

Abstract
Infectious Spleen and Kidney Necrosis Virus (ISKNV) is linked to severe infections that cause significant financial losses in global aquaculture. ISKNV enters the host cell through its major capsid protein (MCP), and the resulting infection can lead to mass mortality of fish. Even though several drugs and vaccines are at various stages of clinical testing, none are currently available. Thus, we sought to assess the potential of seaweed compounds to block viral entrance by inhibiting the MCP. The Seaweed Metabolite Database (1110 compounds) was assessed for potential antiviral activity against ISKNV using high throughput virtual screening. Forty compounds with docking scores of ≥8.0 kcal/mol were screened further. The inhibitory molecules BC012, BC014, BS032, and RC009 were predicted by the docking and MD techniques to bind the MCP protein significantly with binding affinities of −9.2, −9.2, −9.9, and −9.4 kcal/mol, respectively. Also, ADMET characteristics of the compounds indicated drug-likeness. According to this study, marine seaweed compounds may operate as viral entrance inhibitors. For their efficacy to be established, in-vitro and in-vivo testing is required.
Keywords: ADMET, Molecular docking, Dynamic simulation, ISKNV, DFT
Graphical abstract

Highlights
-
•
ISKNV causes serious infections and financial losses in aquaculture worldwide.
-
•
CADD approaches may identify natural compounds that modulate protein activities in pathogenesis-related pathways.
-
•
High-throughput virtual screening revealed four potential seaweed metabolites against ISKNV major capsid protein.
-
•
ADMET, molecular docking, and MD simulations study showed the drug-like features and binding stability of the compounds.
1. Introduction
The ISKNV (Infectious Spleen and Kidney Necrosis Virus) produces disease and results in severe mortality in Nile tilapia, Rainbow trout, Atlantic Salmon, Asian sea bass, and mandarin fishes. The ISKNV infectious epidemic is widespread, affecting various locations in aquafarm in Southeast Asia, Australia, and Japan and causing significant commercial losses in production and export [1]. ISKNV infection causes cell hypertrophy in several organs, such as the kidney, spleen, endocardium, and cranial connective tissue [2]. Until now, more than one hundred distinct iridovirus strains have been discovered, and the genomes of twenty of these strains have been completely sequenced. The ISKNV virions are icosahedral with an average diameter of 150 nm. They have vital methylation at CpGs and circular permutation of their linear dsDNA at the center [1]. In 2001, the whole genome sequence was found, which was 111,362 bp long and comprised 124 possible ORFs [3]. According to Wang et al. the ISKNV virus may taint over 50 freshwater and marine fish species in China, including Clupeiformes, Myctophiformes, Mugiliformes, Perciformes, Pleuronectiformes, and Tetraodontiformes [4]. Previous studies have documented financial loss, but no effective preventative strategies are in place to limit the disease's onset and development [4,5]. Due to its presence across viral pathogens, the major capsid protein (MCP) of this virus is one of the essential genes for analyzing genetic connections among iridoviruses [6]. According to the results of a proteomic study of ISKNV, the virions of this virus include a total of 38 viral structural proteins [7]. Among them, the major capsid protein (MCP-ORF006) is a crucial protein that allows viruses to penetrate the host cell wall [8]. The MCP is a component of the iridovirus responsible for up to 45% of all virion proteins in infected cells [7,9]. In the process of viral infection, envelope proteins are essential structural proteins that play a role in contact between the virus and its host. These interactions include an attachment to host receptors and fusion with host cell membranes [7]. In addition, MCP is also renowned for having the highest protein localization between the outermost envelope layer and the inner membrane [7]. As ISKNV infections are difficult to control, inhibiting the role of MCP in viral replication through potential drug-like molecules could have potential benefits in controlling ISKNV in aquaculture.
Standardized methods of introducing new, effective medicines may be time-consuming, costly, and labor-intensive [10]. To better evaluate numerous potential drug-like molecules, high-throughput screening (HTS) is a method that combines multiple-well microplates with automated processing [11]. As executing a specific HTS program is costly and necessitates employing robotic equipment, HTS facilities must be well-equipped with the needed materials [12]. In contrast, computer-aided drug design, also known as in silico drug design, is a cutting-edge method for rapidly sifting through pharmacological libraries in search of promising novel therapeutics [13]. Finding drug treatments is aided by the in silico virtual screening method, which generates hits for lead compounds in a shorter time frame and at a lower cost [14]. Therefore, the amount of time needed to conceive, create, and optimize a new medicine has been reduced due to advancements in silico drug design. For several decades, virtual screening has been used to identify the best lead compounds with a range of structural features for usage with a specific biological target [15]. Furthermore, computer-aided drug design has been used to identify, validate and explore various biological properties of marine metabolites including anti-inflammatory, anti-infectious, anti-cancer, antiviral, and anti-microbial effects via virtual screening, molecular docking, and dynamics modelling [16,17].
The objective of the current investigation was to conduct a high-throughput virtual screening of seaweed metabolites targeting the MCP protein of ISKNV to find high-affinity inhibitors. A wide range of in silico tools and techniques were used to explore pharmacokinetic, pharmacodynamic, and toxicity properties of the lead candidates, stability of the docked complexes, and molecular reactivity of the lead compounds. No efforts have been taken so far to reveal the antiviral potential of seaweed metabolites targeting ISKNV to design potential drug candidates. Therefore, the present study aimed to evaluate seaweed metabolites against the MCP of ISKNV for their drug candidacy using comprehensive computational approaches.
2. Materials and methods
2.1. Retrieving the MCP sequence, prediction, refinement, and validation of 3D structure
The NCBI database (https://www.ncbi.nlm.nih.gov) was used to retrieve the amino acid (aa) sequence of the major capsid protein (MCP) (Accession No.: AAL72276) found in Infectious Spleen and Kidney Necrosis Virus and downloaded in FASTA format. AlphaFold 2 V.2.1.0 google colab server was used to predict the three-dimensional structure of the target protein [18]. The protein's 3D structure was then modified using the GalaxyWeb server (https://galaxy.seoklab.org). The validity of the structure is an essential step in homology modeling, which is based on the structural analysis of three-dimensional protein that has been experimentally confirmed. Its sequence is aligned with the template structure to create a 3D model of the target protein [19]. In addition, essential confirmation of the suggested MCP protein model has been submitted to ProSA-web [20]. Using the z-score, the server correctly predicted the essential characteristics of the model. If the expected model's z-scores exceed the range of the feature for proteins, the structure is most likely erroneous [20]. Moreover, the Ramachandran Plot Server (https://zlab.umassmed.edu/bu/rama/) was used to conduct a Ramachandran plot analysis, which provided an overall assessment of the efficacy of the proposed protein model [21].
2.2. Preparation of protein macromolecule
After refining the initial protein model, the 3D structure was processed further to prepare for docking studies. The protein had only one chain with a total of 453 amino acids. Afterward, the protein was opened in AutoDockTools v.1.5.7 in a Linux environment using Ubuntu v.18.04.6 LTS through Windows Subsystem for Linux [22,23]. Polar hydrogen and Kollman charges were added to prepare the final receptor [24].
2.3. Preparation of ligands
The Seaweed Metabolite Database was used to retrieve 1110 unique compounds belonging to 811 taxa of red algae (Rhodophyta), 266 taxa of brown algae (Phaeophyta), and 33 taxa of green algae (Chlorophyta) [25]. All the seaweed compounds were retrieved in three-dimensional Protein Data Bank (PDB) format. Afterward, all the PDB files of ligands were converted into PDBQT format using the Open Babel v.2.3.1 software on the Linux command line. All of the PDBQT files were run through the MMFF94 (Merck Molecular Force Field) force field's steepest descent method in 2000 iterations inside the Open Babel v.2.3.1 program in order to get the lowest possible energy state [26,27].
2.4. High-throughput virtual screening
AutoDock Vina v.1.2.0 was used for high-throughput virtual screening in the Linux command line along with the Perl programming script “Vina_windows.pl” [28,29]. Out of 1110 unique ligands, 1046 ligands were docked successfully following blind docking protocol where the whole surface of the receptor macromolecule was targeted for docking with seaweed metabolites without having any prior knowledge of the functional pockets. The target protein was considered rigid, and the ligands were considered flexible while performing docking. Grid box was generated before running the Perl script, and the size coordinates were 64.33 × 84.88 × 77.30 (for X, Y, and Z axes, respectively), and the center coordinates were 17.61 × 0.07 × 0.02 (for X, Y, and Z axes, respectively). Ligands that scored equal to or better than (−8.0 kcal/mol) were selected for further screening following ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) analyses and molecular dynamics simulation.
2.5. ADMET test drug profile assessment
After the high-throughput virtual screening, 40 compounds were selected for their excellent binding affinities (-Kcal/mol) to proceed with ADMET analyses. The absorptions, distribution, metabolism, and excretion qualities of a drug were investigated with the use of the SwissADME server since they are the four most important aspects that influence the pharmacological activity and performance of medicine [30]. Using Open Babel v.2.3.1 on the Linux command line, a canonical SMILES (Simplified Molecular Input Line Entry System) format was created for the top 40 compounds, which were then uploaded to the SwissADME server for determining ADME features. The compounds were assessed considering factors such as physicochemical qualities, lipophilicity, water solubility, drug-likeness, pharmacokinetic properties, and medicinal chemistry features. Furthermore, the BOILED-Egg model was utilized to determine the compounds' performance to pass the Blood Brain Barrier (BBB) [31]. The ProToxII server was used to examine several toxicity endpoints such as acute toxicity, cytotoxicity, hepatotoxicity, mutagenicity, carcinogenicity, unfavorable outcomes (Tox21), pathways, and toxicity targets [32].
2.6. Quantum mechanics (QM)-Based calculation
It is essential to conduct a conformation analysis of the ligand in relation to the binding site to ascertain the possible active conformation of the ligand, the binding affinity, and the strain control involved in the binding process. In this situation, structural optimization, and lowest energy conformations, which need gas-phase energy and the solution phase, are viable options. The classic molecular mechanics (MM) process does not work well in a ligand-protein complex system containing metal ions [33]. Scoring functions have been developed as a consequence of quantum mechanical calculations to characterize the development of the electronic structure and electronic charges inside a system as a reaction progresses. Density functional theory (DFT) is presently used in many quantum mechanical calculations [34]. This effort aimed to do QM calculations for the top four docking compounds using DFT techniques. The ORCA quantum chemistry software tool (Version 4.1.1) was used to optimize bond lengths, bond angles, and dihedral angles for possible compounds, and to print the molecular orbitals of those compounds [35,36]. We utilized ORCA in our analysis since it has evolved into a relatively complete general-purpose program for theoretical research in many fields of chemistry and several adjacent disciplines, such as materials sciences and biological sciences. ORCA includes certain techniques that are either unique to ORCA or were initially developed during the development of ORCA. The ORCA CASSCF module can handle any number of various multiplicities and irreps in state-averaged CASSCF (SA-CASSCF) computations, a capability not present in many other software packages. Besides ORCA is completely free to use for academic researchers where many other similar packages are not completely free [37].
To compute DFT, Becke's three parameters (B3LYP) and Lee-Yang-Parr functionals (B3LYP-D3) were combined with the dispersion correction energy term D3. The typical arrangement of capabilities B3LYP-D3 was selected for this study and 6-31G(d,p) was chosen to characterize the electronic wave function of the molecule [15,38,39]. Conventional B3LYP method have shown remarkable accuracy but one of the major limitations is its failure to accurately represent London Dispersion Interactions. Some other difficulties are B3LYP degrades as the system becomes larger, underestimates reaction barrier heights, yields too low bond dissociation enthalpies, gives improper isomer energy differences and so on. As a mix of conventional functionals and an add-on energy term, modern B3LYP-D3 can provide corrections to the molecular total energy, energy gradient and frequencies. The main advantage of B3LYP-D3 is its computational efficiency and robustness across the periodic table, which makes this approach particularly valuable for the treatment of large systems [[40], [41], [42]].
2.7. Frontier molecular orbital HOMO/LUMO calculation and redocking
The Fukui functions, which are based on Kenichi Fukui's frontier molecular orbital (FMO) hypothesis from the 1950s, are the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). FMOs may be utilized to determine the distance between the HOMO and LUMO orbitals by comparing their energies. The interaction between an electron donor and an electron acceptor pair has the potential to change the other chemical reactivity of a molecule. This is because, in nature, HOMO is mostly a nucleophilic electron donor, whilst LUMO is largely an electron acceptor [43]. During electrophilic-nucleophilic reactions, HOMO electrons move to LUMO, generating an energy gap. Photochemistry and the durability and stability of organic transition metal complexes are both shown by the HOMO-LUMO gap, the energy difference between two molecular orbitals. To fully explain an atom's propensity to electrophilic and nucleophilic reactions, the HOMO and LUMO energies were calculated and displayed using Avogadro [36], and the following Equation (1) was used to determine the energy gap between two molecular orbital HOMOs and LUMOs [44]:
| (1) |
here, ΔE is the HOMO-LUMO gap, ELUMO is the energy of the lowest unoccupied molecular orbital, and EHOMO is the energy of the highest occupied molecular orbital. After geometry optimization in the gas phase, the lead candidates were redocked with the target receptor to evaluate the changes in binding affinities.
2.8. Molecular dynamics simulation
Molecular dynamics simulation (MDS) was carried out for the top four lead compounds for 100 ns using Desmond v.6.3 programs in Schrödinger 2020-3 under the Linux framework to evaluate the thermodynamic stability of the ligand-receptor complexes. The system was solved using a standard SPC (Simple Point Charge) water model with orthorhombic periodic boundary boxes and box distances of 10 Å on all sides to ensure a fixed volume. The physiological states were imitated by adding 0.15 mM NaCl. To minimize the energy of the whole system OPLS3e force field was employed in default settings of the Desmond module after generating the solvated system comprising protein in complex with the seaweed compound as ligand. NPT ensembles were utilized, setting the temperature as 300 K and the pressure as 1.01325 bar (one atmospheric pressure) using the Nose-Hoover temperature coupling and isotropic scaling approach, followed by 20 ps trajectory recording intervals, with an energy value of 1.2. The Desmond module provided raw data in DAT format, which were plotted later using Microsoft Excel v.2013 to evaluate Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Solvent Accessible Surface Area (SASA), and protein-ligand contacts (interaction fractions) for the top four lead compounds.
2.9. Molecular mechanics/Generalized born surface area (MM/GBSA) calculation
In order to determine free binding energies via the MM/GBSA method, this study used the fastDRH server [45]. The AutoDock Vina and AutoDock-GPU docking engines, as well as the structure-truncated MM/PB(GB)SA free energy calculation module, are used on this web server. In the benchmark, this combined rescoring procedure has a success rate of more than 80%, which is much greater than AutoDock Vina (70%). The GAFF2 force field was employed for ligands in this study, whereas the ff14SB force field was used for macromolecules. For pose-rescoring, the truncation radius was fixed to 30 Å.
3. Results
3.1. Retrieval of sequence, 3D structures prediction, and validation
Accession number AAL72276 was used to retrieve the amino acid sequence of the major capsid protein (MCP) from the NCBI database. The protein is comprised of 453 different amino acids. The anticipated three-dimensional structure of the protein model, as generated by the AlphaFold 2 V.2.1.0 google colab server [18]. The Galaxy Refine server refined the protein's predicted tertiary structure, resulting in five improved models and more favored amino acid residues. The preceding rankings demonstrate the superiority of the new model over its peers. Pymol was used to create a visualization of the crude model as well as the refined model 3 with an RMSD value of 0.415 (Fig. S1). The Ramachandran Plot Server in conjunction with the ProSA-Web web server were used to validate the before and after improved MCP protein model. According to the Ramachandran plot server, the study of the structure before it was refined showed that 96.7500% of the structure was in the highly preferred zone. After the refinement process, the server delivered a superior result, with 98.936% of the residues located in the preferred regions as presented in Table 1 and Fig. S2. Using the ProSA-web server, an analysis is done to determine the validation quality and any possible errors in a basic tertiary structure model. A Z-score of −6.76 was found during the validation of the complete MCP protein model as shown in Table 1 and Fig. S3.
Table 1.
Validation of selected protein model by Ramachandran and Z-score studies.
| Parameters | Crude Model | Refine Model | Remarks | |
|---|---|---|---|---|
| Ramachandran | Highly Preferred | 96.750% | 98.936% | Significant |
| Preferred | 2.750% | 1.064% | Significant | |
| Questionable | 0.500% | 0.000% | Significant | |
| ProSA Web | Z-Score | −6.71 | −6.76 | Significant |
3.2. High throughput virtual screening
Molecular docking was carried out successfully for 1110 seaweed compounds. The binding affinity range was −4.3 kcal/mol to −10.8 kcal/mol. Two compounds, RP062 and RL393, were the most minor scoring (−4.3 kcal/mol), where the compound BE004 scored highest, i.e., −10.8 kcal/mol. A total of 40 compounds were selected after the high-throughput virtual screening that scored equal to −8.0 kcal/mol or higher. Among these top-scoring candidates, docking scores ranged from −8.0 kcal/mol to −10.8 kcal/mol (Table 2). All these 40 compounds went through an ADMET assay, and finally, four compounds were selected as the best lead candidates such as BC012 (−9.2 kcal/mol), BC014 (−9.2 kcal/mol), BS032 (−9.9 kcal/mol), and RC009 (−9.4 kcal/mol) as shown in Fig. 2, Fig. 3.
Fig. 1.

Schematic workflow of the present investigation showing stepwise high-throughput virtual screening (HTVS) in silico.
Table 2.
Binding affinities (Kcal/mol) of the top 40 screened compounds after molecular docking analysis.
| Sl. No. | Accession Number | Molecular weight (g/mol) | Source | Binding affinity (Kcal/mol) | 2D structure |
|---|---|---|---|---|---|
| 1 | BC006 | 470.64 | Cystophora fibrosa | −9.4 | 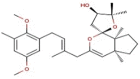 |
| 2 | BC012 | 484.66 | Cystophora fibrosa | −9.2 |  |
| 3 | BC014 | 456.61 | Cystophora fibrosa | −9.2 |  |
| 4 | BD053 | 362.50 | Dictyota divaricata | −8.0 | 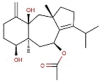 |
| 5 | BE004 | 602.45 | Ecklonia stolonifera | −10.8 | 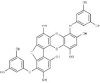 |
| 6 | BE010 | 496.37 | Ecklonia cava | −9.3 | 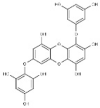 |
| 7 | BE015 | 602.45 | Ecklonia stolonifera | −9.3 | 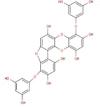 |
| 8 | BS028 | 412.60 | Stypopodium flabelliforme | −10.0 | 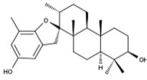 |
| 9 | BS029 | 412.60 | Stypopodium flabelliforme | −10.0 | 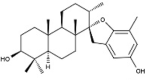 |
| 10 | BS032 | 412.61 | Stypopodium flabelliforme | −9.9 |  |
| 11 | BS057 | 454.64 | Stypopodium flabelliforme | −9.6 |  |
| 12 | BS058 | 554.71 | Stypopodium flabelliforme | −9.4 | 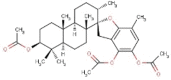 |
| 13 | BS059 | 510.66 | Stypopodium flabelliforme | −9.9 | 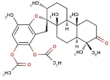 |
| 14 | BS067 | 496.67 | Stypopodium flabelliforme | −10.0 |  |
| 15 | BS069 | 496.67 | Stypopodium flabelliforme | −9.8 | 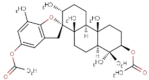 |
| 16 | BS070 | 554.71 | Stypopodium flabelliforme | −10.3 |  |
| 17 | BS071 | 589.15 | Stypopodium flabelliforme | −10.3 | 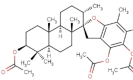 |
| 18 | BT002 | 408.57 | Taonia atomaria | −9.6 | 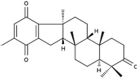 |
| 19 | RC003 | 602.39 | Callophycus serratus | −9.2 | 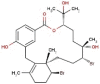 |
| 20 | RC004 | 665.29 | Callophycus serratus | −9.2 |  |
| 21 | RC005 | 584.38 | Callophycus serratus | −9.1 | 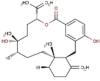 |
| 22 | RC006 | 521.48 | Callophycus serratus | −9.1 | 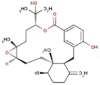 |
| 23 | RC007 | 602.39 | Callophycus serratus | −9.4 | 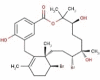 |
| 24 | RC008 | 665.29 | Callophycus serratus | −9.3 | 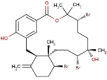 |
| 25 | RC009 | 602.39 | Callophycus serratus | −9.4 | 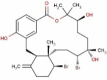 |
| 26 | RC010 | 665.29 | Callophycus serratus | −9.2 | 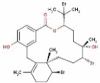 |
| 27 | RC012 | 440.57 | Callophycus serratus | −8.9 | 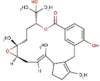 |
| 28 | RC014 | 584.38 | Callophycus serratus | −9.3 |  |
| 29 | RC023 | 584.38 | Callophycus serratus | −8.9 |  |
| 30 | RC034 | 521.48 | Callophycus serratus | −9.1 |  |
| 31 | RC035 | 503.46 | Callophycus serratus | −9.0 |  |
| 32 | RC036 | 584.38 | Callophycus serratus | −9.1 | 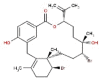 |
| 33 | RC037 | 584.38 | Callophycus serratus | −9.1 | 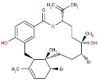 |
| 34 | RC038 | 665.29 | Callophycus serratus | −9.2 | 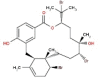 |
| 35 | RC039 | 584.38 | Callophycus serratus | −10.0 |  |
| 36 | RC040 | 584.38 | Callophycus serratus | −10.0 |  |
| 37 | RL015 | 588.41 | Laurencia microcladia | −9.7 | 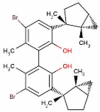 |
| 38 | RL259 | 376.12 | Laurencia cartilage | −8.0 | 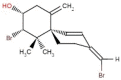 |
| 39 | RL385 | 222.36 | Laurencia majuscula | −8.1 | 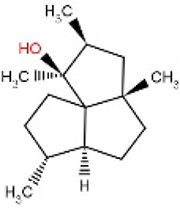 |
| 40 | RS006 | 464.27 | Sphaerococcus coronopifolius | −8.1 | 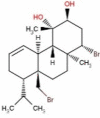 |
Fig. 2.

Two-dimensional chemical structures of the selected four lead compounds from the seaweed metabolite database- (A) BC012; (B) BC014; (C) BS032, and (D) RC009.
Fig. 3.

Docked complexes of the top selected candidates BC012, BC014, BS032, and RC009 with the target protein.
Molecular interactions were analyzed after molecular docking to observe different types of hydrogen bonds and hydrophobic interactions as presented in Table 3. BC012 showed interaction with Try341, Arg346, Tyr376, Thr379, Thr391, Asn392, and Leu396 amino acid residues. Among these residues, only two were involved with conventional hydrogen bonding, such as Thr379 and Asn392, with a bond distance of 2.05 Å and 2.11 Å, respectively, as shown in Fig. 4, Fig. 5.
Table 3.
Interacting amino acid residues for the top selected seaweed metabolites.
| Ligands | Ligands binding sites | Residues involved in conventional hydrogen bond formation (Distance in Å) | Number of conventional hydrogen bonds formed | Residues involved in hydrophobic interactions | Binding affinity (Kcal/mol) |
|---|---|---|---|---|---|
| BC012 | Tyr341, Arg346, Tyr376, Thr379, Thr391, Asn392, Leu396 | Thr379(2.05), Asn392(2.11) | 2 | Tyr341, Arg346, Tyr376, Thr391, Leu396 | −9.2 |
| BC014 | Tyr341, Phe363, Tyr376, Leu380, Leu396 | Tyr376(2.23) | 1 | Tyr341, Phe363, Leu380, Leu396 | −9.2 |
| BS032 | Tyr341, Arg346, Phe363, Cys377, Leu380 | – | 0 | Tyr341, Arg346, Phe363 | −9.9 |
| RC009 | Arg99, Asp102, Ile210, Arg306 | Arg99(2.37), Asp102(2.20) | 2 | Ile210, Arg306 | −9.4 |
Fig. 4.

Two-dimensional molecular interactions of (A) BC012, (B) BC014, (C) BS032, and (D) RC009 with various amino acid residues of the macromolecular receptor.
Fig. 5.

Three-dimensional interactions between the protein and four lead compounds - (A) BC012, (B) BC014, (C) BS032, and (D) RC009 show different interacting residues.
BC014 interacted with Tyr341, Phe363, Tyr376, Leu380, and Leu396, where only Tyr376 formed a conventional hydrogen bond with the protein with a bond distance of 2.23 Å. BS032 formed no conventional hydrogen bonds with the target protein. It interacted with Tyr341, Arg346, Phe363, Cys377, and Leu380. All of these residues were involved in hydrophobic interactions. RC009 formed both conventional hydrogen bonds and hydrophobic interactions. It showed interactions with Arg99, Asp102, Ile210, and Arg306 residues where Arg99 and Asp102 were involved in conventional hydrogen bonding with a bond distance of 2.37 Å and 2.20 Å, respectively. Ile210 and Arg306 were involved in hydrophobic interactions (Fig. 4, Fig. 5). Among the top four candidates, BS032 showed the best docking score (−9.9 kcal/mol) compared to the other three candidates. Fig. 6 for scoring via molecular docking, only the best pose at zero RMSD was selected in AutoDock Vina.
Fig. 6.

Hydrogen-bonds donating and accepting regions during three-dimensional interactions between the macromolecular receptor and four lead compounds – (A) BC012, (B) BC014, (C) BS032, and (D) RC009.
3.3. ADMET test drug profile assessment
ADME analyses unveiled promising pharmacokinetic and pharmacodynamic properties (Table 4). Among the top four metabolites, RC009 showed the highest molecular weight with a value of 590.30 g/mol, whereas BS032 showed the lowest molecular weight with a value of 392.45 g/mol. Several hydrogen bond acceptors were 5 for all the top candidates except BS032. All the compounds were found with high gastrointestinal absorption properties. BC012 was predicted to be impermeable to the Blood Brain Barrier (BBB), whereas BC014, BS032, and RC009 were predicted as permeable to the BBB.
Table 4.
Drug profile and ADME analyses of the best four seaweed metabolites predicted through the SwissADME server.
| Parameters | BC012 | BC014 | BS032 | RC009 | |
|---|---|---|---|---|---|
| Physicochemical properties | Formula | C30H30O5 | C28H24O5 | C27H20O3 | C27H26Br2O5 |
| Molecular weight | 470.56 g/mol | 440.49 g/mol | 392.45 g/mol | 590.30 g/mol | |
| No. H-bond acceptor | 5 | 5 | 3 | 5 | |
| No. H-bond donors | 0 | 1 | 2 | 2 | |
| Molar refractivity | 133.27 | 117.23 | 106.53 | 137.96 | |
| TPSA | 46.15 Å2 | 57.15 Å2 | 49.69 Å2 | 71.45 Å2 | |
| Lipophilicity | Log Po/w (XLOGP3) | 4.75 | 2.01 | 2.47 | 2.47 |
| Log Po/w (WLOGP) | 5.88 | 4.19 | 4.05 | 6.08 | |
| Log Po/w (MLOGP) | 3.72 | 3.53 | 4.60 | 4.60 | |
| Log Po/w (SILICOS-IT) | 6.21 | 4.55 | 4.84 | 4.84 | |
| Consensus Log Po/w | 4.11 | 2.86 | 3.19 | 3.19 | |
| Pharmacokinetics | GI absorption | High | High | High | High |
| BBB permeant | No | Yes | Yes | Yes | |
| P-gp substrate | Yes | Yes | Yes | Yes | |
| CYP1A2 inhibitor | No | No | No | No | |
| CYP2C19 inhibitor | Yes | No | No | No | |
| CYP2C9 inhibitor | No | No | No | No | |
| CYP2D6 inhibitor | No | No | No | No | |
| CYP3A4 inhibitor | Yes | No | No | No | |
| Log Kp (skin permeation) | −5.80 cm/s | −7.56 cm/s | −6.94 cm/s | −6.94 cm/s | |
| Water Solubility | Log S (ESOL) | −5.70 | −3.91 | −3.98 | −3.98 |
| Solubility | 9.45e-04 mg/ml; 2.01e-06 mol/l | 5.47e-02 mg/ml; 1.24e-04 mol/l | 4.14e-02 mg/ml; 1.05e-04 mol/l | 4.14e-02 mg/ml; 1.05e-04 mol/l | |
| Class | Moderately soluble | Soluble | Soluble | Soluble | |
| Drug likeness | Lipinski | Yes; 0 Violation | Yes; 0 Violation | Yes; 1 Violation: MLOGP >4.15 | Yes; 1 Violation: MLOGP >4.15 |
| Veber | Yes | Yes | Yes | Yes | |
| Medicinal Chemistry | Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 |
| PAINS | 0 alert | 0 alert | 0 alert | 0 alert | |
| Synthetic accessibility | 6.84 | 7.38 | 6.76 | 6.76 | |
Table 4Drug profile and ADME analyses of the best four seaweed metabolites predicted through the SwissADME server.
Cytochrome P450 inhibitory property was analyzed where BC014, BS032, and RC009 were found not to inhibit any of the isoforms of Cytochrome P450 such as CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. BC012 was predicted to inhibit only two isoforms, i.e., CYP2C19 and CYP3A4. The Cytochrome P450 inhibitory properties were in the acceptable range for all the top candidates. BC014, BS032, and RC009 were predicted to be soluble in the water solubility criterion, whereas BC012 was found to be moderately soluble. Both BC012 and BC014 followed Lipinski's rule of five without any violation. BS032 and RC0009 showed one violation each in the Lipinski parameter, which was acceptable. All the compounds followed Veber's rules without any violation which was satisfactory. Pan-Assay Interference Compounds (PAINS) criterion revealed zero alerts for all the top candidates. The bioavailability score was found as 0.55 for all four compounds and considering synthetic accessibility, both BS032 and RC009 scored the lowest (6.76) than BC012 (6.84) and BC014 (7.38). ADME results have been depicted in Fig. S4.
ProToxII server predicted toxicity results with a mean accuracy of 58.25% for all four candidates (Table S1). BC014 and BS032 were predicted to have toxicity class 4 while RC009 and BC012 were predicted to have toxicity class 5 and 6 respectively. Considering toxicity class, the BC012 compound showed the best results among the top candidates. Only RC009 was predicted to be active in the carcinogenicity parameter with 50% probability. Except that all the compounds were found inactive in organ toxicity parameters, i.e., hepatotoxicity and toxicity endpoints criteria. Tox21-Nuclear receptor signaling pathways revealed promising results where BC012, BC014, and RC009 were inactive in all the sub-criteria. BS032 showed inactive status in all the sub-criteria except the Estrogen Receptor Alpha sub-criterion, which was found to be active with 53% probability. Tox21-Stress response pathways further corroborated the selection of the top four compounds as BC012, BC014, BS032, and RC009 were found inactive in all the sub-criteria under the Tox21-Stress response pathways.
3.4. Theoretical calculation
3.4.1. Geometry optimization
Most computational biologists, chemists, academics, and researchers employ geometry optimization, a quantum chemical approach, to discover the configuration with the least energy and most stable chemical properties. This method allows for the refinement of weak geometric approximations [46]. Molecular states with the lowest energy emit photons, making the geometry with the lowest energy the most stable. The Avogadro default basis set 6-31G(d,p) has been used to optimize the molecular shape with the lowest energy value. The 2D and 3D structures of the compounds BC012, BC014, BS032, and RC009 were optimized prior to the HOMO-LUMO energy calculation.
3.4.2. Frontier molecular orbital HOMO/LUMO calculation
Equation (1) was used to calculate the HOMO, LUMO, and HOMO-LUMO gap energies, shown in Table S2, and can be utilized to examine the chemical reactivity and kinetic stability of the four selected compounds. The calculated FMO energy band gap values found for the compounds BC012, BC014, BS032, and RC009 were 5.831 eV, 4.807 eV, 4.361 eV, and 3.508 eV, respectively. The graphical view of the HOMO-LUMO orbital formation in the top selected four compounds are given below in Fig. S5.
3.4.3. Redocking of the lead candidates
Re-docking was conducted using pre-defined protein binding sites to find viable docking poses in a confined space. The geometry-optimized structures have been docked, and the scores were −9.4, −9.4, −10.0 and −9.6 kcal/mol for BC012, BC014, BS032 and RC009, respectively, which were better than the previously obtained binding scores as found in Table 3. As a result, it is possible to assume that the QM-based optimization of the compounds became successful for the selected three compounds.
3.5. Molecular dynamics simulation
3.5.1. Root Mean Square Deviation (RMSD)
The four lead compounds showed significant structural stability and compactness during a 100-ns molecular dynamics simulation. RMSD was calculated for ligand fitness on protein and alpha carbon atoms (Cα) of protein complexed with ligands. RMSD of Protein (Cα) revealed initial fluctuations for all the compounds. Among the candidates, BC014 showed the best stability with a mean RMSD (Cα) of 5.93 Å. BC014 became stable after 10 ns, showed a few fluctuations from 40 ns to 70 ns, and became stable again up to 100 ns BC012, with a mean RMSD (Cα) of 6.73 Å showed some initial fluctuations from 0 ns to 30 ns; afterward, it became stable. The mean RMSD (Cα) of BS032 and RC009 was 9.09 Å and 8.26 Å, respectively. RC009 became stable after 20 ns up to 100 ns, and BS032 became stable after 32 ns Ligand fitness RMSD was found satisfactory where the mean RMSD was calculated as 0.62 Å, 0.66 Å, 1.65 Å, and 2.26 Å for RC009, BS032, BC014, and BC012, respectively. RC009 compound showed the best ligand fitness RMSD on the target protein (Fig. 7).
Fig. 7.

Molecular dynamics simulation for 100 ns to analyze RMSD (Root Mean Square Deviation) (Å) of the top selected compounds BC012, BC014, BS032, and RC009 to determine their conformational stability.
3.5.2. Root Mean Square Fluctuation (RMSF)
RMSF analysis was performed with alpha carbon atoms (Cα) of a protein complex with ligands. The mean RMSF values were found as 2.01 Å, 2.11 Å, 2.14 Å, and 2.37 Å for BS032, BC014, BC012, and RC009, respectively. The fluctuation pattern was similar for all four compounds. Significant peaks were observed from 10 to 45, 210 to 230, 310 to 320, 370 to 390, 400 to 425, and 430 to 450 residues. Among the top four lead compounds, RC009 showed the best stability with protein, considering the RMSF analysis (Fig. S6).
3.5.3. Solvent Accessible Surface Area (SASA)
SASA was calculated for all the four protein-ligand complexes where mean SASA values were 26.16 Å2, 13.56 Å2, 88.51 Å2, and 125.028 Å2 for BC012, BC014, BS032, and RC009 complexes. The compound BC014-protein complex showed the lowest value and better structural compactness than the other three compounds (Fig. S7).
3.5.4. Protein-ligand interaction analysis
Protein-ligand contacts of the stable ligand-protein complexes were analyzed for all the top four lead complexes, as demonstrated in Figs. S8 and S9. Of the four interaction types, only three were observed, i.e., hydrogen bonds, hydrophobic interactions, and water bridges. No ionic interactions were observed. BC012-protein complex histogram revealed that Tyr341, Arg346, and Asn392 interacted with the ligand for 100% of the simulation time with three types of bonds. For Tyr341, hydrophobic interactions and water, bridges were observed; for Arg346, hydrogen and hydrophobic interactions were found; and for Asn392 hydrophobic interactions were observed with a very brief exposure of hydrogen bonding. Therefore, these residues were capable of holding the ligand BC012 in the binding pocket. Similarly, the BC014-protein complex histogram revealed Tyr341, Phe363, and Tyr376 as interacting residues for 100 ns. BS032-protein complex histogram unveiled Tyr341, Cys377, and Thr379 as interacting residues for 100 ns. Finally, the RC009-protein complex histogram unleashed Asp102, Ile210, Gln212, Ser213, Arg306, Gln309, and Asn325 as interacting residues for 100 ns? Among these four potential lead compounds, the best protein-ligand contacting pattern was observed for the RC009-protein complex for 100 ns MDS trajectory.
3.6. MM/GBSA calculation
MM/GBSA calculation revealed free binding energies as −38.04 kcal/mol, −26.93 kcal/mol, −29.02 kcal/mol and −32.34 kcal/mol for BC012-protein complex, BC014-protein complex, BS032-protein complex, and RC009-protein complex, respectively. Among the top-selected compounds, BC012 showed the highest amount of free binding energy and BC014 showed the lowest amount of free binding energy.
4. Discussion
The Infectious Spleen and Kidney Necrosis Virus (ISKNV) is the type species of the Megalocytivirus genus, which is responsible for infecting a variety of marine and freshwater fishes and resulting in significant financial losses to the aquaculture industry worldwide [47]. ISKNV infections in tissues, like those caused by other megalocytiviruses, result in the enlargement of infected cells, which is much greater than normal cells [48,49]. Some fishes that are infected with ISKNV and other megalocytiviruses have a death rate of up to 100% [50]. Thus, the development of effective treatment solutions for infectious ailments brought on by these viral pathogens is required.
The major capsid protein (MCP) (ORF006) is an essential structural component of ISKNV that mediates virus entry into the host cell [51]. According to a study, ISKNV employs MCP to connect with the caveolin-1-protein (Cav-1) of the host cell to trigger caveolin endocytosis [52]. Additionally, the MCP is well known for having the maximum protein concentration between the outermost envelope layer and the inner membrane of the virus. Because the major capsid protein (MCP) of ISKNV plays a crucial role in immunological invasion and the transmission of the virus from fish to fish, our objective was to find out potential drug inhibitors from the marine seaweed database that targets explicitly the MCP. Previously, a marine seaweed database (1110 compounds) has shown a potential inhibitory effect on viral pathogens [53]. Therefore, in this study, we screen out the marine seaweed database associated with computer-aided drug design procedures to select the best MCP-inhibited molecules for combating ISKNV disease in fish.
Computer-aided drug design (CADD) is one of the most promising technologies for picking novel compounds to utilize against a specific protein. CADD integrates a broad range of cutting-edge features and approaches, making it one of the most powerful tools available today [54]. Virtual screening, which encompasses molecular docking, molecular dynamic simulation, ADMET, etc., has become a fundamental aspect of drug development courtesy of the CADD techniques, which have reduced the time and money needed for the complete drug development process [46]. In this study, the three-dimensional structure of MCP was hypothesized, the models that were found were improved, and the optimal model was chosen (based on the lowest energy score) followed by previous in-silico studies [55,56]. The research discovered a decent number of Z-scores (−6.76) and the superior features of the most preferred, approved, and disallowed areas for the Ramachandran plot in the 3D structure validation test (Table 1) which allowed the model to use for further validation [57]. As an added step, we used molecular docking and other methods to find possible drug targets. The compounds were first screened using a molecular docking procedure, and the top 4 compounds with binding affinities between −9.2 and −9.9 kcal/mol were selected for further in-silico process. All of the pharmacological analogs tested positive for significant hydrogen bond interactions with the MCP protein, showing that the drugs had an inhibitory activity [15]. For the chosen compounds, RO5 revealed their drug-like qualities [58]. Four of the compounds were determined to have drug-like properties according to all five of Lipinski's standards. Compounds having favorable ADME profiles have been put through further testing to determine their toxicity to either humans or animals [38,39]. All four compounds chosen for further study were determined to be non-toxic.
In the field of organic chemistry, FMO is being used to describe the structure and reactivity of molecules [59]. The HOMO-LUMO bandgap energy is used by the theory to order to explain both the electrical and optical features of molecules [60]. The difference in energy level that exists between the HOMO and LUMO orbitals has a significant impact on a molecule's chemical kinetic stability, chemical hardness, and chemical softness. This difference also plays a role in the susceptibility of atoms to electrophilic and nucleophilic charges [61]. Even though electrons in the LUMO orbital are involved in electrophilic activities, electrons in the HOMO orbital have more freedom to participate in nucleophilic reactions. A soft molecule is one that possesses low HOMO-LUMO gap energy, a high amount of chemical reactivity, and both features together. Because an electron has a low probability of entering the high-energy LUMO, a molecule with a high frontier (HOMO-LUMO) orbital gap should have limited chemical or biological activity and high kinetic stability in this process [62]. Due to their low chemical reactivity and high kinetic stability, molecules that have a high FMO energy gap are more energetically stable than those that have a low FMO energy gap [46]. According to the findings of this research, the HOMO-LUMO gap energy of the compounds BC012, BC014, BS032, and RC009 that were chosen to be examined was much greater than expected, which indicates that the molecules are kinetically stable and have a low level of chemical reactivity [15,39].
In addition, molecular dynamics modelling is utilized to validate the hypothesis that a protein may keep its stability while bound to ligands [46,63]. Additionally, it can ascertain the stiffness and stability of protein-ligand complexes in a particular enclosed environment, such as the human body. The RMSD values of the complex systems point to the most stable compounds, and the RMSF values, which quantify mean fluctuation and define the compactness of the protein-ligand complex, show how firmly the complex is compacted together [64]. The protein-ligand contact also showed a good interacting pattern of the compound after 100 ns of molecular dynamics simulation. Therefore, in this research, after the molecular dynamic modeling, each of the four compounds BC012, BC014, BS032, and RC009 from the database demonstrated stabilities against the MCP protein. In addition, the potential for predicting the relative binding free energies for four ligands against the same target MCP was shown by the MM/GBSA approach used in this work. Therefore, we can draw the conclusion that these molecules can act as an inhibitor against the ISKNV in fish.
Despite the comprehensive computational approach to characterize and validate the lead candidates against ISKNV MCP, the current investigation has some limitations. We have used several in silico tools for molecular docking, ADMET analyses, MD simulation, MM/GBSA and DFT calculation but the accuracy of these tools are not perfect. It is not fully clear that how well the lead candidates will inhibit the MCP in vitro and in vivo for which further wet-lab based studies are required. In addition, Structure Based Drug Design (SBDD) methods have various challenges, such as standard benchmarking, constrained prediction approaches, and a paucity of datasets for different computational investigations [65]. However, computational drug design approach is highly beneficial in its merit to reduce time, labor and cost for the effective discovery of new drugs along with repurposing some existing drugs for therapeutic proposes and this technique has been utilized for the successful discovery of many drugs including Captopril, Dorzolamide, Indinavir, Ritonavir, Aliskiren and so on [66].
5. Conclusions
This is the first study to our knowledge to use a standardized in-silico approach to identify potential antiviral drug candidates that target the major capsid protein. Using integrative molecular modeling, virtual screening, molecular docking, ADMET, and MD simulation approaches, the BC012, BC014, BS032, and RC009 compounds were identified as potential drug candidates from the marine seaweed database. These compounds can inhibit the activity of the major capsid protein, which will aid in the fight against ISKNV in fish. In addition, researchers will be able to develop alternate strategies for the treatment of ISKNV infections by evaluating the activity of the molecule via a range of lab-based tests.
Data availability statement
Data included in article/supp. material/referenced in article.
Authors’ contributions
Sk Injamamul Islam; Sheikh Sunzid Ahmed; Saloa Sanjida; Moslema Jahan Mou: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed materials, analysis tools or data; Wrote the manuscript. Nasim Habib; Md. Akib Ferdous: Analyzed and interpreted the data; Wrote the manuscript.
Funding statement
No funding
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Sk Injamamul Islam reports statistical analysis was provided by Jashore University of Science and Technology. Sk Injamamul Islam reports a relationship with Jashore University Of Science and Technology that includes: non-financial support. Sk Injamamul Islam has patent pending to Assignee. Not applicable.
Acknowledgments
All the authors of this manuscript are thankful to Dr. Foysal Ahmed Sagore and BioSoL center for the technical support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2023.e16383.
Contributor Information
Sk Injamamul Islam, Email: injamamulislam017@gmail.com.
Sheikh Sunzid Ahmed, Email: sunzid79@gmail.com.
Nasim Habib, Email: nhalvee@gmail.com.
Md Akib Ferdous, Email: akibferdousniloy@gmail.com.
Saloa Sanjida, Email: saloa.sanjida94@gmail.com.
Moslema Jahan Mou, Email: moslemajahanmou@gmail.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Fu X., Li N., Liu L., Lin Q., Wang F., Lai Y., Jiang H., Pan H., Shi C., Wu S. Genotype and host range analysis of infectious spleen and kidney necrosis virus (ISKNV) Virus Gene. 2011;42:97–109. doi: 10.1007/s11262-010-0552-x. [DOI] [PubMed] [Google Scholar]
- 2.Fu X., Li K., Niu Y., Lin Q., Liang H., Luo X., Liu L., Li N. The mTOR/PGC-1α/SIRT3 pathway drives reductive glutamine metabolism to reduce oxidative stress caused by ISKNV in CPB cells. Microbiol. Spectr. 2022;10 doi: 10.1128/spectrum.02310-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He J.G., Deng M., Weng S.P., Li Z., Zhou S.Y., Long Q.X., Wang X.Z., Chan S.-M. Complete genome analysis of the Mandarin fish infectious spleen and kidney necrosis iridovirus. Virol. 2001;291:126–139. doi: 10.1006/viro.2001.1208. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y., Lü L., Weng S., Huang J., Chan S.-M., He J. Molecular epidemiology and phylogenetic analysis of a marine fish infectious spleen and kidney necrosis virus-like (ISKNV-like) virus. Arch. Virol. 2007;152:763–773. doi: 10.1007/s00705-006-0870-4. [DOI] [PubMed] [Google Scholar]
- 5.Guo C.-J., Wu Y.-Y., Yang L.-S., Yang X.-B., He J., Mi S., Jia K.-T., Weng S.-P., Yu X.-Q., He J.-G. Infectious spleen and kidney necrosis virus (a fish iridovirus) enters Mandarin fish fry cells via caveola-dependent endocytosis. J. Virol. 2012;86:2621–2631. doi: 10.1128/JVI.06947-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tidona C.A., Schnitzler P., Kehm R., Darai G. Is the major capsid protein of iridoviruses a suitable target for the study of viral evolution? Virus Gene. 1998;16:59–66. doi: 10.1023/A:1007949710031. [DOI] [PubMed] [Google Scholar]
- 7.Dong C.-F., Xiong X.-P., Shuang F., Weng S.-P., Zhang J., Zhang Y., Luo Y.-W., He J.-G. Global landscape of structural proteins of infectious spleen and kidney necrosis virus. J. Virol. 2011;85:2869–2877. doi: 10.1128/JVI.01444-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong C., Xiong X., Luo Y., Weng S., Wang Q., He J. Efficacy of a formalin-killed cell vaccine against infectious spleen and kidney necrosis virus (ISKNV) and immunoproteomic analysis of its major immunogenic proteins. Vet. Microbiol. 2013;162:419–428. doi: 10.1016/j.vetmic.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Li N., Fu X., Guo H., Lin Q., Liu L., Zhang D., Fang X., Wu S. Protein encoded by ORF093 is an effective vaccine candidate for infectious spleen and kidney necrosis virus in Chinese perch Siniperca chuatsi. Fish Shellfish Immunol. 2015;42:88–90. doi: 10.1016/j.fsi.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Lim S.M., Xie T., Westover K.D., Ficarro S.B., Tae H.S., Gurbani D., Sim T., Marto J.A., Jänne P.A., Crews C.M., Gray N.S. Development of small molecules targeting the pseudokinase Her3. Bioorg. Med. Chem. Lett. 2015;25:3382–3389. doi: 10.1016/j.bmcl.2015.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes J.P., Rees S., Kalindjian S.B., Philpott K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szymański P., Markowicz M., Mikiciuk-Olasik E. Adaptation of high-throughput screening in drug discovery—toxicological screening tests. Int. J. Mol. Sci. 2011;13:427–452. doi: 10.3390/ijms13010427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar V., Jung Y.-S., Liang P.-H. Anti-SARS coronavirus agents: a patent review (2008–present) Expert Opin. Ther. Pat. 2013;23:1337–1348. doi: 10.1517/13543776.2013.823159. [DOI] [PubMed] [Google Scholar]
- 14.Wichapong K., Nueangaudom A., Pianwanit S., Sippl W., Kokpol S. Identification of potential hit compounds for Dengue virus NS2B/NS3 protease inhibitors by combining virtual screening and binding free energy calculations. Trop. Biomed. 2013;30:388–408. [PubMed] [Google Scholar]
- 15.Islam S.I., Saloa S., Mahfuj S., Islam M.J., Mou M.J. Computer-aided drug design of Azadirachta indica compounds against nervous necrosis virus by targeting grouper heat shock cognate protein 70 (GHSC70): quantum mechanics calculations and molecular dynamic simulation approaches. Genom. Inform. 2022;20 doi: 10.5808/gi.21063. e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallimore W. In: Marine Metabolites: Oceans of Opportunity. Badal S., Delgoda R., editors. Elsevier; 2017. pp. 377–400. (Pharmacognosy). [DOI] [Google Scholar]
- 18.Skolnick J., Gao M., Zhou H., Singh S. AlphaFold 2: why it works and its implications for understanding the relationships of protein sequence, structure, and function. J. Chem. Inf. Model. 2021;61:4827–4831. doi: 10.1021/acs.jcim.1c01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heo L., Park H., Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:W384–W388. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramachandran G.N., Ramakrishnan C., Sasisekharan V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 22.Singh B., Gupta G. Advances in Digital Forensics XV: 15th IFIP WG 11.9 International Conference. Springer; 2019. Analyzing windows subsystem for linux metadata to detect timestamp forgery; pp. 159–182. [DOI] [Google Scholar]
- 23.Housani B.A., Mutrib B., Jaradi H. 2009 International Conference on the Current Trends in Information Technology (CTIT) United Arab Emirates, IEEE; Dubai: 2009. The Linux review - Ubuntu desktop edition - version 8.10; pp. 1–6. [DOI] [Google Scholar]
- 24.Morris G.M., Huey R., Olson A.J. Using Autodock for ligand‐receptor docking. Curr. Protoc. Bioinformatics. 2008;24:8–14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 25.Davis G.D.J., Vasanthi A.H.R. Seaweed metabolite database (SWMD): a database of natural compounds from marine algae. Bioinformation. 2011;5:361–364. doi: 10.6026/97320630005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17:490–519. doi: 10.1002/(SICI)1096-987X. (199604)17:5/6%3C490::AID-JCC1%3E3.0.CO;2-P. [DOI] [Google Scholar]
- 27.Dallakyan S., Olson A.J. In: Chemical Biology: Methods in Molecular Biology. Hempel J., Williams C., Hong C., editors. Humana Press.; New York: 2015. Small-molecule library screening by docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 28.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tisdall J. O'Reilly Media, Inc.; 2003. Mastering Perl for Bioinformatics: Perl Programming for Bioinformatics; pp. 1–400. [Google Scholar]
- 30.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daina A., Zoete V. A boiled‐egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee P., Eckert A.O., Schrey A.K., Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Z., Zhang Y. Interfacing ab initio quantum mechanical method with classical drude osillator polarizable model for molecular dynamics simulation of chemical reactions. J. Chem. Theor. Comput. 2008;4:1237–1248. doi: 10.1021/ct800116e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hussain M.U., Khan M., Ibrahim M., Khalid A., Ali S., Hussain M., Saleem N., Ahmad S., Muhammad A.G., Sehemi Al, Sultan A. Structural parameters, electronic, linear and nonlinear optical exploration of thiopyrimidine derivatives: a comparison between DFT/TDDFT and experimental study. J. Mol. Struct. 2020;1201 doi: 10.1016/j.molstruc.2019.127183. [DOI] [Google Scholar]
- 35.Maity A., Samanta S., Biswas D., Chakravorty D. Studies on nanoconfinement effect of NiO-SiO2 spin glass within mesoporous Al2O3 template. J. Alloys Compd. 2021;887 doi: 10.1016/j.jallcom.2021.161447. [DOI] [Google Scholar]
- 36.Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012;4:1–17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neese F. Wennmohs, Becker U., Riplinger C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020;152 doi: 10.1063/5.0004608. [DOI] [PubMed] [Google Scholar]
- 38.Sanjida S., Mou M.J., Islam S.I., Mahfuj S. An In-silico approaches for identification of potential natural antiviral drug candidates against Erythrocytic necrosis virus (Iridovirus) by targeting Major capsid protein: a Quantum mechanics calculations approach. Int. J. Life Sci. Biotechnol. 2022;5:294–315. doi: 10.38001/ijlsb.1074392. [DOI] [Google Scholar]
- 39.Islam S.I., Mou M.J., Sanjida S., Mahfuj S. An in-silico approach for identifying phytochemical inhibitors against nervous necrosis virus (NNV) in Asian sea bass by targeting capsid protein. Genet. Aquat. Org. 2022;6:GA487. doi: 10.4194/GA487. [DOI] [Google Scholar]
- 40.Zhang I.Y., Wu J., Xu X. Extending the reliability and applicability of B3LYP. Chem. Commun. 2010;46:3057–3070. doi: 10.1039/C000677G. [DOI] [PubMed] [Google Scholar]
- 41.Schneebeli S.T., Bochevarov A.D., Friesner R.A. Parameterization of a B3LYP specific correction for noncovalent interactions and basis set superposition error on a gigantic data set of CCSD (T) quality noncovalent interaction energies. J. Chem. Theor. Comput. 2011;7:658–668. doi: 10.1021/ct100651f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganji M.D., Hosseini-Khah S.M., Amini-Tabar Z. Theoretical insight into hydrogen adsorption onto graphene: a first-principles B3LYP-D3 study. Phys. Chem. Chem. Phys. 2015;17:2504–2511. doi: 10.1039/C4CP04399E. [DOI] [PubMed] [Google Scholar]
- 43.Li Y., Evans J.N. The Fukui Function: a key concept linking frontier molecular orbital theory and the hard-soft-acid-base principle. J. Am. Chem. Soc. 1995;117:7756–7759. doi: 10.1021/ja00134a021. [DOI] [Google Scholar]
- 44.Majeed A., Hussain W., Yasmin F., Akhtar A., Rasool N. Virtual screening of phytochemicals by targeting HR1 domain of SARS-CoV-2 S protein: molecular docking, molecular dynamics simulations, and DFT studies. BioMed Res. Int. 2021;2021:1–19. doi: 10.1155/2021/6661191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z., Pan H., Sun H., Kang Y., Liu H., Cao D., Hou T. fastDRH: a webserver to predict and analyze protein–ligand complexes based on molecular docking and MM/PB (GB) SA computation, Brief. Bioinformatics. 2022;23 doi: 10.1093/bib/bbac201. bbac201. [DOI] [PubMed] [Google Scholar]
- 46.Bharadwaj S., Dubey A., Yadava U., Mishra S.K., Kang S.G., Dwivedi V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro, Brief. Bioinformatics. 2021;22:1361–1377. doi: 10.1093/bib/bbaa382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y., Fu H.-C., Wang Y.-Y., Huang H.-Z., Fu X.-Z., Li N.-Q. The dynamic immune responses of Mandarin fish (Siniperca chuatsi) to ISKNV in early infection based on full-length transcriptome analysis and weighted gene co-expression network analysis. Fish Shellfish Immunol. 2022;122:191–205. doi: 10.1016/j.fsi.2022.02.017. [DOI] [PubMed] [Google Scholar]
- 48.He J., Zeng K., Weng S., Chan S.-M. Experimental transmission, pathogenicity and physical–chemical properties of infectious spleen and kidney necrosis virus (ISKNV) Aquaculture. 2002;204:11–24. doi: 10.1016/S0044-8486(01)00639-1. [DOI] [Google Scholar]
- 49.Liu L., Yu L., Fu X., Lin Q., Liang H., Niu Y., Li N. First report of megalocytivirus (iridoviridae) in cultured bluegill sunfish, Lepomis macrochirus, in China. Microb. Pathog. 2019;135 doi: 10.1016/j.micpath.2019.103617. [DOI] [PubMed] [Google Scholar]
- 50.Johnson S.J., Hick P.M., Robinson A.P., Rimmer A.E., Tweedie A., Becker J.A. The impact of pooling samples on surveillance sensitivity for the megalocytivirus Infectious spleen and kidney necrosis virus. Transbound. Emerg. Dis. 2019;66:2318–2328. doi: 10.1111/tbed.13288. [DOI] [PubMed] [Google Scholar]
- 51.Throngnumchai B., Jitrakorn S., Sangsuriya P., Unajak S., Khunrae P., Dong H.T., Saksmerprome V., Rattanarojpong T. Refolded recombinant major capsid protein (MCP) from Infectious Spleen and Kidney Necrosis Virus (ISKNV) effectively stimulates serum specific antibody and immune related genes response in Nile tilapia (Oreochromis niloticus), Protein Expr. Purif. 2021;184 doi: 10.1016/j.pep.2021.105876. [DOI] [PubMed] [Google Scholar]
- 52.Guo C.-J., Yang X.-B., Wu Y.-Y., Yang L.-S., Mi S., Liu Z.-Y., Jia K.-T., Huang Y.-X., Weng S.-P., Yu X.-Q., He J.-G. Involvement of caveolin-1 in the Jak–Stat signaling pathway and infectious spleen and kidney necrosis virus infection in Mandarin fish (Siniperca chuatsi) Mol. Immunol. 2011;48:992–1000. doi: 10.1016/j.molimm.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muteeb G., Alshoaibi A., Aatif M., Rehman M., Qayyum M.Z. Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic. Appl. Biol. Chem. 2020;63:1–12. doi: 10.1186/s13765-020-00564-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 55.Islam S.I., Mou M.J. Analysis of a hypothetical protein from Vibrio harveyi identified possible connection with biopolymer metabolism: an in-silico approach. J. Appl. Biol. Sci. 2022;16:191–205. [Google Scholar]
- 56.Islam S.I., Jahan M.M. Functional annotation of uncharacterized protein from Photobacterium damselae subsp. piscicida (Pasteurella piscicida) and comparison of drug target between conventional medicine and phytochemical compound against disease treatment in fish: an in-silico approach. Genet. Aquat. Org. 2022;6:GA453. doi: 10.4194/GA453. [DOI] [Google Scholar]
- 57.Islam S.I., Mou M.J., Sanjida S., Mahfuj S., Alam M.A., Yeasmin A. An In-silico analysis of the molecular interactions between PmCBP-VP24 and PmCBP-VP28 protein complex to understand the initial initiating events of shrimp WSSV infection. Int. J. Life Sci. Biotechnol. 2022;5:235–246. doi: 10.38001/ijlsb.1055840. [DOI] [Google Scholar]
- 58.Lipinski C.A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 59.Seeman J.I., Fukui Kenichi. Frontier molecular orbital theory, and the Woodward‐Hoffmann rules. Part II. A sleeping beauty in chemistry. Chem. Rec. 2022;22 doi: 10.1002/tcr.202100300. [DOI] [PubMed] [Google Scholar]
- 60.Senna M. Materials design through mechanochemical processing. High-Energy Ball Milling. 2010:63–91. doi: 10.1533/9781845699444.1.63. [DOI] [Google Scholar]
- 61.Huang Y., Rong C., Zhang R., Liu S. Evaluating frontier orbital energy and HOMO/LUMO gap with descriptors from density functional reactivity theory. J. Mol. Model. 2017;23:1–12. doi: 10.1007/s00894-016-3175-x. [DOI] [PubMed] [Google Scholar]
- 62.Miar M., Shiroudi A., Pourshamsian K., Oliaey A.R., Hatamjafari F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects. J. Chem. Res. 2021;45:147–158. doi: 10.1177/1747519820932091. [DOI] [Google Scholar]
- 63.Aljahdali M.O., Molla M.H.R., Ahammad F. Compounds identified from marine mangrove plant (Avicennia alba) as potential antiviral drug candidates against WDSV, an in-silico approach. Mar. Drugs. 2021;19:253. doi: 10.3390/md19050253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krupanidhi S., Peele K.A., Venkateswarulu T., Ayyagari V.S., Bobby M.N., Babu D.J., Narayana A.V., Aishwarya G. Screening of phytochemical compounds of Tinospora cordifolia for their inhibitory activity on SARS-CoV-2: an in silico study. J. Biomol. Struct. Dyn. 2021;39:5799–5803. doi: 10.1080/07391102.2020.1787226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saidani N., Grando D., Valadie H., Bastien O., Maréchal E. Potential and limits of in silico target discovery—Case study of the search for new antimalarial chemotherapeutic targets. Infect. Genet. Evol. 2009;9:359–367. doi: 10.1016/j.meegid.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 66.Baig M.H., Ahmad K., Roy S., Ashraf J.M., Adil M., Siddiqui M.H., Khan S., Kamal M.A., Provazník I., Choi I. Computer aided drug design: success and limitations. Curr. Pharmaceut. Des. 2016;22:572–581. doi: 10.2174/1381612822666151125000550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supp. material/referenced in article.