

Abstract
Von Hippel–Lindau (VHL) disease is an autosomal dominant, inherited syndrome with variants in the VHL gene, causing predisposition to multi-organ neoplasms with vessel abnormality. Germline variants in VHL can be detected in 80–90% of patients clinically diagnosed with VHL disease. Here, we summarize the results of genetic tests for 206 Japanese VHL families, and elucidate the molecular mechanisms of VHL disease, especially in variant-negative unsolved cases. Of the 206 families, genetic diagnosis was positive in 175 families (85%), including 134 families (65%) diagnosed by exon sequencing (15 novel variants) and 41 (20%) diagnosed by multiplex ligation-dependent probe amplification (MLPA) (one novel variant). The deleterious variants were significantly enriched in VHL disease Type 1. Interestingly, five synonymous or non-synonymous variants within exon 2 caused exon 2 skipping, which is the first report of exon 2 skipping caused by several missense variants. Whole genome and target deep sequencing analysis were performed for 22 unsolved cases with no variant identified and found three cases with VHL mosaicism (variant allele frequency: 2.5–22%), one with mobile element insertion in the VHL promoter region, and two with a pathogenic variant of BAP1 or SDHB. The variants associated with VHL disease are heterogeneous, and for more accuracy of the genetic diagnosis of VHL disease, comprehensive genome and DNA/RNA analyses are required to detect VHL mosaicism, complicated structure variants and other related gene variants.
Introduction
von Hippel–Lindau (VHL) disease (MIM ID # 193300) takes the form of autosomal dominant inheritance and causes multiple neoplastic or cystic lesions in multiple organs. The central nervous system (CNS) and retinal hemangioblastoma (HGB), pancreatic neuroendocrine tumor (PNET) and cyst, pheochromocytoma of the adrenal grand (PCC), clear cell renal cell carcinoma (ccRCC) and cyst, cystadenoma of epididymis, endolymphatic sac tumor and cystadenoma of uterine broad ligament have been reported as related lesions of VHL disease (1). VHL has a prevalence of ~1 in 36 000 (2). VHL disease is classified into four types according to the presence or absence of major pathological conditions (PCC, RCC, CNS and retinal HGB). First, VHL disease Type 1 (without PCC) and VHL disease Type 2 (with PCC) are classified according to the presence or absence of PCC. Furthermore, VHL disease Type 2 is classified into three types, Type 2A, Type 2B and Type 2C, depending on the presence or absence of RCC, CNS and retinal HGB. Type 2C develops only PCC (1).
VHL disease is caused by a germline pathogenic variant in the tumor suppressor gene VHL, and germline variants in the VHL gene can be detected in 80–90% of patients clinically diagnosed with VHL disease. A study of 945 VHL families (3) found the majority of individuals have a missense variant (52%), followed by frameshift, large deletion (11%), nonsense (11%), splice site (7%) and in-frame deletion/insertion (6%). It is believed that ⁓5% of individuals with a clinical diagnosis of VHL have negative genetic test results from germline deoxyribonucleic acid (DNA) analysis as a case with no variant identified (NVI). These NVI cases are likely to have a mosaic variant or complicated structure variant (SV) of VHL or a germline variant of other responsible genes in VHL-hypoxia-inducible factor (HIF) pathway, such as Elongin C (4), which is a component of the E3 ubiquitin ligase complex and directly binds to VHL protein (pVHL). However, these issues are still questionable, and it is required to find any variants associated with VHL diseases to improve the accuracy of the genetic testing and diagnosis for VHL disease.
The pVHL functions as an E3 ubiqutin ligase complex and controls the degradation of the transcription factor HIF (5,6). VHL-inactivated cells cannot degrade HIFα even under normal oxygen pressure. It is assumed that constitutive and non-physiological expressions of target genes such as vascular endothelial growth factor, platelet-derived growth factor (PDGF) and glucose transporter 1, linked to tumorigenesis, angiogenesis and low-oxygen metabolic pathway (7). Targeting or blocking the VHL-HIF hypoxia pathway and angiogenesis have been attempted as treatment of VHL-related tumors (8–10). Recently the US Food and Drug Administration has approved belzutifan, an oral HIF-2α inhibitor, for treating adults with RCC, CNS HGB or PNET linked to VHL disease. Their use may be limited to VHL patients with positive genetic tests (companion diagnostics), and detection of VHL variants and evaluation or annotation of each variant become more important for treatment strategy of VHL disease.
We have been conducting genetic tests [direct sequencing and multiplex ligation-dependent probe amplification (MLPA)] on patients with clinically diagnosed or suspected VHL disease in Japan for 25 years from 1997. In this report, we summarize VHL variants, which were identified in 206 Japanese families that were diagnosed or highly suspected as VHL disease, and (i) report novel pathogenic variants of VHL and (ii) elucidate responsible variants of VHL and other genes, which were identified in NVI cases by comprehensive genome analysis.
Results
Variant spectrum of VHL in Japanese VHL disease
In this cohort, we collected blood DNAs from 206 families of VHL disease and analyzed VHL exons by the direct sequencing and MLPA. VHL disease Type 1 was 146 families (71%), Type 2 was 56 families (27%) and 4 families (2%) had no clinical information (Table 1). Among them, 175 families were found to have a variant of VHL exons, indicating positive for this genetic testing. The detection rate of VHL variants was 85%. All variants of VHL are listed in Supplementary Material, Table S1, including 16 novel VHL variants (Supplementary Material, Table S2). Among them, 134 families (65%) had a small variant in the exons and exon-intron junctions of VHL, which are 84 missense, 20 nonsenses, 16 frameshifts, nine in-frame indels, three splice-site variants and two duplications (Fig. 1A). MLPA detected 12 large deletions in 41 families, which are spanning FANCD2, BRAK1 (upstream), VHL, IRAK2 and GHRL (downstream) genes at chromosome 3p25.3 (Supplementary Material, Table S3). No other genetic disorders, such as Lynch syndrome (MLH1, 3p22.2), were found to coexist with VHL disease. There was no contiguous gene deletion syndrome in our clinical information.
Table 1.
Summary of the genetic test for VHL disease
| Total VHL families | 206 | |||
|---|---|---|---|---|
| Subtype of VHL disease | ||||
| 1 | 146 | |||
| 2A | 29 | |||
| 2B | 27 | |||
| 2C | 0 | |||
| NA | 4 | |||
| Results of genetic test | ||||
| Direct sequencing | 134 | |||
| MLPA | 41 | |||
| Negative (NVI) | 31 | |||
| Mutation type of VHL | ||||
| Missense | 84 | |||
| Nonsense | 20 | |||
| Large deletion | 41 | |||
| Frameshift | 16 | |||
| In-frame indel | 9 | |||
| Splice site | 3 | |||
| Others | 2 | |||
| 175 | ||||
Figure 1.
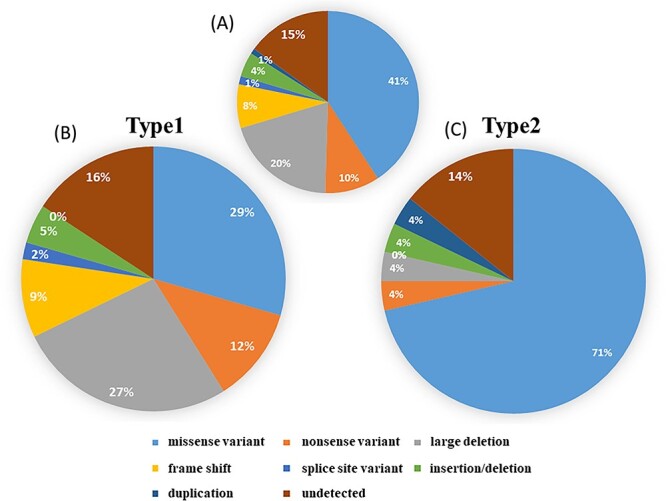
Clinical types and VHL variants. (A) All families (n = 206), regardless of type. (B) Families whose described phenotype does not include PCC (Type 1, n = 146). (C) Families whose described phenotype includes PCC (Type 2, n = 56).
Clinical types and VHL variants
Of the 146 families diagnosed as Type 1, point variants were found in 60 families, of which 43 had missense variants and 17 had nonsense variants. This was followed by 39 large deletions, 14 frameshift variants, seven in-frame indels, three splice-site variants and 23 families were undetected (Fig. 1B). Totally, 50% (73/146) of Type1 cases had a deleterious variant of VHL. On the other hands, of the 56 families diagnosed with Type 2, missense variants were found in 40 families (71%) (Fig. 1C). Large deletions (27%) and truncating variants (23%) including nonsense variants (12%), frameshift variants (9%) and splice-site variants (2%) were significantly enriched in Type 1 phenotype, whereas missense variants (71%) were enriched in Type 2. The amino-acid substitution at codon 167 (p.Arg167Trp or p.Arg167Gln) were particularly common in Type 2 (Fig. 2A).
Figure 2.

Missense variants and exon 2 skipping phenomenon. (A) Distribution of missense variants for Type1 and Type2 VHL. Codon 167 in Exon 3 is known as a one of the hotspots of VHL variants. The three coding exons are indicated by boxes. (B) RT-PCR analysis showed the up-regulation of exon 2 skipping splice variants (E1-E3) in one synonymous variant (c.342T>C) and four missense variants (c.351G>T, c.362A>G, c.370A>C, c.463G>T).
Table 1 shows variants that have not been reported in previous reports and ClinVar. The variants in families with a confirmed clinical diagnosis of VHL disease were determined to be pathogenic variants. Four families who did not develop HGB in CNS or retina were not clinically diagnosed with VHL disease. But our genetic testing demonstrated that these four families had VHL variants (p.Ser72fs, p.Arg108Cys + p.Tyr112His, p.Thr124Pro, p.Arg167dup), respectively. The p.Ser72fs is obviously pathogenic, and the p.Tyr112His is known to be associated with VHL Type 2A (without RCC) (11), but RCC developed in this family. The p.Arg108Cys may have caused RCC. Four individuals in one family had c.370A>C (p.Thr124Pro), who developed PCC, RCC and PNET, without HGB. Reverse transcriptase polymerase chain reaction (RT-PCR) analysis showed exon 2 skipping in this family, described subsequently. Codon 167 is known as a one of the hotspots of VHL variants, and this study also found 22 missense variants in codon 167 (26% of missense variants). These new VHL variants were annotated according to ACMG guidelines. In our study, variant of unknown significance (VUS) of VHL gene did not exist. One family with large deletions of BRK1 and VHL (exons 1 and 3) detected by MLPA was diagnosed as Type 1 (Supplementary Material, Table S1). Deletion of all or part of the VHL gene as well as the near-by gene BRK1 leads to CNS HGB, retinal HGB with a low risk of RCC, which are sometimes called VHL Type 1B.
Exon 2 variants affect exon 2 skipping
One family (Family ID 168) with c.342T>C had no amino-acid changes (p.Gly114Gly). To examine whether this synonymous variant affects splicing and gene expression, we performed RT-PCR of ribonucleic acid (RNAs) from the patient leucocytes. Interestingly, RT-PCR revealed that expression of exon 2 skipping splice variants (E1-E3) was up-regulated, relative to expression of VHL full-length (E1-E2-E3) (Fig. 2B). Previously, there were some reports of synonymous variant without amino-acid change (c.414A>G, p.Pro138Pro), which were related with up-regulation of exon 2 skipping splice variants (E1-E3) (12,13). We also investigated for exon 2 skipping in blood RNAs from six individuals with a missense variant in exon 2 as well as five controls with no variants of VHL. Figure 2B also shows the up-regulation of exon 2 skipping splice variants (E1-E3) in four missense variants [c.351G>T (p.Trp117Cys), c.362A>G (p.Asp121Gly), c.370A>C (p.Thr124Pro), c.463G>T (p.Val155Leu)]. On the other hand, c.344A>G (p.His115Arg) and c.358A>G (p.Arg120Gly) did not up-regulate the exon 2 skipping splice variant (E1-E3). In addition, six missense variants in the exon 1 and exon 3 regions did not up-regulate the exon 2 skipping splice variant (E1-E3). These results indicating a possibility that many variants of VHL exon 2 can be related with splicing change. Of the four families with exon 2 skipping missense variants, two families were diagnosed with Type 2B. The remaining two families were diagnosed as Type 1. In our study, the diagnosis of VHL subtype is imprecise because of lack of follow-up. As shown in the results of the MLPA method (Supplementary Material, Table S3), exon 2 deletion (DNA level) predicts Type 1, whereas exon 2 skipping (RNA level) is thought to increase the risk of developing pheochromocytoma (Type 2).
Whole genome sequencing analysis of VHL disease
Among 206 VHL families, 31 families (15%) were negative for both the direct sequencing and MLPA. To identify novel variants of VHL and other genes, we performed whole genome short-read sequencing for 22 cases with NVI, whose DNAs were available (Table 2). The standard analysis pipeline called germline SNVs and indels in coding and non-coding regions and SVs. As a results, Case04 was found to have a splicing variant of BRCA1 Associated Protein 1 (BAP1) (c.67 + 2T>C), which is likely to affect splicing and pathogenic. This patient developed mesothelioma and gastric cancer as well as RCC and CNS HGB, but did not have retinal HGB, indicating that Case04 should have been diagnosed as BAP1 tumor predisposition syndrome (Table 2). Case11 was found to have a 10-bp deletion of Succinate dehydrogenase subunit B (SDHB) which affected the exon-intron junction of SHDB and obviously pathogenic. The father of this case developed PCC and RCC, whereas the case did not have PCC, nor HGB in CNS or retina (Table 2). This case should have been diagnosed as hereditary pheochromocytoma/paraganglioma (PPGL). We also detected several VUSs of other RCC-related genes, TSC1, TSC2, FLCN, SDHB, SDHA and SDHD, which were validated in the target deep sequencing later (Supplementary Material, Table S4). However, these family members with VUS did not show any other phenotype related with tuberous sclerosis, Birt–Hogg–Dube (BHD) syndrome, nor PPGL and we cannot conclude that they could be responsible for their VHL-related phenotypes.
Table 2.
Summary of variants detected in 22 unsolved cases with NVI
| Phenotype | Responsible variant | VUS and others | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID | Patient ID | Case ID | VHL type | PCC | CNS HGB | Retina HGB | RCC (histology) | Other features | Family history | ||
| 12 | 32 | 1 | 1 | − | + | − | + (NA) | PNET, bilateral adrenal tumors | − | − | − |
| 26 | 85 | 2 | 1 | − | + | − | + (NA) | renal cyst, lung meningioma | mother:RCC | − | − |
| 48 | 137 | 3 | 1 | − | + | − | + (NA) | colon cancer | − | − | − |
| 85 | 237 | 4 | 1 | − | + | − | + (ccRCC) | gastric cancer, mesothelioma | father, sisrer, niece:gastric cancer | BAP1 splicing c.67 + 2 T > C | − |
| 87 | 242 | 5 | 1 | − | + | − | + (NA) | − | − | − | TSC1 p.T778S |
| B-10 | 269 | 6 | 1 | − | + | − | + (ccRCC) | pancreatic lesion | − | VHL p.Leu169Pro mosaicism:AF8% | FLCN p.G299E |
| 107 | 308 | 7 | 1 | − | + | − | + (NA) | − | − | − | − |
| 122 | 346 | 8 | 1 | − | + | − | + (ccRCC) | renal cyst | daughter:CNS HGB | − | TSC1 SV |
| 126 | 355 | 9 | 1 | − | − | + | + (NA) | renal cyst | mother:lung cancer, uncle:gastric cancer | − | TSC2 p.S802G, SDHD p.V72I |
| 137 | 390 | 10 | 1 | − | + | − | + (NA) | − | − | − | − |
| 140 | 399 | 11 | 2B | − | − | − | + (NA) | renal cyst | father:PCC, RCC | SDHB: exon5:c.424_427del:p.D142fs | − |
| 154 | 437 | 12 | 1 | − | + | − | + (ccRCC) | − | − | − | − |
| 155 | 439 | 13 | 2B | + | + | − | + (ccRCC) | renal cyst | − | − | − |
| 3 | 3 | 14 | 1 | − | + | + | − | − | − | − | − |
| 10 | 27 | 15 | 2B | + | + | + | + (NA) | pancreatic cyst | − | VHL p.Arg161X mosaicism: AF 21% | − |
| 16 | 53 | 16 | 1 | − | + | − | − | renal cyst, erythrocytosis, multiple liver cysts | − | − | − |
| 20 | 70 | 17 | 1 | − | + | − | − | renal cyst | − | − | − |
| 32 | 104 | 18 | 2A | + | + | − | − | renal cyst | − | VHL p.Arg167Trp mosaicism: AF 2.5% | − |
| 60 | 174 | 19 | 1 | − | + | − | − | renal cyst | − | − | − |
| 90 | 248 | 20 | 1 | − | + | + | − | epididymal cystadenoma | − | − | SDHB p.S163F |
| 99 | 279 | 21 | 2A | + | − | − | − | paraganglioma, pancreatic lesion, erythrocytosis | − | − | − |
| 152 | 432 | 22 | 1 | − | + | − | − | − | mother:CNS HGB, Retina HGB, Renal cyst | ME insertion in VHL intron 1 | SDHA p.T338I, MET p.N1131I |
Next, to search for novel mutated genes related with VHL phenotype, we extracted rare deleterious variants (nonsense, frameshift or splice site) from variants of the 22 NVI cases in whole genome level (371 variants, 310 genes, Supplementary Material, Table S5). All of genes were deleteriously mutated as a singleton among the 22 NVI cases. The pathway or gene ontology (GO) analysis on these genes with deleterious variants found that 10 genes related with VHL gene are mutated (Supplementary Material, Fig. S1) such as PDGFB and SLC3A2, which are involved with angiogenesis and may be associated with VHL phenotype.
SVs and mobile elements insertion
In this cohort, we performed MLPA, and for these 22 NVI cases we did not have any evidence of copy number changes of VHL gene. However, they possibly may have other complicated SVs of VHL gene or SVs of other cancer predisposing genes. We attempted to call SVs from whole genome sequencing (WGS) data by combining several software and compared the frequency of each SVs in 22 VHL cases and BBJ 3258 WGS control data. The 22 VHL data set (VHL-SV) yielded a total number of 26 815 SVs, including 9179 deletions (DELs), 3063 duplications (DUPs), 14 344 insertions (INSs) and 229 inversions (INVs). In the BBJ data (BBJ-SV), a total number of 133 841 SVs, including 55 284 DELs, 15 222 DUPs, 61 750 INSs, and 1585 INVs, were detected (Kosugi et al., manuscript in submission). After filtering by IGV analysis and comparison of the SV frequency between 22 unsolved VHL and BBJ controls, Case8 was found to have a large insertion in TSC1 exon 7, as shown in its Integrative Genomics Viewer (IGV) picture (Supplementary Material, Fig. S2). However, PCR did not confirm this SV, which may be a complicated SV.
Recently, several reports suggested that mobile elements (ME) insertion such as Alu or LINE affected a number of tumor suppressor gene or cancer predisposing gene, although its detection was difficult from short-read WGS. We used a new tool MEGAnE, which precisely detect and genotypes ME variants from short-read WGS data (14), and searched for ME insertions in VHL and other cancer predisposition genes. As a result, in Case22 we found a ME insertion in intron 1 of VHL gene and long-range PCR validated a 4 kb insertion in this case (Fig. 3). Nanopore long-read sequencing analysis validated this ME insertion, which was ⁓4 kb and included 3 kb SVA transposon (Supplementary Material, Fig. S3). This transposon was almost identical to the transposon located in the intron of PHKA1 gene at chromosome X (Supplementary Material, Fig. S4). The affected VHL intron [chr3: 10142630-10 142 660 (GRCh38)] has several regulatory elements annotated by ENCODE around this region and this insertion can affect the transcriptional activity of VHL gene (Fig. 3). Unfortunately, RNAs of this case is not available and we could not validate VHL expression.
Figure 3.

(A) Whole genome analysis found one ME insertion in the VHL promoter region and long-PCR confirmed a 4 kb insertion. (B) Long-read Nanopore sequencing analysis targeting VHL gene body also confirmed this ME insertion (red arrow).
Target deep sequencing of VHL coding regions for mosaic variants
To confirm variants detected by WGS analysis and to detect mosaic variants, we performed amplicon deep sequencing of all coding exons of VHL gene and RCC-related genes. This method covered 99.9% of all target regions and median depth of the target regions were 3577x. In these unsolved VHL cases, we confirmed heterozygous variants of RCC-related genes (Table 2), and in addition to two deleterious variants BAP1 and SDHB, 7 VUSs of TSC1/2, SDHA, SDHB, SDHD, FLCN and MET were confirmed. Direct sequencing and WGS are not sensitive to detect mosaic variants with low-variant allele frequency (VAF). For mosaic variant with low-VAF, we compared the deep target sequencing data of the 22 NVI VHL cases, 12 non-VHL cases and 5996 of non-cancer controls and extracted low-VAF variants with 1% and more VAF. The peaks of VAF distribution of the controls which mean the background error were 0–1.87% in almost all of the target regions, and Grubbs’s test detected three mosatic variants of VHL gene in three cases as outliers (Fig. 4). Case06 showed p.Leu169Pro (likely pathogenic annotated by ClinVar) of VHL with 8.4% VAF, case15 showed pArg161* (pathogenic) of VHL with 21.9% VAF, and case18 showed pArg167Trp (pathogenic annotated by ClinVar) of VHL with 2.5% VAF (Table 2). We confirmed these low-VAF variants in case06 and case15 by direct Sanger sequencing (Supplementary Material, Fig. S5).
Figure 4.

Mosaic variants in three NVI cases detected by deep sequencing of VHL cases and 6000 controls. Histogram depicts the frequency of chr3:10191506C>T (A), chr3:10191513T>C (B) and chr3:10191488C>T (C) because of the basic error. Arrow shows the VAF of each mosaic variant, and the frequency of the VAF in the control population is 0–1.61% (chr3:C10191488T), 0–1.87% (chr3:T10191513C) and 0–1.54% (chr3: C10191506T). P-value was measured by Grubbs’s test.
Discussion
The VHL gene is composed of three exons, from which mRNA with a total length of ⁓4.5 kb is transcribed and composed of three exons (15). The protein translation region of mRNA is 639 bases. Translation is initiated from two methionine sites, codons 1 and 54 and VHL proteins of 213 and 160 amino acids (pVHL213, pVHL160) are produced, both of which have tumor-suppressing functions (16,17). Another VHL protein of 172 amino acids (pVHL172) is also known that lacks exon 2 because of alternative splicing but has no function as a tumor suppressor gene (18). In our study, we identified 134 intragenic variants, all of which were located between codons 55 and 198. In our data and previous reports (19,20), no VHL disease patient had a pathogenic variant between 1st methionine and 2nd methionine (Codon 1-54, Fig. 2A). Even if there is a genetic variant between them, the occurrence of VHL disease might be prevented if the protein initiated from second methionine is functioning normally. Interestingly, in this study several missense variants (including one synonymous variant) in the exon 2 region were found to up-regulate the VHL exon 2 skipping splice variant (E1-E3) expression relative to VHL full-length (E1-E2-E3) expression in mRNA level. These missense variants in exon 2 are likely to affect the formation of exon 2 splicing, but further verification and experimental evidence are required.
Regarding the genotype–phenotype correlation of VHL disease, Type 1 is characterized by large deletions, and truncating variants, associated with VHL instability and high HIF activity (21–23). In Type 2, regardless of the subtype, missense variants, which retain partial functionality of pVHL, are predominantly found (21,23). In our study, variants and their frequency of each type of VHL disease were largely similar to those of previous reports (21–23), but there were some unexplained results. PCC occurred in four families with large deletions and nonsense variants associated with Type 1. Contrary to PCC did not develop in 5 of 22 families with a missense variant (codon 167) associated with Type 2. A possibility of developing PCC in the future cannot be ruled out because we did not conduct follow-up surveys for these five families.
Furthermore, our data show that ⁓15% of families are undetectable by VHL genetic testing (NVI cases). We here demonstrated that one of the NVI factors is mosaicism. Several studies (24,25) have identified mosaic variants in ⁓5–8.5% of patients with clinically diagnosed or suspected VHL disease. Although it is widely accepted that mosaicism leads to milder phenotypes, we could not observe milder phenotypes. Because VHL disease is a neoplastic disease, it may be difficult to detect mosaicism from clinical manifestations. In our data, whole genome sequencing and deep-targeted sequencing revealed that the NVI cases include three with mosaicism for VHL pathogenic variants, one with ME in the VHL promoter region, and two who genetically belong to other syndromes; BAP1 tumor predisposition syndrome and PPGL.
Especially when a case has no HGB and developed PCC or RCC, it is necessary to differentiate not only VHL disease but also other hereditary renal cancers. Hereditary renal cancers include VHL disease, BHD syndrome, hereditary papillary renal carcinoma type 1, hereditary leiomyomatosis and renal cell cancer, BAP1 tumor predisposition syndrome, succinate dehydrogenase deficient RCC, Cowden syndrome and tuberous sclerosis. Because VHL disease develops only clear cell RCC, clinical information regarding the pathological diagnosis of renal tumors is useful for differential diagnosis of hereditary renal cancers. In our data, of the 22 NVI cases, 14 cases developed RCC. Of these, five cases had clinical information regarding the pathological diagnosis of renal tumors, all of which were clear cell RCC. The remaining nine cases had no information. Given that VHL disease has a high incidence of HGB, the presence of HGB is considered to be essential for clinical diagnostic criteria for VHL disease in Japan as well. If clinical diagnostic criteria are not met, genetic testing is performed for definitive diagnosis. The finding of BAP1 or SDHB variants in two cases indicates that clinical diagnosis still has room for improvement. We suggest that a differential diagnosis of hereditary renal cancer is necessary when RCC is present, and that obtaining a detailed family history is important. At present, genetic testing and immunohistochemistry using tumor specimens may be important tools to complement the diagnosis of VHL mosaic cases or other hereditary tumor cases (26). In any case, establishment of mosaicism diagnosis is important for more accurate genetic diagnosis. At present deep target (panel) sequencing for several genes is common for the genetic test for many types of hereditary diseases and mosaic variants can be called from these genetic testing accurately, and we should use these panel sequencing for VHL-suspected cases as well as other hereditary disease.
Finally, even after whole genome and target sequencing of 22 NVI cases, still 16 cases with VHL disease were not found to have a responsible gene/variant (Table 2). We performed haplotype analysis by using variants information from WGS and examined shared haplotypes, especially at 3p25 among the NVI cases. But we did not detect any shared haplotypes of these Japanese VHL patients with NVI (data not shown). In addition, a cryptic exon called E1’ was recently found in the intron region between exon 1 and exon 2, and several variants in this region were reported to cause polycythemia or VHL disease (28,29). We performed direct sequencing of the E1’ region, but no variants were found. Hence, any common unknown variants of VHL are not responsible for these Japanese cases and rare variants of other genes might be related with this VHL phenotype.
In summary, (1) we show VHL variants data of Japanese VHL disease and (2) report novel pathogenic variants. (3) Several missense variants (including synonymous one) in exon 2 of VHL are likely to cause exon 2 skipping. (4) Genetic test-negative cases included somatic mosaicism, structural abnormalities in the VHL promoter region, and other hereditary tumors. On the basis of these results, it is important to establish new complementary diagnostic methods, such as genetic testing and immunohistochemistry using tumor samples, and RNA sequencing using blood samples. In the future, it is expected that therapeutics on the basis of synthetic lethality associated with VHL gene variants will be approved, although drug use may be indicated for VHL pathogenic variant-positive patients. Therefore, there is an urgent need to clarify the pathogenesis of VHL disease patients whose genetic cause has not been identified.
Materials and Methods
Sample collection and DNA extraction
Between 1997 and 2022, total 532 blood samples were transferred from 114 hospitals in Japan to Kochi University for the genetic testing (direct sequencing or MLPA). Ultimately, 206 families (443 people) were clinically diagnosed or suspected as VHL diseases. We provide clinical information of 299 patients (Supplementary Material, Table S2). The remaining 144 people were ruled out for VHL disease by the genetic testing. Department of Urology, Kochi University is the core laboratory. The diagnostic criteria for isolated cases of VHL disease are the existence of two HGBs (CNS or retina) or a HGB coupled with a visceral manifestation. In familial cases, the presence of only one manifestation is enough for the diagnosis (1). By type of VHL disease, Type 1 was 146 families (71%), Type 2 was 56 families (27%) and four families (2%) had no clinical information. DNAs was extracted from whole blood by outsourcing to SRL, Inc. (Tokyo, Japan). This study was approved by the Ethics Committee in Kochi University and RIKEN. Informed consent was obtained from patients or legal guardians.
Direct sequencing
Exons 1–3 and flanking intronic sequences of the VHL gene were PCR amplified using AmpliTaq Gold DNA Polymerase (Themo Fisher scientific). Briefly, 100 ng of genomic DNA, were amplified using 200 μm of each deoxyribonucleotide triphosphate, 0.5 μm of forward and reverse primers (primers available upon request) and 0.5 U of Taq DNA polymerase, in a final volume of 50 μL. Cycling conditions were as follows: initial denaturation at 95°C for 10 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 60 s and extension at 72°C for 60 s and a final extension at 72°C for 9 min. PCR products were purified with FastGene Dye Terminator Removal kit (NIPPON Genetics Co., Ltd, Tokyo, Japan) and sequenced with Big Dye Terminator v1.1 Cycle Sequencing Kit according to manufacturer’s instructions (Applied Biosystems, Foster City, California, USA). All sequences were analyzed in forward and reverse directions on an ABI3130 DNA analyzer. Primer sequences were as follows: VHL Exon 1 (5′-TGGTCTGGATCGCGGAGGGAAT-3′ and 5′-GACCGTGCTATCGTCCCTGC-3′), exon 2 (5′-GTGGCTCTTTAACAACCTTTGC-3′ and 5′-CCTGTACTTACCACAACAACCTTATC-3′), Exon 3 (5′-TTCCTTGTACTGAGACCCTAGT-3′ and 5′-AGCTGAGATGAAACAGTGTAAGT-3′).
Multiplex ligation–dependent probe amplification
MLPA analysis to detect copy number changes in the VHL gene was carried out using the SALSA P016-C2VHL probe kit (MRC-Holland, Amsterdam, Netherlands). The kit contains nine probes to the VHL gene (four in exon 1, three in exon 2 and two in exon 3), an additional eight probes to six near-by genes (CNTN6, FANCD2, BRK1, IRAK2, GHRL and MLH1) on 3p22.2–26.3 and two probes on 3p, which are further telomeric or centromeric from VHL, and 12 reference probes detecting sequences on other chromosomes. Briefly, 50 ng DNA was denatured at 98°C for 5 min, the MLPA probe cocktail was added to a total volume of 8 μL and allowed to hybridize for 16 h at 60°C. Following addition of Ligase-65 and ligation at 54°C for 15 min, the ligase was inactivated at 98°C for 5 min. PCR primers, deoxynucleotide triphosphates and polymerase mix were then added and PCR was carried out for 33 cycles of (95°C for 30 s, 60°C for 30 s and 72°C for 60 s). Products were then analyzed using an ABI PRISM 3130 Genetic Analyzer with GeneScan 500 LIZ dye size standard. Data was generated using GeneMapper software and relative probe signals were calculated by dividing each measured peak height by the sum of all peak heights in that sample and then normalizing the result to that obtained on control DNA samples. The possibility of copy number changes for control probes in aneuploid tumors, differences in reaction efficiency between samples and the presence of relatively small but variable amounts of normal cells in the tumor samples complicated the analysis, and accordingly, each sample was compared with multiple controls and those reproducibly giving the best fit were used for analysis.
Reverse transcriptase polymerase chain reaction
Total RNA was extracted from blood using the Direct-zol RNA MiniPrep Kit (ZYMO TESEARCH, CA, USA) according to the manufacturer’s instructions, treated with DNase I (Roche, Basel, Switzerland), and reverse transcribed to single-stranded cDNA using Random Hexamers and Superscript reverse transcriptase IV (Invitrogen, Carlsbad, CA, USA). PCR reactions were then performed using the single-stranded cDNA as the PCR template. The primer sequences used were: GAPDH (5′-CGGATTTGGTCGTATTGG-3′, 5′-TCCTGGAAGATGGTGATG-3′) and VHL (5′- GCGTCGTGCTGCCCGTATG-3′, 5′-TTCTGCACATTTGGGTGGTCTT-3′). The design of the VHL primers was on the basis of the article by Liu et al. (12). The PCR conditions were as follows: initial denaturation at 95°C for 10 min, followed by numerous cycles of denaturation at 95°C for 60 s, annealing at 58°C for 60 s, and elongation at 72°C for 60 s on a C1000 Thermal Cycler (Bio-RAD, CA, USA) (30 cycles for GAPDH, 30 cycles for VHL).
Whole genome sequencing and variant call
The libraries were prepared using the TruSeq Nano DNA Library Prep Kit (Illumina) following the manufacturer’s protocol from DNAs of 22 NVI cases. Paired-end sequencing of 150-bp reads was performed using NovaSeq5000 (Illumina). The median sequencing depth is ×30. Sequence reads were mapped to the human reference genome GRCh37 using BWA-0.7.8, and the PCR duplicates were removed using the Picard tool. Variant call was performed by GATK HaplotyperCaller (ver. 3.7-0). For SV detection, we used a computational tool (MOPline) that incorporates multiple SV detection algorithms (Kosugi et al., manuscript in submission). MOPline iteratively merges optimized overlapping calls from multiple algorithms in each SV type and size-range to increase precision and genotypes SV sites including reference alleles to increase recall using short-read WGS data. Overlap calls were selected using seven existing algorithms (CNVnator, GRIDSS, inGAP-sv, Manta, MATCHCLIP, MELT and Wham). SVs were detected using MOPline from the VHL 22 WGS dataset and the BioBank Japan (BBJ) high coverage WGS dataset, respectively. BBJ WGS datasets were obtained from 3258 individuals with any of seven different diseases, that were enrolled in the BBJ Project. The VHL data set (VHL-SV) yielded a total number of 26 815 SVs, and in the BBJ data (BBJ-SV), a total number of 133 841 SVs were detected (Kosugi et al., manuscript in submission). The median number of SVs per individual for VHL-SV and BBJ-SV were 9174 and 16 122, respectively. The low number of SVs per individual for VHL is because of the small sample size of the VHL WGS data because MOPline can effectively recover ‘Missing Calls’ (SVs missed in the selection step of overlap calls) from WGS data sets of with large sample sizes. For ME insertion detection, we used a new tool MEGAnE, which precisely detect and genotypes ME variants from short-read WGS data (14).
GO analysis
To identify the relationship between the variant genes found in each patient and the VHL-related genes, the variant genes found in each patient were exported to ‘IPA [http://www.ingenuity.com]’ (Qiagen) for Ingenuity Pathway Analysis (IPA) built path explorer analysis and pathdesigner. Genes with variants found are shown in black, VHL-related genes in red, and VHL-related signals in yellow highlight.
Long-read sequencing for SVs of VHL gene
To characterize the SV and TE insertion of the VHL cases, we designed guide RNAs targeting VHL gene body and after CRISPR/Cas cutting and the adaptor ligation of DNA by using Cas9 Sequencing Kit (cat# SQK-CS9109, we sequenced VHL regions by MinION system of Oxford Nanopore technologies. Base call was performed by Guppy-6.0.6 with high accuracy condition and long-read sequences were aligned to GRCh38 by minimap2-2.24.
Target deep sequencing of VHL gene and RCC-related genes
We selected 14 RCC-related genes (BAP1, CDC73, FH, FLCN, MET, MITF, PTEN, SDHA, SDHB, SDHC, SDHD, TSC1, TSC2 and VHL), and sequenced all the coding regions with the addition of 2-bp flanking intronic sequences. A multiplex PCR-based targeted sequencing method was employed to sequence the target region, using an Illumina HiSeq2500 instrument with 125 bp ×2 paired-end reads. The detail method was described by Sekine et al. (27). Sequence reads generated for each individual were aligned to the human reference sequence (hg19) using the Burrows-Wheeler Aligner (ver. 0.7.17). As a quality control, individuals whose target region covered ˃98% with ˃20 sequence reads were selected for further analysis. We used HaplotypeCaller and UnifiedGenotyper of GATK (ver. 3.7-0) to separately call the variants for each individual. Genotypes for all individuals were determined for each variant on the basis of the sequencing read ratios of the reference and alternative alleles.
Mosaic variant call
SNV/Indel variants were called using VarScan2 germline mode (mpileup2cns) with parameters require reporting only variant positions (--variants), at least 20× coverage (--min-coverage 20) and 2%~ alternative allele frequency (--min-var-freq 0.02).Variants found in the 12 non-VHL cases were filtered out to exclude false positive and 44 variants found in the only 22 NVI VHL cases were picked up as candidates of mosaic variant. These SNVs in exon were annotated with ANNOVAR. To validate lower frequency SNV, variants were called at 1% cutoff with VarScan2 (--min-var-freq 0.01) and 245 candidates were also selected with same filtering. Then, VAF of the 250 candidates under 40% frequency, which are unlikely germline variants, were compared with 5996 samples (27) as controls to further exclude false positive. Five candidates which show the highest VAF compared with the 5996 controls were selected and Grubbs’s test was performed to detect outliers and mark candidates as mosaic variant or not.
Supplementary Material
Acknowledgements
We thank Research Instrument and Radio-isotope Research, Division of Biological Research, Science Research Center, Kochi University for the use of research instruments and technical staff in RIKEN IMS for their technical assistances. And we also acknowledge all of the VHL patients and their families who participated in this study. This study was partially supported by The Kidney Foundation, Japan and RIKEN internal fund.
Contributor Information
Kenji Tamura, Department of Urology, Kochi Medical School, Kochi University, Nankoku, Japan.
Yuki Kanazashi, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Chiaki Kawada, Department of Urology, Kochi Medical School, Kochi University, Nankoku, Japan.
Yuya Sekine, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan; Department of Urology, Akita University Graduate School of Medicine, Akita 010-8543, Japan.
Kazuhiro Maejima, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Shingo Ashida, Department of Urology, Kochi Medical School, Kochi University, Nankoku, Japan.
Takashi Karashima, Department of Urology, Kochi Medical School, Kochi University, Nankoku, Japan.
Shohei Kojima, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Nickolas F Parrish, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Shunichi Kosugi, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Chikashi Terao, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Shota Sasagawa, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Masashi Fujita, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Todd A Johnson, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Yukihide Momozawa, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Keiji Inoue, Department of Urology, Kochi Medical School, Kochi University, Nankoku, Japan.
Taro Shuin, Department of Urology, Kochi Medical School, Kochi University, Nankoku, Japan.
Hidewaki Nakagawa, RIKEN Center for Integrative Medical Sciences, Yokohama, Japan.
Conflict of Interest statement
None declared.
Authors’ Contribution
Conceptualization: T.S., H.N.; data curation: K.T., Y.K., H.N.; formal analysis: K.T., Y.K., C.K., Y.S., K.M, S.K., N.P., S.K., C.T., S.S., M.F., T.A.J, Y.M., H.N.; funding acquisition: T.S., H.N.; project administration: K.T., C.K., S.A.,T.K.,T.S.; investigation: K.T., T.S., H.N.; resources: K.T., C.K., S.A., T.K., K.I., T.S.; writing: K.T., Y.K., C.K., H.N.
References
- 1. Lonser, R.R., Glenn, G.M., Walther, M., Chew, E.Y., Libutti, S.K., Linehan, W.M. and Oldfield, E.H. (2003) von Hippel-Lindau disease. Lancet, 361, 2059–2067. [DOI] [PubMed] [Google Scholar]
- 2. Maher, E.R., Iselius, L., Yates, J.R., Littler, M., Benjamin, C., Harris, R., Sampson, J., Williams, A., Ferguson-Smith, M.A. and Morton, N. (1991) Von Hippel-Lindau disease: a genetic study. J. Med. Genet., 28, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nordstrom-O'Brien, M., van der Luijt, R.B., van Rooijen, E., van den Ouweland, A.M., Majoor-Krakauer, D.F., Lolkema, M.P., van Brussel, A., Voest, E.E. and Giles, R.H. (2010) Genetic analysis of von Hippel-Lindau disease. Hum. Mutat., 31, 521–537. [DOI] [PubMed] [Google Scholar]
- 4. Andreou, A., Yngvadottir, B., Bassaganyas, L., Clark, G., Martin, E., Whitworth, J., Cornish, A.J., Houlston, R.S., Rich, P., Egan, C. et al. (2022) Elongin C (ELOC/TCEB1)-associated von Hippel-Lindau disease. Hum. Mol. Genet., 31, 2728–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J.M., Lane, W.S. and Kaelin, W.G., Jr. (2001) HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science, 292, 464–468. [DOI] [PubMed] [Google Scholar]
- 6. Jaakkola, P., Mole, D.R., Tian, Y.M., Wilson, M.I., Gielbert, J., Gaskell, S.J., von Kriegsheim, A., Hebestreit, H.F., Mukherji, M., Schofield, C.J. et al. (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2 regulated prolyl hydroxylation. Science, 292, 468–472. [DOI] [PubMed] [Google Scholar]
- 7. Pouysségur, J., Dayan, F. and Mazure, N.M. (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature, 441, 437–443. [DOI] [PubMed] [Google Scholar]
- 8. Wallace, E.M., Rizzi, J.P., Han, G., When, P.M., Cao, Z., Du, X., Cheng, T., Czerwinski, R.M., Dixon, D.D., Goggin, B.S. et al. (2016) A small-molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res., 76, 5491–5500. [DOI] [PubMed] [Google Scholar]
- 9. Cho, H., Du, X., Rizzi, J.P., Liberzon, E., Chakraborty, A.A., Gao, W., Carvo, I., Signoretti, S., Bruick, R.K., Josey, J.A. et al. (2016) On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature, 539, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen, W., Hill, H., Christie, A., Kim, M.S., Holloman, E., Pavia-Jimenez, A., Homayoun, F., Ma, Y., Patel, N., Yell, P. et al. (2016) Targeting renal cell carcinoma with a HIF-2 antagonist. Nature, 539, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bradley, J.F., Collins, D.L., Schimke, R.N., Parrott, H.N. and Rothberg, P.G. (1999) Two distinct phenotypes caused by two different missense mutations in the same codon of the VHL gene. Am. J. Med. Genet., 87, 163–167. [DOI] [PubMed] [Google Scholar]
- 12. Liu, F., Calhoun, B., Alam, M.S., Sun, M., Wang, X., Zhang, C., Haldar, K. and Lu, X. (2020) Case report: a synonymous VHL mutation (c.414A > G, p.Pro138Pro) causes pathogenic familial hemangioblastoma through dysregulated splicing. BMC Med Genet., 21, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flores, S.K., Cheng, Z., Jasper, A.M., Natori, K., Okamoto, T., Tanabe, A., Gotoh, K., Shibata, H., Sakurai, A., Nakai, T. et al. (2019) A synonymous VHL variant in exon 2 confers susceptibility to familial pheochromocytoma and von Hippel-Lindau disease. J. Clin. Endocrinol. Metab., 104, 3826–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kojima, S., Koyama, S., Ka, M., Saito, Y., Parrish, E.H., Endo, M., Takata, S., Mizukoshi, M., Hikino, K., Takeda, A. et al. (2022) Mobile elements in human population-specific genome and phenotype divergence. bioRxiv. 10.1101/2022.03.25.485726. [DOI] [Google Scholar]
- 15. Renbaum, P., Duh, F.M., Latif, F., Zbar, B., Lerman, M.I. and Kuzmin, I. (1996) Isolation and characterization of the full-length 3′ untranslated region of the human von Hippel-Lindau tumor suppressor gene. Hum. Genet., 98, 666–671. [DOI] [PubMed] [Google Scholar]
- 16. Iliopoulos, O., Kibel, A., Gray, S. and Kaelin, W.G., Jr. (1995) Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med., 1, 822–826. [DOI] [PubMed] [Google Scholar]
- 17. Schoenfeld, A., Davidowitz, E.J. and Burk, R.D. (1998) A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc. Natl. Acad. Sci. USA., 95, 8817–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hascoet, P., Chesnel, F., Jouan, F., Le Goff, C., Couturier, A., Darrigrand, E., Mahe, F., Rioux-Leclercq, N., Le Goff, X. and Arlot-Bonnemains, Y. (2017) The pVHL172 isoform is not a tumor suppressor and up-regulates a subset of pro-tumorigenic genes including TGFB1 and MMP13. Oncotarget, 8, 75989–76002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zbar, B., Kishida, T., Chen, F., Schmidt, L., Maher, E.R., Richards, F.M., Crossey, P.A., Webster, A.R., Affara, N.A., Ferguson-Smith, M.A. et al. (1996) Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum. Mutat., 8, 348–357. [DOI] [PubMed] [Google Scholar]
- 20. Yoshida, M., Ashida, S., Kondo, K., Kobayashi, K., Kanno, H., Shinohara, N., Shitara, N., Kishida, T., Kawakami, S., Baba, M. et al. (2000) Germ-line mutation analysis in patients with von Hippel-Lindau disease in Japan: an extended study of 77 families. Jpn. J. Cancer Res., 91, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chou, A., Toon, C., Pickett, J. and Gill, A.J. (2013) von Hippel-Lindau syndrome. Front. Horm. Res., 41, 30–49. [DOI] [PubMed] [Google Scholar]
- 22. Aronow, M.E., Wiley, H.E., Gaudric, A., Krivosic, V., Gorin, M.B., Shields, C.L., Shields, J.A., Jonasch, E.W., Singh, A.D. and Chew, E.Y. (2019) VON HIPPEL-LINDAU DISEASE: update on pathogenesis and systemic aspects. Retina, 39, 2243–2253. [DOI] [PubMed] [Google Scholar]
- 23. Maher, E.R. and Sandford, R.N. (2019) von Hippel-Lindau disease: an update. Curr. Genet. Med. Rep., 7, 227–235. [Google Scholar]
- 24. Sgambati, M.T., Stolle, C., Choyke, P.L., Walther, M.M., Zbar, B., Linehan, W.M. and Glenn, G.M. (2000) Mosaicism in von Hippel-Lindau disease: lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am. J. Hum. Genet., 66, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coppin, L., Plouvier, P., Crépin, M., Jourdain, A.S., Ait Yahya, E., Richard, S., Bressac-de Paillerets, B., Cardot-Bauters, C., Lejeune, S., Leclerc, J. and Pigny, P. (2019) Optimization of next-generation sequencing technologies for von Hippel Lindau (VHL) mosaic mutation detection and development of confirmation methods. J Mol Diagn., 21, 462–470. [DOI] [PubMed] [Google Scholar]
- 26. Oldfield, L.E., Grzybowski, J., Grenier, S., Chao, E., Downs, G.S., Farncombe, K.M., Stockley, T.L., Mete, O. and Kim, R.H. (2022) VHL mosaicism: the added value of multi-tissue analysis. NPJ Genom Med., 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sekine, Y., Iwasaki, Y., Aoi, T., Endo, M., Hirata, M., Kamatani, Y., Matsuda, K., Sugano, K., Yoshida, T., Murakami, Y. et al. (2022) Different risk genes contribute to clear cell and non-clear cell renal cell carcinoma in 1532 Japanese patients and 5996 controls. Hum. Mol. Genet., 31, 1962–1969. [DOI] [PubMed] [Google Scholar]
- 28. Lenglet, M., Robriquet, F., Schwarz, K., Camps, C., Couturier, A., Hoogewijs, D., Buffet, A., Knight, S.J.L., Gad, S., Couvé, S. et al. (2018) Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood, 132, 469–483. [DOI] [PubMed] [Google Scholar]
- 29. Buffet, A., Calsina, B., Flores, S., Giraud, S., Lenglet, M., Romanet, P., Deflorenne, E., Aller, J., Bourdeau, I., Bressac-de Paillerets, B. et al. (2020) Germline mutations in the new E1' cryptic exon of the VHL gene in patients with tumours of von Hippel-Lindau disease spectrum or with paraganglioma. J. Med. Genet., 57, 752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.