

Abstract
Immune cells and skin and mucosal epithelial cells recognize invasive microbes and other signs of danger to sound alarms that recruit responder cells and initiate an immediate “innate” immune response. An especially powerful alarm is triggered by cytosolic sensors of invasive infection that assemble into multimolecular complexes, called inflammasomes, that activate the inflammatory caspases, leading to maturation and secretion of proinflammatory cytokines and pyroptosis, an inflammatory death of the infected cell. Work in the past year has defined the molecular basis of pyroptosis. Activated inflammatory caspases cleave Gasdermin D (GSDMD), a cytosolic protein in immune antigen-presenting cells and epithelia. Cleavage separates the autoinhibitory C-terminal fragment from the active N-terminal fragment, which moves to the cell membrane, binds to lipids on the inside of the cell membrane, and oligomerizes to form membrane pores that disrupt cell membrane integrity, causing death and leakage of small molecules, including the proinflammatory cytokines and GSDMD itself. GSDMD also binds to cardiolipin on bacterial membranes and kills the very bacteria that activate the inflammasome. GSDMD belongs to a family of poorly studied gasdermins, expressed in the skin and mucosa, which can also form membrane pores. Spontaneous mutations that disrupt the binding of the N- and C-terminal domains of other gasdermins are associated with alopecia and asthma. Here, we review recent studies that identified the roles of the inflammasome, inflammatory caspases, and GSDMD in pyroptosis and highlight some of the outstanding questions about their roles in innate immunity, control of infection, and sepsis.
1. INTRODUCTION
As the first line of host defense, innate immunity initiates immediate host responses to contain invading microbes and sets the stage for activating delayed adaptive immune responses to continue and enhance control of pathogenic infection and establish immunological memory to protect against subsequent exposures. The innate immune response is triggered by recognition of conserved microbial features by host pattern recognition receptors (PRRs) that are prominently expressed by immune antigen-presenting myeloid cells of the monocyte/macrophage and dendritic cell lineages and by epithelial cells in the skin and mucosa, the body’s barrier surfaces where infection is first encountered. These microbe-recognizing receptors distinguish infectious agents from the organism’s own proteins, nucleic acids, lipids, and sugars by their molecular features and intracellular localization. The PRRs can be divided into receptors on the cell surface or within endosomes that recognize signs of infection in the environment, such as Toll-like receptors (TLRs) and C-type lectin-like receptors, and intracellular sensors of invasive pathogens, such as RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), AIM2-like receptors (ALRs), pyrin, cyclic GMP-AMP synthase (cGAS), and STING (Burdette et al., 2011; Sun, Wu, Du, Chen, & Chen, 2013; Takeuchi & Akira, 2010). The environmental PRRs initiate downstream signaling that activates the nuclear translocation of the nuclear factor-κB and/or interferon (IFN) regulatory factors to induce transcription of inflammatory genes, including inflammatory caspases and proinflammatory cytokines, IFNs, and IFN-regulated genes (IRG), that prime the cell and tissue to respond to infection or other dangers. The cytosolic PRRs can be divided into two groups—the RLRs, cGAS, and STING that recognize foreign cytosolic RNA, DNA, and cyclic dinucleotides, respectively, and trigger an IFN antiinfectious response and the NLRs, ALRs, and pyrin that trigger the assembly of large cytosolic inflammatory complexes, called the canonical inflammasome, that activate caspase-1. Activated caspase-1 results in processing and release of inflammatory cytokines (IL-1β, IL-18) and an inflammatory cell death, pyroptosis, caused by loss of plasma membrane integrity (Lamkanfi & Dixit, 2014). A recent addition to the list of cytosolic sensors are the inflammatory caspases, caspase-4 and -5 in humans and caspase-11 in mice, which bind cytosolic lipopolysaccharide (LPS), a sign of invasive gram− bacterial infection. The complex of LPS with caspase-4/5/11 is termed the noncanonical inflammasome. The noncanonical inflammasome also activates pyroptosis, but only causes processing of proinflammatory cytokines indirectly by activating the canonical inflammasome through a not well-defined mechanism.
Inflammasome activation plays a crucial role in host immune defense against infection. The proinflammatory cytokines recruit immune cells to sites of infection and also reduce the threshold for stimulating adaptive immune responses. Pyroptosis expels invading bacteria from the intracellular niche that is favorable for their survival and replication, reducing pathogen loads (Miao et al., 2010). Multiple mouse studies suggest that deficient inflammasome activation enhances bacterial virulence (summarized in Section 5.1). Some pathogenic intracellular bacteria, including Listeria monocytogenes and Salmonella typhimurium, turn off expression of gene products that are recognized by NLR sensors when they invade (Miao et al., 2010; Sauer et al., 2010; Zhao et al., 2016), and others, such as Yersinia pseudotuberculosis, produce virulence factors that inhibit activation of specific inflammasomes (Chung et al., 2016; LaRock & Cookson, 2012). In addition mice genetically deficient in specific canonical or noncanonical inflammasome components are more susceptible to various bacterial pathogens, including S. typhimurium, Mycobacterium tuberculosis, Legionella pneumophila, and Francisella tularensis (Cerqueira, Pereira, Silva, Cunha, & Zamboni, 2015; Fernandes-Alnemri et al., 2010; Knodler et al., 2014; Saiga et al., 2012; Sellin et al., 2014). Several studies show that both the canonical and the noncanonical inflammasome are important in maintaining normal gut microbiota, since mice deficient in Casp11 are more susceptible to DSS-induced colitis and mice deficient in NLRP6 are predisposed to develop inflammatory bowel disease (Demon et al., 2014; Elinav et al., 2011). However, despite the evidence in mouse models for the importance of inflammasome activation in protection from bacterial infection and microbial homeostasis, as of now there are no known human immunodeficiency syndromes caused by genetic deficiencies of the inflammasome.
By contrast, there are many examples where genetic changes that cause overactivation of the inflammasome lead to autoimmune and autoinflammatory diseases in mice and humans (Chae et al., 2011; French, 1997; Kastner, Aksentijevich, & Goldbach-Mansky, 2010). Many of these rare genetic diseases in humans can now be treated by inhibiting the proinflammatory cytokines, suggesting that these cytokines are the key mediators of inflammatory pathology in these autoinflammatory genetic diseases, such as familial Mediterranean fever or Muckle–Wells syndrome (Dinarello, Simon, & van der Meer, 2012). In addition, poorly controlled infection leads to overexuberant and continuous inflammasome activation that results in the severe, vascular leak, and multiorgan failure sepsis syndrome, which is often fatal and is the leading cause of childhood death in the world and a contributing factor in about a third of adult hospital deaths (Liu, Escobar, et al., 2014; Singer et al., 2016). The recently discovered noncanonical inflammasome may be especially important in the etiology of sepsis since mice genetically deficient in caspase-11 (but not in just caspase-1) are resistant to death from LPS-induced sepsis (Kayagaki et al., 2015, 2013). Clinical trials using antagonists of the proinflammatory cytokines have been ineffective for treating sepsis (Marshall, 2014), which suggests that pyroptosis, the other major event triggered by inflammasome activation, is the key pathogenic mediator of sepsis.
Until the past year, the molecular basis for pyroptosis was unknown. Recent studies identified Gasdermin D (GSDMD) as the critical mediator of pyroptosis (Kayagaki et al., 2015; Shi, Zhao, et al., 2015). GSDMD is the only known experimentally validated substrate of all the inflammatory caspases and seems to be their most important substrate in pyroptosis (except possibly for the caspases themselves, which become activated by auto-proteolysis within the inflammasome). Indeed, mice that are genetically deficient in Gsdmd, like caspase-11-deficient mice, survive an otherwise lethal LPS challenge (Kayagaki et al., 2015). The activated inflammatory caspases cleave GSDMD to generate an N-terminal cleavage product (GSDMD-NT) that causes pyroptosis by oligomerizing in the plasma membrane to form pores (Aglietti et al., 2016; Ding et al., 2016; Liu & Lieberman, 2017; Liu et al., 2016; Sborgi et al., 2016). Pore formation by the N-terminal fragment is inhibited by the C-terminal fragment within full-length GSDMD when inflammatory caspases are inactive (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). GSDMD belongs to a family of homologous proteins, the gasdermins (GSDMs), which arose sometime in vertebrates by gene duplication, that are expressed in the same cells as the inflammasome (Tamura et al., 2007). Cleavage of other GSDM family members also activates pore formation, but other GSDMs are not substrates of the inflammatory caspases (Ding et al., 2016). Whether they are cleaved in vivo and what activates them are largely unknown. However, spontaneous mutations that appear to disrupt the N-terminal–C-terminal domain interface of mouse GSDMA3 are associated with alopecia (Ding et al., 2016; Porter et al., 2002; Tanaka et al., 2007).
This review describes our current understanding of the mechanism and role of pyroptosis in innate immunity and bacterial defense. We also discuss the many questions raised by the recent pyroptosis papers about the role of pyroptosis and GSDMs in immunity, pathogen defense, and diseases caused by overactivation of pyroptosis.
2. INFLAMMASOMES AND INFLAMMATORY CELL DEATH
2.1. Canonical Inflammasomes
Inflammasomes are formed in response to invasive microbial pathogens or signs of intracellular danger. Nonpathogenic microbes that coexist without causing harm do not trigger the inflammasome. Inflammasome triggers include cytosolic bacterial or viral DNA, bacterial proteins injected into the cytosol by bacterial secretion systems, and cytosolic entry of bacterial toxins. The concept of the inflammasome was proposed by Tschopp in 2002, as he and his colleagues identified and characterized a multiprotein complex, consisting of an NLR named NALP1 (NACHT, leucine-rich repeat (LRR), and pyrin domain (PYD)-containing protein, also known as NLRP1), the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD), also known as PYCARD), and the inflammatory caspase, caspase-1 (Martinon, Burns, & Tschopp, 2002). Inflammasomes are formed by association of PYD or CARD domains in one protein with corresponding domains in other proteins within the complex. The inflammasome forms a platform to activate the inactive caspase-1 zymogen by antiparallel dimerization that brings the catalytic domain of one caspase-1 molecule into proximity with one of the two sites that need to be cleaved to activate the enzyme (Elliott, Rouge, Wiesmann, & Scheer, 2009). Proximity-induced proteolysis cuts caspase-1 twice into noncovalently linked p10 and p20 subunits of the active enzyme.
The NLR family proteins contain an N-terminal-binding region (such as a CARD, PYD, or baculovirus inhibitor repeat domain) needed for multimerization, a conserved nucleotide binding and oligomerization domain (NOD or NACHT) and C-terminal leucine rich repeats (LRRs), responsible for recognizing a pathogen-associated molecular pattern (Fritz, Ferrero, Philpott, & Girardin, 2006). 22 and 34 NLR genes have been identified by bioinformatics analysis in the human and mouse genomes, respectively. Each NLR protein (NLRP1, NLRP3, NLRP6, NLRP7, NLRP12, and the NAIP/NLRC4 complex) senses a specific type of danger signal to initiate its assembly into an inflammasome (Chen, Shaw, Kim, & Nunez, 2009; Kanneganti, Lamkanfi, & Nunez, 2007; Fig. 1). The NLRP1 inflammasome is activated by cytosolic lethal toxin (LT), a key virulence factor of Bacillus anthracis (Cordoba-Rodriguez, Fang, Lankford, & Frucht, 2004) when NLRP1 is cleaved by the LT metalloprotease (Chavarria-Smith & Vance, 2013; Levinsohn et al., 2012). The NLRP3 inflammasome, which has been extensively studied, is activated by a broad spectrum of stimuli, including ATP, microbial toxins, and crystals (alum, silica, urate, cholesterol) (Elliott & Sutterwala, 2015). The underlying mechanism by which NLRP3 senses these diverse danger signs is uncertain. It is generally believed that NLRP3 senses a common cytosolic perturbation, such as a change in intracellular K+, but the underlying mechanism is unknown. The NAIPs are so far the only NLR family members that sense and bind to well-defined microbial ligands to initiate the NAIP/NLRC4 inflammasome. The mouse genome encodes seven NAIPs (NAIP1–7), while the human genome only has one NAIP gene (Endrizzi, Hadinoto, Growney, Miller, & Dietrich, 2000). Human NAIP and mouse NAIP1 recognize the needle protein of the bacterial type III secretion system (T3SS), mouse NAIP2 detects the T3SS rod protein, and mouse NAIP5 and NAIP6 detect bacterial flagellin (Kofoed & Vance, 2011; Yang, Zhao, Shi, & Shao, 2013; Zhao et al., 2011). The ligands of the NLRP6, NLRP7, and NLRP12 inflammasomes are unknown, but they are activated by mucosal microbiota, microbial acylated lipopeptides, and Yersinia pestis infection, respectively (Elinav et al., 2011; Khare et al., 2012; Vladimer et al., 2012).
Fig. 1.
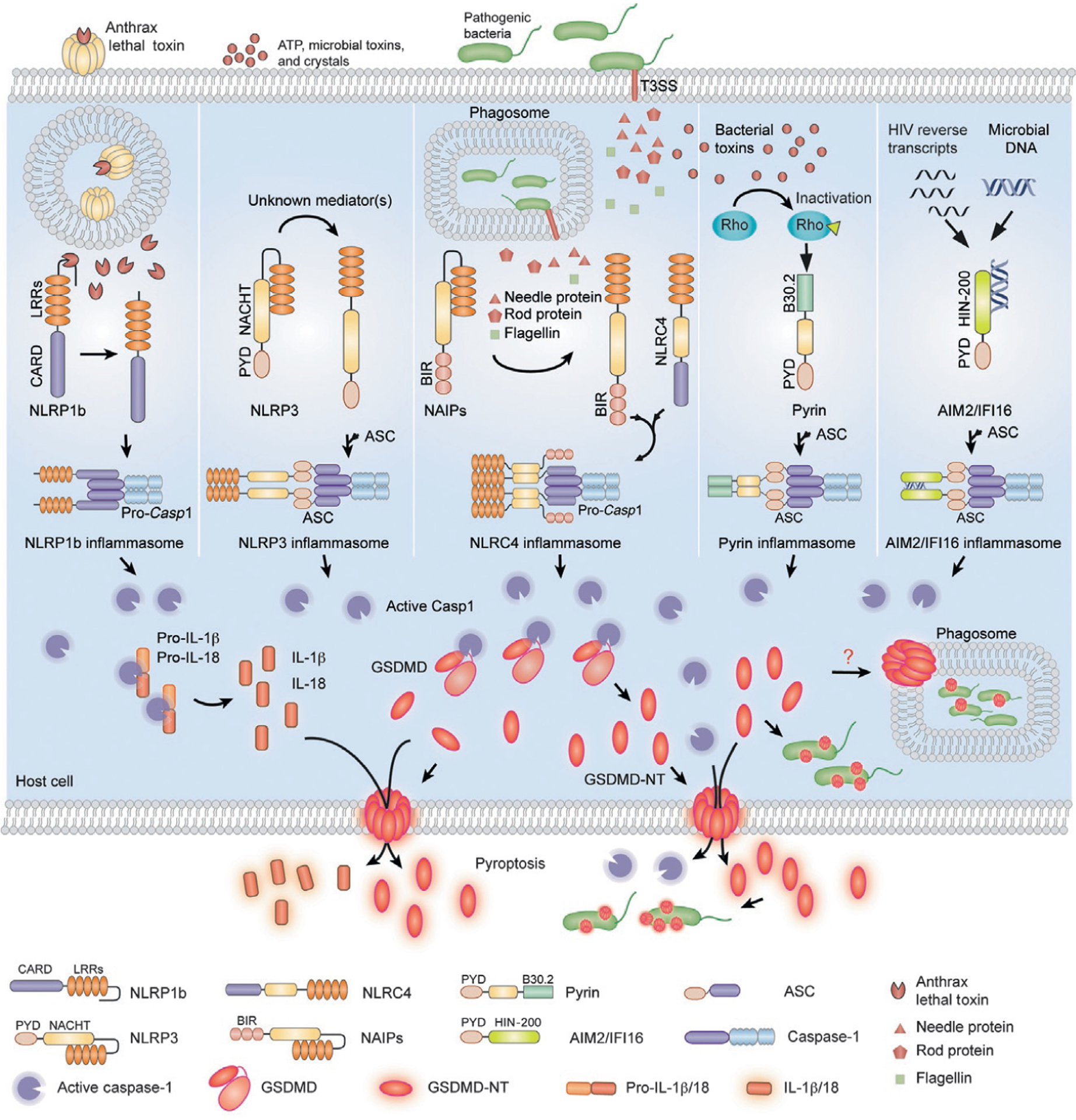
Model of canonical inflammasome activation and inflammatory cell death. NLR proteins (NLRP1, NLRP3, NAIP/NLRC4), pyrin and ALR proteins (AIM2 and IFI16) sense danger signs and initiate assembly of canonical inflammasomes. NLRP1b is activated by the metalloprotease activity of Bacillus anthracis lethal toxin (LT). The NLRP3 inflammasome is activated by a broad spectrum of stimuli, such as ATP, microbial toxins, and crystals, by an unknown mechanism. NAIPs recognize and directly bind to bacterial flagellin or bacterial type III secretion system components to initiate the NLRC4-dependent inflammasome. Pyrin detects the inactivation of Rho GTPases by bacterial toxins. AIM2 and IFI16 sense DNA via their HIN domain. The canonical inflammasome complexes form a platform to activate caspase-1. Activated caspase-1 processes proinflammatory cytokines (pro-IL-1β, pro-IL-18) and cleaves GSDMD, releasing its N-terminal fragment GSDMD-NT, which forms pores in the plasma membrane of the infected cell, resulting in pyroptotic cell death during which bacteria, inflammatory cytokines caspase-1, and GSDMD-NT are released. GSDMD-NT also directly kills intracellular and cell-free bacteria. It is not known whether GSDMD-NT permeabilizes the phagosome membrane and accesses bacteria that replicate within phagosomes.
The ALRs, also known as the PYHIN family, contain an N-terminal PYD for binding to the PYD-containing adaptor protein ASC, and one or two C-terminal HIN-200 (hematopoietic IFN-inducible nuclear antigen with 200 amino acid repeats) domains that sense DNA, produced by DNA viruses and retroviruses (either infectious or endogenous), invasive bacteria, or cytosolic self-DNA that can be found in the cytosol during certain pathological situations (Fernandes-Alnemri, Yu, Datta, Wu, & Alnemri, 2009; Hornung et al., 2009; Kerur et al., 2011; Roberts et al., 2009; Fig. 1). AIM2 and IFI16 are the best studied ALRs.
Another canonical inflammasome-activating sensor is pyrin, a PYD-containing protein encoded by the MEFV (Mediterranean fever) gene that indirectly recognizes bacterial toxins/effector proteins from Clostridium difficile, Vibrio parahaemolyticus, and Histophilus somni and other enteropathogenic bacteria (Aubert et al., 2016; Ratner et al., 2016; Xu et al., 2014). These bacterial products inactivate Rho GTPases, causing pyrin dephosphorylation, dimerization, and ASC recruitment through a heterotypic PYD:PYD interaction (Fig. 1).
The canonical inflammasome has mostly been studied in myeloid professional antigen-presenting cells, where caspase-1 is constitutively expressed. However, the extent of caspase-1 expression in the skin and mucosal epithelia under normal homeostatic circumstances where these epithelia are in constant contact with commensal microbiota is unclear. The proinflammatory cytokine IL-1β and other proinflammatory genes are upregulated in both professional antigen-presenting cells and barrier epithelia by danger signals that activate NF-κB. These cells act as hubs for establishing host inflammatory status, through both processing inflammatory cytokines to their mature form and driving pyroptotic cell death. Caspase-1 activation is required for all the inflammatory sequelae of canonical inflammasome assembly (Fig. 1).
2.2. The Noncanonical Inflammasome
The other inflammatory caspases, caspase-11 in mice and caspase-4 and -5 in humans, are expressed more broadly than caspase-1. The importance of these other caspases was obscured by many of the early studies of inflammasome activation in Casp1−/− mice, which were generated by targeting embryonic stem cells from 129 strain mice, because this strain contains a splice site deletion in Casp11 that results in a prematurely terminated transcript that does not produce caspase-11 protein (Kang et al., 2000; Kayagaki et al., 2011). Because the Casp1 and Casp11 genes are adjacent, backcrossing into other strains that encode for functional caspase-11 does not restore caspase-11 expression. Hence these Casp1−/− mice (Casp1−/−Casp11129mt/129mt) were effectively doubly knocked out for both inflammatory caspase genes. In fact, mice genetically deficient in just caspase-11, but not just caspase-1, are resistant to LPS-induced septic death (Li et al., 1995; Wang et al., 1998). Moreover, LPS-induced caspase-1 activation and IL-1β secretion are markedly decreased in caspase-11-deficient mice (Kayagaki et al., 2013; Wang et al., 1998). Thus, caspase-11 plays a critical role in the LPS response and sepsis. However, how LPS activates caspase-11 was unknown. In 2011 Kayagaki et al. found that IL-1β maturation and secretion and pyroptosis in response to infection with gram− bacteria (Escherichia coli, Citrobacter rodentium, and Vibrio cholerae) were blocked in Casp11−/− macrophages from C57BL/6 mice and WT macrophages from 129 mice, whereas activation of canonical NLRP1, NLRP3, and NAIP–NLRC4 inflammasomes was intact (Kayagaki et al., 2011). This study called the caspase-11-dependent inflammasome the “noncanonical inflammasome” (Kayagaki et al., 2011). Follow-up studies identified intracellular LPS derived from gram− bacterial cell walls as the trigger for noncanonical inflammasome activation and found it to be independent of Toll-like receptor 4 (TLR4), the receptor for extracellular LPS (Hagar, Powell, Aachoui, Ernst, & Miao, 2013; Kayagaki et al., 2013). Electroporation of LPS into the cytosol activates caspase-11, whereas infection with LPS-deficient bacteria fails to activate caspase-11. Soon thereafter, the lipid A component of LPS was found to bind to caspase-11 and human caspase-4 and -5 directly by a high-affinity interaction with its CARD domain to cause inflammatory caspase oligomerization and activation and pyroptosis (Shi et al., 2014; Fig. 2). This study suggested a novel model for a simplified noncanonical inflammasome in which immune sensing and caspase activation are combined in a single inflammatory caspase molecule without needing any adaptors. This elegant model is not universally accepted. Structural studies of an LPS–caspase-11 complex are needed to confirm the model and define the features (i.e., size, number of monomers, conformational changes that induce caspase-11 autoactivation) of the noncanonical inflammasome. A structure could also provide a basis for identifying specific caspase-11 inhibitors, which are not currently available, but would be invaluable for probing the significance of caspase-11 activation and might be useful for developing therapies to treat sepsis. Although LPS is the only known PAMP recognized by caspase-11, it would be worthwhile to investigate whether any lipids or other products of gram+ bacteria or other intracellular pathogens, such as parasites, bind to and activate caspase-11.
Fig. 2.
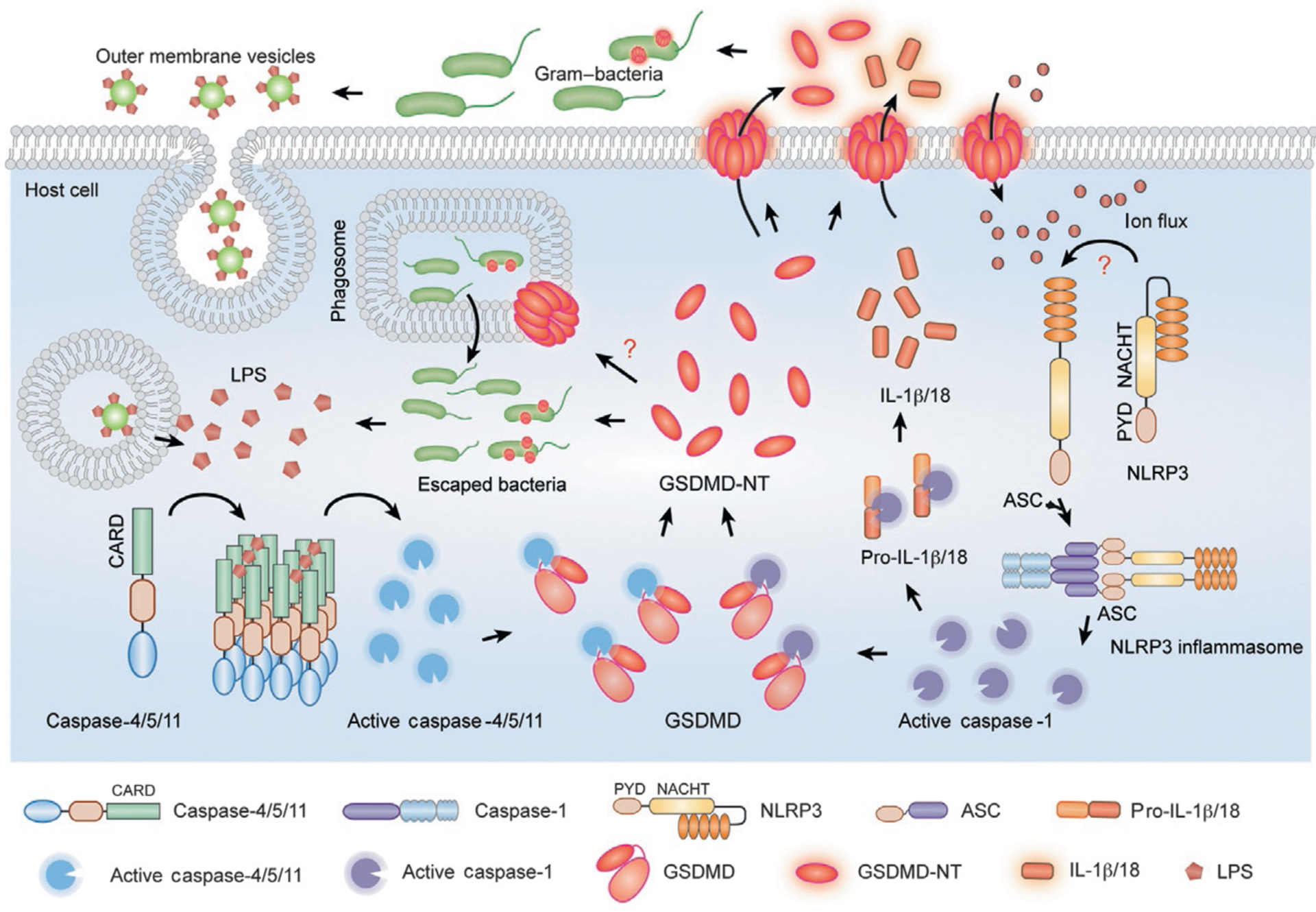
Cytosolic LPS activates noncanonical inflammasome activation and pyroptosis. LPS, from intracellular gram− bacteria or from outer membrane vesicles produced by cell-free gram− bacteria that are taken up by cells, binds with high affinity to caspase-4/5/11, causing self-assembly of the noncanonical inflammasome. These caspases are then activated and trigger pyroptosis by cleaving GSDMD. Active caspase-4/5/11 do not directly cleave the proinflammatory cytokine precursors, but cause them to be processed indirectly by activating the NLRP3 inflammasome and caspase-1. The NLRP3 inflammasome and caspase-1 may be activated by ion fluxes caused by damage to the plasma membrane.
Although activated caspase-11 induces pyroptosis, it does not directly cleave the proinflammatory cytokine precursors. However, noncanonical inflammasome activation leads to secondary activation of the canonical inflammasome and caspase-1 and inflammatory cytokine processing and secretion (Kayagaki et al., 2011; Fig. 2). The underlying mechanism of caspase-1 activation is not completely clear, although it does not occur in cells genetically deficient in NLRP3 or ASC, suggesting that it is mediated by NLRP3 activation in response to loss of cell membrane integrity (Kayagaki et al., 2011). However, there is currently no direct evidence for the assembly of the canonical inflammasome in the dying cell or understanding of how it is triggered.
2.3. Pyroptosis
Infection of macrophages with invasive Shigella flexneri and S. typhimurium induces death of the host cell. Death was initially attributed to apoptosis since host cell DNA fragmentation and membrane blebbing are observed and caspase-3 is cleaved as a late event (Chen, Kaniga, & Galan, 1996; Lindgren, Stojiljkovic, & Heffron, 1996; Monack, Raupach, Hromockyj, & Falkow, 1996; Zychlinsky, Prevost, & Sansonetti, 1992). However, further analysis revealed that macrophage death uniquely depends on caspase-1 rather than apoptotic caspases and is also accompanied by disruption of the cell membrane and release of inflammatory cytokines and other small molecules (Brennan & Cookson, 2000; Chen, Kaniga, & Galan, 1996; Chen, Smith, Thirumalai, & Zychlinsky, 1996; Fernandez-Prada, Hoover, Tall, & Venkatesan, 1997; Hersh et al., 1999; Hilbi et al., 1998; Zychlinsky, Fitting, Cavaillon, & Sansonetti, 1994). Inflammasome-mediated cell death is inflammatory, in striking contrast to effector caspase-mediated apoptosis, but depends on caspase activation, which distinguishes it from unprogrammed necrosis and programmed cell death by necroptosis, caused by RIPK3-MLKL activation. In 2001, Cookson and his colleagues termed this inflammasome-dependent type of cell death “pyroptosis” from the Greek roots “pyro” relating to fire or fever, and “ptosis” meaning falling off (Cookson & Brennan, 2001).
Cells undergoing pyroptosis appear to have unique morphological features (Table 1). In necroptosis, MLKL membrane binding forms plasma membrane oligomers that generate ion channels that cause osmotic swelling and sudden bursting of the cell membrane (Newton & Manning, 2016). In contrast during pyroptosis, whose mechanism will be described in Section 3, plasma membrane pores are formed that allow release of small molecules, including ATP and small-molecular-weight proteins, including HMGB1 and proinflammatory cytokines (Chen et al., 2016; Fink & Cookson, 2007). Because proteins are released, the cell likely maintains its osmolarity and does not burst. Moreover, the cell appears to flatten as cell contents are released (Chen et al., 2016). However, plasma membrane pores likely cause an influx of Ca++, which is high in extracellular fluids but low within the cell, which could activate a Ca++-dependent damaged cell membrane repair response, like that induced by the killer cell pore-forming protein perforin and bacterial pore-forming proteins (Idone et al., 2008; Keefe et al., 2005). The stereotypic plasma membrane repair response typically involves multiple mechanisms to restore plasma membrane integrity, including patching by fusion of intracellular vesicular membranes with the plasma membrane, blebbing to remove damaged membrane, and endocytosis of the damaged membrane (Andrews, Almeida, & Corrotte, 2014). Membrane blebbing and release of membrane-delimited blebs, which also occur during apoptosis, are a well-described feature of pyroptosis. However, it will be worth studying whether other features of plasma membrane repair are induced during pyroptosis and whether there may be circumstances under which cell death is averted after inflammasome activation by cellular repair responses. Although DNA fragmentation has been reported during pyroptosis (Brennan & Cookson, 2000), other nuclear features that occur during apoptosis, such as chromatin condensation and nuclear fragmentation, do not seem to occur and a mechanism for DNA fragmentation has not been described.
Table 1.
Cell Death Programs
| Type | Morphological Features | Molecular Features | Characteristic | Signaling Pathways | Effectors |
|---|---|---|---|---|---|
| Apoptosis | Cell shrinkage, membrane blebbing, chromatin condensation, nuclear fragmentation | Cell membrane lipid scrambling, mitochondrial outer membrane permeabilization, genomic DNA fragmentation, translation inhibition | Noninflammatory | Initiator and effector caspase activation | Effector caspases |
| Necroptosis | Cell rounding up, cell swelling, explosive cell membrane rupture | Membrane ion channel formation, release of cellular contents | Inflammatory | TNFRSF—RIPK1/RIPK3, DAI—RIPK3, TLR—TRIF/RIPK3 | MLKL |
| Pyroptosis | Membrane blebbing, cell swelling, plasma membrane rupture | Cell membrane pore formation, release of inflammatory intracellular contents, DNA fragmentation | Inflammatory | Canonical/noncanonical inflammasome, inflammatory caspase activation | Inflammatory caspases, GSDMD |
Now that the underlying mechanism of pyroptosis is known, it will be worthwhile to carefully evaluate the features, the kinetics of their appearance, and underlying mechanisms of cellular changes that occur in pyroptosis. For example, DNA fragmentation could be a late event caused by secondary caspase-3 activation rather than an intrinsic feature of pyroptosis. If it occurs early in pyroptosis, then it would be helpful to identify the inflammatory caspase-activated DNase and determine whether DNA fragmentation promotes pyroptotic death. Similarly, it will be useful to examine whether mitochondria or other organelles are damaged (for example, by GSDMD pores) during pyroptosis. It is likely that the inflammatory caspases have additional biologically important substrates besides the proinflammatory cytokines and GSDMD. Identifying other inflammatory caspase substrates that might trigger subroutines of pyroptotic programmed cell death would be a useful strategy for understanding all the molecular features of pyroptosis. There are some hints of other substrates in a proteomics study by Wells and colleagues, which initially identified GSDMD as a substrate of caspase-1, but did not probe the biological significance of its cleavage (Agard, Maltby, & Wells, 2010). However, that study did not find that the other inflammatory caspases cleave GSDMD, suggesting that more sensitive methods may be necessary to uncover new inflammatory caspase substrates.
2.4. Processing and Release of Proinflammatory Cytokines and Inflammatory Mediators
Unlike other proinflammatory cytokines, IL-1β and IL-18 are induced and synthesized as inactive zymogens (pro-IL-1β, pro-IL-18), which need to be proteolytically activated. Mature IL-1β and IL-18 are potent inflammatory cytokines that cause local and systematic inflammation and also function to promote T cell activation and B cell antibody production (Dinarello, 2009; Sims & Smith, 2010). They also promote the polarization of T cells to inflammatory TH17 cells (Chung et al., 2009; Coccia et al., 2012). Both cytokines have been implicated as key mediators of autoimmune, infectious, and degenerative diseases (Dinarello et al., 2012). Activated caspase-1, but not the other inflammatory caspases, cleaves and activates pro-IL-1β and pro-IL-18.
Inflammasome activation causes the release of IL-1β and IL-18 to the extracellular milieu. Because these cytokines lack an N-terminal signal peptide for secretion via the endoplasmic reticulum to Golgi exocytosis pathway, how they are released from inflammasome-activated cells was previously unclear. Pyroptosis-induced disruption of the integrity of the plasma membrane was proposed as the mechanism for their release (Liu, Yamaguchi, et al., 2014). This is very likely, especially as we now know (see Section 3) that the pores formed in the plasma membrane during pyroptosis have an inner diameter of ~15 nm, which is much bigger than processed IL-1β, which has a diameter of ~4.5 nm (Aglietti et al., 2016; Ding et al., 2016; Finzel et al., 1989; Liu et al., 2016; Sborgi et al., 2016; Figs. 1 and 2). Recent studies have shown that active caspase-1 and the cleaved N-terminal fragment of the pore-forming protein GSDMD are also released from pyroptotic cells (Liu & Lieberman, 2017; Liu et al., 2016). It is highly likely that many cytokines and chemokines, which are all low-molecular-weight proteins, other inflammatory mediators, such as HMGB1, small soluble molecules, like ATP, and other proteins are also released and contribute to the inflammation that accompanies pyroptosis. An unbiased analysis of the culture supernatants of pyroptotic macrophages, dendritic cells, and epithelial cells to define what molecules are released and their properties should be very informative. For example, are all molecules below a certain size efficiently released or are there other defining properties that determine what gets released during pyroptosis?
3. THE MOLECULAR BASIS OF PYROPTOSIS
3.1. Cleavage of GSDMD by Inflammatory Caspases Initiates Pyroptosis
The underlying mechanism by which inflammatory caspases initiate pyroptosis was not defined until recently. Because active caspase proteolysis was known to be required for pyroptosis, cleavage of an unknown substrate(s) was a likely initiating event. In 2010 Agard et al. used a proteomics method that is selective for identifying newly cleaved proteins to profile substrates in human monocyte cell line lysates treated with the human inflammatory caspases or in the same cells induced to undergo pyroptosis by treatment with urate crystals, transfection with DNA, or treatment with LPS and ATP (Agard et al., 2010). They identified 82 candidate substrates of caspase-1, only 3 possible substrates of caspase-4 and no caspase-5 substrates. Fewer than 30 newly cleaved proteins were identified in the cells triggered to undergo pyroptosis, but only 6 substrates were common to all 3 methods of inducing pyroptosis. Of those 6, 1 was also identified as a substrate of both caspase-1 and -4 (the splicing factor U2AF2) and 3 others were identified as putative caspase-1 substrates (GSDMD, tumor protein D54 encoded by the TPD52L2 gene, and the eukaryotic translation initiator factor eIF3J). GSDMD, U2AF2, and eIF3J were all experimentally validated in in vitro caspase-1 cleavage assays. Of note, GSDMD was the best substrate—cleaved 50–500 times faster than any other substrate tested. However, this paper did not examine the role of these identified substrates in inflammasome pathways and there was no follow-up. In 2015, two papers independently identified GSDMD as a substrate of the inflammatory caspases by genetic screens designed to identify essential genes for initiating pyroptosis (Kayagaki et al., 2015; Shi, Zhao, et al., 2015). In one paper, an ethyl-N-nitrosourea (ENU) mutagenesis screen in mice identified a novel mutant mouse strain that contained a single-nucleotide variant (an Ile105Asn substitution) in Gsdmd, which failed to produce IL-1β upon cytosolic LPS treatment (Kayagaki et al., 2015). The other paper performed an unbiased genome-wide CRISPR/Cas9 screen in Tlr4−/− immortalized bone marrow-derived macrophages (iBMDMs) to search for factors needed for canonical or non-canonical inflammasome-induced pyroptosis (Shi, Zhao, et al., 2015). Gsdmd was identified as the top hit (Shi, Zhao, et al., 2015). Gsdmd−/− iBMDMs did not secrete inflammatory cytokines or undergo pyroptosis in response to cytosolic LPS and ectopic expression of GSDMD completely restored the response (Kayagaki et al., 2015; Shi, Zhao, et al., 2015). Similarly, GSDMD was essential for caspase-1-mediated pyroptosis and IL-1β release after canonical inflammasome activation (Kayagaki et al., 2015; Shi, Zhao, et al., 2015). As expected, Gsdmd deficiency did not affect apoptosis (Shi, Zhao, et al., 2015). In addition to showing that pyroptosis depends on GSDMD, these results also implied that proinflammatory cytokine secretion depends on pyroptosis. Of note, Gsdmd−/− iBMDMs underwent delayed pyroptosis after canonical inflammasome activation (Kayagaki et al., 2015), suggesting that there may be other caspase-1 substrates that can mediate GSDMD-independent cell death.
After canonical or noncanonical inflammasome assembly, the activated inflammatory caspases cleave GSDMD after Asp276 (mouse)/275 (human) to generate an N-terminal fragment (GSDMD-NT, ~30 kDa) and a C-terminal fragment (GSDMD-CT, ~23 kDa) (Agard et al., 2010; Kayagaki et al., 2015; Shi, Zhao, et al., 2015; Figs. 1 and 2). Mutation of the cleavage site blocks GSDMD cleavage by inflammatory caspases and pyroptosis (Kayagaki et al., 2015; Shi, Zhao, et al., 2015), suggesting that GSDMD cleavage initiates pyroptosis. In fact ectopic overexpression of GSDMD-NT, but not GSDMD or GSDMD-CT, rapidly triggers cell death (Kayagaki et al., 2015; Shi, Zhao, et al., 2015). Thus, GSDMD-NT is the key mediator of pyroptotic death.
3.2. GSDMD-NT Induces Pyroptosis by Forming Membrane Pores
Five papers quickly uncovered the mechanism behind GSDMD-NT-mediated pyroptosis (Aglietti et al., 2016; Chen et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). GSDMD-NT was predicted to share structural homology (but not amino acid homology) with the membrane attack complex perforin-like domain of the killer cell pore-forming protein perforin. In particular, the predicted structure of GSDMD-NT contained homologous regions to the two regions of perforin that are modeled to undergo conformational unwinding into amphipathic β-strands for membrane insertion. This prediction led us to hypothesize that GSDMD-NT might oligomerize to form membrane pores that permeabilize mammalian cell membranes in cells undergoing pyroptosis (Liu & Lieberman, 2017). Indeed, GSDMD-NT migrates in high-molecular-weight oligomers on nonreducing and native polyacrylamide gels, while GSDMD migrates like a monomer (Chen et al., 2016; Liu et al., 2016). The oligomers fall apart in the presence of reducing agents, suggesting that intermolecular or intra-molecular disulfide bonds are critical to oligomerization. Indeed mutation of either Cys39 or Cys192 to Ala blocks oligomerization (Liu et al., 2016). While GSDMD localizes to soluble cytosolic fractions of cells, high-molecular-weight GSDMD-NT fractionates with heavy membranes and localizes to the plasma membrane by confocal microscopy (Aglietti et al., 2016; Chen et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016).
To define the lipid requirements for GSDMD-NT oligomerization, binding of GSDMD-NT, GSDMD, and GSDMD-CT to membrane lipids spotted on membrane strips was assessed (Chen et al., 2016; Ding et al., 2016; Liu et al., 2016). Neither GSDMD nor GSDMD-CT binds to any lipid, but GSDMD-NT binds to the phosphatidylinositol phosphates (PIPs), PtdIns(4)P and PtdIns(4,5)P2, and phosphatidic acid as well as phosphatidylserine (PS), all of which are found exclusively on the inner leaflet of mammalian cell membranes. GSDMD-NT does not bind to phosphatidylcholine or phosphatidylethanoloamine (PE) present on both leaflets. GSDMD-NT also strongly binds to the mitochondrial and bacterial lipid, cardiolipin. In comparison, perforin, which permeabilizes target cell plasma membranes from the outside, binds to PE, but not PS, and the same phospholipids as GSDMD-NT, while granulysin, which permeabilizes microbial membranes, binds to cardiolipin (Liu et al., 2016). These phospholipid-binding properties determine whether GSDMD-NT binds to liposomes— GSDMD-NT only binds to liposomes constructed with the lipids it binds on membranes (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). Importantly, GSDMD-NT permeabilizes liposomes containing PS, PIPs, or cardiolipin, suggesting that GSDMD-NT forms pores in the lipid bilayer (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016). In fact, several groups were able to visualize GSDMD-NT membrane pores formed on liposomes by negative staining electron microscopy (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016; Fig. 3). The pores are relatively homogenous in size, composed of ~16 subunits with a ~10–16-nm inner diameter. These pores are big enough to allow small molecules and small-medium-molecular-weight proteins, like cytokines, to pass through readily. Some of the lipids that GSDMD-NT binds are also found on the cytosolic side of intracellular organelles, especially endosomes and phagosomes that pinch off from the plasma membrane. Although there is currently no evidence that GSDMD-NT binds to any intracellular vesicles or that these vesicles are damaged during pyroptosis, this would be worth investigating.
Fig. 3.
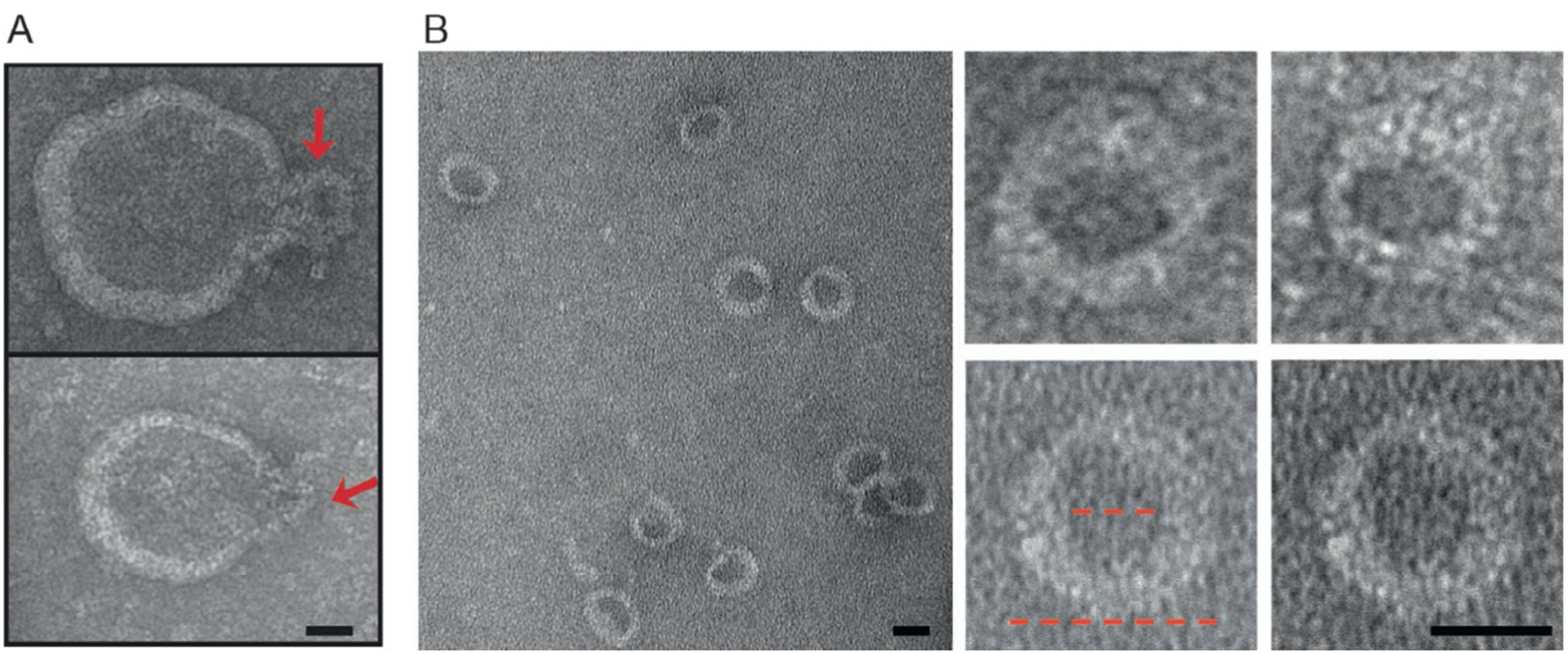
GSDMD-NT forms pores in liposomes. Shown are representative negative staining electron microscopy images of side views (A, indicated by arrows) and top-down views (B) of GSDMD-NT pores formed in phosphatidyl serine-containing liposomes. The inner and outer diameters of GSDMD-NT pores (red dotted lines) are approximately 15 and 32 nm, respectively. Scale bars, 20 nm. Figure is taken from Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature, 535, 153.
So far no one has been able to visualize GSDMD-NT pores in cells undergoing pyroptosis or to obtain high-resolution structures of GSDMD-NT pores using cryoelectron microscopy. Although GSDMD is a reasonably well-behaved protein, attempts to crystallize it have also failed. A 1.9-Å structure of the full-length mouse ortholog GSDMA3 was obtained by Shao’s group (Ding et al., 2016). The N- and C-termini each form separate domains, which are dissimilar from other known structures, that are separated by a long exposed loop, a likely cleavage site. The structure contains a large interface between the N- and C-terminal domains. The loop connecting the two domains of GSDMA3 has no caspase cleavage site, and GSDMA3 is not cleaved by any of the caspases. However, when the loop of GSDMA3 is engineered to be cleaved, it separates from the C-terminal domain and, like GSDMD-NT, causes pyroptosis (Shi, Zhao, et al., 2015). Because GSDMD is about 70% homologous to GSDMA3, the structure of full-length GSDMD can be modeled based on the crystal structure of GSDMA3. GSDMD and GSDMA3 have similar structures (Fig. 4). Caspase cleavage of GSDMD to GSDMD-NT produces a protein that aggregates, loses activity, and is difficult to purify and work with. Therefore, detailed structural information about GSDMD-NT (or any of the N-terminal GSDMs) or the pores they form, which would be very informative about mechanism and be helpful for finding inhibitors of pyroptosis, is not available. The current model is that binding of the C-terminal fragment inhibits the N-terminal fragment from binding to lipids, which may be the first step in pore formation. Lipid binding likely causes a radical conformational change of the N-terminal domain, allowing it to oligomerize in membranes and form pores. Because GSDMD-NT on its own forms pores in vitro in liposomes, pore formation does not seem to require any auxiliary factors. Moreover, pore formation occurs in Ca++-free medium and is thus independent of Ca++ (Liu et al., 2016).
Fig. 4.
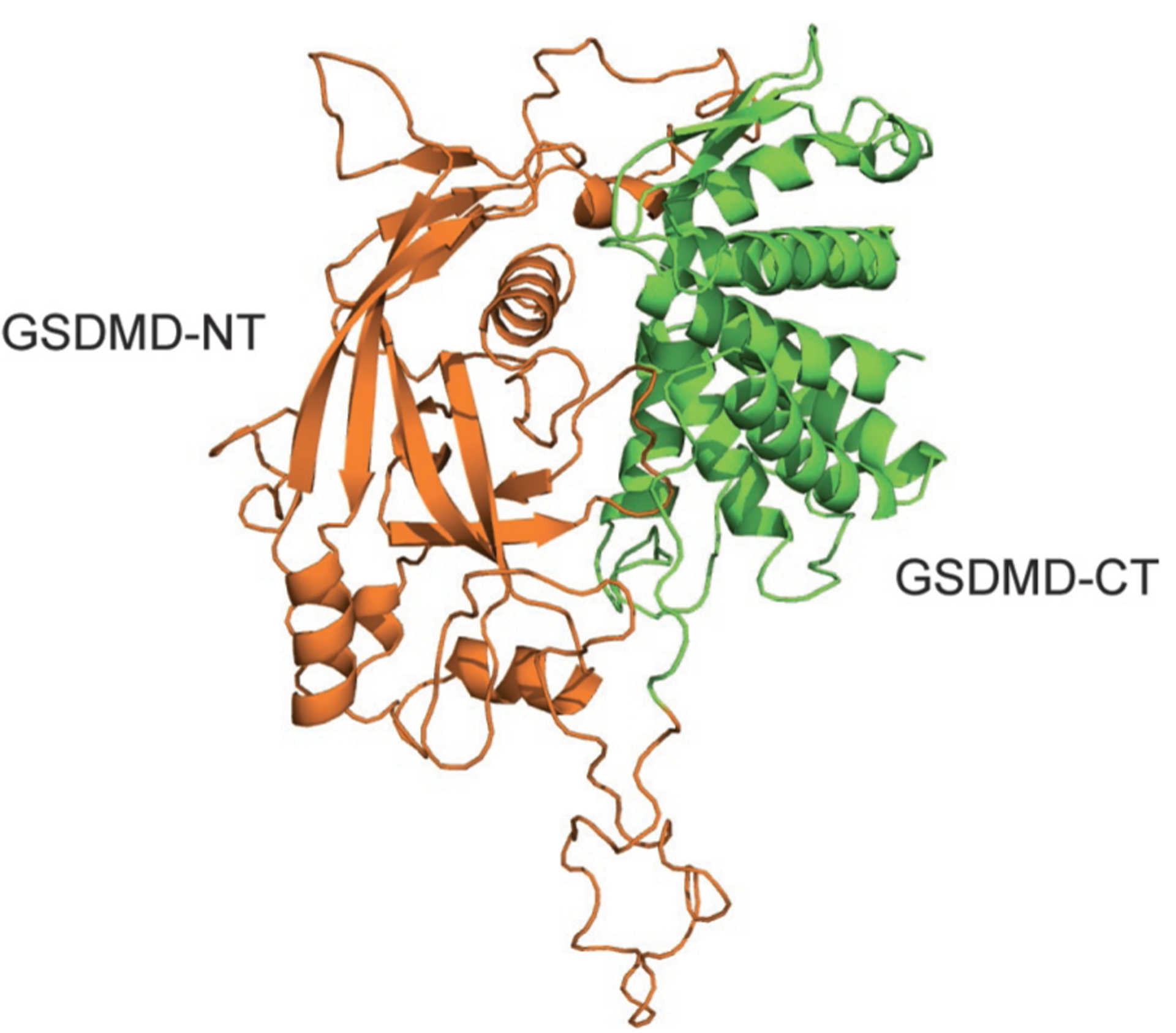
Structure of GSDMD modeled based on the crystal structure of GSDMA3. The structural model of GSDMD was generated based on the structure of GSDMA3 (PDB: 5B5R) described in Ding et al. (2016) using the Swiss Model server. Orange, GSDMD-NT; green, GSDMD-CT.
GSDMD-NT, but not full-length GSDMD, is released from pyroptotic cells (Liu et al., 2016; Figs. 1 and 2). Because it does not bind to plasma membrane outer leaflet lipids, it does not injure bystander resting cells (Ding et al., 2016; Liu et al., 2016). Thus, although the pyroptotic cell releases inflammatory mediators, pyroptosis does not spread, which limits the extent of inflammation and tissue damage.
3.3. N-Terminal GSDMD Kills Bacteria
Pyroptosis was previously thought to protect against bacterial infection indirectly by forcing intracellular bacteria from their favorable replicative niche inside cells to the extracellular milieu, where they replicate less and are phagocytosed and killed by neutrophils recruited to sites of inflammation. However, the experimental support for this idea is limited (Miao et al., 2010). Because GSDMD-NT binds to cardiolipin, a component of bacterial membranes, we investigated whether GSDMD-NT might bind to bacteria and kill them by damaging their cell membranes. Indeed expression of GSDMD-NT in bacteria is cytotoxic. More importantly, GSDMD-NT binds to, permeabilizes, and causes the death of both gram− (E. coli) and gram+ (Staphylococcus aureus) bacteria in vitro at low nanomolar concentrations, as verified by assays for bacterial colony-forming unit and growth and propidium iodide uptake (Liu et al., 2016). GSDMD-NT treatment also causes lysis of gram+ Bacillus megaterium protoplasts (Ding et al., 2016). GSDMD-NT, but not full-length GSDMD, is released from pyroptotic cells, and pyroptotic cell culture supernatants kill free bacteria (Liu et al., 2016). Killing is abrogated by depletion of GSDMD-NT, showing it depends on GSDMD-NT (Liu et al., 2016). Intracellular Listeria appears to also be directly killed by GSDMD-NT since overexpression of GSDMD-NT in mammalian cells suppresses intracellular bacterial growth, while knockdown of endogenous GSDMD does the opposite (Liu et al., 2016). These data suggest a direct, and likely powerful, antibacterial defense triggered by GSDMD cleavage—formation of pores in bacterial membranes that directly kill bacteria (Figs. 1 and 2). Thus, the innate immune pyroptotic response to invasive microbes may directly destroy them.
However, more work is needed to confirm direct bacterial killing by GSDMD-NT and many questions remain. So far a direct protective effect of GSDMD-NT on bacteria has only been shown in vitro. GSDMD-NT binding and pore formation is presumed to be mediated by cardiolipin, but this has not been shown. It is also unclear if and how GSDMD-NT penetrates the cell wall of bacteria or crosses the outer membrane of gram− bacteria to access the inner membrane. Until recently cardiolipin was thought to be restricted to the inner bacterial membrane, but a recent study shows that cardiolipin is transported from the inner to outer membrane, making the outer membrane also a possible target of GSDMD-NT (Dalebroux et al., 2015). Electron microscopy studies in GSDMD-NT-treated free bacteria and infected cells should be able to visualize the membranes GSDMD-NT binds and whether it assembles into discrete pores and characterizes the morphological changes in attacked bacteria. In infected cells, we need to know whether GSDMD-NT just attacks cytosolic bacteria or also bacteria within phagosomes. Cytosolic bacteria may be more vulnerable than those residing in intracellular vacuoles, based on the observation that S. typhimurium ΔsifA mutant strain which escapes the phagosome and replicates in the cytosol is attenuated compared to the vacuolar wild-type strain (Aachoui et al., 2013). Again immunofluorescence microscopy and electron microscopy with wild-type and mutant bacteria that do or do not escape the phagosome (such as hlo mutant L. monocytogenes) should be able to begin to answer this question. Experiments in infected mice lacking Gsdmd or bearing nonfunctional mutants are needed to confirm the role of GSDMD in protection from different types of pathogenic bacteria. However, since there is currently no experimental way to separate host cell and bacterial membrane damage by GSDMD-NT, these in vivo experiments will not be able to distinguish whether bacterial control is due to host cell pyroptosis or direct bacterial killing. An intriguing recent single-cell study showed that cytosolic Salmonella replication is inhibited by canonical and noncanonical inflammasome activation, by a mechanism that depends on proinflammatory caspase enzymatic activity, but occurs in Gsdmd−/− cells and before host cell pyroptosis (Thurston et al., 2016). This finding suggests that there may be other unknown direct antibacterial mechanisms mediated by other inflammatory caspase substrates.
Some protozoan parasites also activate the inflammasome (Zamboni & Lima-Junior, 2015). It will be worth exploring whether GSDMD-NT has activity against other classes of microbial pathogens including parasites and fungi.
Only invasive bacteria that replicate within cells or inject bacterial mediators, including toxins or secretion system-delivered effector proteins, into the cytosol have been thought to activate the inflammasome and pyroptosis. However, the noncanonical inflammasome may also be activated in both phagocytic and nonphagocytic cells by noninvasive gram− bacteria. A recent study showed that gram− bacteria secrete LPS-coated outer membrane vesicles (OMVs) that are taken up by clathrin-mediated endocytosis and are released from endosomes into the cytosol, where they activate caspase-11 and pyroptosis (Vanaja et al., 2016; Fig. 2). Mice injected with a mutant strain of E. coli that produces fewer OMVs have much lower levels of circulating proinflammatory cytokines than mice infected with wild-type bacteria, suggesting that noninvasive bacteria can activate pyroptosis. Therefore, GSDMD may play an important role in bacterial defense more broadly or in maintaining the homeostasis of the microbiota, especially since GSDMD-NT can kill free bacteria. However, if this mechanism proves physiologically important, one wonders how commensal and pathogenic bacteria are distinguished.
4. OTHER GSDM FAMILY MEMBERS AND GSDM-RELATED GENES
GSDMD belongs to a poorly characterized novel gene family, the GSDMs (Tamura et al., 2007). In humans, this gene family consists of GSDMA, GSDMB, GSDMC, and GSDMD on chromosomes 17q21.1, 17q12, 8q24.21, and 8q24.3, respectively. There are also two more distant genes, DFNA5 and DFNB59 (also known as pejvakin) that share common N-terminal domains with GSDMs and are considered part of the GSDM superfamily (Fig. 5 and Table 2). Mice have three GSDMA counterparts (Gsdma1–3) and four GSDMC counterparts (Gsdmc1–3) clustered on mouse chromosomes 11 D and 15 D1, respectively, and a GSDMD homologue (Gsdmd) on chromosome 15D3-E1, but no GSDMB homologue (Tamura et al., 2007). All family members share highly conserved N- and C-terminal domains, while the linker region is variable. Not much is known about the physiological functions of GSDMs other than GSDMD, but the recent GSDMD studies should spark renewed interest in this gene family. Their name comes from their selective expression in the gastrointestinal tract and dermis (Saeki, Kuwahara, Sasaki, Satoh, & Shiroishi, 2000). However, they are also expressed in immune cells and other mucosal epithelia, including lung and genital tract (Carl-McGrath, Schneider-Stock, Ebert, & Rocken, 2008; Das et al., 2016). Although not much is known about the normal expression of the GSDMs or how it is regulated, aberrant expression of some of these genes has been reported in tumors (both increased and decreased expression of different family members) and in immune disorders.
Fig. 5.
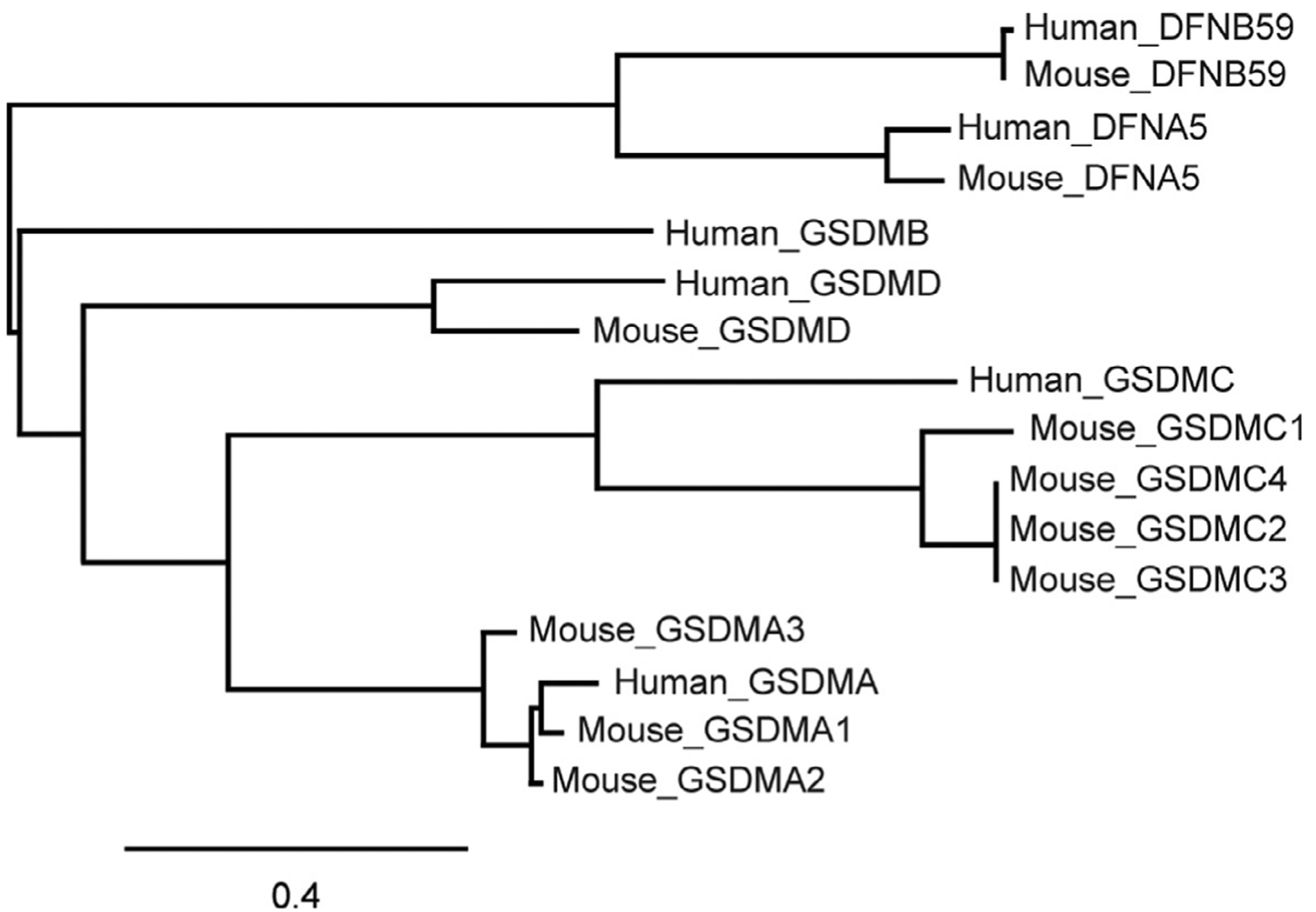
Phylogenetic relationship of the human and mouse gasdermin superfamily. Phylogenetic tree showing the inferred evolutionary relationships of human and mouse gasdermin superfamily members generated using MUSCLE for amino acid sequence alignment and “maximum likelihood” method for phylogeny. Scale bar indicates the number of substitutions per amino acid.
Table 2.
Human Gasdermin Genes
| Gene Name (Chromosomal Location) | Mouse Orthologues (Chromosomal Location) | Extent of Homology to GSDMD | Expression Pattern | Physiological Functions | Disease Links |
|---|---|---|---|---|---|
| GSDMA (17q21.1) | Gsdma1, Gsdma2, Gsdma3 (11 D) | 63.4% | Upper gastrointestinal tract (esophagus, stomach), skin | Unknown | Hair follicle and sebaceous gland developmental defects, alopecia |
| GSDMB (17q21.1) | — | 56.2% | Esophagus, stomach, colon, hepatocytes, bronchial epithelial cells | Unknown | Asthma, cancer |
| GSDMC (8q24.21) | Gsdmc1, Gsdmc2, Gsdmc3, Gsdmc4 (15 D1) | 61.7% | Esophagus, small intestine, colon, trachea, spleen | Unknown | — |
| GSDMD (8q24.3) | Gsdmd (15 D3) | — | Esophagus, stomach, small intestine, colon, lymphocytes | Pyroptosis | — |
| DFNA5 (7p15.3) | Dfna5 (6 B2.3) | 56.1% | Placenta, cochlea, brain, kidney | Unknown | Autosomal dominant nonsyndromic hearing loss |
| DFNB59 (2q31.2) | Dfnb59 (2 C3) | 54% | Brain, eye, inner ear, auditory pathway, heart, lung, kidney, liver, intestine, testis | Hair cell and auditory neuron homeostasis | Recessive nonsyndromic hearing impairment |
Polymorphisms or mutations in GSDMs are associated with skin disease and asthma. Gsdma3 was originally identified as a mutation linked to developmental defects in hair follicle and sebaceous gland development, and post developmental activation of Gsdma3 causes alopecia. Three spontaneous (Dfl, Re-den, Rim3) and six ENU mutagenesis-induced Gsdma3 (Fgn, Bsk, Rco2, Ae, Gsdma3M1Btlr, Gsdma3I359N) mutant mouse lines have been reported to be associated with skin abnormalities (epidermal hyperplasia, hyperkeratosis, and abnormal hair development), all of which are due to mutations in or loss of the C-terminal domain of Gsdma3 (Kumar et al., 2012; Li et al., 2010; Lunny et al., 2005; Porter et al., 2002; Runkel et al., 2004; Tanaka et al., 2007). However, no skin defects are observed in Gsdma3−/− mice, suggesting that the N-terminal domain of GSDMA3 is responsible for the disease phenotypes of the mutant mice and that the mutations are gain-of-function mutants in which the C-terminal domain loses its ability to inhibit the N-terminal domain (Shi, Tang, et al., 2015). Indeed the Ae mutant and Gsdma3-NT both are reported to induce autophagy and cause cell death (Shi, Tang, et al., 2015). In the Gsdma3 structure, the C-terminal domain mutations abolish contacts between the N- and C-terminal domains that likely cause constitutive activation of Gsdma3 (Ding et al., 2016). Although plausible, pore formation by Gsdma3 mutants has not yet been demonstrated. Gsdma3-NT binds to specific phospholipids, oligomerizes, and forms pores on cell membranes, resulting in cell death (Ding et al., 2016). In fact, ectopic expression of the N-terminal domains of all GSDMs is cytotoxic (Ding et al., 2016). So far there is no evidence that the GSDMs, other than GSDMD, are inflammatory caspase substrates. In fact most do not have any potential Asp caspase cleavage sites in their connecting loop. Whether, under what circumstances, and how each GSDM is activated to generate functional N-terminal fragments in response to physiological or pathological cues awaits further exploration. A recent paper, however, shows that DFNA5, which has a conserved DxxD sequence in its connecting loop, is a caspase-3, but not caspase-1, substrate during apoptosis that causes “secondary necrosis” and distinct morphological changes in cells undergoing apoptosis, even in Casp1−/−Casp11−/− mouse macrophages (Rogers et al., 2017). It is not yet clear whether this “secondary necrosis” is actually pyroptosis and what molecules get released from these dying cells. Also the kinetics of DFNA5 cleavage and plasma membrane damage, especially in relation to PS externalization (annexin V staining), would be important to know to understand whether its effects might be physiologically important in vivo when macrophages quickly recognize and phagocytose annexin V+ cells.
GSDMB is highly expressed in lung bronchial epithelia and its polymorphisms are associated with asthma (Kang et al., 2012). However, asthma promotion by GSDMB is probably not related to cell death. In fact GSDMB is predominantly a nuclear protein, which causes airway hyperresponsiveness and airway remodeling by activating the transcription of the TGF-β1 and 5-lipoxygenase genes (Das et al., 2016).
GSDMA is not expressed in esophageal and gastric cancers, while GSDMB expression is upregulated in many types of cancers including hepatocarcinomas, gastric, breast, and uterine cervix cancers (Carl-McGrath et al., 2008; Hergueta-Redondo et al., 2014; Saeki et al., 2007, 2000, 2009; Sun et al., 2008). GSDMC expression is increased in metastatic melanoma (Watabe et al., 2001). The correlation of GSDM expression with cancer progression suggests that GSDMA may function as a tumor suppressor gene to inhibit malignant transformation of cells, while GSDMB may promote tumor cell proliferation and metastasis. Different splice variants may have distinct functions. How the GSDMs function in tumorigenesis is not well defined.
DFNA5 and DFNB59, which share conserved N-terminal domains with GSDMs, are expressed in different types of cells—DFNA5 is mainly expressed in placenta, cochlea, brain, and kidney, and DFNB59 is expressed in brain, eye, inner ear, auditory pathway, heart, lung, kidney, liver, intestine, and testis (Delmaghani et al., 2006; Van Laer et al., 1998). DFNA5 and DFNB59 genetic variants are responsible for autosomal dominant and recessive nonsyndromic hearing impairment, respectively (Delmaghani et al., 2006; Van Laer et al., 1998). DFNA5-induced hearing loss is attributed to a gain-of-function mutation that causes a truncated protein that presumably triggers cell death. DFNB59 defends against noise-induced oxidative stress by modulating peroxisome function to maintain hair cells and auditory neurons (Delmaghani et al., 2015).
5. THE ROLE OF PYROPTOSIS IN IMMUNE RESPONSES TO INFECTION
5.1. Antibacterial Defense
Stimulation of the canonical or noncanonical inflammasome during bacterial infection defends against invading bacteria in mouse models. The inflammasome appears to play a role in maintaining normal gut microbial homeostasis. Genetic deficiency of the NLRP6 inflammasome in colonic epithelial cells leads to overgrowth of intestinal Prevotellaceae bacteria and a predisposition to inflammatory bowel disease (Elinav et al., 2011), and Casp11−/− mice are more susceptible to DSS-induced colitis, which is triggered by gut microbiota (Demon et al., 2014).
S. typhimurium virulence depends on its ability to avoid NAIP/NLRC4-mediated immune sensing via repressing flagellin expression and modifying its T3SS effectors (Miao et al., 2010; Zhao et al., 2016). Clearance of mutant strains that activate the NAIP/NLRC4 canonical inflammasome is mediated by pyroptosis rather than by proinflammatory cytokine secretion (Miao et al., 2010). In the murine Salmonella diarrhea model, two studies showed that NAIP–NLRC4 inflammasome and the caspase-11 noncanonical inflammasome drive pyroptotic host cell death, which causes extrusion of Salmonella-infected cells into the gut lumen, restricting Salmonella replication in enterocytes independently of secretion of the proinflammatory cytokines (Knodler et al., 2014; Sellin et al., 2014). A recent study suggests that there are additional unknown antibacterial defenses triggered by activated inflammatory caspases—cytosolic Salmonella replication is inhibited by canonical and noncanonical inflammasome activation, which depends on caspase-1/11 activity, but is independent of secreted inflammatory cytokines and mammalian cell death (Thurston et al., 2016).
L. monocytogenes suppresses flagellin expression when it invades cells, and bacteria engineered to continue to express flagellin are rapidly cleared (Sauer et al., 2010; Warren et al., 2011). In L. pneumophila infection, the NLRC4-mediated inflammasome, but not the noncanonical inflammasome, is responsible for restricting replication in macrophages and in mice by recognizing flagellin (Cerqueira et al., 2015).
The AIM2 inflammasome is activated by F. tularensis and M. tuberculosis, but not S. typhimurium, infection when bacterial DNA leaks into the cytosol (Fernandes-Alnemri et al., 2010; Rathinam et al., 2010; Saiga et al., 2012). AIM2 deficiency increases bacterial burden in tissues and leads to higher mortality (Fernandes-Alnemri et al., 2010). Similarly, ASC−/− and Casp1−/− mice infected with F. tularensis have a worse outcome (Mariathasan, Weiss, Dixit, & Monack, 2005). Treatment of wild-type mice with neutralizing antibodies against IL-1β and IL-18 also inhibits bacterial clearance, but not as much as in Casp1−/− mice (Mariathasan et al., 2005), suggesting that both pyroptosis and inflammatory cytokine processing and secretion contribute to Francisella control. Aim2−/− mice are also more susceptible to M. tuberculosis infection (Saiga et al., 2012). In some cases, inflammasome activation is detrimental. For example, activation of the NLRP1 inflammasome by B. anthracis LT is responsible for LT-mediated lung injury and death, which is independent of IL-1β and likely depends on pyroptosis triggered by caspase-1 (Kovarova et al., 2012).
In examples of antibacterial defense by the noncanonical inflammasome, Casp11−/− mice are susceptible to Burkholderia thailandensis, which is nonpathogenic in wild-type mice. Protection is independent of inflammatory cytokines, suggesting that it is mediated by caspase-11-initiated pyroptosis (Aachoui et al., 2013). Caspase-11-mediated bacterial defense is important for other invasive bacteria, which replicate in the cytosol (Aachoui et al., 2013). Caspase-11 only recognizes LPS that has 5 or 6 acylated lipid A chains. Francisella novicida and Y. pestis maintain their virulence by avoiding caspase-11 activation by removing lipid side chains from lipid A to produce molecules with only 4 acylated chains (Hagar et al., 2013).
Additional evidence for a protective role of the inflammasome in bacterial infection comes from the idenification of bacterial virulence factors that inhibit inflammasome activation. Examples are Y. pseudotuberculosis YopM, a pyrin and caspase-1 inhibitor (Chung et al., 2016; LaRock & Cookson, 2012) and S. flexneri OspC3, a caspase-4 inhibitor (Kobayashi et al., 2013). The YopM mutant only colonizes Casp1−/−Casp11−/− mice, and the OspC3 mutant strain causes profound intestinal inflammation in guinea pigs that inhibits Shigella infection.
5.2. Sepsis
Although inflammasome activation helps protect against invasive pathogens, the strength and duration of inflammasome signaling must be tightly controlled, because too much inflammation is dangerous. Sepsis, an inflammatory syndrome associated with disseminated infection, can lead to a severe, often fatal, vascular leak syndrome with multiorgan failure (septic shock). It is the leading cause of mortality in children globally and contributes to ~30% of deaths of hospitalized patients in the United States (Liu, Escobar, et al., 2014; Singer et al., 2016). There are approximately 30 million cases of sepsis per year in the world, of which about a fifth are fatal (Fleischmann et al., 2016). Patients who survive the acute illness often have a long-lasting impaired response to LPS and infection. Although hundreds of clinical studies to control sepsis have been attempted based on promising results in mouse models, these have all failed and there is still no effective way to treat sepsis (Hennessy, Parker, & O’Neill, 2010; Marshall, 2014). The difference in responses in mice and humans suggests that humans are more susceptible to sepsis than mice. However, the failure to identify effective therapies for human sepsis could also be due to the fact that most human patients are only identified once sepsis is already well under way, when it may be difficult to reverse. Many of these failed studies attempted to block proinflammatory cytokines. Our incomplete understanding of the molecular events occurring during sepsis may have interfered with the development of effective therapies.
Gram− bacterial cell wall LPS, also known as endotoxin, is a key trigger of sepsis (Sweet & Hume, 1996). The cell surface TLR4 senses LPS, activates NF-κB, and induces transcription of many proinflammatory genes. Tlr4 knockout or TLR4-blocking antibodies protect mice from lethal gram−bacterial sepsis (Roger et al., 2009). However, blocking TLR4 or proinflammatory cytokines was ineffective at combatting sepsis in clinical trials (Hennessy et al., 2010). These failures imply that TLR4 and the proinflammatory cytokines may not be the major mediators of septic shock in humans. Recent studies uncovered caspase-11 as a new LPS sensor that detects cytosolic LPS and then self-assembles into the noncanonical inflammasome (Shi et al., 2014). Caspase-11 and pyroptosis may be the critical mediators of LPS-induced sepsis, since mice genetically deficient only in caspase-11 or its downstream mediator GSDMD survive an otherwise lethal LPS challenge (Kayagaki et al., 2015, 2011). However, sepsis can also be triggered independently of LPS. In a polymicrobial mouse model of sepsis, cecal ligation and puncture, only blockade of IL-1β and IL-18 protected against sepsis; knocking out Casp1 and/or Casp11 did not confer protection (Aziz, Jacob, & Wang, 2014). It is likely that depending on the pathogen and tissues involved different pathways may be critical in the delicate balance between combatting infection and inflammatory morbidities that are unleashed by inflammasome activation.
Expression of some canonical inflammasome components needs to be induced by danger signals. The same is true for caspase-11 in mice. This feature explains why TLR4 is needed for LPS to induce sepsis in mice. However, the requirement for TLR4 can be bypassed by priming with other danger signals. For example, Tlr4−/− mice develop lethal gram− bacterial sepsis when pretreated with poly(I:C) (TLR3 agonist) to prime caspase-11 expression (Aachoui et al., 2013; Kayagaki et al., 2013). In contrast to the canonical inflammasome, the noncanonical inflammasome in humans does not need to be primed—it is standing ready to respond to invasive gram− bacteria as caspase-4/5 are constitutively expressed (Rathinam et al., 2012; Shi et al., 2014).
It is too early to know whether the recent discovery of the noncanonical inflammasome and of the critical role of GSDMD in pyroptosis will lead to effective treatments of sepsis. So far there are no specific inhibitors of either caspase-11 or GSDMD-NT to try. Moreover, if pyroptosis not only causes inflammation, but is also an important antibacterial agent that directly kills bacteria, inhibiting caspase-11 or GSDMD could be both helpful and harmful.
5.3. HIV Infection
The mechanism responsible for CD4 T cell depletion in human immunodeficiency virus (HIV) infection has been a long-standing puzzle. Although productively infected cells die from cytopathic effects of the virus, only a very small proportion of CD4 T cells (usually ~1 in a million) is productively infected, making it difficult to explain why CD4 T cell numbers decline so dramatically in untreated infection. A few recent papers from the W. Greene laboratory attribute CD4 T cell depletion, the underlying cause of immunodeficiency in HIV infection, to canonical inflammasome activation (Doitsh et al., 2014; Monroe et al., 2014). They provide evidence that HIV enters resting CD4 T cells from lymphoid tissues, but not blood, and begins reverse transcription, but later stages of viral replication are aborted. The reverse transcripts are recognized by the DNA sensor IFI16, which assembles an ASC-containing canonical inflammasome, that activates caspase-1 and pyroptosis, leading to CD4+ T cell depletion. Caspase-1 inhibitors preserve CD4+ T cell viability during in vitro HIV infection. So far these results have not been confirmed by another laboratory and the composition of the IFI16 inflammasome is not yet defined. A role for GSDMD in CD4 T cell depletion in HIV infection has also not yet been shown. However, if these results are confirmed, they may provide a new way to treat HIV immunodeficiency by inhibiting the canonical inflammasome or pyroptosis.
6. CONCLUDING REMARKS AND PERSPECTIVES
Recent studies have changed our understanding of innate immune recognition of invasive microbes. Previous studies focused on NLR and ALR recognition of PAMPs within immune cells to trigger the canonical inflammasome, activate caspase-1, and trigger proinflammatory cytokine processing and release and activate pyroptosis. The canonical pathway needs to be primed by TLR sensing of environmental microbes to activate expression of the inflammasome and caspase genes. We now know that a simplified noncanonical inflammasome that combines an LPS sensor and inflammatory caspase within the same molecule is constitutively expressed in human mucosal, skin, and immune cells and activates a potent inflammatory response to invasive gram− bacteria. This newly discovered pathway may be more important than the canonical pathway in human protection from infection and the uncontrolled inflammatory response responsible for sepsis. However, that remains to be seen. Importantly in the past year the mechanism responsible for pyroptosis has been identified. GSDMD, a cytosolic protein constitutively expressed in the same cells as the inflammatory caspases, is a critical substrate of the inflammatory caspases. When cleaved, it forms pores in the host cell membrane that cause pyroptosis and the release of the processed inflammatory cytokines. A structural model for how GSDMD pores are formed is still lacking.
Cleaved GSDMD also kills the bacteria that trigger pyroptosis presumably by forming pores in bacterial, as well as mammalian, membranes. Further work is needed to better understand the circumstances under which GSDMD-mediated bacterial killing is important. GSDMD-NT should be a reliable biomarker for detecting pyroptosis, which cannot be easily distinguished from other forms of programmed death. However, so far there are no antibodies that detect GSDMD-NT or its pores. Nor are there specific small molecule inhibitors of the noncanonical inflammasome caspases or GSDMD, which could provide important tools for deciphering the physiological importance of this newly described pathway in infection and potentially provide powerful new therapeutic drugs for treating inflammatory diseases, including sepsis. The next few years should also uncover the roles of the other GSDMs in the skin and mucosa.
ACKNOWLEDGMENTS
We thank our colleagues in our lab and the Hao Wu lab for useful discussions. This work was supported by a Charles A. King Trust Fellowship to X.L.
REFERENCES
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. (2013). Caspase-11 protects against bacteria that escape the vacuole. Science, 339, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard NJ, Maltby D, & Wells JA (2010). Inflammatory stimuli regulate caspase substrate profiles. Molecular & Cellular Proteomics, 9, 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proceedings of the National Academy of Sciences of the United States of America, 113, 7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NW, Almeida PE, & Corrotte M (2014). Damage control: Cellular mechanisms of plasma membrane repair. Trends in Cell Biology, 24, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert DF, Xu H, Yang J, Shi X, Gao W, Li L, et al. (2016). A Burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host & Microbe, 19, 664. [DOI] [PubMed] [Google Scholar]
- Aziz M, Jacob A, & Wang P (2014). Revisiting caspases in sepsis. Cell Death & Disease, 5, e1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan MA, & Cookson BT (2000). Salmonella induces macrophage death by caspase-1-dependent necrosis. Molecular Microbiology, 38, 31. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature, 478, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl-McGrath S, Schneider-Stock R, Ebert M, & Rocken C (2008). Differential expression and localisation of gasdermin-like (GSDML), a novel member of the cancer-associated GSDMDC protein family, in neoplastic and non-neoplastic gastric, hepatic, and colon tissues. Pathology, 40, 13. [DOI] [PubMed] [Google Scholar]
- Cerqueira DM, Pereira MS, Silva AL, Cunha LD, & Zamboni DS (2015). Caspase-1 but not caspase-11 is required for NLRC4-mediated pyroptosis and restriction of infection by flagellated Legionella species in mouse macrophages and in vivo. Journal of Immunology, 195, 2303. [DOI] [PubMed] [Google Scholar]
- Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. (2011). Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity, 34, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarria-Smith J, & Vance RE (2013). Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathogens, 9, e1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Research, 26, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LM, Kaniga K, & Galan JE (1996). Salmonella spp. are cytotoxic for cultured macrophages. Molecular Microbiology, 21, 1101. [DOI] [PubMed] [Google Scholar]
- Chen G, Shaw MH, Kim YG, & Nunez G (2009). NOD-like receptors: Role in innate immunity and inflammatory disease. Annual Review of Pathology, 4, 365. [DOI] [PubMed] [Google Scholar]
- Chen Y, Smith MR, Thirumalai K, & Zychlinsky A (1996). A bacterial invasin induces macrophage apoptosis by binding directly to ICE. The EMBO Journal, 15, 3853. [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. (2009). Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity, 30, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, et al. (2016). The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host & Microbe, 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. (2012). IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. The Journal of Experimental Medicine, 209, 1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson BT, & Brennan MA (2001). Pro-inflammatory programmed cell death. Trends in Microbiology, 9, 113. [DOI] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Fang H, Lankford CS, & Frucht DM (2004). Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1beta and IL-18. The Journal of Biological Chemistry, 279, 20563. [DOI] [PubMed] [Google Scholar]
- Dalebroux ZD, Edrozo MB, Pfuetzner RA, Ressl S, Kulasekara BR, Blanc MP, et al. (2015). Delivery of cardiolipins to the Salmonella outer membrane is necessary for survival within host tissues and virulence. Cell Host & Microbe, 17, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Miller M, Beppu AK, Mueller J, McGeough MD, Vuong C, et al. (2016). GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proceedings of the National Academy of Sciences of the United States of America, 113, 13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmaghani S, Defourny J, Aghaie A, Beurg M, Dulon D, Thelen N, et al. (2015). Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell, 163, 894. [DOI] [PubMed] [Google Scholar]
- Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, et al. (2006). Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nature Genetics, 38, 770. [DOI] [PubMed] [Google Scholar]
- Demon D, Kuchmiy A, Fossoul A, Zhu Q, Kanneganti TD, & Lamkanfi M (2014). Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunology, 7, 1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (2009). Immunological and inflammatory functions of the interleukin-1 family. Annual Review of Immunology, 27, 519. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Simon A, & van der Meer JW (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature Reviews. Drug Discovery, 11, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature, 535, 111. [DOI] [PubMed] [Google Scholar]
- Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. (2014). Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature, 505, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell, 145, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JM, Rouge L, Wiesmann C, & Scheer JM (2009). Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. The Journal of Biological Chemistry, 284, 6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott EI, & Sutterwala FS (2015). Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunological Reviews, 265, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrizzi MG, Hadinoto V, Growney JD, Miller W, & Dietrich WF (2000). Genomic sequence analysis of the mouse Naip gene array. Genome Research, 10, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Datta P, Wu J, & Alnemri ES (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature, 458, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature Immunology, 11, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Prada CM, Hoover DL, Tall BD, & Venkatesan MM (1997). Human monocyte-derived macrophages infected with virulent Shigella flexneri in vitro undergo a rapid cytolytic event similar to oncosis but not apoptosis. Infection and Immunity, 65, 1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, & Cookson BT (2007). Pyroptosis and host cell death responses during Salmonella infection. Cellular Microbiology, 9, 2562. [DOI] [PubMed] [Google Scholar]
- Finzel BC, Clancy LL, Holland DR, Muchmore SW, Watenpaugh KD, & Einspahr HM (1989). Crystal structure of recombinant human interleukin-1 beta at 2.0 A resolution. Journal of Molecular Biology, 209, 779. [DOI] [PubMed] [Google Scholar]
- Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, et al. (2016). Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. American Journal of Respiratory and Critical Care Medicine, 193, 259. [DOI] [PubMed] [Google Scholar]
- French FMFC (1997). A candidate gene for familial Mediterranean fever. Nature Genetics, 17, 25. [DOI] [PubMed] [Google Scholar]
- Fritz JH, Ferrero RL, Philpott DJ, & Girardin SE (2006). Nod-like proteins in immunity, inflammation and disease. Nature Immunology, 7, 1250. [DOI] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, & Miao EA (2013). Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science, 341, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy EJ, Parker AE, & O’Neill LA (2010). Targeting Toll-like receptors: Emerging therapeutics? Nature Reviews. Drug Discovery, 9, 293. [DOI] [PubMed] [Google Scholar]
- Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. (2014). Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS One, 9, e90099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, & Zychlinsky A (1999). The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proceedings of the National Academy of Sciences of the United States of America, 96, 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. (1998). Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. The Journal of Biological Chemistry, 273, 32895. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature, 458, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, & Andrews NW (2008). Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. The Journal of Cell Biology, 180, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, et al. (2000). Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. The Journal of Cell Biology, 149, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Yu HS, Seo JH, Kim HY, Jung YH, Kim YJ, et al. (2012). GSDMB/ORMDL3 variants contribute to asthma susceptibility and eosinophil-mediated bronchial hyperresponsiveness. Human Immunology, 73, 954. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, & Nunez G (2007). Intracellular NOD-like receptors in host defense and disease. Immunity, 27, 549. [DOI] [PubMed] [Google Scholar]
- Kastner DL, Aksentijevich I, & Goldbach-Mansky R (2010). Autoinflammatory disease reloaded: A clinical perspective. Cell, 140, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature, 526, 666. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature, 479, 117. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science, 341, 1246. [DOI] [PubMed] [Google Scholar]
- Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, et al. (2005). Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity, 23, 249. [DOI] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host & Microbe, 9, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. (2012). An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity, 36, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. (2014). Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host & Microbe, 16, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. (2013). The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host & Microbe, 13, 570. [DOI] [PubMed] [Google Scholar]
- Kofoed EM, & Vance RE (2011). Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature, 477, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, et al. (2012). NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. Journal of Immunology, 189, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Rathkolb B, Budde BS, Nurnberg P, de Angelis MH, Aigner B, et al. (2012). Gsdma3(I359N) is a novel ENU-induced mutant mouse line for studying the function of Gasdermin A3 in the hair follicle and epidermis. Journal of Dermatological Science, 67, 190. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, & Dixit VM (2014). Mechanisms and functions of inflammasomes. Cell, 157, 1013. [DOI] [PubMed] [Google Scholar]
- LaRock CN, & Cookson BT (2012). The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host & Microbe, 12, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. (2012). Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathogens, 8, e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, et al. (1995). Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell, 80, 401. [DOI] [PubMed] [Google Scholar]
- Li J, Zhou Y, Yang T, Wang N, Lian X, & Yang L (2010). Gsdma3 is required for hair follicle differentiation in mice. Biochemical and Biophysical Research Communications, 403, 18. [DOI] [PubMed] [Google Scholar]
- Lindgren SW, Stojiljkovic I, & Heffron F (1996). Macrophage killing is an essential virulence mechanism of Salmonella typhimurium. Proceedings of the National Academy of Sciences of the United States of America, 93, 4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, et al. (2014). Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA, 312, 90. [DOI] [PubMed] [Google Scholar]
- Liu X, & Lieberman J (2017). How ICE lights the pyroptosis fire. Cell Death and Differentiation, 24, 197–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K, et al. (2014). Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Reports, 8, 974. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature, 535, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, & Porter RM (2005). Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. The Journal of Investigative Dermatology, 124, 615. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Dixit VM, & Monack DM (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. The Journal of Experimental Medicine, 202, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC (2014). Why have clinical trials in sepsis failed? Trends in Molecular Medicine, 20, 195. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, & Tschopp J (2002). The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell, 10, 417. [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nature Immunology, 11, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Raupach B, Hromockyj AE, & Falkow S (1996). Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proceedings of the National Academy of Sciences of the United States of America, 93, 9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. (2014). IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science, 343, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, & Manning G (2016). Necroptosis and inflammation. Annual Review of Biochemistry, 85, 743. [DOI] [PubMed] [Google Scholar]
- Porter RM, Jahoda CA, Lunny DP, Henderson G, Ross J, McLean WH, et al. (2002). Defolliculated (dfl): A dominant mouse mutation leading to poor sebaceous gland differentiation and total elimination of pelage follicles. The Journal of Investigative Dermatology, 119, 32. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature Immunology, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell, 150, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner D, Orning MP, Proulx MK, Wang D, Gavrilin MA, Wewers MD, et al. (2016). The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathogens, 12, e1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. (2009). HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science, 323, 1057. [DOI] [PubMed] [Google Scholar]
- Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, et al. (2009). Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proceedings of the National Academy of Sciences of the United States of America, 106, 2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, & Alnemri ES (2017). Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nature Communications, 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkel F, Marquardt A, Stoeger C, Kochmann E, Simon D, Kohnke B, et al. (2004). The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics, 84, 824. [DOI] [PubMed] [Google Scholar]
- Saeki N, Kim DH, Usui T, Aoyagi K, Tatsuta T, Aoki K, et al. (2007). GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-beta-dependent apoptotic signalling. Oncogene, 26, 6488. [DOI] [PubMed] [Google Scholar]
- Saeki N, Kuwahara Y, Sasaki H, Satoh H, & Shiroishi T (2000). Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mammalian Genome, 11, 718. [DOI] [PubMed] [Google Scholar]
- Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. (2009). Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes, Chromosomes & Cancer, 48, 261. [DOI] [PubMed] [Google Scholar]
- Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. (2012). Critical role of AIM2 in Mycobacterium tuberculosis infection. International Immunology, 24, 637. [DOI] [PubMed] [Google Scholar]
- Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, & Portnoy DA (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host & Microbe, 7, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. The EMBO Journal, 35, 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. (2014). Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host & Microbe, 16, 237. [DOI] [PubMed] [Google Scholar]
- Shi P, Tang A, Xian L, Hou S, Zou D, Lv Y, et al. (2015). Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. The Biochemical Journal, 468, 325. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature, 514, 187. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature, 526, 660. [DOI] [PubMed] [Google Scholar]
- Sims JE, & Smith DE (2010). The IL-1 family: Regulators of immunity. Nature Reviews. Immunology, 10, 89. [DOI] [PubMed] [Google Scholar]
- Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. (2016). The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA, 315, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, & Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science, 339, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Yang J, Xing G, Sun Q, Zhang L, & He F (2008). Expression of GSDML associates with tumor progression in uterine cervix cancer. Translational Oncology, 1, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet MJ, & Hume DA (1996). Endotoxin signal transduction in macrophages. Journal of Leukocyte Biology, 60, 8. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, & Akira S (2010). Pattern recognition receptors and inflammation. Cell, 140, 805. [DOI] [PubMed] [Google Scholar]
- Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. (2007). Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics, 89, 618. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Tamura M, Aoki A, Fujii T, Komiyama H, Sagai T, et al. (2007). A new Gsdma3 mutation affecting anagen phase of first hair cycle. Biochemical and Biophysical Research Communications, 359, 902. [DOI] [PubMed] [Google Scholar]
- Thurston TL, Matthews SA, Jennings E, Alix E, Shao F, Shenoy AR, et al. (2016). Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nature Communications, 7, 13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. (1998). Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nature Genetics, 20, 194. [DOI] [PubMed] [Google Scholar]
- Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, et al. (2016). Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell, 165, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. (2012). The NLRP12 inflammasome recognizes Yersinia pestis. Immunity, 37, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung YK, Zhu H, Li E, & Yuan J (1998). Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell, 92, 501. [DOI] [PubMed] [Google Scholar]
- Warren SE, Duong H, Mao DP, Armstrong A, Rajan J, Miao EA, et al. (2011). Generation of a Listeria vaccine strain by enhanced caspase-1 activation. European Journal of Immunology, 41, 1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, et al. (2001). Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Japanese Journal of Cancer Research, 92, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. (2014). Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature, 513, 237. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhao Y, Shi J, & Shao F (2013). Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proceedings of the National Academy of Sciences of the United States of America, 110, 14408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamboni DS, & Lima-Junior DS (2015). Inflammasomes in host response to protozoan parasites. Immunological Reviews, 265, 156. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Shi J, Shi X, Wang Y, Wang F, & Shao F (2016). Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. The Journal of Experimental Medicine, 213, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. (2011). The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature, 477, 596. [DOI] [PubMed] [Google Scholar]
- Zychlinsky A, Fitting C, Cavaillon JM, & Sansonetti PJ (1994). Interleukin 1 is released by murine macrophages during apoptosis induced by Shigella flexneri. The Journal of Clinical Investigation, 94, 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zychlinsky A, Prevost MC, & Sansonetti PJ (1992). Shigella flexneri induces apoptosis in infected macrophages. Nature, 358, 167. [DOI] [PubMed] [Google Scholar]