

Abstract
The European Molecular Biology Laboratory (EMBL) Nucleotide Sequence Database (http://www.ebi.ac. uk/embl/index.html ) is maintained at the European Bioinformatics Institute (EBI) in an international collaboration with the DNA Data Bank of Japan (DDBJ) and GenBank (USA). Data is exchanged amongst the collaborative databases on a daily basis. The major contributors to the EMBL database are individual authors and genome project groups. WEBIN is the preferred web-based submission system for individual submitters, whilst automatic procedures allow incorporation of sequence data from large-scale genome sequencing centres and from the European Patent Office (EPO). Database releases are produced quarterly. Network services allow free access to the most up-to-date data collection via Internet and WWW interfaces. EBI’s Sequence Retrieval System (SRS) is a network browser for databanks in molecular biology, integrating and linking the main nucleotide and protein databases plus many specialised databases. For sequence similarity searching a variety of tools (e.g., BLITZ, FASTA, BLAST) are available which allow external users to compare their own sequences against the most currently available data in the EMBL Nucleotide Sequence Database and SWISS-PROT.
THE EMBL NUCLEOTIDE SEQUENCE DATABASE
Scope of the database
The European Molecular Biology Laboratory (EMBL) Nucleotide Sequence Database (http://www.ebi.ac.uk/embl/index.html ) is a comprehensive collection of primary nucleotide sequences maintained at the European Bioinformatics Institute (EBI). Data are received from genome sequencing centres, individual scientists and patent offices. New data are released daily into the EMBLNEW database and are immediately available. The EMBL and EMBLNEW databases are stored and maintained in an ORACLE data management system and can be searched on the Internet with the Sequence Retrieval System (SRS) (1), the EBI search engine for molecular biology databanks.
The international collaboration
DDBJ (Japan), GenBank (USA) and EMBL exchange new and updated data on a daily basis to achieve optimal synchronisation. Users need only submit to one of the collaborating databases, irrespective of where the sequence will be published. The three databases adhere to a set of documented guidelines (The DDBJ/EMBL/GenBank Feature Table Definition) which regulate the content and syntax of the database entries. These guidelines ensure that the data continue to be made available in a format that can be exchanged efficiently between the databases, is compatible with current bioinformatics software and reflects developments in the fields of molecular and general biology.
History and growth
Established in 1980, the database was historically tightly coupled to the publication of sequences in the scientific literature, but quickly electronic submissions became usual practice. Today, the volume of data submitted by direct transfer of data from major sequencing centres, such as the Sanger Centre, overshadows all other input. In recent years the EMBL database has doubled in size nearly every year and on the October 1, 1999 contained 4.7 million entries representing over 3.6 Gigabases of nucleotide sequence. Database statistics are available and can be viewed at http://www3.ebi.ac.uk/Services/DBStats/ . The last 3 years have seen a phenomenal increase in the amount of data submitted by genome sequencing centres and a substantial increase in the number of new and completed genome projects. During the first 8 months of 1999 >1.6 million new entries (1.3 Gigabases) were made public, an average of 6400 entries (5.4 Mbases) per day.
Accession numbers
Accession numbers are unique identifiers which permanently identify sequences in the database. Accession numbers are assigned and communicated to authors within 2 working days of receipt of submission. These accession numbers (e.g. X64011 and AJ000001) are required by many biological journals before manuscripts are accepted. The suggested wording for citing a sequence in a publication is ‘These sequence data have been submitted to the DDBJ/EMBL/GenBank databases under accession number AJ654321’.
Sequence identifiers
In addition to unique and stable accession numbers, EMBL database entries include new sequence identifiers to specify changes in sequence versions:
• nucleotide sequence identifier represented by sequence version line type
Example: SV X99911.3
• protein sequence identifier for valid CDS features represented by ‘/protein_id’ feature qualifier
Example: /protein_id=“CAA45406.1”
The identifiers themselves remain stable within a given entry, whilst the version number increments with every sequence update. Protein identifiers can be used by external databases (such as SWISS-PROT) as an identifier onto which cross-references can be built at the feature level, e.g., to individual CDS features. Protein identifiers are currently assigned to all CDS features in the nucleotide sequence database to identify the exact protein translation for each coding sequence. These protein identifiers can be found in the Feature Table qualifier /protein_id.
Protein translations
Translation of protein coding regions in EMBL entries (represented by CDS features) will automatically be added to the TrEMBL protein database. SWISS-PROT (2) curators draw from this pool to subsequently create the SWISS-PROT database. EMBL nucleotide entries are cross-referenced (via the /db_xref qualifier) to the TrEMBL and SWISS-PROT databases.
Integration with other databases
Interconnectivity between biomolecular databases has become essential for utilising the wealth of information becoming available. Where appropriate, EMBL Database entries are cross-referenced to other databases like the Eukaryotic Promoter database (3) TRANSFAC (4), FlyBase (5), TrEMBL and SWISS-PROT. SWISS-PROT itself is linked to more than 30 different databases thus providing a focal point for database interconnectivity. Cross-references to external databases are represented in the EMBL flat-file line type ‘DR’ and where appropriate, at the feature level via the feature qualifier /db_xref. These cross-references allow access to additional information concerning the entry that is more appropriately stored in other dedicated databases.
Example: DR SWISS-PROT; P28763; SODM_LISIV.
FT CDS 109..717
FT /db_xref=“SWISS-PROT:P28763”
Biological annotation
In the era of high-throughput genome project data, the importance of careful curation of individual sequences directly submitted by individual researchers and discussed in the scientific literature is obvious. It is such sequences which have often been the subject of experimental research elucidating features and function, while genome project submissions in most cases will ‘only’ include preliminary gene annotations based on gene prediction programs. Sequence annotation is an essential part of EMBL sequence records and current database policy is to reject submissions for which no sequence annotation has been provided, unless these describe expressed sequence tag sites (ESTs) or unfinished high throughput genome sequences (HTGs). In particular, it is essential to provide locations of coding regions, even when partial or preliminary, to allow inclusion of the corresponding translated protein sequence in the protein databases (TrEMBL and SWISS-PROT).
WWW guides
Three internet guides have been written by curation staff to help submitters annotate their sequences. The guides are available from the EMBL-EBI WWW site and from within WEBIN.
• WebFeat. A complete list of feature table key and qualifier definitions, providing full explanations of their use.
• EMBL Annotation Examples. A selection of EMBL approved feature table annotations for some common biological sequences (i.e., ribosomal RNA, mitochondrial genome).
• DE Line Standards. Guidelines on how to create a suitable definition for submissions following database conventions.
Database entry structure
Database entries are distributed in EMBL flat-file format which is supported by most sequence analysis software packages and also provides a structure usable by human readers. The EMBL flat-file comprises of a series of strictly controlled line types (for details see User Manual) that are presented in a tabular manner and consists of four major blocks of data.
• Descriptions and identifiers. Entry name, confidential status, molecule type, taxonomic division, and total sequence length (found in the ID line); accession number (AC); sequence version (SV); date of creation and last update (DT); brief description of the sequence (DE); keywords (KW); taxonomic classification (OS, OC) and links to related database entries (DR).
• Citations. The citation details (RX, RA, RT and RL) of the associated publication and the name (RA) and contact details (RL) of the original submitter.
• Features. Detailed source information, biological features, feature locations and feature qualifiers (multiple FT lines).
• Sequence. Total sequence length, base composition (SQ) and sequence.
Taxonomy and top organism statistics
The comprehensive sequence-based collaborative taxonomy includes >50 000 different species. The unified taxonomy was developed and is maintained at the NCBI in collaboration with EMBL and DDBJ and with the assistance of external advisors and curators. The aim is to centralise the classification of all organisms appearing in the nucleotide sequence database. When EMBL receives a sequence with an organism which is not included in the taxonomy database, the details of the submissions are sent to NCBI taxonomy curators who place the organism in the correct place on the tree. Entries are not released into the public domain until the sequenced organism is classified. Organisms are identified at the species level in the taxonomy database. When the scientific name is not available at the time of submission a provisional name (e.g., unidentified soil organism R6-12) can be provided. Submitters should communicate new taxonomic details to NCBI when they are known so that the database can be updated. EMBL database top five organisms in September 1999 are Homo sapiens (53.4%), Mus musculus (8.9%), Drosophila melanogaster (5.5%), Caenorhabditis elegans (5.2%) and Arabidopsis thaliana (4.3%).
Database divisions
Divisions provide subsets of the database which reflect the areas of interest of users. The EMBL Database currently consists of 18 divisions with each entry belonging in exactly one division. In each entry the division is indicated using the three letter codes, e.g., PRO = Prokaryotes, HUM = Human, PHG = Bacteriophages, PLN = Plants etc. The grouping is mainly based on taxonomy with a few exceptions like the HTG, EST, STS (sequence tagged sites) and GSS (genome survey sequences) divisions. For these divisions, grouping is based on the specific nature of the underlying data.
Expressed sequence tags (ESTs). The EST division files contain sequence and mapping data on ‘single-pass’ cDNA sequences or ESTs from a number of organisms. In addition to the EST division files in the EMBL database release, the EBI provides ESTLIB, which includes further information about the libraries from which the EST sequences were derived. The EST division entries in EMBL are cross-referenced to ESTLIB with a /db_xref qualifier on the source feature.
FT source 1..1739
FT /organism=“Magnaporthe grisea″
FT /db_xref=“ESTLIB:863”
High-throughput genome sequences (HTGs). In order to make genome sequences produced by high-throughput sequencing projects available to the user community as soon as possible, the HTG division includes ‘unfinished’ genome project data with annotation for many of these records being generated through computer analyses. Entries in this division all contain keywords to indicate the status of the sequencing (e.g., HTGS_PHASE1 to HTGS_PHASE3). A single accession number is assigned to one clone, and as sequencing progresses and the entry passes from one phase to another, it will retain the same accession number with only the most recent version of a HTG record remaining in EMBL. Once ‘finished’, HTG sequences are moved into the relevant primary EMBL taxonomic division. EMBL Release 60 (September 1999) included >544 Mb of unfinished HTG data.
Patent sequence data. The EMBL database continues to collaborate with the European Patent Office to capture patent sequences from patent applications and integrate US and Japanese patent sequence data provided by our DDBJ and GenBank collaborators. Patent data can be retrieved from the EMBLNEW and EMBL databases and are also available via ftp.
SUBMISSION OF SEQUENCE DATA
Two major sources contribute to the EMBL Database: individual scientists, who submit data directly to the collaborating databases, and genome project groups which produce very large volumes of nucleotide sequence data over an extended period of time, including bulk submissions of ESTs, STSs, GSSs or large genomic records (high-throughput and finished data). Researchers submitting new sequences directly to the EMBL database use either the Internet (WEBIN) or a stand-alone software tool (SEQUIN). Detailed information for submitters is available from the EBI WEB pages (http://www.ebi.ac.uk/Submissions/index.html ) or the reference card ‘Quick Guide to Sequence Submissions’ edited by EMBNET.
Data confidentiality and release dates
Sequences submitted to the database can be released to the public immediately or withheld until an author-specified date. Data are never withheld after publication. A confidential record will not be released into the public database until expiry of the hold date or journal publication, whichever comes first. At any time, the submitter may update information in the record. We encourage authors to notify the EMBL Database of publication so that confidential records may be released and public records can be updated in synchrony with the journal publication.
Direct submission systems
The EBI’s submission tools incorporate facilities for providing and checking biological information.
Vector scanning. A WWW-based interactive vector scanning service is available for submitters to assist in the screening of sequences for vector contamination before submission. The vector screening service uses the latest implementation of the BLAST algorithm and the special sequence databank EMVEC, comprised of an extraction of sequences from the SYNthetic division of EMBL commonly used in cloning and sequencing experiments. EMVEC is updated with each release of EMBL and is available from the EBI’s ftp server.
WEBIN. WEBIN is an Internet based tool for submission of nucleotide sequences to the EMBL database. WEBIN is designed to allow fast submission of either single, multiple or even very large numbers of sequences (bulks). WEBIN is available from the EMBL WWW home page or at URL: http://www.ebi.ac.uk/embl/Submission/webin.html
Sequence annotation in WEBIN is added from the ‘Summary and Sequence Features’ page. Any number of relevant features can be easily added to the sequence feature table from the comprehensive list and by filling out the specific feature forms. To assist submitters in selecting features for their sequence, WebFeat provides a full description of all EMBL features and qualifiers while the EMBL Annotation Examples illustrate how these features and qualifiers should be used within standard EMBL entries.
SEQUIN. Sequin is a stand-alone software tool developed by the NCBI for submitting and updating nucleotide sequences to the GenBank, EMBL or DDBJ databases. Sequin contains a number of built-in validation functions for enhanced quality assurance and runs on Macintosh, PC/Windows and UNIX computers.
Submitters who do not have a reliable connection to the WWW may contact datasubs@ebi.ac.uk . Handwritten forms, disks mailed by post and AUTHORIN submissions are no longer accepted.
Genome project data
At least in sheer quantity, large-scale sequencing projects have become the major sources of new sequence data. A selection of groups submitting to the database is listed below:
• CNS/Genoscope projects (various organisms)
• ESSA Arabidopsis thaliana
• European Drosophila Mapping Consortium
• MIPS human EST
• Max Planck Institute Berlin Human
• MRC/HGMP Fugu GSS
• Oxford MGC/HGMP Mouse X
• Pasteur various microbial genomes
• Sanger Centre Human genome project
• Sanger Centre Caenorhabditis elegans nematode project
• Sanger Centre various micro-organisms
• European IMAGE clone sequencing consortium
• Shanghai NCGR rice genome project
The EMBL database opens submission accounts for groups producing large volumes of nucleotide sequence data over an extended period. Database entries produced at the research site are deposited and updated directly by the genome project submitter using FTP or Email. Full details of the procedure can be found from the EMBL EBI WWW site (http://www3. ebi.ac.uk/Services/GenomeSubm/ ). Each submission account is curated by EBI biologists. Groups that wish to make use of this submission procedure should contact the database at: datasubs@ebi.ac.uk
Sequence data produced at sequencing centres will be included in the database as soon as they become available from the individual sequencing groups, and will immediately become available for homology searches via network services. High-throughput sequence records are included in the HTG division and contain keywords to indicate the finishing status of the sequencing (i.e., HTGS_PHASE1, HTGS_PHASE2 or HTGS_PHASE3).
The progress of a number of large genome sequencing projects is monitored in the Genome MOT (genome monitoring table). A collection of graphs and tables shows the progress of the major eukaryotic genome sequencing project, calculates the total amount of finished and unfinished (draft) genomic DNA sequences deposited per year into the DDBJ/EMBL/GenBank databases for a number of organisms, and is updated on a daily basis. In addition, the Genome MOT website gives direct access to database records and provides mapping information for individual clones.
CON division. Among the database collaboration a new database division (CON) is being developed which will represent complete genomes, or other long sequences, constructed from segmented entries. Each CON division entry will have an accession number and will contain information on how the construct is built from segments. In addition, the complete entry containing the full sequence, features and references will be retrievable through SRS.
Draft human genome. A consortium of five publicly funded sequencing centres (Sanger, Baylor, WashU, Whitehead and DOE) is expected to produce a draft version of the human genome by February 2000. In collaboration with the Sanger Centre, the European Bioinformatics Institute is planning to make the fully analysed human genome accessible through the EnsEMBL project. Figure 1 shows the progress of the Human Genome Project from January 1989 to October 1999.
Figure 1.
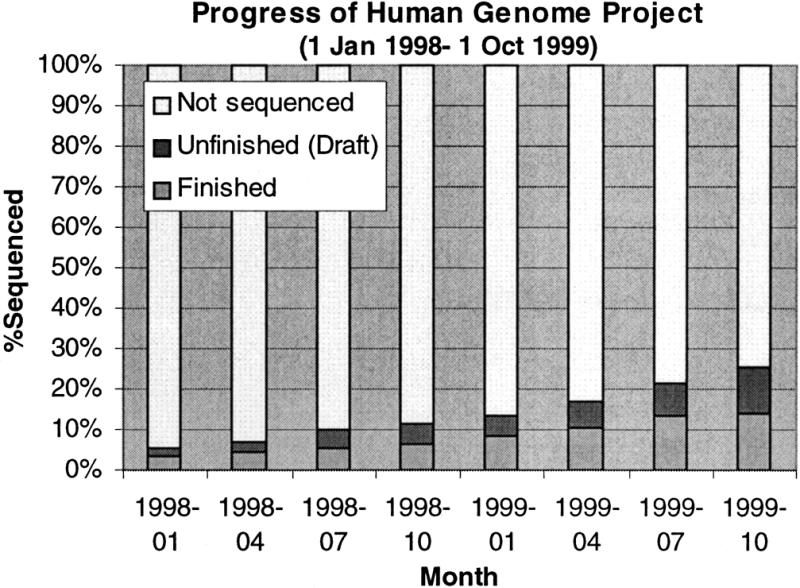
Progress of Human Genome Project (1 Jan 1998–1 Oct 1999).
Updating existing database entries
Researchers wishing to update existing EMBL sequences should use the Internet WEBUP form or contact the database at update@ebi.ac.uk
Sequence alignment submissions
Since 1990, the EBI has accepted the alignment data from phylogenetic and population analysis of either nucleotide or amino acid sequences. With the need to permanently store data in electronic form, the alignment archive has doubled in size in recent years. Alignment data can be retrieved from the EBI’s FTP site at ftp://ftp.ebi.ac.uk/pub/databases/embl/align/ while submission information is available from our home page. We currently accept standard alignment formats (e.g., NEXUS, PHYLIP, CLUSTALW and GCG/MSF) or Sequin output. Unique alignment numbers (e.g., DS32096) are assigned to each alignment and should be included in the published article. The issue of format standardisation is still under discussion by database staff and will result in major enhancements in the acquisition and retrieval of alignment data in the near future.
DATA DISTRIBUTION, SEARCHING AND SEQUENCE ANALYSIS
EBI network services
Database releases are produced quarterly, while network services allow access to the most up-to-date data collection via the Internet. Data access to sequence data at the EBI is also granted via Email using the netserver or interactively via the WWW where the main service is composed of an SRS server. Additionally, databases as well as software can be downloaded from the EBI’s FTP server.
Sequence Retrieval System (SRS)
The SRS server at the EBI integrates and links a comprehensive collection of specialised databanks along with the main nucleotide and protein databases. The SRS system allows the databases to be searched using, for example, sequence, annotations, keywords, author names. Complex, cross-database queries can also be executed and users should refer to the detailed instructions which are available online.
Sequence searching
The EBI provides a comprehensive set of sequence database searching algorithms that can be accessed both interactively from the EMBL EBI WWW site (http://www2.ebi.ac.uk/ ) or by Email. EMBL may be searched as a whole or by individual taxonomic division. The most commonly used algorithms available are Fasta3 (6) and NCBI-Blast2 (7). Fasta3 will find a single high-scoring gapped alignment between the query nucleotide sequence and database sequences. Comparisons between a nucleotide sequence and the protein databases can be made using fastx/y3, whilst tfastx/y3 allows comparisons between a protein sequence and the translated DNA databank. Ssearch3, the generic implementation of the Smith–Waterman algorithm (8) for nucleotide and protein database searches is provided as part of the fasta3 package. BLITZ (Bic_SW) facilitates more sensitive searches using the Smith–Waterman algorithm. WU-Blast2 and NCBI-Blast2 are fast algorithms for sequence searching that allow gaps, but which may find more than one match to the database sequences if multiple domains exist.
Sequence analysis
Specialised sequence analysis programs are available from the EBI. Such services include multiple sequence alignment and inference of phylogenies using CLUSTALW (9), Gene prediction using GeneMark (10), pattern searching and discovery using PRATT, as well as applications which have been developed in-house for various projects.
EMBnet
The European Molecular Biology Network (http://www.embnet. org ) was initiated in 1988 to link European laboratories using biocomputing and bioinformatics in molecular biology research as well as to increase the availability and usefulness of the molecular biology databases within Europe. Remote copies of the nucleotide and protein sequence databases, updated daily, as well as other molecular biology resources, are held at nationally mandated nodes. As bioinformatics grows, EMBnet plays an important role in support, training, research and development for the European bioinformatics research community. A full listing of sites maintaining daily updated copies of the EMBL Database is available from the EBI at http://www.ebi.ac.uk/embl/Access/other_sites.html
CITING THE EMBL DATABASE
The preferred form for citation of the EMBL Nucleotide Sequence Database is Stoesser,G., Baker,W., van den Broek,A.E., Camon,E., Hingamp,P., Sterk,P. and Tuli,M.A. (2000) The EMBL Nucleotide Sequence Database. Nucleic Acids Res., 28, 19–23.
CONTACTING THE EMBL DATABASE
Computer network:
For data submissions datasubs@ebi.ac.uk
For other inquiries datalib@ebi.ac.uk
For updates/publication
notification s update@ebi.ac.uk
Postal address:
EMBL Nucleotide Sequence Submissions, European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK
Telephone:
For data submissions +44 1223 494499
General +44 1223 494444
Fax:
For data submissions +44 1223 494472
General +44 1223 494468
SUPPLEMENTARY MATERIAL
Table of relevant URL links available at NAR Online.
REFERENCES
- 1.Etzold T., Ulyanov,A. and Argos,P. (1996) Methods Enzymol., 266, 114–128. [DOI] [PubMed] [Google Scholar]
- 2.Bairoch A. and Apweiler,R. (1998) Nucleic Acids Res., 26, 38–42. Updated article in this issue: Nucleic Acids Res. (2000), 28, 45–48.9399796 [Google Scholar]
- 3.Périer R.C., Junier,T. and Bucher,P. (1998) Nucleic Acids Res., 26, 353–357. Updated article in this issue: Nucleic Acids Res. (2000), 28, 302–303.9399872 [Google Scholar]
- 4.Heinemeyer T., Wingender,E., Reuter,I., Hermjakob,H., Kel,A.E., Kel,O.V., Ignatieva,E.V., Ananko,E.A., Podkolodnaya,O.A., Kolpakov,F.A., Podkolodny,N.L. and Kolchanov,N.A. (1998) Nucleic Acids Res., 26, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearson W.R. (1994) Methods Mol.Biol., 24, 307–331. [DOI] [PubMed] [Google Scholar]
- 6.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 7.Smith R.F. and Waterman,M.S. (1981) Adv. Appl. Math., 2, 482–489. [Google Scholar]
- 8.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borodovsky M. (1993) Comput. Chem., 17, 123–133. [DOI] [PubMed] [Google Scholar]
- 10.Jonassen I., Collins,J.F. and Higgins,D.G. (1995) Protein Sci., 4, 1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.