

Abstract
Background:
Alcohol consumption has been shown to disrupt hepatic lipid homeostasis. Long-chain acyl-CoA synthetase 1 (ACSL1) critically regulates hepatic fatty acid metabolism and lipid homeostasis by channeling fatty acids to lipid metabolic pathways. However, it remains unclear how ACSL1 contributes to the development of alcohol-associated liver disease (ALD).
Methods:
We performed chronic alcohol feeding animal studies with hepatocyte-specific ACSL1 knockout (ACSL1Δhep) mice, hepatocyte-specific STAT5 knockout (STAT5Δhep) mice, and ACSL1Δhep based-STAT5B overexpression (Stat5b-OE) mice. Cell studies were conducted to define the causal role of ACSL1 deficiency in the pathogenesis of alcohol-induced liver injury. The clinical relevance of the STAT5-ACSL1 pathway was examined using liver tissues from patients with alcoholic hepatitis (AH) and normal subjects (Normal).
Results:
We found that chronic alcohol consumption reduced hepatic ACSL1 expression in AH patients and ALD mice. Hepatocyte-specific ACSL1 deletion exacerbated alcohol-induced liver injury by increasing free fatty acids (FFA) accumulation and cell death. Cell studies revealed that FFA elicited the translocation of BAX and p-MLKL to the lysosomal membrane, resulting in lysosomal membrane permeabilization (LMP) and thereby initiating lysosomal-mediated cell death pathway. Furthermore, we identified that the signal transducer and activator of transcription 5 (STAT5) is a novel transcriptional regulator of ACSL1. Deletion of STAT5 exacerbated alcohol-induced liver injury in association with downregulation of ACSL1, and reactivation of ACSL1 by STAT5 overexpression effectively ameliorated alcohol-induced liver injury. In addition, ACSL1 expression was positively correlated with STAT5 and negatively correlated with cell death was also validated in the liver of AH patients.
Conclusions:
ACSL1 deficiency due to STAT5 inactivation critically mediates alcohol-induced lipotoxicity and cell death in the development of ALD. These findings provide insights into alcohol-induced liver injury.
Keywords: Alcohol-associated liver disease, STAT5, ACSL1, Cell death, Lysosomal membrane permeabilization, Free fatty acids
1. Introduction
Alcohol-associated liver disease (ALD) is a leading cause of morbidity and mortality worldwide. It develops from a simple fatty liver (steatosis) to alcoholic hepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma [1]. About 95,000 people die each year from alcohol-related causes, making alcohol the third-leading preventable cause of death in the United States [2]. However, there are limited Food and Drug Administration (FDA)-approved pharmacological or nutritional therapies for any stage of ALD [3]. Therefore, a better understanding of ALD pathogenesis and identification of therapeutic targets are needed.
Long-chain acyl-CoA synthetase (ACSL) plays a crucial role in fatty acid metabolism and lipid homeostasis by catalyzing free fatty acid (FFA) to fatty acyl-CoA (FA-CoA) [4]. There are five distinct isoforms of ACSLs in the body, including ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6 [5,6]. While all ACSL isoforms except ACSL6 are expressed in the liver, ACSL1 is the most predominant isoform and contributes 50 % of total hepatic ACSLs activity [7]. ACSL1 can effectively utilize saturated fatty acids including 10–16 carbon atoms and unsaturated fatty acids with 20–26 carbon atoms for fatty acid oxidation, triglyceride synthesis, and phospholipid production [8,9]. It has been shown that hepatocyte deletion of ACSL1 reduces, and hepatocyte overexpression of ACSL1 increases, hepatic acyl-CoA content [7,10], so ACSL1 plays crucial roles in adipose tissue [11], heart [12], liver [7] and brain [13] involved in various human diseases. However, the role of ACSL1 in ALD has not been defined.
Signal transducer and activator of transcription (STATs) proteins are transcription factors involved in signal transduction and mediate diverse biological processes [14,15]. Perturbation of STATs has been observed in various diseases, such as cancer [16], immune system disorders [17], and metabolic diseases [18,19]. Alcohol consumption has also been reported to activate STAT1/3 [20–22] but inhibited STAT2 [22]. One of the most extensively investigated STATs is STAT5, which plays a key role in liver metabolism and pathophysiology [23,24]. Hepatocyte-specific knockout of STAT5 induces abnormal lipid metabolism in the liver of mice, causing fatty liver [25], fibrosis, and even cancer [26]. Although both are involved in hepatic lipid metabolism, relationship between STAT5 and ACSL1 remains unknown at either steady state or under disease conditions.
In this study, we provide evidence that ACSL1 reduction is a driving force of alcohol-induced liver injury via activating BAX/p-MLKLlysosomal membrane translocation, which causes lysosomal membrane permeabilization (LMP). Importantly, the study uncovers a novel transcriptional regulatory mechanism of ACSL1 by identifying STAT5 binding to the B promoter of ACSL1.
2. Materials and methods
2.1. Human liver samples
Liver samples from patients (age range 32–49 years old) with severe alcoholic hepatitis (AH) and healthy donors (normal control) were collected at Johns Hopkins University under the support of NIAAA-funded Clinical Resource for Alcoholic Hepatitis Investigations (R24AA025017). Clinical information of patients with severe AH has been published [27]. Collecting tissues from explanted livers or biopsies from normal subjects’ livers had been approved by Institutional Review Boards at Johns Hopkins Medical Institutions (IRB00107893, IRB00021325).
2.2. Mice
ACSL1 flox mice were a gift from Dr. Rosalind Coleman (Acsl1flox) [7]. STAT5 flox mice (Stat5flox, Strain #: 032053-JAX) and Albumin-Cre mice (Alb-cre, Strain #: 003574) were purchased from the Jackson Laboratory (Bar Harbor, ME). Acsl1flox mice or Stat5flox mice were bred with Alb-cre mice to generate mice with hepatocyte-specific knockout of ACSL1 (Acsl1Δhep) or Stat5 (Stat5Δhep). Sibling littermates of floxed mice (Flox) were used as controls. To selectively overexpress Stat5b (Stat5b-OE) in the hepatocytes in mice, AAV8-TBG-Stat5b vectors were used (Vector Biolabs). Stat5b overexpression was induced by the tail vein injection of recombinant adeno-associated viral serotype 8 (AAV8) gene transfer vectors at 1 × 1011 genome copies per mouse. Mice injected with AAV8-Null vectors were used as control (Stat5b-null). All experiments were performed following the protocol approved by the North Carolina Research Campus Institutional Animal Care and Use Committee.
2.3. Chronic alcohol feeding and treatments
Twelve-week-old male mice were fed an alcohol-containing Lieber-DeCarli liquid diet (alcohol-fed; AF) or an isocaloric control liquid diet (pair-fed; PF) for 8 weeks as described previously [28]. Briefly, alcoholfed mice (AF) were fed ad libitum on an ethanol-containing Lieber-DeCarli liquid diet, whereas control mice in the pair-fed group (PF) were fed an isocaloric control lipid diet with the same amount consumed by the alcohol group. Ethanol concentrations in the alcohol diet ranged from 4.0 % to 4.42 % with a 0.14 % increase every 2 weeks. All ingredients used in the liquid diets were purchased from Dyets, Inc. (Bethlehem, PA) except for ethanol (Sigma-Aldrich, St. Louis, MO).
2.4. Establishment of stable STAT5 and ACSL1 knockdown cell lines
In a 6-well tissue culture plate, Hepa-1c1c7 cells (American Type Culture Collection, Rockville, MD) were seeded at a density of 1.5 × 105 in 3 mL of standard growth medium per well. After reaching 60 % confluency, cells were transfected with Stat5b Double Nickase Plasmid or Acsl1 Double Nickase Plasmid following the manufacturer’s guidelines (Santa Cruz Biology). Cells transfected with Control Double Nickase Plasmid were used as control. After 24 h, cells were selected with puromycin at 1–3 mg/mL for 3–5 days. After selection, the singlecell colonies were isolated to confirm complete allelic knockouts by Western blot and Real-time PCR analysis.
2.5. Cell transfections for BAX and MLKL knockdown
In a 12-well tissue culture plate, Hepa-1c1c7 cells were seeded at a density of 1 × 105 in 1 ml standard growth medium per well. After overnight incubation, cells were transfected with BAX siRNA, MLKL siRNA, or control siRNA using lipofectamine 3000 transfection. After 24 h of transfection, cells were treated with palmitic acid (PA) at 100 μM for 8 h or 24 h.
2.6. Biochemical measurements
Serum ALT and AST levels were measured by commercial kits (Cayman Chemical), respectively. Serum LCN2 levels were measured by mouse Lipocalin-2/NGAL ELISA kit (R&D systems). The hepatic levels of TG and FFA were measured with colorimetric assay kits (BioVision). Briefly, lipids were extracted using chloroform/methanol (2:1), vacuumed, and redissolved in 5 % Triton X-100/methyl alcohol mixture (1:1 vol/vol), then FFA and TG contents were determined according to the manufacturer’s instructions.
2.7. Flow cytometry analyses
For the frequency of neutrophils in the liver, antibodies including CD45 (BioLegend), CD11b (BioLegend), and Ly6G (BioLegend) were added to cells in a flow cytometry staining buffer. After 25 min of incubation in dark on ice, 7-AAD (5 μl/per test) was added and incubated for 5 min at room temperature to exclude dead cells. Flow cytometry using a fluorescence-activated cell sorting (FACS) Melody analyzer (BD Life Sciences), and data were analyzed with Flowjo 10.1 software (BD Life Sciences).
2.8. Histopathology, immunohistochemistry (IHC), and immunofluorescence (IF)
Liver tissue paraffin sections were subjected to histopathological hematoxylin and eosin (HE) and IHC staining, and liver tissue cryostat sections were used for IF staining as previously described [29]. For co-localization of lysosomes with BAX or p-MLKL, cells were incubated with 50 nM LysoTracker red DND-99 for 30 min at 37 °C, then fixed and proceeded with IF microscopy of BAX or p-MLKL. The nuclei were counterstained with DAPI.
2.9. TUNEL staining assay and neutral lipid droplets staining with BODIPY
Liver cell death was assessed by the TUNEL method (Millipore Sigma). Neutral lipid droplets in mouse liver were stained using borondipyrromethene (BODIPY). Liver cryostat sections were incubated with 2 μM BODIPY 493/503 for 20 min at room temperature and then fixed in 4 % formaldehyde for 15 min at room temperature and counterstained with DAPI.
2.10. Western blot, immunoprecipitation (IP), and non-reducing SDS-PAGE assay
Whole protein lysates of the liver and Hepa-1c1c7 cells were extracted using lysis buffer supplemented with the protease inhibitor and phosphatase inhibitor (Sigma-Aldrich). Aliquots containing 40 μg of proteins were loaded following standard procedures, all antibodies used are summarized in supplementary Table 2.
For IP studies, total protein (1000 μg) was incubated 20 μL protein A/G agarose beads on a rocker for 1 h at 4 °C and then centrifuged at 14,000 ×g for 30 s at 4 °C to remove the protein A/G. The supernatant was collected and incubated with 2.5 μg BAX antibody overnight at 4 °C on a rocker, and then 20 μL protein A/G agarose beads were added, followed by incubation overnight with mixing, and then centrifuged at 14,000 ×g for 30 s at 4 °C. After washing three times with cold PBS, the pellets were resuspended in 60 μL of electrophoresis sample buffer, boiled for 5 min, and SDS-PAGE was performed following Western blotting procedures.
Non-reducing SDS-PAGE is standard SDS-PAGE, but the sample buffer excludes 2-mercaptoethanol or DTT.
2.11. Isolation and purification of lysosomes from Hepa-1c1c7 cells
Hepa-1c1c7 cells were seeded at a density of 2 × 106 cells/dish in 100 × 17 mm (Corning, NY, USA) in a standard grow medium. After reaching 80 % confluence, cells were treated with PA for 8 h at 100 μM in a serum-free DMEM medium. Then cells were harvested for lysosome isolation using a lysosome enrichment kit (Thermo Fisher Scientific) following the manufacturer’s instructions.
2.12. Dual-luciferase reporter assay
The sequences of ACSL1 were synthesized and cloned into the pGL3-Basic vector by GeneCopoeia, Inc. (Rockville, MD, USA). Generated reporter vectors include: WT clone for mouse Acsl1 with custom 2 kb promoter sequence, MUT1 clone for mouse ACSL1 with custom 2 kb promoter sequence with mutant TTCTAAGAA to AACTAAGAA, MUT2 clone for mouse Acsl1 with custom 2 kb promoter sequence with mutant TTCTGGCAA to AACTGGCAA, MUT3 clone for mouse Acsl1 with custom 2 kb promoter sequence with mutant both TTCTAAGAA to AACTAAGAA and TTCTGGCAA to AACTGGCAA. These vectors were transfected into Hepa-1c1c7 cells using Lipofectamine 3000. After 24 h, the relative luciferase activity was examined using a Dual-luciferase (R) Reporter Assay System by Promega GloMax 96 Microplate Luminometer (Promega).
2.13. RNA isolation and real-time PCR
Total RNA was isolated using TRIzol Reagent (Invitrogen, Oregon, USA) from mouse liver tissue. qRT-PCR was performed using SYBR Green. Primers used for real-time PCR are depicted in Supplementary Key Resources Table 1. The mRNA levels of genes were normalized to that of RPS17 and expressed relative to the control.
2.14. Transcriptome processing and analysis
The gene expression profiles of GSE143318 [27] and GSE28619 [30] were obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo), The raw data were downloaded as MINiML files, it contains the data for all platforms, samples, and GSE records of the GSE. The extracted data were normalized by log2 transformation. The microarray data were normalized by the normalized quantiles function of the preprocess Core package in R software (version 3.4.1). Probes were converted to gene symbols according to the annotation information of the normalized data in the platform. The R software heatmap package was used to draw multi-gene correlation. The expression correlation of multiple genes was analyzed with Spearman, which describes the correlation between quantitative variables without normal distribution. P < 0.05 was considered statistically significant.
2.15. Primary hepatocytes isolation and oxygen consumption rate, real-time ATP rate assay
A two-step perfusion method was used as previously described [31]. Isolated primary hepatocytes were seeded on a 24-well cell culture microplate (1 × 105 cells/well, Agilent Technologies, United States), then the cells were treated with/without 500 μM PA for 24 h. After that, Oxygen consumption rates (OCR) were measured using Seahorse XF Cell Mito Stress Test Kit by Agilent Seahorse XFe24 Analyzers, the cells were incubated in XF assay medium (Seahorse XF-RPMI containing either 1 mM sodium pyruvate, 2 mM l-glutamine and 10 mM glucose). Three measurements were obtained under basal conditions and upon addition of oligomycin (1.5 μM), fluoro-carbonyl cyanide phenylhydrazone (FCCP, 0.5 μM), and rotenone/antimycin A (0.5 μM). ATP production was measured using Seahorse XF Real-time ATP Rate Kit, the cells were incubated in XF assay medium and measured upon addition of oligomycin (1.5 μM) and rotenone/antimycin A (0.5 μM).
2.16. Acyl-CoA analyses
Liver sample preparation and MRM of parent-daughter ion combinations for short and long-chain acyl Coenzymes A were conducted following the standard protocol detailed earlier [32,33]. Chromatographic conditions are as follows: The samples are injected on reverse phase HPLC column (Phenomenex, Hydro RP, 3 μ, 2.1 × 150 mm) and eluted with gradient between water and acetonitrile containing 0.1 % ammonium acetate by weight. The gradient for acetonitrile is 0 min – 0 %, 5 min – 65 %, at a flow rate of 0.2 ml/min. The HPLC eluate is directly introduced to ESI source of QTRAP5500 mass analyzer (ABSCIEX) in the positive ion mode with the following conditions: Curtain gas: 35 psi, GS1: 45 psi, GS2: 45 psi, Temperature: 600 °C, Ion Spray Voltage: 5500 V, Collision gas: low, Declustering Potential: 60 V, Collisional energy: 53 eV, and Entrance Potential: 10 V. The data are collected using Analyst 1.6.2 software and the MRM transition chromatograms are quantitated by MultiQuant software (both from ABSCIEX). The internal standard (C-17 analogs of the sphingolipids) signal in each chromatogram is used for normalization for recovery as well as relative quantitation of each analyte.
Description of additional materials is presented in the Supplementary Data.
2.17. Statistical analysis
Data met with normal distribution were analyzed using the independent-samples t-test or one-way analysis of variance followed by Tukey’s multiple comparison. Data that do not conform to a normal distribution were analyzed using Nonparametric tests followed by the Median test (K samples). Statistical analyses were performed using SPSS 21. Data were expressed as mean ± SD. In all tests, P < 0.05 was taken as significant.
3. Results
3.1. ACSL1 is down-regulated in the liver of patients with alcoholic hepatitis
To establish the role of ACSL1 in ALD development, we used Gene Ontology enrichment analysis and Gene Set Enrichment Analysis to analyze the main enriched signaling pathways of ACSL1 and found that fatty acid degradation pathway was significantly enriched in alcoholic hepatitis (AH) liver tissues (Fig. 1A, B). Subsequently, we investigated its expression by Kyoto Encyclopedia of Genes and Genomes analysis and observed that ACSL1 was significantly decreased in AH liver (Fig. 1C). To verify the observed hepatic ACSL1 reduction in AH, we performed Western bot and IHC staining with liver sections from normal subjects and AH patients. Compared with liver tissues from normal subjects, ACSL1 protein levels were significantly decreased in the liver of AH patients (Fig. 1D, E).
Fig. 1.
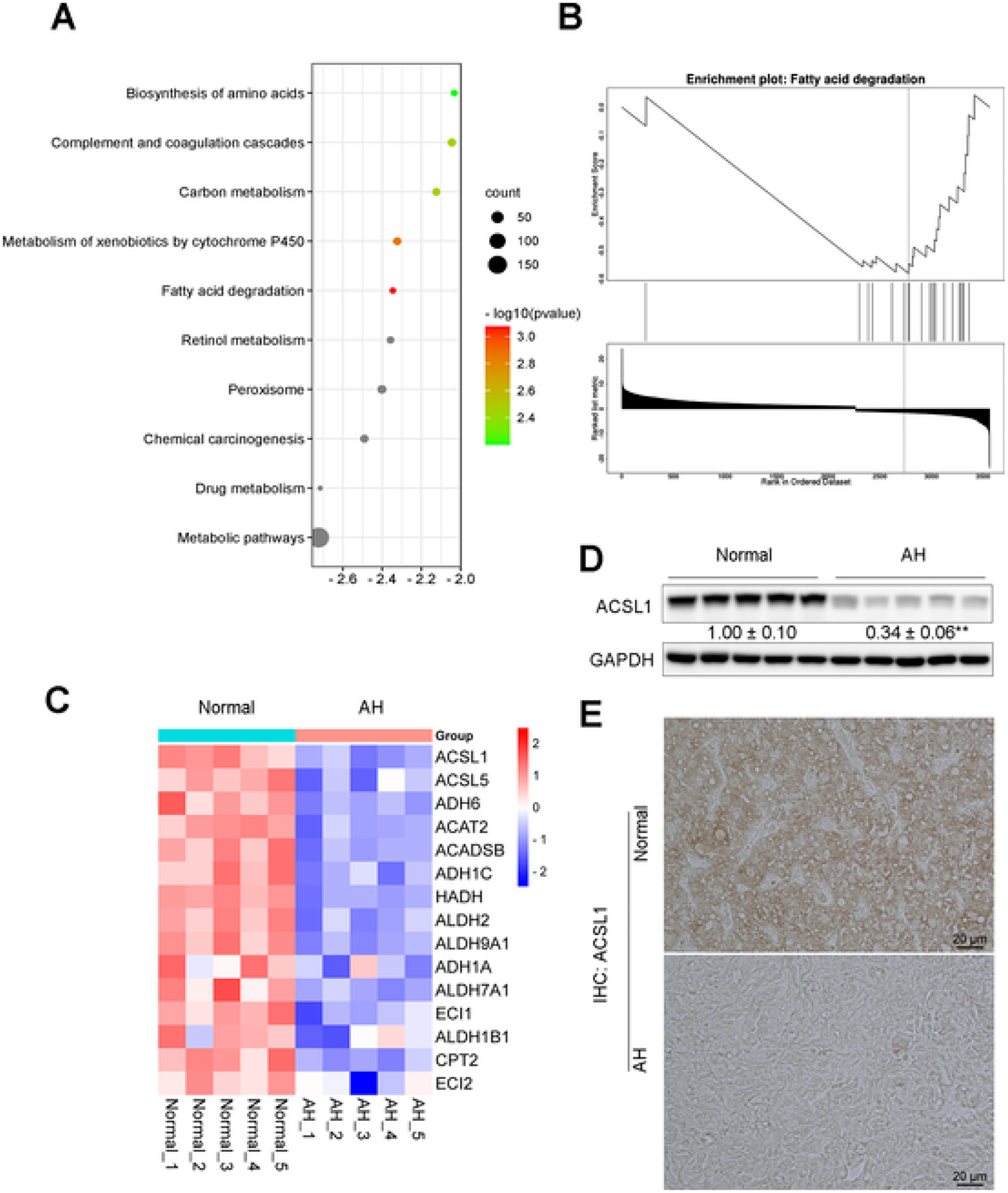
Decreased ACSL1 expression in human patients with alcoholic hepatitis. (A) Gene Ontology enrichment analysis showing the top 10 downregulated cellular pathways in alcoholic hepatitis (AH) patients. Transcriptomic data of livers from patients with AH and normal donors (Normal) was obtained from the publicly available GEO database (GSE 143318), Normal controls, n = 5; AH patients, n = 5. (B) Gene Set Enrichment Analysis showing the cellular pathway enriched in fatty acid degradation. (C) Heatmap showing the expression profiles of fatty acid degradation-associated genes that were down-regulated in AH patient liver tissues. (D) ACSL1 expression in (n = 5/group) in human liver sections with healthy liver and AH patient liver. (E) IHC staining of ACSL1 in human liver sections from AH patients and normal donors. Data are presented as mean ± SD. Values with different superscripts are significantly different.
3.2. Hepatocyte-specific ACSL1 deletion aggravates alcohol-induced liver injury
To investigate the causal role of hepatic ACSL1 in ALD, we generated hepatocyte-specific ACSL1 knockout mice (Fig. 2A, B). ACSL1 deletion increased liver weight (Fig. S1A), liver to body weight ratio (Fig. S1B), and caused higher mortality after alcohol feeding (Fig. 2C), without affecting body weight (Fig. S1C). Further analysis found that ACSL1 deletion exaggerated alcohol-induced liver injury as indicated by increased serum ALT (Fig. 2D) and AST levels (Fig. 2E). TUNEL assay revealed that ACSL1 deletion aggravated alcohol-induced hepatic cell death (Fig. 2F). ACSL1 deletion also exacerbated alcohol-induced hepatic inflammation as indicated by upregulated LCN2 expression (Fig. 2G, H) and neutrophil infiltration (Fig. 2I).
Fig. 2.
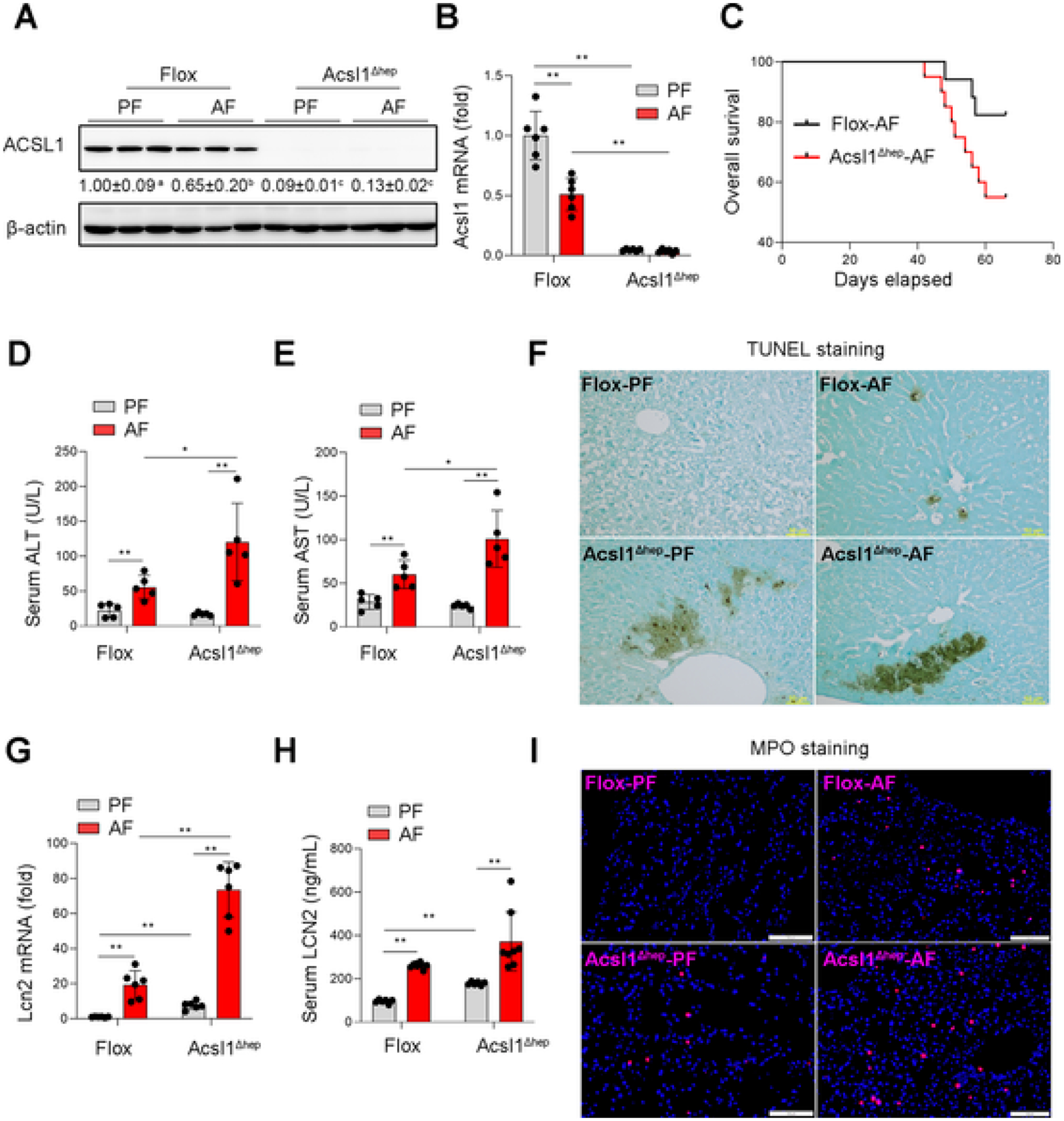
ACSL1 deficiency aggravates alcohol-induced liver injury. Wild-type Flox mice and hepatocyte-specific ACSL1 knockout mice (Acsl1 Δhep) were subjected to alcohol feeding for 8 weeks. (A) Western blot analysis of ACSL1 protein levels (n = 3/group). (B) qPCR assay of hepatic Acsl1 mRNA levels (n = 6/group). (C) Kaplan–Meier survival analysis determined the survival probability between the Flox mice and Acsl1Δhep mice with alcohol feeding for 8 weeks. (D–E) Serum levels of ALT (D) and AST (E) (n = 5/group). (F) TUNEL staining on the liver tissue sections. Scale bars = 50 μm. (G) qPCR assay of hepatic Lcn2 mRNA levels (n = 6/group). (H) Serum levels of LCN2 (n = 6–8/group). (I) IF staining of MPO in liver tissue sections (n = 6/group). Scale bars = 100 μm. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, values with different superscripts are significantly different.
3.3. Hepatocyte-specific ACSL1 deletion reduces the ability of mitochondrial fatty acid β oxidation and induces FFA accumulation in the liver
ACSL1 deletion per se increased hepatic FFA levels and aggravated alcohol-induced hepatic FFA accumulation (Fig. 3A) but did not affect hepatic TG levels (Figs. 3B; S2A, B) as well as expressions of TG synthesis genes (Fig. S2C). To understand how ACSL1 deletion causes hepatic FFA accumulation, genes involved in mitochondrial and peroxisomal β-oxidation as well as endoplasmic reticulum (ER) ω-oxidation were measured. Regardless of alcohol feeding, ACSL1 deletion downregulated mitochondrial CPT1α (Fig. 3C, D), which interacts with ACSL1 at the mitochondrial outer membrane, and is the rate-limiting enzyme for mitochondrial β-oxidation [34]. However, ACSL1 deletion led to compensatory enhancement of peroxisomal β-oxidation and ER ω-oxidation (Fig. 3C, D), which may be associated with a higher level of oxidative stress and ER stress in ACSL1 knockout mice as indicated by increased expression of CYP2E1, ATF4, and 4-HNE (Fig. S2D, E). In line with increased hepatic FFAs levels, lipidomics analysis showed that several FA-CoAs, including myristoyl-CoA (C14:0), palmitoyl-CoA (C16:0), stearoyl-CoA (C18:0), and arachidonyl-CoA (C20:4), were all significantly decreased in ACSL1 knockout mice (Fig. S2F). Subsequently, seahorse assay using isolated primary hepatocytes from floxed mice and ACSL1 deletion mice revealed that ACSL1 deletion reduced mitochondrial oxygen consumption rate (OCR) and ATP generation, which were further worsened upon palmitic acid (C16:0, PA) stimulation (Fig. 3D–F). These data indicate that hepatic ACSL1 reduction primarily impairs mitochondrial fatty acids β oxidation and leads to the accumulation of FFAs in hepatocytes in ALD.
Fig. 3.
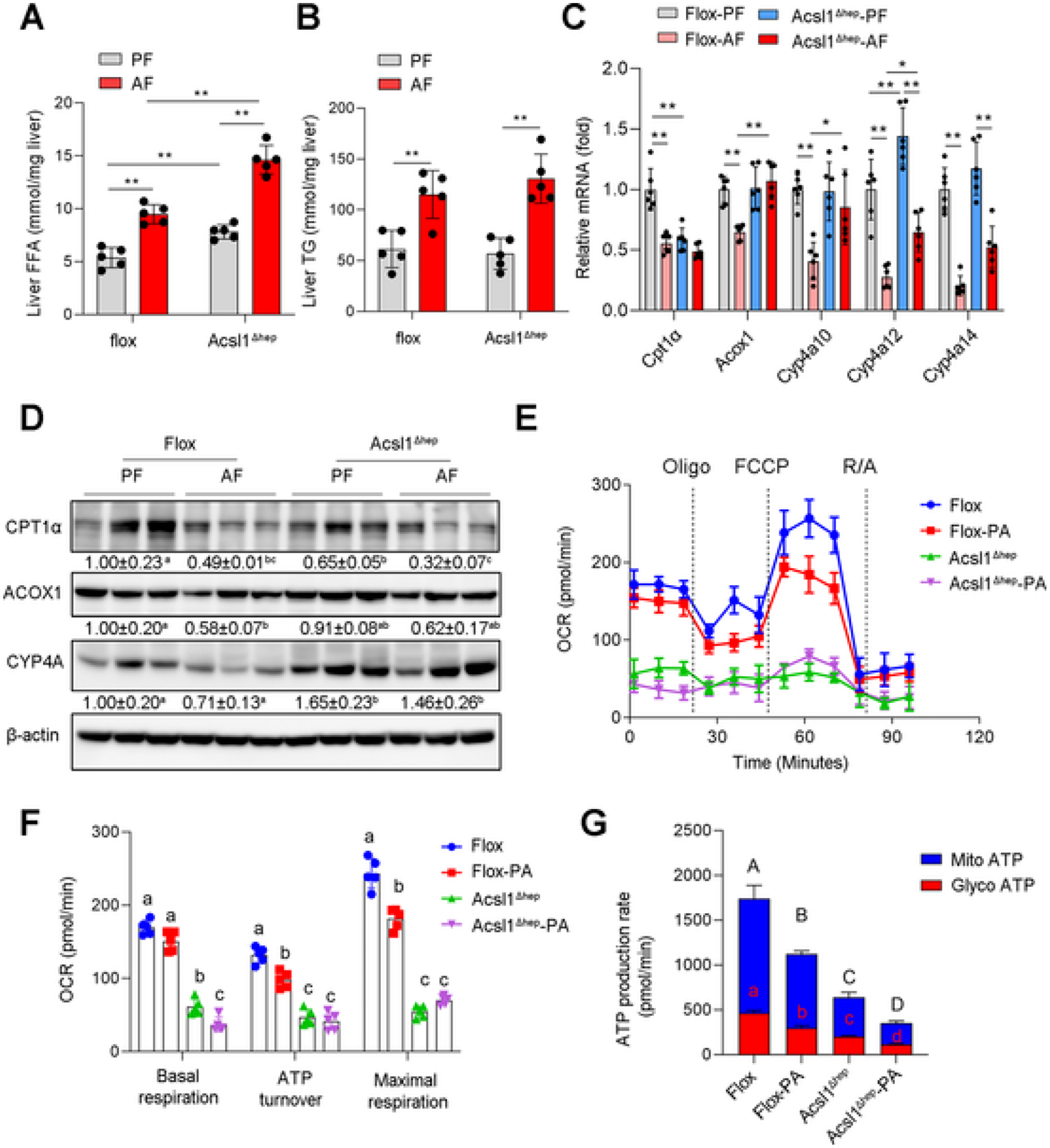
ACSL1 deficiency suppresses mitochondrial fatty acid β oxidation. (A-B) The hepatic FFA (A) and TG (B) levels in Flox mice and Acsl1Δhep mice with or without alcohol feeding for 8 weeks (n = 5/group). (C) The mRNA expression of fatty acids oxidation-related genes in liver tissues (n = 6/group). (D) Western blot analysis of fatty acid oxidation-related proteins. (E–F) Extracellular flux analysis of OCR in primary hepatocytes isolated from Flox mice and Acsl1Δhep mice with/without PA (500 μM) treatment for 24 h, cells were then sequentially treated with oligomycin (Oligo), FCCP, and rotenone/antimycin A (R/A) as indicated (E). n = 5 technical replicates of 1 biological replicate. The mean value ± SD is represented (F). (G) Seahorse XF real-time ATP rate analysis of primary hepatocytes isolated from Flox mice and Acsl1Δhep mice. With/without PA (500 μM) treatment for 24 h, the mitochondrial ATP (mitoATP) and glycolysis-generated ATP (glycoATP) were measured upon addition of oligomycin and rotenone/antimycin A. n = 5 technical replicates of 1 biological replicate. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, values with different superscripts are significantly different.
3.4. Hepatocyte-specific ACSL1 deletion sensitizes hepatocytes to cell death via activating BAX/p-MLKL pathway
Programmed cell death is thought to play a central role in the progression of alcohol-induced liver injury [35]. We next assessed apoptosis and necroptosis, the 2 major types of cell death in the liver of mice. We found that ACSL1 deletion significantly increased necroptotic marker proteins including p-RIPK3 and p-MLKL. Interestingly, among 7 apoptotic markers tested, only BAX was increased after ACSL1 deletion regardless of alcohol feeding, whereas BAK, BAD, BIM, PUMA, BCL2, and BCL-XL were not changed (Figs. 4A, S3A). Similarly, in vitro pharmacological inhibition of ACSL1 by Triacsin C (TC) dose-dependently induced BAX expression along with activation of necroptotic p-MLKL (Fig. S3B). These data suggest that ACSL1 deficiency sensitizes hepatocytes to cell death and may be associated with the activation of BAX and p-MLKL. However, it was still unclear how BAX and p-MLKL cooperate to initiate cell death in ALD. To address this question, we assessed the expression of p-MLKL and BAX in PA-treated Hepa-1c1c7 cells, and found that the expression of p-MLKL and BAX were significantly increased by PA treatment along with LDH release in a time-dependent manner (Fig. S3C–F). Further, Co-IP analysis revealed a protein interaction between BAX and p-MLKL upon PA-induced cell death (Fig. 4B). Next, to understand the mechanisms of the interaction between BAX and p-MLKL, we employed Bax inhibitor peptide V5 (BIP-V5, a Bax-mediated apoptosis inhibitor) and necrosulfonamide (NSA, a specific inhibitor of MLKL). We found that BIP-V5 significantly inhibited p-MLKL expression, but NSA did not affect BAX expression (Fig. 4C). Similarly, BAX knockdown by siRNA decreased the phosphorylation of MLKL without affecting the oligomerization of MLKL, whereas MLKL knockdown did not alter PA-induced BAX (Fig. 4D). Further analysis by IF staining confirmed that interfering with BAX blocked PA-induced p-MLKL plasma membrane translocation (Fig. 4E, F). These data suggest that BAX is an upstream regulator of MLKL.
Fig. 4.
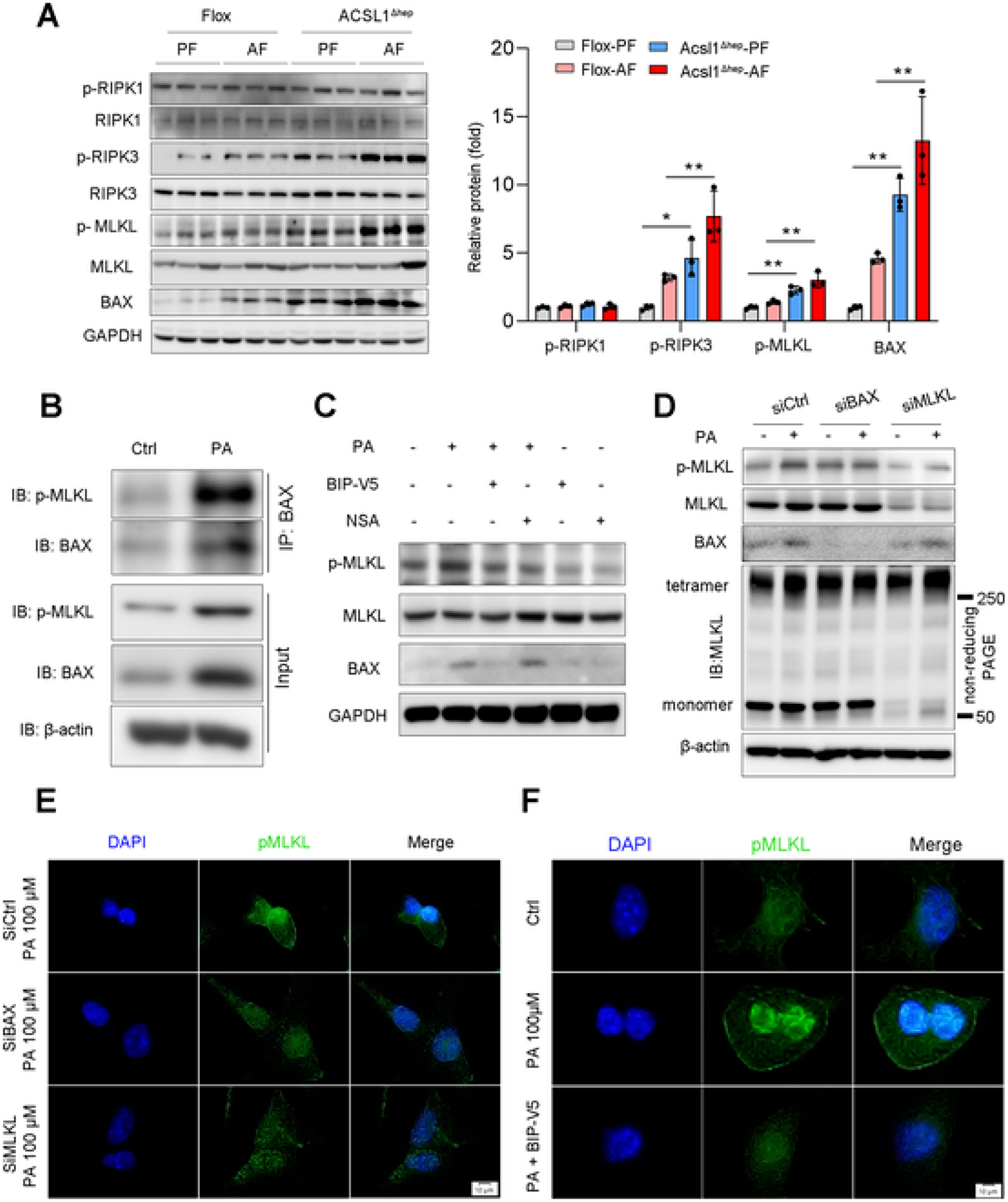
ACSL1 deficiency sensitizes hepatocytes to cell death. (A) Protein levels of apoptotic and necroptotic proteins in the liver from Flox mice and Acsl1Δhep mice with or without alcohol feeding for 8 weeks (n = 3/group). (B) IP analysis of BAX and p-MLKL protein interaction in Hepa-1c1c7 cells treated with/without PA (100 μM) for 24 h. (C) Western blot analysis of p-MLKL, MLKL, and BAX in Hepa-1c1c7 cells treated with BIP-V5 at 100 μM or NSA at 5 μM for 24 h under PA (100 μM) stimulation. (D) Western blot analysis of BAX, p-MLKL, and MLKL in Hepa-1c1c7 cells after BAX and MLKL knockdown by siRNAs with/without PA (100 μM) stimulation for 24 h. p-MLKL oligomerization was analyzed by non-reducing SDS/PAGE. (E) IF analysis of p-MLKL subcellular translocation in Hepa-1c1c7 cells after BAX and MLKL knockdown by siRNAs with/without PA (100 μM) stimulation for 24 h. Scale bars = 10 μm. (F) IF analysis of p-MLKL membrane translocation in Hepa-1c1c7 cells after BAX inhibited by its specific inhibitor BIP-V5 at 200 μM with PA stimulation for 24 h. Scale bars = 10 μm. Data are presented as mean ± SD. *P < 0.05, **P < 0.01.
3.5. ACSL1 deficiency exacerbates lysosomal translocation of BAX and p-MLKL, and blocks BAX/p-MLKL signaling preventing lysosomal membrane permeabilization
Both BAX and MLKL are involved in membrane pore formation when cells undergo programmed cell death [36]. Therefore, we questioned whether BAX-MLKL signaling affects lysosomal membrane in tegrity and if so, whether ACSL1 deficiency promotes an increase in lysosomal membrane permeabilization (LMP), which initiates a cell death pathway [37]. For this purpose, we knocked down ACSL1 in Hepa-1c1c7 cells and performed a galectin-3 puncta assay, which is used for visualization of LMP in live cells. While PA-induced galectin-3 puncta formation, more puncta were found within the leaky lysosomes in ACSL1 knockdown cells, indicating ACSL1 deficiency exacerbates PA-induced LMP (Fig. S4A, B). Next, we performed double IF staining of BAX or p-MLKL with the lysosomal tracker and found that BAX and p-MLKL were colocalized with lysosomes under PA stimulation and ACSL1 knockdown caused more lysosomal translocation of BAX and p-MLKL (Fig. 5A, B). The co-localization of BAX and p-MLKL with lysosome was also determined by Western blot in pure lysosomal fractions isolated from Hepa-1c1c7 cells with or without PA stimulation (Fig. 5C). The effect of BAX on p-MLKL lysosomal translocation was also determined by IF. Similar to blocking plasma membrane translocation, knockdown of BAX also prevented the translocation of p-MLKL to the lysosome (Fig. 5D). Further analysis by IF staining revealed that knockdown of BAX or MLKL by siRNA significantly reduced puncta formation under PA stimulation (Fig. 5E). This is consistent with the observation that cathepsin B, a lysosomal protease released from the disrupted lysosome to cytosol, was reduced in both knockdown cell models with PA treatment (Fig. 5F). These data demonstrate that BAX and MLKL are involved in LMP and BAX-mediated p-MLKL lysosomal translocation plays a key role in ACSL1 deficiency-induced LMP.
Fig. 5.
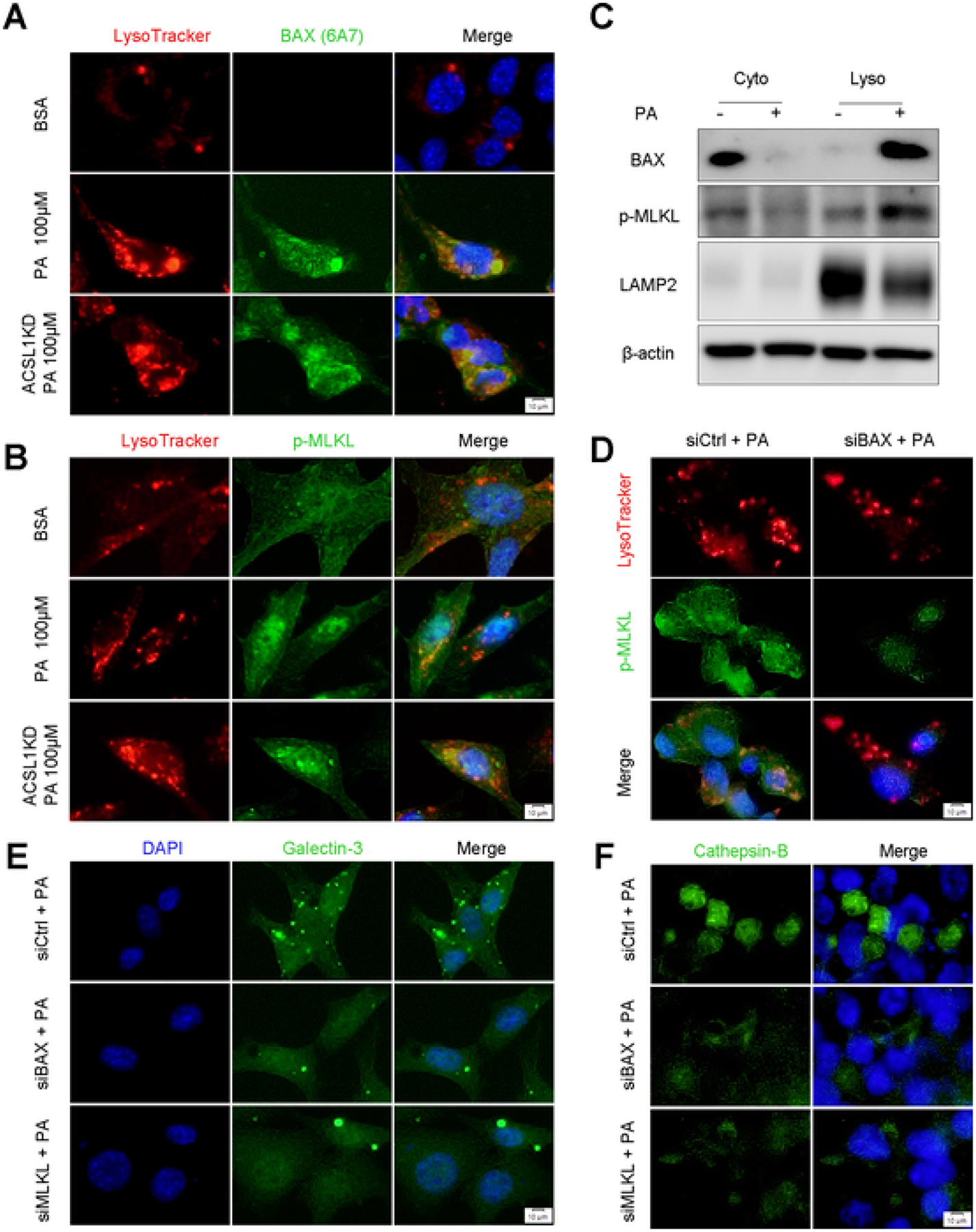
BAX and MLKL Regulate Lysosomal Membrane Permeabilization. (A) Colocalization of activated BAX and Lysosome tracker was examined by immunofluorescence microscopy in Hepa-1c1c7 cells with/without PA (100 μM) stimulation for 8 h. Scale bars = 10 μm. (B) Colocalization of p-MLKL and Lysosome tracker was examined by immunofluorescence microscopy in Hepa-1c1c7 cells with/without PA (100 μM) stimulation for 8 h. Scale bars = 10 μm. (C) Lysosomal BAX and p-MLKL were measured by Western blot using isolated lysosome fractions from Hepa-1c1c7 cells with/without PA (100 μM) stimulation for 8 h. (D) Colocalization of p-MLK and Lysosome tracker was examined by immunofluorescence microscopy in Hepa-1c1c7 cell with BAX knockdown by siRNAs under PA (100 μM) stimulation for 8 h. Scale bars = 10 μm. (E-F) Lysosomal membrane permeabilization was measured by immunofluorescence galectin-3 puncta assay (E) and IF staining of cathepsin B (F) in Hepa-1c1c7 cell with BAX or MLKL knockdown by siRNAs under PA (100 μM) stimulation for 8 h. Scale bars = 10 μm.
Next, we examined the subcellular localization and expression levels of TFEB, a master regulator of lysosomal biogenesis [38]. Unexpectedly, knockdown of BAX or MLKL did not block PA-induced TFEB translocation from the cytoplasm to the nucleus (Fig. S4C). Western blot revealed that knockdown of BAX and MLKL increased TFEB expression but did not restore PA-induced TFEB nuclear translocation. In contrast, BAX and MLKL knockdown reversed PA-reduced LAMP2, which is localized on the lysosomal membrane (Fig. S4D). These results demonstrate that BAX/p-MLKL modulates lysosomal function by altering LMP rather than affecting lysosomal biogenesis.
3.6. STAT5 specifically regulates the B-ACSL1 transcript, and hepatocyte-specific STAT5 deletion exacerbates alcohol-induced liver injury
Next, we explored the mechanism by which ACSL1 is downregulated in ALD models. It has been reported that ACSL1 gene in humans and rats can generate three transcript variants that encode the fulllength ACSL1 protein with 698 amino acids through alternative splicing [39,40]. Similar to humans and rats, the mouse ACSL1 gene also has three transcripts (v1, v2, and v3) that contain the same coding exon 2 but different 5′ untranslated regions. To understand how ACSL1 is transcriptionally regulated, we designated v1 as A-ACSL1, v2 as B-ACSL1, and v3 as C-ACSL1 consistent with the human and rat ACSL1 mRNA nomenclature (Fig. 6A upper) and performed nucleotide sequence analysis within the 2 kb 5′-flanking sequence of all three ACSL1 promoters. We found that there are two putative STAT5 binding sites located at the B promoter of ACSL1 but not the other 2 promoters, one is consensus STAT5 binding motifs (TTCNNNGAA) and the other one is non-consensus motifs (Fig. 6A lower). Next, the dual-luciferase reporter assay confirmed the functionality of these binding sites. Mutation of these binding sites blocked ACSL1 expression compared with wild-type plasmids (Fig. 6A, B).
Fig. 6.
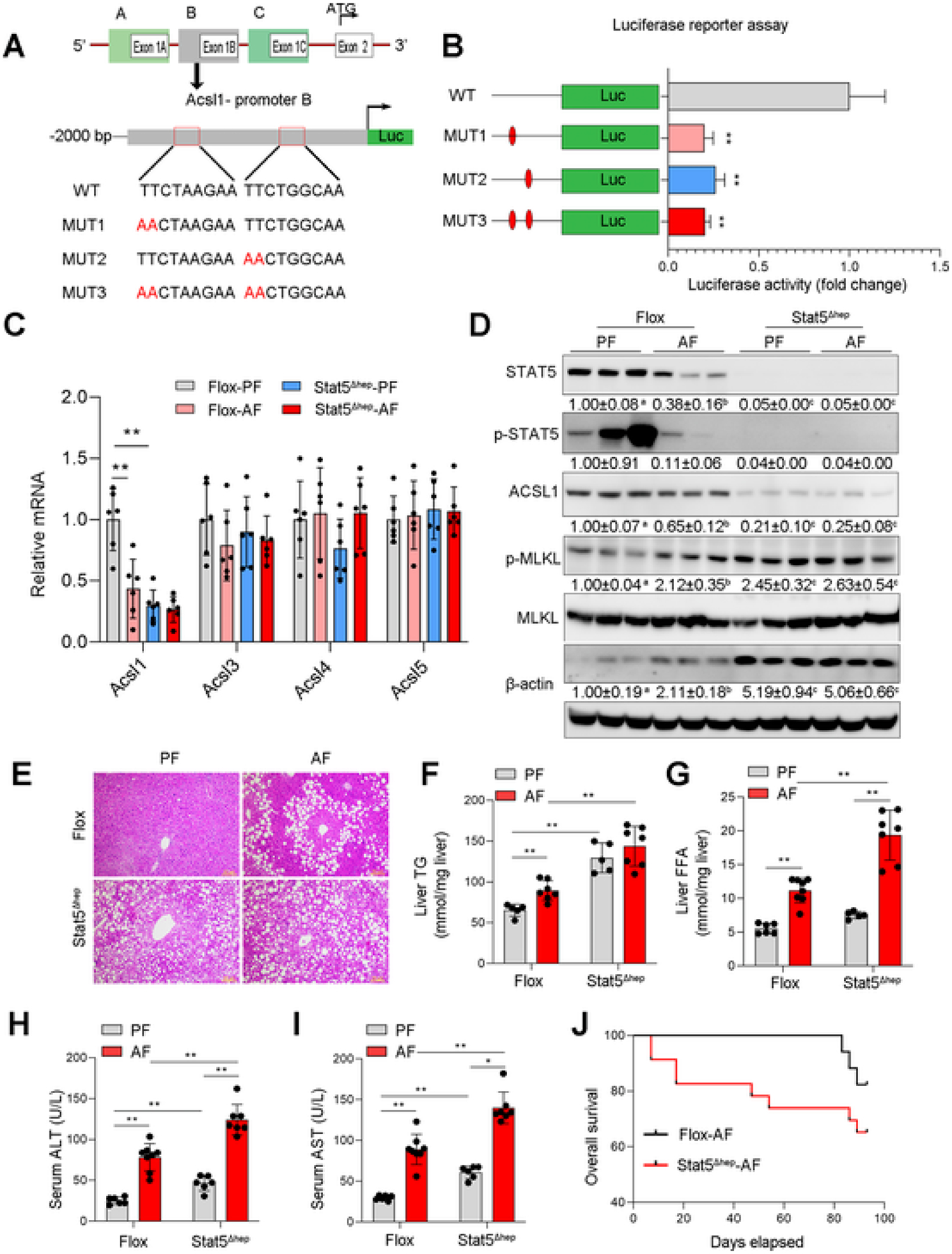
STAT5 Regulates ACSL1 and mediates Alcohol-induced Liver Injury. (A) Diagram of mouse ACSL1 gene structure and schematic of the luciferase construct containing the B-promoter of ACSL1. The wild-type (WT) consensus binding sites are illustrated along with the nucleotides mutated (MUT) in each construct. (B) Hepa-1c1c7 cells were transfected vectors with a luciferase construct containing the wild-type binding sites (WT) or mutated binding sites (MUT1, MUT2, and MUT3), respectively. Luciferase activity was measured by Promega GloMax 96 Microplate Luminometer (n = 3/group). (C) The mRNA levels of ACSL1, ACSL3, ACSL4, and ACSL5 in liver tissues of Flox mice and Stat5Δhep mice with or without alcohol feeding for 8 weeks (n = 6/group). (D) Western blot assay of hepatic proteins related to apoptotic and necroptotic cell death (n = 3/group). (E) H&E staining of liver tissue sections. Scale bars = 50 μm. (F) The hepatic triglycerides (TG) levels (n = 5–7/group). (G) The hepatic free fatty acids (FFA) levels (n = 5–7/group). (H–I) Serum levels of ALT (H) and AST (I) (n = 6–7/group). (J) Kaplan–Meier survival analysis of survival probability during 8 weeks of alcohol feeding. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, values with different superscripts are significantly different.
To further explore the regulatory role of STAT5 in ACSL1 gene expression, we first knocked down STAT5 in Hepa-1c1c7 cells and found that STAT5 knockdown significantly reduced the protein (Fig. S5A) and mRNA (Fig. S5B) levels of ACSL1. Next, we generated hepatocyte-specific STAT5 knockout mice (Fig. S5C, D) and performed alcohol feeding for 8 weeks. Loss of STAT5 did not affect body weight but increased liver weight and liver to body weight ratio and caused liver enlargement (Fig. S5E–H). qRT-PCR analyses showed that STAT5 deletion significantly decreased mRNA levels of ACSL1 but no other ACSL isoforms (Fig. 6C). Western blot analyses revealed that STAT5 deletion exacerbated alcohol-induced cell death along with reduced ACSL1 expression (Fig. 6D). Furthermore, STAT5 deletion aggravated alcohol-induced accumulation of TG (Fig. 6E, F), and elevated alcohol-induced hepatic FFA (Fig. 6G). Accordingly, alcohol-induced serum ALT and AST levels (Fig. 6H, I), as well as mortality (Fig. 6J), were significantly increased by STAT5 deletion. Moreover, deletion of STAT5 also caused a more severe inflammatory response in the liver (Fig. S5I, J). Previous studies showed that SREBP2 [39] and PPARa [40] are involved in the regulation of ACSL1. Thus, we further examined whether they also cooperate with STAT5 in ACSL1 regulation in the present study. The data showed that although alcohol exposure reduced the expression of SREBP2 and PPARa in the floxed mice, STAT5 knockout increased their expression after alcohol feeding (Fig. S5K–M). Moreover, STAT5 knockout did not reduce the expressions of other STATs (Fig. S5N). These results provided the first evidence that STAT5 regulates ACSL1 at a transcriptional level independent of SREBP2/PPARα and suppression of the STAT5-ACSL1 signaling pathway is implicated in alcohol-induced liver injury.
3.7. ACSL1 is required for STAT5-mediated protection against alcohol-induced liver injury
To further determine whether the STAT5-ACSL1 signaling pathway serves as a potential target for ALD, STAT5b gene was delivered to ACSL1 floxed and ACSL1 knockout mice via injection of either AAV8-TBG-null or AAV8-TBG-Stat5b vectors before chronic alcohol feeding. As shown in Supplementary Fig. S6, hepatic STAT5b mRNA levels were increased after AAV8-TBG-Stat5b injection (Fig. S6A). STAT5 overexpression did not affect body weight, liver weight, and liver to body weight ratio after alcohol feeding (Fig. S6B–D), but strongly reduced TG accumulation in the liver (Fig. S6E, F). STAT5 overexpression prevented alcohol-reduced hepatic ACSL1 expression and reversed BAX as well as p-MLKL in ACSL1 floxed mice but not in ACSL1 knockout mice (Fig. 7A, B). STAT5 overexpression alleviated serum ALT and AST levels (Fig. 7C, D), and mortality (Fig. 7E) in ACSL1 floxed mice but these beneficial effects were diminished in ACSL1 knockout mice. Furthermore, STAT5 overexpression suppressed alcohol-induced inflammatory responses in floxed mice but not in ACSL1 deletion mice (Fig. 7F–I). These results indicate that ACSL1 is a critical downstream molecule of STAT5 and activation of the STAT5-ACSL1 signaling pathway can effectively protect against alcohol-induced liver injury.
Fig. 7.
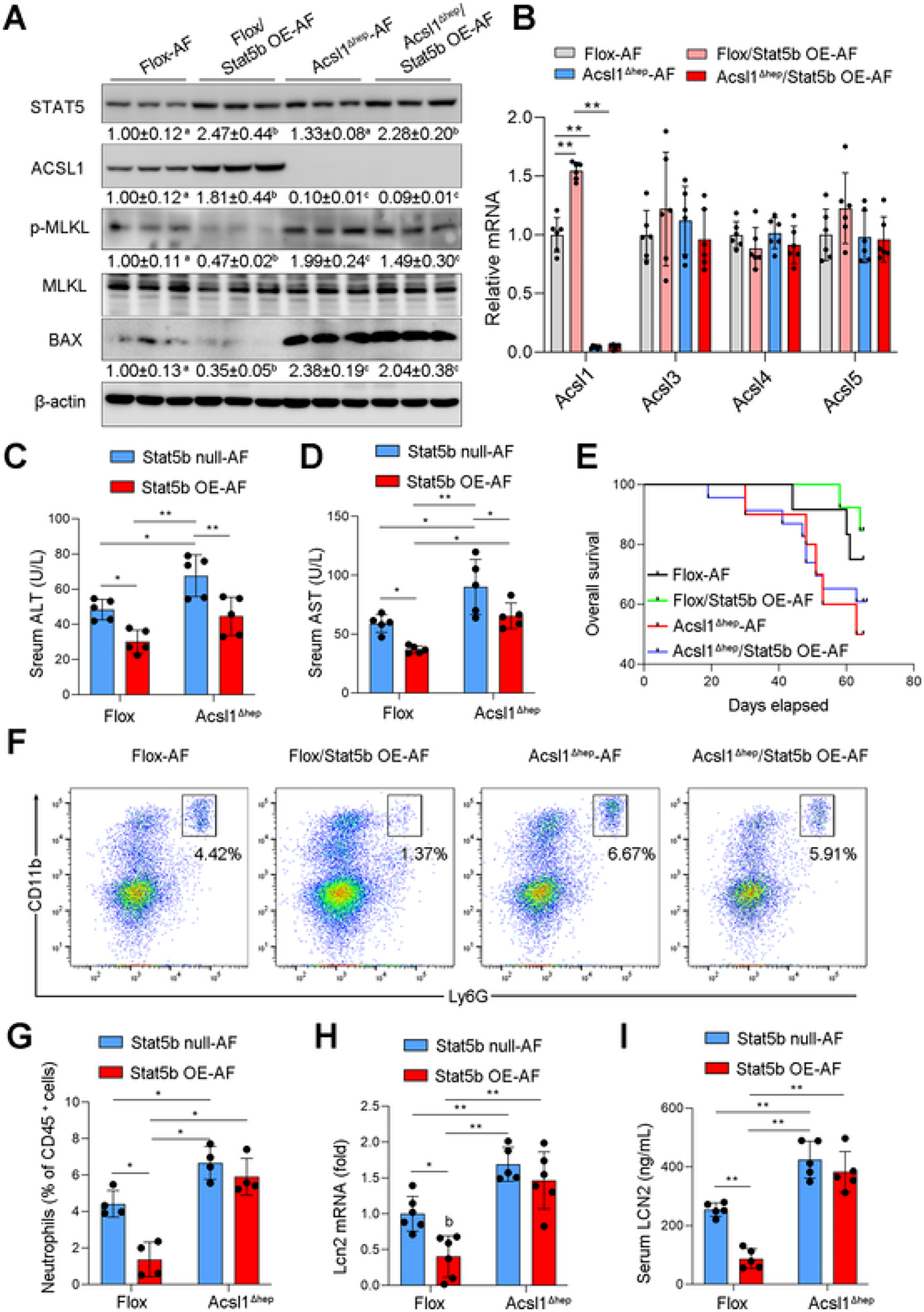
Activation of STAT5-ACSL1 signaling pathway protects against alcohol-induced liver injury. Wild-type Flox mice and Acsl1Δhep mice with or without Stat5b overexpression (Stat5b OE) were subjected to alcohol feeding for 8 weeks. (A) Western blot assay of proteins related to apoptotic and necroptotic cell death (n = 3/group). (B) qPCR analysis of hepatic mRNA levels of ACSL1, ACSL3, ACSL4, and ACSL5 (n = 6/group). (C–D) Serum levels of ALT (C) and AST (D) (n = 5/group). (E) Kaplan–Meier survival analysis of survival probability during 8 weeks of alcohol feeding. (F-G) Flow cytometry analysis of hepatic infiltration of Ly6G+CD1+ neutrophils (f) and quantification (g), gated on singlet 7AAD− CD45+ cells. (n = 4/group). (H) qPCR analysis of hepatic mRNA levels of Lcn2 (n = 6/group). (I) Serum levels of LCN2 (n = 5/group). Data are presented as mean ± SD. *P < 0.05, **P < 0.01, values with different superscripts are significantly different.
3.8. STAT5-ACSL1 signaling is down-regulated and negatively correlates with liver injury in AH patients
To validate the clinical relevance of hepatic STAT5-ACSL1 signaling discovered in the mouse model, we analyzed genes related to STAT5-ACSL1 signaling pathway and cell death markers using another independent GEO dataset (GSE28619). Compared to the normal subjects, the mRNA levels of STAT5b, but not STAT5a, were significantly decreased in the liver of AH patients. In accordance, ACSL1 mRNA was significantly decreased, while the mRNA levels of cell death markers, including BAX, RIPK3, and MLKL, were significantly increased in the liver of AH patients (Fig. 8A), which is consistent with the protein levels of these markers measured by Western blot (Fig. 8B). IF staining and IHC staining (Fig. 8C) further confirmed the protein levels of STAT5 and cell death markers in AH patients. Correlation analysis revealed that ACSL1 was positively correlated with STAT5b, but negatively correlated with cell death (Fig. 8D). Collectively, the results suggest that the suppression of STAT5-ACSL1 signaling pathway contributes to alcohol-induced liver injury in patients.
Fig. 8.
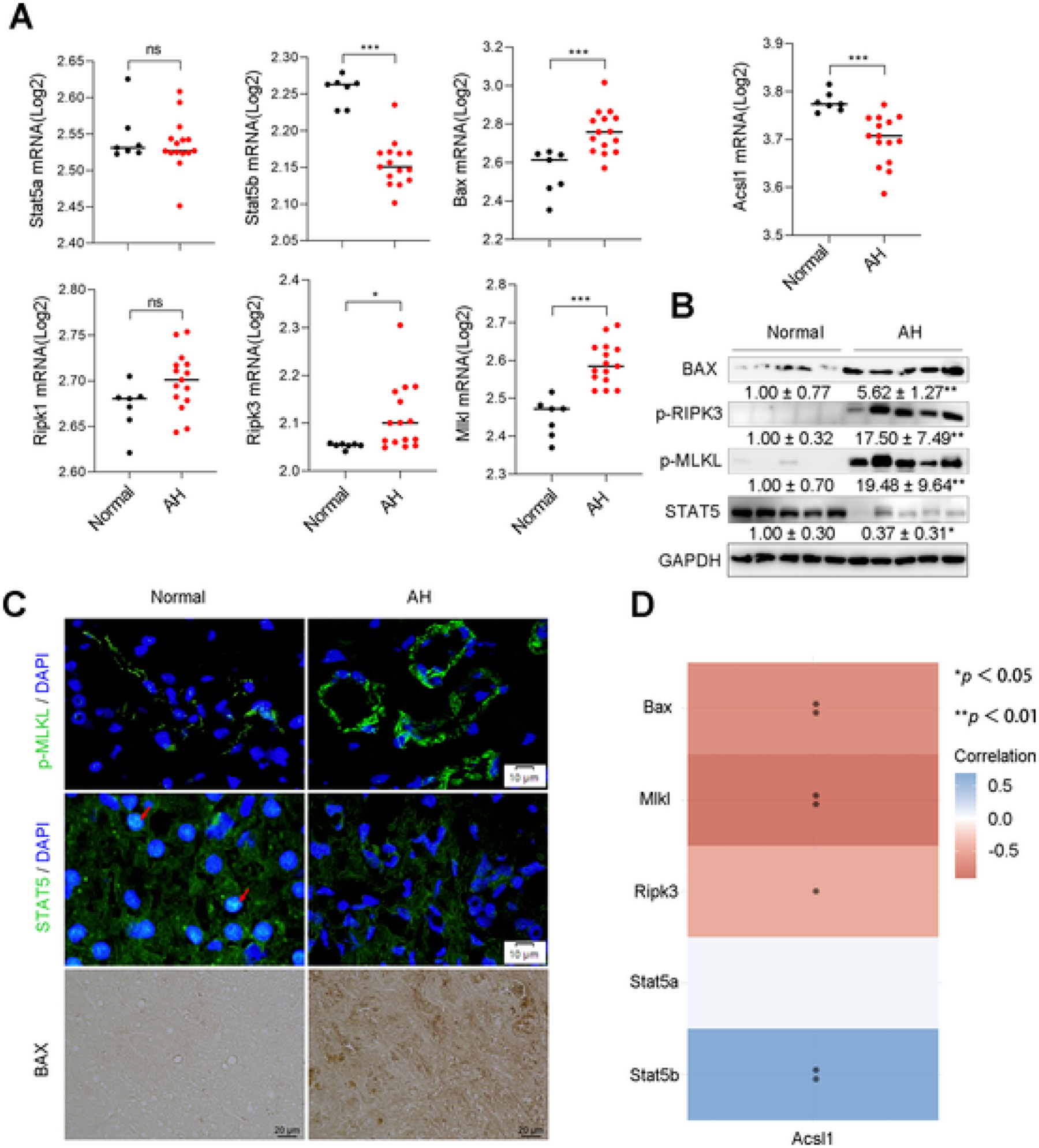
STAT5-ACSL1 signaling mediated liver injury in patients with alcoholic hepatitis. (A) The RNA-seq analysis of Stat5a, Stat5b, Acsl1, Bax, Ripk1, Ripk3, and Mlkl in liver tissues from normal subjects (Normal, n = 7) and patients with alcoholic hepatitis (AH, n = 15). GEO accession number: GSE28619, *P < 0.05, **P < 0.01, ***P < 0.001. (B) Western blot assay of hepatic proteins related to STAT5-ACSL1signaling and cell death markers in the liver of normal subjects (Normal, n = 5) and AH patients (AH, n = 5). (C) IF staining of p-MLKL and SATAT5 (Scale bars = 10 μm.), and IHC staining of BAX (Scale bars = 20 μm.) on liver tissue sections from normal subjects and AH patients. Arrows in red indicate the nuclear localization of STAT5. (D) A heatmap of correlative analysis of mRNA levels of Acsl1 with Stat5, apoptotic gene Bax and necroptotic genes including Mlkl and Ripk3. The abscissa and ordinate represent genes, different colors represent different correlation coefficients (blue represents positive correlation whereas red represents negative correlation), the darker the color, the stronger the relation. *P < 0.05, **P < 0.01, values with different superscripts are significantly different.
4. Discussion
The present study provides multiple lines of evidence supporting that downregulation of hepatic expression of ACSL1 represents a pathophysiological factor in the development of alcohol-induced liver injury. Loss of ACSL1 leads to mitochondrial dysfunction and excessive accumulation of FFAs, which drives lysosomal translocation of BAX/p-MLKL, resulting in LMP and ultimately initiating the lysosomal cell death program. Furthermore, we revealed, for the first time, that STAT5 transcriptionally regulates ACSL1 by directly binding to the B-promoter of ACSL1. Furthermore, reactivation of ACSL1 by STAT5 overexpression effectively reduced alcohol-induced liver injury. These findings reinforce that STAT5-ACSL1 signaling pathway may serve as a promising molecular therapeutic target for ALD.
ACSL1 plays critical roles in lipid metabolism and homeostasis, dysfunction of ACSL1 related to obesity [11], cardiac dysfunction [12,41], sepsis [42], NAFLD [43], and even cancers [44,45]. In the liver, ACSL1 is located on both the outer mitochondrial membrane and ER [46], suggesting a role of ACSL1 in the regulation of fatty acid β-oxidation and TG synthesis. However, reports on the effects of ACSL1 on hepatic TG synthesis are still controversial. ACSL1 overexpression has been shown to increase oleate incorporation into triglyceride in primary hepatocytes as well as triglyceride content in the liver [10]. On the other hand, hepatocyte ACSL1 deletion mice displayed a 25 %–35 % reduction in acyl-CoA content in the liver, but surprisingly, hepatic triglyceride contents were not changed in the liver of ACSL1 deletion mice fed either control or high-fat diet [7]. In this study, our mouse model of ALD showed that loss of ACSL1 reduced FA-CoA and increased hepatic FFA levels. Although FA-CoA can be directed into different metabolic pathways, ACSL1 deletion reduced mitochondrial FA β-oxidation but did not affect hepatic triglyceride content as well as key genes involved in TG synthesis, including Pparγ, Dgat1 and Dgat2. These results suggest that ACSL1 may primarily direct FA-CoA into the mitochondria for FA β-oxidation rather than into the ER for TG synthesis. A recent study found that ACSL4 knockout can alleviate high fat diet-induced hepatic lipid accumulation [47], indicating a role of ACSL4 in TG synthesis. These results suggest that ACSL1 plays a major role in directing FA-CoAs into the mitochondrial fatty acid β-oxidation pathway, while other ACSL isoforms such as ACSL4 may direct FA-CoAs into the ER TG synthesis pathway. In addition, ACSL1 deficiency-induced compensatory enhancement of peroxisomal β-oxidation and endoplasmic reticulum ω-oxidation may also contribute to alcohol-induced liver injury by promoting ROS generation and oxidative stress [48]. Loss of ACSL1 exacerbates the expression of CYP2E1, a key enzyme in ethanol oxidation, generating ROS in the process of ethanol metabolism. Accordingly, oxidative stress (4-HNE) and ER stress (ATF4), are strongly induced after ACSL1 deletion. These results suggest a causal role of ACSL1 deficiency in alcohol-induced liver injury.
Cell death plays a central role in the progression of alcohol-induced liver injury [35]. Although studies have demonstrated the existence of multiple types of cell death in ALD, mechanisms underlying alcohol-induced programmed cell death remain largely unclear. For example, blocking apoptotic signaling by inhibition of caspase-8 did not completely protect the liver from alcohol-induced liver injury [49] due to necroptosis being implicated after alcohol feeding [50]. Correspondingly, blocking RIPK3-mediated necroptosis did not prevent alcohol-induced apoptosis in the liver [50]. In this study, we observed that ACSL1 deficiency, regardless of alcohol exposure, induced FFA accumulation in the liver along with a significant increase in p-MLKL and BAX. IP analysis revealed a protein-protein interaction between BAX and p-MLKL, indicating a possible role of the apoptotic molecule, BAX, in the signaling of necroptosis under FFA lipotoxicity conditions. Indeed, our in vitro studies revealed that BAX is an upstream regulator of MLKL. Knockdown of BAX significantly reduced the phosphorylation level of MLKL, thus preventing the translocation of p-MLKL to the plasma membrane. Both BAX and MLKL are involved in membrane pore formation during cell death [36], however, whether MLKL mediates LMP remains unclear. Previous studies have shown that FFAs induce translocation of apoptotic protein BAX from the cytosol to lysosomal membranes and induce LMP [51]. Therefore, we speculate that MLKL may be involved in the changes of LMP. Indeed, in vitro study showed that MLKL knockdown protected cells from PA-induced LMP in Hepa-1c1c7 cells. While plasma membrane translocation of p-MLKL has been well documented in necroptosis [52–54]. The data from the current study suggest a novel function of MLKL in LMP activation, and a possible BAX-MLKL cell death pathway in FFA-induced necroptosis.
Down-regulation of ACSL1 has been reported in ALD, but the regulatory mechanism of ACSL1 expression is poorly understood. Previous studies have demonstrated that ACSL1 is regulated by PPARα with a PPAR-responsive element (PPRE) [40] or by SREBP2 with a sterol regulatory element (SRE) [39] in the C-promoter, which are master regulators of lipid metabolism in the liver. STAT5 also has been shown critically regulate lipid metabolism in the liver. Liver-specific STAT5 knockout mice developed hepatic steatosis, and aggravated liver damage [55]. In this study, we revealed that unlike the transcriptional regulatory mechanism of PPARα and SREBP2 on ACSL1 expression, STAT5 is executed by binding directly to the B-promoter of ACSL1. In addition, we found that STAT5 did not affect the gene expression of other ACSL isoforms, indicating a specific regulation of ACSL1. Furthermore, we observed that STAT5-mediated ACSL1 regulation is independent of SREBP2 and PPARα since SREBP2 and PPARα were not affected by STAT5 knockout. Therefore, these results uncover a novel transcriptional regulatory mechanism of ACSL1.
STAT5 belongs to a family of STAT proteins, which also includes STAT1, STAT2, STAT3, STAT4, and STAT6 [24,56]. In this study, we found that among all STATs, only STAT5 was decreased after alcohol feeding. Interestingly, loss of STAT5 led to the upregulation of STAT1 and STAT4 regardless of alcohol exposure, indicating a possible compensatory regulation among STATs. This indicates that STAT5 may have crosstalk with other STATs to a certain extent. In addition, STATs participate in different signaling pathways by linking to the Janus kinase (JAK) family of proteins (including JAK1, JAK2, JAK3 and Tyk2) [22]. A previous study reported that Tyk2 is involved in the transcriptional regulation of ACSL4 in macrophages, suggesting that JAK-STATs may also interact with other ACSLs in different cell types, and further exploration of STATs, ACSLs, and their interactions in different cell types are needed in the future studies.
In conclusion, we demonstrate that loss of ACSL1 leads to hepatic FFAs accumulation, which causes LMP activation and cell death via lysosomal translocation of BAX/p-MLKL. In addition, we uncovered STAT5 as a novel transcriptional regulator of ACSL1, and activation of ACSL1 by STAT5 effectively ameliorated alcohol-induced liver injury. Thus, this study provides novel insights into the role of STAT5-ACSL1 signaling pathway in regulating alcohol-induced liver injury and offers a potential therapeutic target for treating ALD.
4.1. Strengths/weaknesses
ACSL1 plays a critical role in regulating hepatic fatty acid metabolism and lipid homeostasis, and dysfunction of ACSL1 is closely related to many diseases, however, its role in the development of ALD remains unclear. Our work demonstrates a causal role of ACSL1 deficiency in alcohol-induced liver injury. The study also uncovers that STAT5 is a new transcriptional regulator of ACSL1, and activation of STAT5-ACSL1 signaling effectively ameliorates alcohol-induced liver injury, suggesting a potential therapeutic target for ALD treatment. In addition, our in vitro study reveals that Bax, as a functional apoptotic molecule, mediates p-MLKL plasma/lysosomal membrane translocation upon fatty acids treatment, although this novel BAX-MLKL pathway needs further validation in vivo. The AH patients we studied is a late stage of liver injury characterized by marked hepatocellular damage, steatosis, and pericellular fibrosis. However, our mouse model of ALD only produces an early stage of liver injury, which does not fully mimic AH patients. Therefore, whether targeting STAT5-ACSL1 applies to the late stages of ALD remains to be further explored.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health grants R01AA018844 (Zhanxiang Zhou), R01AA020212 (Zhanxiang Zhou), and R24AA025017 (Zhaoli Sun). We also thank Dr. Rosalind Coleman of the University of North Carolina at Chapel Hill for providing the ACSL1 floxed mice.
Abbreviations
- AAV8
recombinant adeno-associated viral serotype 8
- ACSL
long-chain acyl-CoA synthetase
- AH
alcoholic hepatitis
- ALT
alanine aminotransferase
- ALD
alcohol-associated liver disease
- AST
aspartate aminotransferase
- BAX
BCL2 associated X
- BIP-V5
Bax inhibitor peptide V5
- CPT1a
carnitine palmitoyltransferase 1A
- FFA
free fatty acids
- LAMP2
lysosomal associated membrane protein 2
- LCN2
Lipocalin 2
- LMP
lysosomal membrane permeabilization
- MLKL
mixed lineage kinase domain-like pseudokinase
- NSA
necrosulfonamide
- MPO
Myeloperoxidase
- OCR
oxygen consumption rate
- PA
palmitate
- STAT5
signal transducer and activator of transcription 5
- TG
triglyceride
- TUNEL
terminal deoxynucleotidyl transferase uridine triphosphate nick end labeling
Footnotes
Ethics declarations
All the animal experiments were performed following the protocol approved by the North Carolina Research Campus Institutional Animal Care and Use Committee. All human tissues collected from explanted livers or biopsies from donor’s livers had been approved by Institutional Review Boards at Johns Hopkins Medical Institutions (IRB00107893, IRB00021325).
CRediT authorship contribution statement
Haibo Dong : Conceptualization, Methodology, Investigation, Formal analysis, Writing – original draft. Wei Zhong : Methodology, Investigation, Writing – review & editing. Wenliang Zhang : Methodology, Investigation. Liuyi Hao : Formal analysis, Investigation, Writing – review & editing. Wei Guo : Investigation, Writing – review & editing. Ruichao Yue : Investigation, Writing – review & editing. Xinguo Sun : Investigation. Zhaoli Sun : Resources, Writing – review & editing. Ramon Bataller : Resources, Writing – review & editing. Zhanxiang Zhou : Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Declaration of competing interest
All the authors declared no competing interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.metabol.2022.155334.
Data availability
Materials for transcriptome analysis in this paper are publicly available at Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/) with accession numbers described in Table 2. Any additional information required to reanalyze the data reported in this paper is available from the corresponding author upon reasonable request.
References
- [1].Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141(5):1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mokdad AH, Marks JS, Stroup DF, Gerberding JL Actual causes of death in the United States, 2000. JAMA 2004;291(10):1238–45. [DOI] [PubMed] [Google Scholar]
- [3].Vuittonet CL, Halse M, Leggio L, Fricchione SB, Brickley M, Haass-Koffler CL, et al. Pharmacotherapy for alcoholic patients with alcoholic liver disease. AmJHealthSystPharm 2014;71(15):1265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mashek DG, Li LO, Coleman RA Long-chain acyl-CoA synthetases and fatty acid channeling. Future Lipidol 2007;2(4):465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yan S, Yang XF, Liu HL, Fu N, Ouyang Y, Qing K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol 2015;21(12):3492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klett EL, Chen S, Yechoor A, Lih FB, Coleman RA. Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J Lipid Res 2017;58(5):884–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li LO, Ellis JM, Paich HA, Wang S, Gong N, Altshuller G, et al. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem 2009; 284(41):27816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Iijima H, Fujino T, Minekura H, Suzuki H, Kang MJ, Yamamoto T. Biochemical studies of two rat acyl-CoA synthetases, ACS1 and ACS2. Eur J Biochem 1996;242 (2):186–90. [DOI] [PubMed] [Google Scholar]
- [9].Li LO, Mashek DG, An J, Doughman SD, Newgard CB, Coleman RA. Overexpression of rat long chain acyl-coa synthetase 1 alters fatty acid metabolism in rat primary hepatocytes. J Biol Chem 2006;281(48):37246–55. [DOI] [PubMed] [Google Scholar]
- [10].Parkes HA, Preston E, Wilks D, Ballesteros M, Carpenter L, Wood L, et al. Overexpression of acyl-CoA synthetase-1 increases lipid deposition in hepatic (HepG2) cells and rodent liver in vivo. Am J Physiol Endocrinol Metab 2006;291 (4):E737–44. [DOI] [PubMed] [Google Scholar]
- [11].Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, et al. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab 2010;12(1):53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, et al. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol Cell Biol 2011;31(6):1252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fernandez RF, Ellis JM Acyl-CoA synthetases as regulators of brain phospholipid acyl-chain diversity. Prostaglandins Leukot Essent Fatty Acids 2020; 161:102175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ihle JN STATs: signal transducers and activators of transcription. Cell 1996;84 (3):331–4. [DOI] [PubMed] [Google Scholar]
- [15].Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 2015;194(1):21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Loh CY, Arya A, Naema AF, Wong WF, Sethi G, Looi CY. Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication. Front Oncol 2019;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 2015;66:311–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mishra N, Mishra S. Signal transducer and activator of transcriptions (STATs)-at the crossroads of obesity-linked non-alcoholic steatohepatitis and hepatocellular carcinoma. Hepatobiliary SurgNutr. 2019;8(4):407–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. MetabClinExp 2016;65(8):1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li Y, Wei M, Yuan Q, Liu Y, Tian T, Hou L, et al. MyD88 in hepatic stellate cells promotes the development of alcoholic fatty liver via the AKT pathway. J Mol Med 2022;100(7):1071–85. [DOI] [PubMed] [Google Scholar]
- [21].Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 2008;134(4):1148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nguyen VA, Gao B Expression of interferon alfa signaling components in human alcoholic liver disease. Hepatology 2002;35(2):425–32. [DOI] [PubMed] [Google Scholar]
- [23].Baik M, Yu JH, Hennighausen L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann N Y Acad Sci 2011;1229: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol 2005;2(2):92–100. [PubMed] [Google Scholar]
- [25].Barclay JL, Nelson CN, Ishikawa M, Murray LA, Kerr LM, McPhee TR, et al. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 2011;152(1):181–92. [DOI] [PubMed] [Google Scholar]
- [26].Hosui A, Kimura A, Yamaji D, Zhu BM, Na R, Hennighausen L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation. J Exp Med 2009;206(4):819–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hyun J, Sun Z, Ahmadi AR, Bangru S, Chembazhi UV, Du K, et al. Epithelial splicing regulatory protein 2-mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. J Clin Invest 2020;130(4):2129–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, et al. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J Hepatol 2015;62(6):1375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dong H, Hao L, Zhang W, Zhong W, Guo W, Yue R, et al. Activation of AhR-NQO1 signaling pathway protects against alcohol-induced liver injury by improving redox balance. Cell Mol Gastroenterol Hepatol 2021;12(3):793–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Affo S, Dominguez M, Lozano JJ, Sancho-Bru P, Rodrigo-Torres D, Morales-Ibanez O, et al. Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut 2013;62(3):452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Charni-Natan M, Goldstein I. Protocol for primary mouse hepatocyte isolation. STAR Protocols 2020;1(2):100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Armando JW, Boghigian BA, Pfeifer BA LC-MS/MS quantification of short-chain acyl-CoA’s in Escherichia coli demonstrates versatile propionyl-CoA synthetase substrate specificity. Lett Appl Microbiol 2012;54(2):140–8. [DOI] [PubMed] [Google Scholar]
- [33].Blachnio-Zabielska AU, Koutsari C, Jensen MD. Measuring long-chain acyl-coenzyme a concentrations and enrichment using liquid chromatography/tandem mass spectrometry with selected reaction monitoring. Rapid CommunMass Spectrom 2011;25(15):2223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem 2011;286(29):25655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Miyata T, Nagy LE. Programmed cell death in alcohol-associated liver disease. Clin Mol Hepatol 2020;26(4):618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Flores-Romero H, Ros U, Garcia-Saez AJ. Pore formation in regulated cell death. EMBO J 2020;39(23):e105753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 2008;27(50):6434–51. [DOI] [PubMed] [Google Scholar]
- [38].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332(6036):1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Singh AB, Kan CF, Dong B, Liu J. SREBP2 activation induces hepatic long-chain acyl-CoA synthetase 1 (ACSL1) expression in vivo and in vitro through a sterol regulatory element (SRE) motif of the ACSL1 C-promoter. J Biol Chem 2016;291 (10):5373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schoonjans K, Watanabe M, Suzuki H, Mahfoudi A, Krey G, Wahli W, et al. Induction of the acyl-coenzyme a synthetase gene by fibrates and fatty acids is mediated by a peroxisome proliferator response element in the C promoter. J Biol Chem 1995;270(33):19269–76. [DOI] [PubMed] [Google Scholar]
- [41].Paul DS, Grevengoed TJ, Pascual F, Ellis JM, Willis MS, Coleman RA. Deficiency of cardiac acyl-CoA synthetase-1 induces diastolic dysfunction, but pathologic hypertrophy is reversed by rapamycin. Biochim Biophys Acta 2014; 1841(6):880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Roelands J, Garand M, Hinchcliff E, Ma Y, Shah P, Toufiq M, et al. Long-chain acyl-CoA synthetase 1 role in sepsis and immunity: perspectives from a parallel review of public transcriptome datasets and of the literature. Front Immunol 2019; 10:2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huh JY, Reilly SM, Abu-Odeh M, Murphy AN, Mahata SK, Zhang J, et al. Green CR et al. : TANK-binding kinase 1 regulates the localization of acyl-CoA synthetase ACSL1 to control hepatic fatty acid oxidation. Cell Metab 2020;32(6):1012–1027 e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ma Y, Zha J, Yang X, Li Q, Zhang Q, Yin A, et al. Long-chain fatty acyl-CoA synthetase 1 promotes prostate cancer progression by elevation of lipogenesis and fatty acid beta-oxidation. Oncogene 2021;40(10):1806–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Guo L, Lu J, Gao J, Li M, Wang H, Zhan X. The function of SNHG7/miR-449a/ACSL1 axis in thyroid cancer. J Cell Biochem 2020;121(10):4034–42. [DOI] [PubMed] [Google Scholar]
- [46].Young PA, Senkal CE, Suchanek AL, Grevengoed TJ, Lin DD, Zhao L, et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J Biol Chem 2018;293(43): 16724–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Duan J, Wang Z, Duan R, Yang C, Zhao R, Feng Q, et al. Therapeutic targeting of hepatic ACSL4 ameliorates NASH in mice. Hepatology 2022;75(1):140–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park EC, Kim SI, Hong Y, Hwang JW, Cho GS, Cha HN, et al. Inhibition of CYP4A reduces hepatic endoplasmic reticulum stress and features of diabetes in mice. Gastroenterology 2014;147(4):860–9. [DOI] [PubMed] [Google Scholar]
- [49].Hao F, Cubero FJ, Ramadori P, Liao L, Haas U, Lambertz D, et al. Inhibition of Caspase-8 does not protect from alcohol-induced liver apoptosis but alleviates alcoholic hepatic steatosis in mice. Cell Death Dis 2017;8(10):e3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013;57(5):1773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol 2006;290(6):G1339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014;54(1):133–46. [DOI] [PubMed] [Google Scholar]
- [53].Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2014;24(1):105–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014;16(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cui Y, Hosui A, Sun R, Shen K, Gavrilova O, Chen W, et al. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 2007;46(2):504–13. [DOI] [PubMed] [Google Scholar]
- [56].Ihle JN The Stat family in cytokine signaling. Curr Opin Cell Biol 2001;13(2): 211–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Materials for transcriptome analysis in this paper are publicly available at Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/) with accession numbers described in Table 2. Any additional information required to reanalyze the data reported in this paper is available from the corresponding author upon reasonable request.