitation; NC, normal control; sh, short hairpin; CHX, cycloheximide.
SIRT1 deacetylates CPT1A to suppress its ubiquitination degradation
Acetylation is a critical post-translational modification that regulates protein stability by affecting the level of protein ubiquitination [18]. SIRT1, as a deacetylase, may affect its ubiquitination level by deacetylating CPT1A. To confirm this conjecture, HepG2 cells were transfected with either SIRT1 wild-type (WT) or SIRT1 catalytic mutant plasmids (SIRT1-H363Y) [19]. Western blot indicated that SIRT1-WT, but not catalytic mutant, markedly elevated the CPT1A protein level (Figure 8A). Accordingly, SIRT1 deacetylase activity was necessary for CPT1A stability. In addition, to observe the effects of BBR and SIRT1 on the acetylation and ubiquitination levels of CPT1A, co-IP assays were performed in HepG2 cells. SIRT1 overexpression and BBR reduced the acetylation and ubiquitination levels of CPT1A. In contrast, SIRT1 knock-down and PA treatment increased the acetylation and ubiquitination of CPT1A (Figure 8B–F). Furthermore, the acetylation levels of CPT1A in liver tissues were detected by using co-IP and the results disclosed that HFD increased the acetylation levels of CPT1A and BBR treatment decreased its acetylation levels (Figure 8G and H). The results showed that SIRT1 reduced the acetylation and ubiquitination levels of CPT1A. We speculated that SIRT1 may decrease the acetylation level of CPT1A, leading to a reduction in its ubiquitination level, and consequently affecting the stability of CPT1A. To validate this hypothesis, we co-transfected HA-CPT1A-WT plasmids with SIRT1-WT or catalytic mutant plasmids (SIRT1-H363Y) in HEK293T cells, which was followed by immunoprecipitation using anti-HA or anti-IgG antibody, and blotting with anti-acetylated-lysine and anti-ubiquitin antibodies. The results indicated that SIRT1-WT, but not SIRT1-H363Y, markedly increased the protein levels of HA-CPT1A. As predicted, compared with the control group, SIRT1-WT decreased the acetylation level of exogenous CPT1A alongside its ubiquitination level. However, SIRT1-H363Y did not significantly influence CPT1A acetylation and ubiquitination compared with the control group (Figure 8I). Together, these results indicated that SIRT1 reduced the ubiquitination level of CPT1A by deacetylating CPT1A, which increased the protein stability.
Figure 8.

SIRT1 deacetylates CPT1A to suppress its ubiquitination degradation. (A) SIRT1 and CPT1A protein expression levels in HepG2 cells transfected with normal control (NC), SIRT1-WT, or mutant plasmids (SIRT1-H363Y). (B) co-IP experiments in HepG2 cells exposed to vehicle or PA with or without BBR treatment, using an anti-CPT1A antibody for IP and anti-acetyl-lysine antibody or anti-ubiquitin antibody for immunoblotting. (C) co-IP experiments in SIRT1 knock-down or SIRT1 overexpression HepG2 cells, utilizing an anti-CPT1A antibody for IP and an anti-acetyl-lysine antibody or anti-ubiquitin antibody for immunoblotting. (D) Quantitative analysis of CPT1A acetylation levels in HepG2 cells exposed to vehicle or PA with or without BBR treatment. (E) and (F) Quantitative analysis of CPT1A acetylation levels in (E) SIRT1 knock-down or (F) SIRT1 overexpression HepG2 cells. (G) CPT1A acetylation levels in livers of C57BL/6J mice fed a chow diet or HFD with or without BBR treatment. (H) Quantitative analysis of CPT1A acetylation levels in livers of C57BL/6J mice. (I) co-IP experiments in HEK293T cells co-transfected with HA-CPT1A-WT and SIRT1-WT or catalytic mutant plasmids (SIRT1-H363Y), employing an anti-HA antibody for IP and anti-acetyl-lysine antibodies and anti-ubiquitin antibodies for immunoblotting. All results are representative of three independent experiments. Values are presented as means ± SD; *P < 0.05 (compared with the control group or chow diet group); #P < 0.05 (compared with the PA group or HFD group). WT, wild-type; NC, normal control; sh, short hairpin; Lv, lentivirus; co-IP, co-immunoprecipitation; IP, immunoprecipitation; PA, palmitate; BBR, berberine; HFD, high-fat diet; HEK, human embryonic kidney; Ac, acetylation; Ub, ubiquitin.
SIRT1 deacetylates CPT1A at the Lys675 site
Acetylation prediction tools and research literature were employed to predict the deacetylation sites of CPT1A [20]. We selected four possible acetylation sites (Lys195, Lys292, Lys580, and Lys675) and mutated the lysine (K) to arginine (R) to generate four deacetylated mimetic mutants (K195R, K292R, K580R, K675R). HA-CPT1A-WT and four mutant plasmids were transfected in SIRT1 knock-down or control HEK293T cells, respectively, followed by immunoprecipitation utilizing HA antibody and blotting with anti-acetylated-lysine antibody. The findings demonstrated that CPT1A-K195R, CPT1A-K292R, CPT1A-K580R, and CPT1A-WT acetylation levels were considerably increased in SIRT1 knock-down cells, whereas no difference was observed in CPT1A-K675R acetylation levels, which indicated that CPT1A could be acetylated at Lys675 (Figure 9A and B). To further explore whether acetylation mutation modulates CPT1A protein stability, HEK293T cells transfected with HA-CPT1A-WT or the mutant HA-CPT1A-K675R plasmids were treated with CHX. The findings revealed that the CPT1A in the mutant group was more stable than that in the WT group, which indicated CPT1A stability was the result of protein acetylation at the Lys675 residue (Figure 9C). To investigate the effect of Lys675 mutation on the ubiquitination level of CPT1A, HEK293T cells were transfected with HA-CPT1A-WT or the mutant HA-CPT1A-K675R plasmids. Cell lysates were immunoprecipitated with anti-HA antibody and blotted with anti-acetyl-lysine and anti-ubiquitin antibodies. Compared with the WT group, the ubiquitination level of CPT1A-K675R was significantly reduced (Figure 9D). These data implied that SIRT1 deacetylated the Lys675 site on CPT1A, which suppressed the ubiquitination degradation of CPT1A.
Figure 9.

SIRT1 deacetylates CPT1A at the Lys675 site. (A) shNC or shSIRT1 HEK293T cells were transfected with either HA-CPT1A-WT or different mutant plasmids including K195R, K292R, K580R, and K675R. Cell lysates were immunoprecipitated with an anti-HA antibody and blotted with an anti-acetyl-lysine antibody. (B) Quantitative analysis of CPT1A acetylation levels in HEK293T cells transfected with either HA-CPT1A-WT or different mutant plasmids as indicated. (C) CPT1A protein stability time course in HEK293T cells transfected with HA-CPT1A-WT and HA-CPT1A-K675R mutants following treatment with 100 μg/μL of CHX. (D) HEK293T cells transfected with HA-CPT1A-WT or HA-CPT1A-K675R mutant plasmids; cell lysates were immunoprecipitated with anti-HA antibody and blotted with anti-acetyl-lysine and anti-ubiquitin antibodies. All results are representative of three independent experiments. Values are provided as means ± SD; *P < 0.05 (compared with the control group). NC, normal control; sh, short hairpin; HEK, human embryonic kidney; CHX, cycloheximide; Ac, acetylation; Ub, ubiquitin.
Discussion
This study disclosed that BBR alleviated non-alcoholic hepatic steatosis in vivo and in vitro. BBR increased the expression of SIRT1, SIRT1 deacetylated CPT1A at the Lys675 site, which suppressed the ubiquitination degradation of CPT1A. The increased CPT1A promoted FAO in hepatocytes, thereby alleviating hepatic steatosis. A schematic diagram of BBR in ameliorating non-alcoholic hepatic steatosis is shown in the Graphical Abstract.
Studies have suggested that the BBR drug concentration in the liver was significantly higher than that in other tissues and blood, which may partly explain the advantageous effect of BBR on NAFLD [21]. Recent studies have shown that BBR is distributed primarily in the nucleus, cytoplasm, and mitochondria at the cellular level [22, 23]. Furthermore, it has been found to inhibit mitochondrial respiratory complex I and increase the intracellular NAD+/NADH ratio [24]. A series of studies disclosed that BBR promoted the expression of SIRT1 in many cells such as hepatocytes, fibroblasts, spinal nerve cells, skeletal muscle cells, and cardiomyocytes [16, 25, 26]. As a metabolic sensor, SIRT1 extensively participates in the process of glucose and lipid metabolism in the liver [27]. Liver SIRT1 overexpression attenuated TG accumulation in mice, whereas hepatic SIRT1 deficiency aggravated liver TG deposition in HFD-induced mice [28–30]. Our study revealed that BBR effectively alleviated non-alcoholic hepatic steatosis in vivo and in vitro. The expression of SIRT1 was considerably decreased in HFD-fed mice and hepatocytes exposed to PA compared with the control group, and BBR treatment significantly increased SIRT1 expression. Furthermore, SIRT1 overexpression mimicked the impact of BBR in reducing the TG level, while SIRT1 knock-down weakened the effect of BBR. These findings indicated that SIRT1 mediated the therapeutic effect of BBR on NAFLD.
The major metabolic process of lipids in hepatocytes includes fatty acid uptake, lipid synthesis, FAO, and secretion through very low-density lipoprotein [31]. We performed Western blot to evaluate the expression of SIRT1 and proteins involved in lipid metabolism, such as lipid uptake (CD36), lipid synthesis (SCD1, SREBP1, FASN, ACC, p-ACC), and FAO (CPT1A), in both liver tissues and cell models. We discovered that the HFD challenge reduced the protein level of SIRT1, but increased CPT1A. BBR significantly increased the protein levels of SIRT1 and CPT1A in animal and cell models, which indicated that CPT1A may also participate in the therapeutic effects of BBR on NAFLD. Further experiment demonstrated that HFD challenge triggered the expression of CPT1A at the transcriptional level, which could be a compensatory response to fatty acid stimulation, but inhibited the activity of CPT1A, while BBR treatment increased the protein level of CPT1A alongside the activity. This may help explain why elevated levels of CPT1A were insufficient in preventing hepatic steatosis in HFD-fed mice, whereas BBR-induced upregulation of CPT1A effectively improved hepatic steatosis. Our findings are consistent with the research conducted by Softic et al. [32], which reported that although HFD triggered an increase in CPT1A expression, it also elevated the acetylation level of CPT1A and impaired its activity in mice. CPT1A is a rate-limiting enzyme that regulates FAO in hepatocytes, majorly distributed in the outer mitochondrial membrane of hepatocytes. Research validated that mice with liver-specific CPT1A deletion exhibited severe liver steatosis, and liver CPT1A overexpression improved lipid deposition induced by HFD [33, 34]. Therefore, hepatic CPT1A is essential for maintaining lipid homeostasis and functions as a protective role in the development of NAFLD. Our experiments showed that BBR elevated the protein levels of SIRT1 and CPT1A. To investigate the relationship among BBR, SIRT1, and CPT1A, we performed a series of experiments on SIRT1 and CPT1A in HepG2 cells. The results indicated that CPT1A was a downstream molecule of SIRT1 and SIRT1 regulated the protein level of CPT1A at the post-transcriptional level. BBR promoted the protein level of CPT1A by increased expression of SIRT1 and hence enhanced FAO in hepatocytes, which ultimately alleviated non-alcoholic hepatic steatosis.
Lysine acetylation is an evolutionarily conserved post-translational modification of proteins generated by lysine acetyltransferase and reversed by lysine deacetylases [18, 35]. Acetylation modification regulates various biological effects, such as protein stability, enzymatic activity, subcellular localization, and crosstalk with other post-translational modifications [36, 37]. Studies revealed that sirtuins regulated the levels or activity of metabolic enzymes by deacetylating them. For instance, acetylation suppressed the enzymatic activity of acetyl-CoA synthetase 1 (AceCS1), whereas deacetylation by SIRT1 restored the activity [38]. Acetylation of long-chain acyl-coenzyme A dehydrogenase (LCAD) inhibited its activity and deacetylation of LCAD by SIRT3 led to an increase in its enzymatic activity [39]. As a metabolic enzyme, CPT1A also undergoes acetylation modifications [32, 40]. Our research disclosed that SIRT1 interacted with CPT1A and regulated the protein level of CPT1A by deacetylating CPT1A. co-IP and subsequent experiments in HEK293T cells demonstrated that SIRT1 deacetylated CPT1A at the Lys675 site, which reduced the ubiquitination level of CPT1A and increased the protein stability. Based on relevant studies, lysine acetylation may influence ubiquitination by two mechanisms. One is that acetylation and ubiquitination compete for the same lysine site, in which case acetylation inhibits ubiquitination [41]. Another situation is that acetylation of certain lysine sites reduces the electrostatic repulsion between ubiquitin and protein, which increases the affinity of ubiquitin. In this case, lysine acetylation promotes ubiquitination [42]. In this research, the acetylation of CPT1A may affect its ubiquitination through the second mechanism but the specific mechanism remains to be clarified. Our research disclosed an interesting phenomenon: SIRT1 inhibited the degradation of CPT1A but the reduced SIRT1 did not decrease the protein level of CPT1A in HFD-fed mice. This may be due to the increased mRNA level of CPT1A induced by HFD, which could be a compensatory response to fatty acid stimulation. Of course, it cannot be excluded that HFD promoted the expression of CPT1A through other mechanisms.
Our study revealed a new mechanism of BBR in the treatment of NAFLD. We first confirmed that SIRT1 deacetylated CPT1A at the Lys675 site, which stabilized CPT1A by inhibiting the ubiquitin-dependent degradation. Nonetheless, this research has several shortcomings. The direct target of BBR, the specific mechanism of BBR increasing the expression of SIRT1, and the mechanism of the acetylation of CPT1A influencing its ubiquitination remain to be clarified.
Conclusions
Berberine increased the expression of SIRT1; SIRT1 deacetylated CPT1A at the Lys675 site, which suppressed its ubiquitin-dependent degradation, thereby promoting FAO and alleviating non-alcoholic liver steatosis in mice.
Supplementary Material
{kind=link}
Acknowledgements
We would like to thank Prof. Dongsheng Yao of Jinan University for his support and help in this study.
Contributor Information
Peng Wang, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Ruikai Li, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Yuqi Li, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Siwei Tan, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Jie Jiang, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Huiling Liu, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Xiuqing Wei, Department of Gastroenterology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong, P. R. China.
Supplementary data
Supplementary data are available at Gastroenterology Report online.
Authors’ Contributions
P.W. and X.W. designed the project. P.W., Y.L., R.L., and S.T. conducted the experiments and data analysis. H.L. and J.J. supplied essential reagents. P.W. drafted the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Science and Technology Foundation of Guangzhou, China [No. 201903010099].
Conflict of Interest
None declared.
References
- 1. Goh GB, Kwan C, Lim SY. et al. Perceptions of non-alcoholic fatty liver disease-an Asian community-based study. Gastroenterol Rep (Oxf) 2016;4:131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Le MH, Yeo YH, Li X. et al. 2019 Global NAFLD prevalence: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2022;20:2809–17.e28. [DOI] [PubMed] [Google Scholar]
- 3. Zhou J, Zhou F, Wang W. et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology 2020;71:1851–64. [DOI] [PubMed] [Google Scholar]
- 4. Tillhon M, Guaman Ortiz LM, Lombardi P. et al. Berberine: new perspectives for old remedies. Biochem Pharmacol 2012;84:1260–7. [DOI] [PubMed] [Google Scholar]
- 5. Sun Y, Xia M, Yan H. et al. Berberine attenuates hepatic steatosis and enhances energy expenditure in mice by inducing autophagy and fibroblast growth factor 21. Br J Pharmacol 2018;175:374–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee YS, Kim WS, Kim KH. et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 2006;55:2256–64. [DOI] [PubMed] [Google Scholar]
- 7. Zhu X, Bian H, Wang L. et al. Berberine attenuates nonalcoholic hepatic steatosis through the AMPK-SREBP-1c-SCD1 pathway. Free Radic Biol Med 2019;141:192–204. [DOI] [PubMed] [Google Scholar]
- 8. Nassir F, Ibdah JA.. Sirtuins and nonalcoholic fatty liver disease. World J Gastroenterol 2016;22:10084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pulla VK, Battu MB, Alvala M. et al. Can targeting SIRT-1 to treat type 2 diabetes be a good strategy? A review. Expert Opin Ther Targets 2012;16:819–32. [DOI] [PubMed] [Google Scholar]
- 10. Kwon S, Seok S, Yau P. et al. Obesity and aging diminish sirtuin 1 (SIRT1)-mediated deacetylation of SIRT3, leading to hyperacetylation and decreased activity and stability of SIRT3. J Biol Chem 2017;292:17312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mulligan P, Yang F, Di Stefano L. et al. A SIRT1-LSD1 corepressor complex regulates notch target gene expression and development. Mol Cell 2011;42:689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajamohan SB, Pillai VB, Gupta M. et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol 2009;29:4116–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ponugoti B, Kim DH, Xiao Z. et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem 2010;285:33959–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Purushotham A, Schug TT, Xu Q. et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009;9:327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu J, Zhang Y, Yu Z. et al. Berberine mitigates hepatic insulin resistance by enhancing mitochondrial architecture via the SIRT1/Opa1 signalling pathway. Acta Biochim Biophys Sin (Shanghai) 2022;54:1464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shan MY, Dai Y, Ren XD. et al. Berberine mitigates nonalcoholic hepatic steatosis by downregulating SIRT1-FoxO1-SREBP2 pathway for cholesterol synthesis. J Integr Med 2021;19:545–54. [DOI] [PubMed] [Google Scholar]
- 17. Nagarajan SR, Paul HM, Krycer JR. et al. Lipid and glucose metabolism in hepatocyte cell lines and primary mouse hepatocytes: a comprehensive resource for in vitro studies of hepatic metabolism. Am J Physiol Endocrinol Metab 2019;316:E578–e89. [DOI] [PubMed] [Google Scholar]
- 18. Narita T, Weinert BT, Choudhary C.. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 2019;20:156–74. [DOI] [PubMed] [Google Scholar]
- 19. Xu C, Cai Y, Fan P. et al. Calorie restriction prevents metabolic aging caused by abnormal SIRT1 function in adipose tissues. Diabetes 2015;64:1576–90. [DOI] [PubMed] [Google Scholar]
- 20. Weinert BT, Schölz C, Wagner SA. et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 2013;4:842–51. [DOI] [PubMed] [Google Scholar]
- 21. Tan XS, Ma JY, Feng R. et al. Tissue distribution of berberine and its metabolites after oral administration in rats. PLoS One 2013;8:e77969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serafim TL, Oliveira PJ, Sardao VA. et al. Different concentrations of berberine result in distinct cellular localization patterns and cell cycle effects in a melanoma cell line. Cancer Chemother Pharmacol 2008;61:1007–18. [DOI] [PubMed] [Google Scholar]
- 23. Jin M, Ji X, Stoika R. et al. Synthesis of a novel fluorescent berberine derivative convenient for its subcellular localization study. Bioorgan Chem 2020;101:104021. [DOI] [PubMed] [Google Scholar]
- 24. Turner N, Li JY, Gosby A. et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-Activated protein kinase and improve insulin action. Diabetes 2008;57:1414–8. [DOI] [PubMed] [Google Scholar]
- 25. Zhu X, Yue H, Guo X. et al. The preconditioning of berberine suppresses hydrogen peroxide-induced premature senescence via regulation of sirtuin 1. Oxid Med Cell Longev 2017;2017:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X, Liu XD, Xian YF. et al. Berberine enhances survival and axonal regeneration of motoneurons following spinal root avulsion and re-implantation in rats. Free Radic Biol Med 2019;143:454–70. [DOI] [PubMed] [Google Scholar]
- 27. Ding RB, Bao J, Deng CX.. Emerging roles of SIRT1 in fatty liver diseases. Int J Biol Sci 2017;13:852–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang RH, Li C, Deng CX.. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int J Biol Sci 2010;6:682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang RH, Kim HS, Xiao C. et al. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest 2011;121:4477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pfluger PT, Herranz D, Velasco MS. et al. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA 2008;105:9793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen JC, Horton JD, Hobbs HH.. Human fatty liver disease: old questions and new insights. Science 2011;332:1519–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Softic S, Meyer JG, Wang GX. et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab 2019;30:735–53.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weber M, Mera P, Casas J. et al. Liver CPT1A gene therapy reduces diet-induced hepatic steatosis in mice and highlights potential lipid biomarkers for human NAFLD. Faseb J 2020;34:11816–37. [DOI] [PubMed] [Google Scholar]
- 34. Sun W, Nie T, Li K. et al. Hepatic cpt1a facilitates liver-adipose cross-talk via induction of FGF21 in mice. Diabetes 2022;71:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choudhary C, Weinert BT, Nishida Y. et al. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 2014;15:536–50. [DOI] [PubMed] [Google Scholar]
- 36. Verdin E, Ott M.. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol 2015;16:258–64. [DOI] [PubMed] [Google Scholar]
- 37. Yang XJ, Seto E.. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 2008;31:449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hallows WC, Lee S, Denu JM.. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA 2006;103:10230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu X, Zhu XP, Bai JY. et al. Berberine alleviates nonalcoholic fatty liver induced by a high-fat diet in mice by activating SIRT3. Faseb J 2019;33:7289–300. [DOI] [PubMed] [Google Scholar]
- 40. Schlaepfer IR, Joshi M.. CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology 2020;161:1–14. [DOI] [PubMed] [Google Scholar]
- 41. Guarani V, Deflorian G, Franco CA. et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 2011;473:234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. You Z, Jiang WX, Qin LY. et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat Commun 2019;10:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.