

Abstract
Background
To understand the shared genetic basis between colorectal cancer (CRC) and other cancers and identify potential pleiotropic loci for compensating the missing genetic heritability of CRC.
Methods
We conducted a systematic genome-wide pleiotropy scan to appraise associations between cancer-related genetic variants and CRC risk among European populations. Single nucleotide polymorphism (SNP)-set analysis was performed using data from the UK Biobank and the Study of Colorectal Cancer in Scotland (10 039 CRC cases and 30 277 controls) to evaluate the overlapped genetic regions for susceptibility of CRC and other cancers. The variant-level pleiotropic associations between CRC and other cancers were examined by CRC genome-wide association study meta-analysis and the pleiotropic analysis under composite null hypothesis (PLACO) pleiotropy test. Gene-based, co-expression and pathway enrichment analyses were performed to explore potential shared biological pathways. The interaction between novel genetic variants and common environmental factors was further examined for their effects on CRC.
Results
Genome-wide pleiotropic analysis identified three novel SNPs (rs2230469, rs9277378 and rs143190905) and three mapped genes (PIP4K2A, HLA-DPB1 and RTEL1) to be associated with CRC. These genetic variants were significant expressions quantitative trait loci in colon tissue, influencing the expression of their mapped genes. Significant interactions of PIP4K2A and HLA-DPB1 with environmental factors, including smoking and alcohol drinking, were observed. All mapped genes and their co-expressed genes were significantly enriched in pathways involved in carcinogenesis.
Conclusion
Our findings provide an important insight into the shared genetic basis between CRC and other cancers. We revealed several novel CRC susceptibility loci to help understand the genetic architecture of CRC.
Introduction
Globally, colorectal cancer (CRC) is one of the three common malignancies and the second cause of cancer death, with an estimated 1.9 million new CRC cases and 0.9 million deaths in 2020, resulting in a heavy disease burden (1). Genetic factors play an important role in the occurrence of CRC, supported by the evidence that siblings of CRC patients have over 2-fold higher CRC risk, and the heritability of CRC has been estimated to be around 12–40% (2,3). Already conducted genome-wide association studies (GWASs) have identified more than 150 CRC-related single nucleotide polymorphisms (SNPs) (4) and only a small proportion of CRC heritability is explained by the reported genetic variants (5). Much of the heritable risk of CRC remains unexplained, and current studies indicate that further common risk variants remain to be discovered (3,4).
Notably, plentiful genetic pleiotropy has been observed among human complex diseases with 23% of reported genetic variants to be associated with more than one trait (6), and this phenomenon is particularly predominant among the risk loci related to cancers (7). The discovery of pleiotropic effects may allow for the identification of shared genes and pathways that influence carcinogenesis across different cancers. For instance, some of the genetic susceptibility regions of CRC, such as 5p15.33, 8q24, 10p14 and 11q23.1, have been found to be associated with lung cancer, bladder cancer, lymphoma, glioma, prostate cancer and basal cell carcinoma (4,8–15). Several studies have shown the shared heritability of CRC with other cancers (16–18). In addition, a study that examined the genetic pleiotropy of other cancer-related SNPs identified several novel genetic variants for CRC, and other studies also found cross-cancer pleiotropic variants for CRC (19–22), indicating the potential of shared genetic basis between CRC and other cancers (23). Given that an increasing number of genetic variants have been identified for different types of cancers by numerous GWASs in the recent decade (24), examining the pleiotropic effect of these genetic variants on CRC risk would provide insights into understanding the heritable risk of CRC and dissecting the biological mechanisms that underlie their shared etiology.
Additionally, environmental exposure also plays an etiologic role in CRC, and some environmental factors, such as smoking, alcohol consumption, processed meat consumption, abnormal body mass index (BMI), physical inactivity and vitamin D deficiency, have been well linked to CRC risk (25). Exploration of the interplay of genetic variants with environmental factors on CRC may contribute to explaining the missing heritability of CRC and identify a subpopulation with a higher risk of CRC and the potential to benefit most from health intervention (26).
Here, we performed a systematic analysis to test for any potential pleiotropic associations of GWAS-identified risk variants of other cancers with CRC risk, and then explore the interaction effects of novel CRC susceptibility variants with well-established environmental factors for CRC. Specifically, a systematical genome-wide pleiotropy scan was first performed to appraise the associations between other cancer-related SNPs and CRC risk among a large population of European ancestry. Gene-based, co-expression and pathway enrichment analyses were carried out to explore the possible biological processes and pathways of these identified pleiotropic signals on CRC. Then, we further examined the interaction effects of novel CRC susceptibility variants with environmental factors (smoking, alcohol drinking, processed meat consumption, BMI, physical activity and serum vitamin D) on CRC risk.
Results
An overview of common susceptibility regions between CRC and other cancers
From the NHGRI-EBI GWAS catalog, we identified a total of 2941 genetic variants associated with different types of cancer with P-value ≤ 5 × 10−8. Of them, 279 SNPs had already been reported as genetic risk variants for CRC (Supplementary Material, Table S1). We excluded SNPs that were previously reported to be associated with CRC (whatever CRC, colon cancer or rectal cancer) or SNPs that were in linkage disequilibrium (LD) with them. The remaining 2411 genetic variants associated with 16 different types of cancer [i.e. lung cancer, breast cancer, gastric cancer, esophageal cancer, prostate cancer, ovarian cancer, leukemia/lymphoma, skin cancers, hepatocellular carcinoma, bladder/renal cancer, glioma/neuroblastoma, pancreatic cancer, head/neck cancer, cervical/endometrial cancer, cross cancers (variants previously reported to be associated with two or more types of cancer were classified into the ‘cross cancers’ group) and other cancers] were included in subsequent analysis.
The identified 279 CRC genetic variants were mapped into 116 genomic regions, and 81 of them overlapped with the regions of other cancers. The overview of 81 susceptibility regions across each cancer type is shown in Figure 1. There were five CRC susceptibility regions (5p15.33, 6p21.32, 6p21.33, 6p22.1 and 8q24.21) that were shared by more than eight cancer types. SNP-set analysis indicated that CRC genomic susceptibility regions were associated with other cancers, including leukemia/lymphoma, cervical/endometrial cancer, hepatocellular carcinoma, gastric cancer, head/neck cancer, glioma/neuroblastoma and bladder/renal cancer, at a nominal threshold of P < 0.05 or false discovery rate (FDR) threshold of < 0.1 (Supplementary Material, Table S2).
Figure 1.

Heatmap for a general overview of susceptibility regions across each cancer type. For non-colorectal cancers, only susceptibility regions overlapped with that of colorectal cancer were included. The intensity of color represents the number of GWAS susceptibility variants in the region, with darker color indicating more susceptibility variants.
Three novel cross-cancer pleiotropic variants associated with CRC risk
The associations between cancer-related genetic variants and CRC risk were examined based on a meta-analysis of CRC GWAS datasets. We identified five independent SNPs that were significantly associated with CRC risk (FDR < 0.05) (Supplementary Material, Table S3). Rs2230469 [odds ratio (OR): 1.07, 95% confidence interval (CI): 1.04 to 1.10] and rs7953330 (OR: 0.93, 95% CI: 0.90 to 0.96) were located in novel susceptibility regions (10p12.2 and 12p13.33); rs9277378 (OR: 0.92, 95% CI: 0.88 to 0.95), rs143190905 (OR: 0.89, 95% CI: 0.83 to 0.94) and rs116846195 (OR: 0.75, 95% CI: 0.66 to 0.87) were located in known CRC susceptibility regions but were independent of already published genetic variants (Supplementary Material, Table S4). In validation analysis, three (rs9277378, rs2230469 and rs143190905) of five SNPs were significantly associated with CRC risk in UK Biobank (UKBB) after multiple testing corrections (FDR < 0.05), and the direction of these associations was consistent with the discovery set (Supplementary Material, Table S5). The cross-cancer pleiotropic analysis showed significant pleiotropic associations of the three novel variants with CRC and their previously reported cancer (P-pleiotropy < 0.008) (Table 1).
Table 1.
Three novel cross-cancer pleiotropic variants were identified to associated with colorectal cancer risk
| SNP | Chr | Region | Located gene | Effect/ref allele | Discovery stage | Validation stage | Reported cancer | P -pleiotropy a | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P-value b | FDR b | OR (95% CI) | P-value c | FDR c | |||||||
| rs9277378 | 6 | 6p21.32 | HLA-DPB1 | G/A | 0.92 (0.88 to 0.95) | 7.86 × 10−6 | 0.010 | 0.95 (0.92–0.98) | 0.002 | 0.011 | Lymphoma | 8.31 × 10−12 |
| rs2230469 | 10 | 10p12.2 | PIP4K2A | C/T | 1.07 (1.04 to 1.10) | 9.70 × 10−6 | 0.010 | 1.04 (1.01–1.08) | 0.009 | 0.022 | Leukemia | 1.01 × 10−4 |
| rs143190905 | 20 | 20q13.33 | RTEL1 | T/G | 0.89 (0.83 to 0.94) | 5.37 × 10−5 | 0.020 | 0.94 (0.89–0.99) | 0.023 | 0.038 | Cutaneous melanoma | 2.95 × 10−6 |
aThe Ppleiotropy was derived from the pleiotropy analysis via PLACO utilizing GWAS summary data of CRC and each of the reported cancers.
bThe P-value and FDR were derived from the CRC GWAS meta-analysis.
cThe P-value and FDR were derived from the validation analysis in UK Biobank population.
Functional annotation and gene-based analysis verified three CRC susceptibility genes
The functional characteristics of the three novel variants were assessed by silico annotation methods. We found that rs9277378 located in HLA-DPB1, rs143190905 located in RTEL1 were intronic and rs2230469 located in PIP4K2A was missense variant (Supplementary Material, Table S6). These genetic variants were predicted to play a regulatory role in gene expression by HaploReg v4.1 and RegulomeDB, and one (rs2230469) of them was annotated as a deleterious variant (Combined Annotation-Dependent Depletion (CADD) PHRED-scaled score = 19.23).
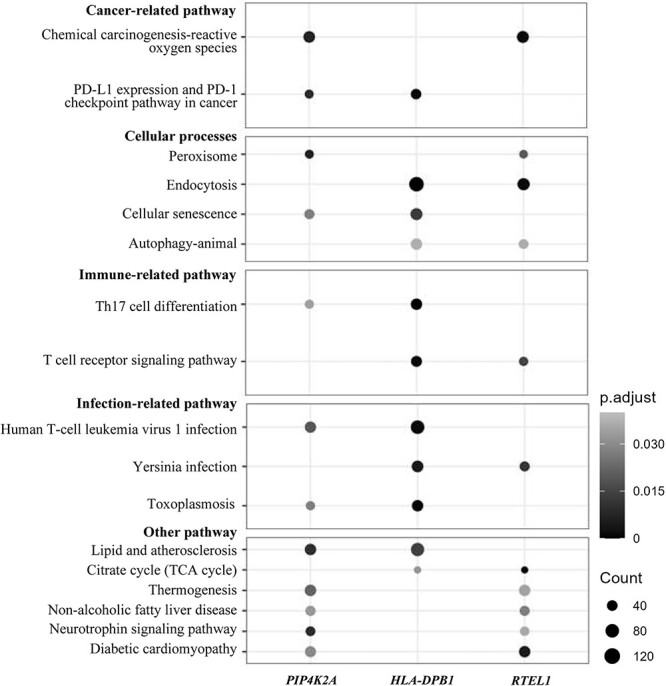
The expression quantitative trait loci (eQTL) analysis further found that all three variants were significant eQTL in the colon tissue, influencing the expression of multiple genes (Supplementary Material, Table S7). Among them, rs9277378 was associated with the expression of six genes in the colon-sigmoid and/or colon-transverse tissue, with the most significant association being with HLA-DPB2 in the colon-sigmoid tissue (β = 0.88, P = 2.00 × 10−37) (Supplementary Material, Fig. S1a). Rs2230469 was most significantly associated with PIP4K2A expression in the colon-sigmoid tissue (β = −0.27, P = 7.90 × 10−11) (Supplementary Material, Fig. S1b). Rs143190905 was most significantly associated with the expression of STMN3 in the colon-sigmoid tissue (β = −0.21, P = 4.80 × 10−5) (Supplementary Material, Fig. S1c). For their located genes, all of them (HLA-DPB1, PIP4K2A and RTEL1) were protein-coding genes (Supplementary Material, Table S6). Gene-based analysis verified these mapped genes were significantly associated with CRC risk (P = 6.10 × 10−6−1.70 × 10−4) (Table 2). Co-expression and pathway enrichment analysis of the mapped genes (PIP4K2A, HLA-DPB1 and RTEL1) showed that these genes were significantly aggregated in pathways related to cancer, cellular processes, immunity and infection (PBH < 0.05) (Supplementary Material, Table S8). The main enrichment pathways are shown in Figure 2.
Table 2.
The associations of mapped genes with colorectal cancer risk from gene-based analysis
| Gene | Chr | Start | Stop | Z value | P-valuea |
|---|---|---|---|---|---|
| PIP4K2A | 10 | 22 823 766 | 23 003 503 | 4.374 | 6.10 × 10−6 |
| HLA-DPB1 | 6 | 33 043 703 | 33 057 473 | 4.318 | 7.86 × 10−6 |
| RTEL1 | 20 | 62 289 163 | 62 327 606 | 3.582 | 1.70 × 10−4 |
aThe statistically significant threshold is a P-value < 0.017 (0.05/number of genes tested).
Figure 2.

The enrichment KEGG pathways of three mapped genes and their co-expressed genes in colon tissue. The enrichment pathways overlapped by two or over mapped genes are shown. The size of the dots represents the number of genes in a pathway, and the darker color represents the smaller P-value of a pathway.
Gene–environment interaction effects on CRC risk
We identified significant interaction effects of PIP4K2A rs2230469 with smoking and alcohol intake and HLA-DPB1 rs9277378 with alcohol intake after accounting for multiple testing (FDR < 0.05) (Table 3). The results of stratification analyses for these significant G × E interactions are shown in Supplementary Material, Tables 9 and 10. For rs2230469 × E interactions (Supplementary Material, Table S9), smoking was more strongly associated with increased CRC risk for participants with the CC genotype (HR: 1.47, 95% CI: 1.24 to 1.75) than for participants with TC or TT genotype. Alcohol intake was more strongly associated with increased CRC risk for participants with the TC genotype (>50 g/day vs. < 12.5 g/day, HR: 1.51, 95% CI: 1.32 to 1.73). For rs9277378 × E interactions (Supplementary Material, Table S10), alcohol intake was more strongly associated with increased CRC risk for participants with the GG genotype (>50 g/day vs. < 12.5 g/day, HR: 1.50, 95% CI: 1.10 to 2.06) than for participants with AG or AA genotype.
Table 3.
Gene–environment (G × E) interactions for colorectal cancer risk based on incident cases and controls from the UK Biobank
| Environmental factor | PIP4K2A rs2230469 × E interaction | HLA-DPB1 rs9277378 × E interaction | RTEL1 rs143190905 × E interaction | ||||||
|---|---|---|---|---|---|---|---|---|---|
| β | P-value | FDR | β | P-value | FDR | β | P-value | FDR | |
| Smoking | 0.135 | 2.10 × 10−5 | 1.26 × 10−4 | 0.082 | 0.016 | 0.057 | −7.62 × 10−5 | 0.999 | 0.999 |
| Alcohol intake | 0.118 | 1.41 × 10−6 | 2.54 × 10−5 | 0.117 | 8.35 × 10−6 | 7.51 × 10−5 | 0.104 | 0.063 | 0.095 |
| Processed meat consumption | 0.072 | 0.032 | 0.082 | 0.076 | 0.036 | 0.082 | −0.007 | 0.930 | 0.984 |
| BMI | 0.069 | 3.25 × 10−4 | 0.047 | 0.067 | 0.075 | 0.104 | 0.162 | 0.052 | 0.084 |
| Physical activity | 0.014 | 0.254 | 0.737 | 0.025 | 0.572 | 0.687 | −0.094 | 0.311 | 0.400 |
| Serum vitamin D | -0.056 | 2.10 × 10−4 | 0.014 | -0.050 | 0.044 | 0.085 | −0.118 | 0.027 | 0.081 |
Adjusted for age at enrollment, sex, genotyping array, the first 10 genetic principal components and other five environment risk factors.
Discussion
In this study, we conducted a systematic genome-wide pleiotropy scan to appraise associations between 2411 cancer-related genetic variants and CRC risk. We identified three novel SNPs (rs2230469, rs9277378 and rs143190905) and three mapped genes (PIP4K2A, HLA-DPB1 and RTEL1) to be associated with CRC risk. The functional analysis found that these variants were eQTLs for gene expression in colon tissue, and the mapped genes and their co-expression genes were significantly enriched in pathways involved in carcinogenesis. We additionally identified significant interactions of PIP4K2A rs2230469 and HLA-DPB1 rs9277378 with environmental factors on CRC risk.
We found that 81 of 116 CRC susceptibility regions were shared with other cancers, and SNP-set analysis also indicated the potential genetic overlap between CRC and other cancers. Similarly, a familial clustering investigation found a reliable association between multiple myeloma and CRC risk, indicating shared genetic susceptibility between multiple myeloma and CRC (27). Chen et al. found that glioma was locally genetically correlated with CRC in 5p15.33, and there were significant local genetic correlations between prostate and CRC in 4q24 and 8q24 (17). However, another study based on whole GWAS summary statistics observed that the genetic correlation of head/neck cancer with CRC was not significant (16). It was explained that shared genetic susceptibility among cancers might only exist in specific regions and not uniformly distributed on a genome-wide scale, which might partly contribute to the divergence of results (17,28).
Three novel pleiotropic variants and their mapped genes were identified to be associated with CRC risk, and these pleiotropic associations were further validated by cross-cancer pleiotropic analysis. We found 10p12.2 (rs2230469) as a novel CRC susceptibility region, which has not ever been reported in previous studies. Rs2230469, a known leukemia susceptibility variant (29), is located in PIP4K2A. The eQTL analysis showed that rs2230469 was also significantly associated with PIP4K2A expression in colon tissue. Significant interactions of PIP4K2A rs2230469 with smoking and alcohol intake on CRC risk were observed, indicating potential effect modifications. PIP4K2A was reported to participate in the regulation of cell proliferation, differentiation and apoptosis and control the activation of PI3K/Akt in cancer (30). Consistently, we found that PIP4K2A and its co-expressed genes in colon tissue were significantly enriched in the PI3K-Akt signaling pathway, which is closely involved in cancers (31).
For rs9277378 (6p21.32) and rs143190905 (20q13.33), although they were located in known CRC susceptibility regions, they were independent of published variants of CRC. Rs9277378 (HLA-DPB1), a known lymphoma susceptibility variant (32), was identified as a CRC risk locus in the current study and could influence the expression of six genes (including HLA-DPB1) in colon tissue. We also observed potential effect modifications of HLA-DPB1 rs9277378 with alcohol intake on CRC risk. The HLA-DPB1 gene belongs to the HLA class II beta chain paralogues and plays an important part in the immune system (33). Evidence has shown that the HLA class II antigen expression is lacking in one-third of CRC cases with high-level microsatellite instability, and the lack of HLA class II antigen expression mediated by RFX5 gene mutation may contribute to immune evasion in CRC cases (34). Consistently, we found that HLA-DPB1 and its co-expressed genes in colon tissue were significantly enriched in immune-related and cancer-related pathways. Rs143190905, previously reported to be a cutaneous melanoma susceptibility variant (35), is located in RTEL1. RTEL1 encodes the DNA helicase that plays a role in the stability and protection of telomeres and genome, which may affect human diseases, comprising cancer (36). Evidence has demonstrated that telomere shortening plays a pivotal role in CRC carcinogenesis by promoting the instability of chromosomes (37). We also found that RTEL1 and its co-expressed genes in colon tissue were significantly enriched in cellular processes and cancer-related pathways. However, the specific mechanism of its effect on CRC carcinogenesis remains to be further investigated.
The strength of the current study is the fact that we performed a systematic pleiotropy analysis utilizing the candidate-SNPs strategy based on robust prior evidence from cancer GWASs, which provided an excellent opportunity to understand the shared genetic basis between CRC and other cancers. Second, the large number of participants from multiple CRC GWASs with well-design elevated the statistical power and the reliability of results. The identified novel susceptibility loci for CRC risk could account for a part of the missing heritability of CRC. Furthermore, we examined the presence of potential effect modifications for novel susceptibility CRC variants and environmental factors to provide insights into CRC etiology. However, some limitations should also be considered. Firstly, the current findings were based on participants with European ancestry, which may partly limit the generalizability to the population with other ancestries. Secondly, the strict significance threshold of P-value ≤ 5 × 10−8 was utilized in the SNP selection process, which may result in missing some SNPs with weaker associations with other cancers. Thirdly, although co-expression and pathway analyses were performed to explore potential biological processes and pathways of these identified signals on CRC, exact mechanisms still need to be further clarified by molecular and animal studies.
In summary, our study identified three novel cross-cancer pleiotropic variants to be associated with CRC risk and revealed significant G × E interactions between their mapped genes and environmental factors on CRC risk. Our findings provide an important insight into the shared genetic basis of CRC with other cancers, which helps us understand the genetic architecture of CRC from the shared genetic components. Further validation studies on the identified genes and ascertainment of their underlying biological mechanisms via molecular and animal experiments are needed.
Materials and Methods
Summary of GWAS-identified genetic variants related to cancer risk
We first searched the NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas/ accessed in July 2021) to retrieve GWAS-identified variants (P < 5 × 10−8) associated with any type of cancer. Genetic variants previously reported to be associated with CRC and those in LD with them (r2 > 0.1) were excluded. Figure 3 presents an outline of the overall design and analysis steps of this study.
Figure 3.
Flowchart of the study design and analysis steps.
Study populations and quality control
A nested CRC case–control study from UKBB (38) and the Study of Colorectal Cancer in Scotland (SOCCS) (39) were used to estimate the overall shared genetic basis between CRC and other cancers. Then, we made use of a meta-analysis of 11 previously published CRC GWASs of European ancestry (40) to examine the pleiotropic associations between GWAS-identified risk variants of other cancers and CRC risk. Validation analysis was performed among UKBB CRC cases (prevalence and incidence) and controls to verify the effect of identified pleiotropic variants on CRC risk. For further interpreting the possibility of pleiotropy, we conducted a pleiotropy analysis using the PLACO (41) based on GWAS summary statistics of CRC and three other cancers from the FinnGen cohort of European ancestry (42). Lastly, gene–environment interaction analyses were performed based on incident CRC cases and controls from UKBB. Standard quality control (QC) measures were applied to each of these datasets. Specifically, SNPs with a minor allele frequency < 0.5% or Hardy–Weinberg equilibrium significance <1 × 10−5 were excluded, and for imputed variants, only genetic variants with an imputation quality value of ≥0.8 were used. Participants with a low SNP call rate (<0.95), as well as those identified to be of non-European ancestry were left out. For apparent first-degree relative pairs, the control was excluded from a case–control pair. More details of the study populations, genotyping, QC and imputation information have been described previously (39).
After the QC process, a total of 10 039 CRC cases and 30 277 controls from UKBB and SOCCS were included in the overall association analysis of each cancer type with CRC risk; a meta-GWAS of 16 871 CRC cases and 26 328 controls were used to identify cross-cancer pleiotropic associations with CRC; a total of 9276 CRC cases (prevalence and incidence) and 440 089 controls from UKBB were used to verify the effect of identified pleiotropic variants on CRC; GWAS summary data of three other cancers (955 cases and 271 463 controls for lymphoma, 1299 cases and 271 463 controls for leukemia, and 2705 cases and 259 583 controls for cutaneous melanoma) were used to validate the cross-cancer pleiotropy; and a total of 6742 incident CRC cases and 440 089 controls from UKBB were included in the gene–environment interaction analysis. The ethics approval was obtained from the relevant authorities, and all participants provided informed consent. The basic characteristics of these datasets are displayed in Supplementary Material, Table S11.
Genome-wide scan of cross-cancer pleiotropic associations with CRC
We first scanned the overlapped regions mapped by previously reported CRC susceptibility variants and other cancer-related variants to overview the common susceptibility regions between CRC and other cancers. Specifically, SNPs were mapped into a region by searching NCBI Variation Viewer (https://www.ncbi.nlm.nih.gov/variation/view). When CRC-related SNPs and other cancer-related SNPs were located in the same region, we defined that they had overlapped regions. Then, SNP-set analysis was performed among study populations of UKBB and SOCCS using the ‘SKAT’ package. This package was designed to test the overall association between a group of SNPs and a phenotype by aggregating the weighted variance-component score statistics for each SNP within a group utilizing the kernel function (43). In this case, we divided the selected variants into different groups by cancer type and tested the overall association between each group of variants and CRC risk. Sex, age and the first 20 genetic principal components (PCs) were adjusted in the model, and P < 0.05 was considered the nominal significance level. The computing details of the PCs have been described previously (38).
Logistic regression with an additive effect model was used to estimate the association between GWAS-identified cancer variants and CRC risk. The ORs (95%, CIs) of each SNP for CRC risk were combined across 11 GWAS datasets (40) using a meta-analysis of the random effects model in R version 4.1.0. The index of heterogeneity (I2) was calculated, and SNPs with I2 > 0.75 were removed. FDR by Benjamini-Hochberg (BH) method was utilized for multiple testing correction, and FDR < 0.05 was defined as the significance level. To identify independent signals, only the SNP with the smallest P-value in each region was retained, whereas those in high LD (r2 > 0.1) were excluded. The LD was estimated by PLINK 2.0 using the 1000 Genomes Project phase 3 (EUR) as reference data. After the identification of new CRC susceptibility variants, a comprehensive literature search for these variants was conducted to confirm novelty.
To verify the effect of identified pleiotropic variants on CRC risk, validation analysis was performed in the UKBB population. Cancer cases of UKBB were identified through linkage to Hospital Episode Statistics and national cancer and death registries. CRC cases were defined as malignant neoplasms of the colon, rectum, and rectosigmoid junction using the International Classification of Diseases (ICD), ICD-9 or ICD-10. After excluding controls with other cancers, a total of 9276 CRC cases and 440 089 controls remained. The effects of identified pleiotropic variants on CRC risk were estimated using the R function ‘snp.logistic’ of the ‘CGEN’ package (44) with adjustment of age at enrollment, sex, genotyping array and the first 10 genetic PCs. FDR was utilized for multiple testing corrections, and an FDR < 0.05 was defined as the significance level.
For further interpreting the possibility of pleiotropy, we used the pleiotropic analysis under composite null hypothesis (PLACO) (41) to conduct a pleiotropy analysis based on GWAS summary statistics of CRC and three other cancers that were previously reported to be associated with the identified three CRC susceptibility variants. PLACO is a powerful method for detecting pleiotropic variants between two phenotypes under a composite null hypothesis of no pleiotropy that a genetic variant is associated with only one or none of the phenotypes. Specifically, we used PLACO to evaluate the pleiotropic association between each novel CRC susceptibility variant and two phenotypes (CRC and previously reported other cancer of this variant). Using GWAS summary statistics as input (e.g. CRC and lymphoma), it tested the null hypothesis based on the product of the Z statistics of the SNPs from the two summary statistics and derived a null distribution of the test statistic in the form of a mixture distribution, allowing for fractions of SNPs to be associated with only one or none of the phenotypes. To reduce false-positive findings, we used a strict Bonferroni correction method with a P-value < 0.017 (0.05/3) as the significant threshold.
Functional annotation of the novel pleiotropic variants
eQTL analysis was further performed to explore whether these pleiotropic variants could regulate gene expression in colon tissue using data from the GTEx portal (version 8) (45). HaploReg v4.1 (46) and RegulomeDB (47) were applied to annotate and predict the regulatory potential of pleiotropic variants. The functional role of these pleiotropic variants was annotated based on the following criteria: (i) conservation (Siphy and/or GERP); (ii) presence in the DNase hypersensitivity, promoter, or enhancer region or (iii) with the RegulomeDB rank of ≤ 3 (48). The potential deleteriousness of genetic mutation of these variants was evaluated using CADD (49), which combined more than 60 diverse annotations to identify proxy-deleterious. A CADD PHRED-scaled score greater than 10 indicated the top 10% of most deleterious variants of all reference genome single-nucleotide variants.
Gene-based, co-expression and pathway enrichment analyses
To further understand the possible biological processes and pathways in which these pleiotropic variants were involved, we first mapped these independent signals into genes based on the database of NCBI GRCh37. To test whether these identified genes were associated with CRC susceptibility, a gene-based analysis was performed using summary statistics from the current meta-GWAS analysis in the MAGMA software (50). A P-value < 0.017 (0.05/3) was defined as the significance level. Then, co-expression and pathway enrichment analysis were performed to explore the potential biological functions and pathways of these identified genes. Specifically, gene expression data in colon tissue were downloaded from the GTEx portal (version 8) (45), and a linear regression model was applied to identify co-expressed genes for each identified gene. Each mapped gene and its co-expressed genes were combined to perform pathway enrichment analysis utilizing the ‘clusterProfiler’ package (51) based on KEGG. An adjusted P-value by the BH method of < 0.05 was defined as the significance level.
Gene–environment (G × E) interaction analysis in UKBB
A systematic analysis of interactions between the novel CRC susceptibility variants and common environmental risk factors, specifically, smoking (never smokers; smokers), alcohol intake (light: <12.5 g/day; moderate: 12.5–50 g/day; heavy: >50 g/day), processed meat consumption (≤1 time/week; >1 time/week), BMI (normal: 18.5 to <25.0 kg/m2; overweight or obesity: ≥25.0 kg/m2), physical activity (regular physical activity or not) and serum vitamin D (which reflects the level of vitamin D in the body, mainly derived from ultraviolet-B radiation) (52) (<25 nmol/L; 25–50 nmol/L; >50 nmol/L) was performed to explore their combined effect on CRC risk in UKBB population. Information on demographic characteristics, lifestyle factors and dietary was collected via a self-administered touchscreen questionnaire and nurse-led interviews in UKBB. Regular physical activity was considered as having met ≥150 min/week of moderate activity, ≥75 min/week of vigorous activity, ≥5 days/week of moderate physical activity, ≥1 day/week of vigorous activity or an equivalent combination of moderate and vigorous activity (53).
The interactions of environmental factors with novel CRC susceptibility variants in UKBB (6742 incident CRC cases and 440 089 controls) on CRC risk were estimated. Age at enrollment, sex, genotyping array, the first 10 genetic PCs and other five environmental factors (e.g. when evaluating the interaction of smoking with variants on CRC risk, alcohol intake, processed meat consumption, BMI, physical activity and serum vitamin D were also selected as covariates) were adjusted in the model to correct for potential confounding effects. FDR was utilized for multiple testing corrections, and an FDR < 0.05 was defined as the significance level. For significant G × E interactions, we further examined the associations of environmental factors with CRC risk stratified by SNPs genotypes using Cox proportional-hazards regression models, which considered both the occurrence of CRC and the duration from exposure to onset of CRC. All statistical analyses were performed using R version 4.1.0 unless otherwise noted.
Supplementary Material
Acknowledgements
This study was conducted using CRC summary statistics form 11 previous CRC GWASs and individual data from the UK Biobank (application number 66345). The authors thank all the participants, institutions and their staff for providing data.
Conflict of Interest statement. All authors declare that they have no conflict of interest.
Contributor Information
Jing Sun, Department of Big Data in Health Science School of Public Health, and Center of Clinical Big Data and Analytics of The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Lijuan Wang, Department of Big Data in Health Science School of Public Health, and Center of Clinical Big Data and Analytics of The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; Centre for Global Health, Usher Institute, University of Edinburgh, Edinburgh EH8 9AG, UK.
Xuan Zhou, Department of Big Data in Health Science School of Public Health, and Center of Clinical Big Data and Analytics of The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Lidan Hu, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou 310005, China.
Shuai Yuan, Unit of Cardiovascular and Nutritional Epidemiology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm 171 77, Sweden.
Zilong Bian, Department of Big Data in Health Science School of Public Health, and Center of Clinical Big Data and Analytics of The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Jie Chen, Department of Big Data in Health Science School of Public Health, and Center of Clinical Big Data and Analytics of The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China.
Yingshuang Zhu, Colorectal Surgery and Oncology, Key Laboratory of Cancer Prevention and Intervention, Ministry of Education, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China.
Susan M Farrington, Cancer Research UK Edinburgh Centre, Medical Research Council Institute of Genetics and Cancer, University of Edinburgh, Edinburgh EH4 2XU, UK.
Harry Campbell, Centre for Global Health, Usher Institute, University of Edinburgh, Edinburgh EH8 9AG, UK.
Kefeng Ding, Colorectal Surgery and Oncology, Key Laboratory of Cancer Prevention and Intervention, Ministry of Education, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China.
Dongfeng Zhang, Department of Epidemiology and Health Statistics, The School of Public Health of Qingdao University, Qingdao 266071, China.
Malcolm G Dunlop, Cancer Research UK Edinburgh Centre, Medical Research Council Institute of Genetics and Cancer, University of Edinburgh, Edinburgh EH4 2XU, UK.
Evropi Theodoratou, Centre for Global Health, Usher Institute, University of Edinburgh, Edinburgh EH8 9AG, UK; Cancer Research UK Edinburgh Centre, Medical Research Council Institute of Genetics and Cancer, University of Edinburgh, Edinburgh EH4 2XU, UK.
Xue Li, Department of Big Data in Health Science School of Public Health, and Center of Clinical Big Data and Analytics of The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310058, China; The Key Laboratory of Intelligent Preventive Medicine of Zhejiang Province, Hangzhou, Zhejiang 310058, China.
Funding
The Natural Science Fund for Distinguished Young Scholars of Zhejiang Province (LR22H260001 to X.L.); the National Nature Science Foundation of China (82204019 to X.L.); CRUK Career Development Fellowship (C31250/A22804 to E.T.). The work is supported by Programme Grant funding from Cancer Research UK (C348/A12076) and by funding for the infrastructure and staffing of the Edinburgh CRUK Cancer Research Centre. This work is funded by a grant to MGD as Project Leader with the MRC Human Genetics Unit Centre Grant (U127527198). This study is also supported by the project of the regional diagnosis and treatment center of the Health Planning Committee (No. JBZX-201903) to K.D.
Role of the funder
The funders had no role in the design of the study; the collection, analysis and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
Author contributions
JS: formal analysis, software, methodology, visualization and writing—original draft. LW and XZ: data curation, software and methodology. SMF, HC, LH, SY, ZB, JC and YZ: project administration and investigation. XL, ET, MD, DZ and KD: conceptualization, resources, supervision and writing—review & editing. All authors critically reviewed the manuscript and contributed important intellectual content. All authors have read and approved the final manuscript as submitted.
Data Availability
The results of this study are included in this published article and its supplementary information files. The UK Biobank is an open-access resource and bona fide researchers can apply to use the UK Biobank dataset by registering and applying at http://ukbiobank.ac.uk/register-apply/. Further information is available from the corresponding author upon request.
References
- 1. Sung, H., Ferlay, J., Siegel, R.L., Laversanne, M., Soerjomataram, I., Jemal, A. and Bray, F. (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin., 71, 209–249. [DOI] [PubMed] [Google Scholar]
- 2. Graff, R.E., Möller, S., Passarelli, M.N., Witte, J.S., Skytthe, A., Christensen, K., Tan, Q., Adami, H.O., Czene, K., Harris, J.R. et al. (2017) Familial risk and heritability of colorectal cancer in the Nordic twin study of cancer. Clin. Gastroenterol. Hepatol., 15, 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiao, S., Peters, U., Berndt, S., Brenner, H., Butterbach, K., Caan, B.J., Carlson, C.S., Chan, A.T., Chang-Claude, J., Chanock, S. et al. (2014) Estimating the heritability of colorectal cancer. Hum. Mol. Genet., 23, 3898–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Montazeri, Z., Li, X., Nyiraneza, C., Ma, X., Timofeeva, M., Svinti, V., Meng, X., He, Y., Bo, Y., Morgan, S. et al. (2020) Systematic meta-analyses, field synopsis and global assessment of the evidence of genetic association studies in colorectal cancer. Gut, 69, 1460–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schubert, S.A., Morreau, H., de Miranda, N. and van Wezel, T. (2020) The missing heritability of familial colorectal cancer. Mutagenesis, 35, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Novo, I., López-Cortegano, E. and Caballero, A. (2021) Highly pleiotropic variants of human traits are enriched in genomic regions with strong background selection. Hum. Genet., 140, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sivakumaran, S., Agakov, F., Theodoratou, E., Prendergast, J.G., Zgaga, L., Manolio, T., Rudan, I., McKeigue, P., Wilson, J.F. and Campbell, H. (2011) Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet., 89, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rafnar, T., Sulem, P., Stacey, S.N., Geller, F., Gudmundsson, J., Sigurdsson, A., Jakobsdottir, M., Helgadottir, H., Thorlacius, S., Aben, K.K. et al. (2009) Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet., 41, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yeager, M., Orr, N., Hayes, R.B., Jacobs, K.B., Kraft, P., Wacholder, S., Minichiello, M.J., Fearnhead, P., Yu, K., Chatterjee, N. et al. (2007) Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet., 39, 645–649. [DOI] [PubMed] [Google Scholar]
- 10. Rothman, N., Garcia-Closas, M., Chatterjee, N., Malats, N., Wu, X., Figueroa, J.D., Real, F.X., Van Den Berg, D., Matullo, G., Baris, D. et al. (2010) A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet., 42, 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shete, S., Hosking, F.J., Robertson, L.B., Dobbins, S.E., Sanson, M., Malmer, B., Simon, M., Marie, Y., Boisselier, B., Delattre, J.Y. et al. (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet., 41, 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen, H., Zhu, M. and Wang, C. (2019) Precision oncology of lung cancer: genetic and genomic differences in Chinese population. NPJ. Precis. Oncol., 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sud, A., Thomsen, H., Law, P.J., Försti, A., Filho, M., Holroyd, A., Broderick, P., Orlando, G., Lenive, O., Wright, L. et al. (2017) Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nat. Commun., 8, 1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stacey, S.N., Helgason, H., Gudjonsson, S.A., Thorleifsson, G., Zink, F., Sigurdsson, A., Kehr, B., Gudmundsson, J., Sulem, P., Sigurgeirsson, B. et al. (2015) New basal cell carcinoma susceptibility loci. Nat. Commun., 6, 6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sud, A., Thomsen, H., Orlando, G., Försti, A., Law, P.J., Broderick, P., Cooke, R., Hariri, F., Pastinen, T., Easton, D.F. et al. (2018) Genome-wide association study implicates immune dysfunction in the development of Hodgkin lymphoma. Blood, 132, 2040–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang, X., Finucane, H.K., Schumacher, F.R., Schmit, S.L., Tyrer, J.P., Han, Y., Michailidou, K., Lesseur, C., Kuchenbaecker, K.B., Dennis, J. et al. (2019) Shared heritability and functional enrichment across six solid cancers. Nat. Commun., 10, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, H., Majumdar, A., Wang, L., Kar, S., Brown, K.M., Feng, H., Turman, C., Dennis, J., Easton, D., Michailidou, K. et al. (2021) Large-scale cross-cancer fine-mapping of the 5p15.33 region reveals multiple independent signals. HGG Adv., 2, 100041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hung, R.J., Ulrich, C.M., Goode, E.L., Brhane, Y., Muir, K., Chan, A.T., Marchand, L.L., Schildkraut, J., Witte, J.S., Eeles, R. et al. (2015) Cross cancer genomic investigation of inflammation pathway for five common cancers: lung, ovary, prostate, breast, and colorectal cancer. J. Natl. Cancer Inst., 107, djv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fehringer, G., Kraft, P., Pharoah, P.D., Eeles, R.A., Chatterjee, N., Schumacher, F.R., Schildkraut, J.M., Lindström, S., Brennan, P., Bickeböller, H. et al. (2016) Cross-cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res., 76, 5103–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karami, S., Han, Y., Pande, M., Cheng, I., Rudd, J., Pierce, B.L., Nutter, E.L., Schumacher, F.R., Kote-Jarai, Z., Lindstrom, S. et al. (2016) Telomere structure and maintenance gene variants and risk of five cancer types. Int. J. Cancer, 139, 2655–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Toth, R., Scherer, D., Kelemen, L.E., Risch, A., Hazra, A., Balavarca, Y., Issa, J.J., Moreno, V., Eeles, R.A., Ogino, S. et al. (2017) Genetic variants in epigenetic pathways and risks of multiple cancers in the GAME-ON consortium. Cancer Epidemiol. Biomark. Prev., 26, 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rashkin, S.R., Graff, R.E., Kachuri, L., Thai, K.K., Alexeeff, S.E., Blatchins, M.A., Cavazos, T.B., Corley, D.A., Emami, N.C., Hoffman, J.D. et al. (2020) Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun., 11, 4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng, I., Kocarnik, J.M., Dumitrescu, L., Lindor, N.M., Chang-Claude, J., Avery, C.L., Caberto, C.P., Love, S.A., Slattery, M.L., Chan, A.T. et al. (2014) Pleiotropic effects of genetic risk variants for other cancers on colorectal cancer risk: PAGE, GECCO and CCFR consortia. Gut, 63, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buniello, A., MacArthur, J.A.L., Cerezo, M., Harris, L.W., Hayhurst, J., Malangone, C., McMahon, A., Morales, J., Mountjoy, E., Sollis, E. et al. (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res., 47, D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keum, N. and Giovannucci, E. (2019) Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol., 16, 713–732. [DOI] [PubMed] [Google Scholar]
- 26. Yang, T., Li, X., Farrington, S.M., Dunlop, M.G., Campbell, H., Timofeeva, M. and Theodoratou, E. (2020) A systematic analysis of interactions between environmental risk factors and genetic variation in susceptibility to colorectal cancer. Cancer Epidemiol. Biomark. Prev., 29, 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frank, C., Fallah, M., Chen, T., Mai, E.K., Sundquist, J., Försti, A. and Hemminki, K. (2016) Search for familial clustering of multiple myeloma with any cancer. Leukemia, 30, 627–632. [DOI] [PubMed] [Google Scholar]
- 28. van Rheenen, W., Peyrot, W.J., Schork, A.J., Lee, S.H. and Wray, N.R. (2019) Genetic correlations of polygenic disease traits: from theory to practice. Nat. Rev. Genet., 20, 567–581. [DOI] [PubMed] [Google Scholar]
- 29. Vijayakrishnan, J., Studd, J., Broderick, P., Kinnersley, B., Holroyd, A., Law, P.J., Kumar, R., Allan, J.M., Harrison, C.J. et al. (2018) Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat. Commun., 9, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thapa, N., Tan, X., Choi, S., Lambert, P.F., Rapraeger, A.C. and Anderson, R.A. (2016) The hidden conundrum of phosphoinositide signaling in cancer. Trends Cancer, 2, 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vivanco, I. and Sawyers, C.L. (2002) The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer, 2, 489–501. [DOI] [PubMed] [Google Scholar]
- 32. Li, Z., Xia, Y., Feng, L.N., Chen, J.R., Li, H.M., Cui, J., Cai, Q.Q., Sim, K.S., Nairismägi, M.L., Laurensia, Y. et al. (2016) Genetic risk of extranodal natural killer T-cell lymphoma: a genome-wide association study. Lancet Oncol., 17, 1240–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Benacerraf, B. (1981) Role of MHC gene products in immune regulation. Science, 212, 1229–1238. [DOI] [PubMed] [Google Scholar]
- 34. Michel, S., Linnebacher, M., Alcaniz, J., Voss, M., Wagner, R., Dippold, W., Becker, C., von Knebel Doeberitz, M., Ferrone, S. and Kloor, M. (2010) Lack of HLA class II antigen expression in microsatellite unstable colorectal carcinomas is caused by mutations in HLA class II regulatory genes. Int. J. Cancer, 127, 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landi, M.T., Bishop, D.T., MacGregor, S., Machiela, M.J., Stratigos, A.J., Ghiorzo, P., Brossard, M., Calista, D., Choi, J., Fargnoli, M.C. et al. (2020) Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat. Genet., 52, 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Björkman, A., Johansen, S.L., Lin, L., Schertzer, M., Kanellis, D.C., Katsori, A.M., Christensen, S.T., Luo, Y., Andersen, J.S., Elsässer, S.J. et al. (2020) Human RTEL1 associates with Poldip3 to facilitate responses to replication stress and R-loop resolution. Genes Dev., 34, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bertorelle, R., Rampazzo, E., Pucciarelli, S., Nitti, D. and De Rossi, A. (2014) Telomeres, telomerase and colorectal cancer. World J. Gastroenterol., 20, 1940–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L.T., Sharp, K., Motyer, A., Vukcevic, D., Delaneau, O., O'Connell, J. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li, X., Timofeeva, M., Spiliopoulou, A., McKeigue, P., He, Y., Zhang, X., Svinti, V., Campbell, H., Houlston, R.S., Tomlinson, I.P.M. et al. (2020) Prediction of colorectal cancer risk based on profiling with common genetic variants. Int. J. Cancer, 147, 3431–3437. [DOI] [PubMed] [Google Scholar]
- 40. Law, P.J., Timofeeva, M., Fernandez-Rozadilla, C., Broderick, P., Studd, J., Fernandez-Tajes, J., Farrington, S., Svinti, V., Palles, C., Orlando, G. et al. (2019) Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun., 10, 2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ray, D. and Chatterjee, N. (2020) A powerful method for pleiotropic analysis under composite null hypothesis identifies novel shared loci between type 2 diabetes and prostate cancer. PLoS Genet., 16, e1009218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kurki, M.I., Karjalainen, J., Palta, P., Sipilä, T.P., Kristiansson, K., Donner, K., Reeve, M.P., Laivuori, H., Aavikko, M., Kaunisto, M.A. et al. (2022) FinnGen: unique genetic insights from combining isolated population and national health register data. in press., 2022.2003.2003.22271360.
- 43. Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J.D. and Lin, X. (2013) Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet., 92, 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song, M., Wheeler, W., Caporaso, N.E., Landi, M.T. and Chatterjee, N. (2018) Using imputed genotype data in the joint score tests for genetic association and gene-environment interactions in case-control studies. Genet. Epidemiol., 42, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.GTEx Consortium (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ward, L.D. and Kellis, M. (2016) HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res., 44, D877–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dong, S. and Boyle, A.P. (2019) Predicting functional variants in enhancer and promoter elements using RegulomeDB. Hum. Mutat., 40, 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chung, C.C., Kanetsky, P.A., Wang, Z., Hildebrandt, M.A., Koster, R., Skotheim, R.I., Kratz, C.P., Turnbull, C., Cortessis, V.K., Bakken, A.C. et al. (2013) Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet., 45, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Leeuw, C.A., Mooij, J.M., Heskes, T. and Posthuma, D. (2015) MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol., 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu, G., Wang, L.G., Han, Y. and He, Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barrea, L., Savastano, S., Di Somma, C., Savanelli, M.C., Nappi, F., Albanese, L., Orio, F. and Colao, A. (2017) Low serum vitamin D-status, air pollution and obesity: a dangerous liaison. Rev. Endocr. Metab. Disord., 18, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Piercy, K.L., Troiano, R.P., Ballard, R.M., Carlson, S.A., Fulton, J.E., Galuska, D.A., George, S.M. and Olson, R.D. (2018) The physical activity guidelines for Americans. JAMA, 320, 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The results of this study are included in this published article and its supplementary information files. The UK Biobank is an open-access resource and bona fide researchers can apply to use the UK Biobank dataset by registering and applying at http://ukbiobank.ac.uk/register-apply/. Further information is available from the corresponding author upon request.