

Abstract
Executive functioning in cocaine/polydrug (marijuana, alcohol, tobacco) exposed infants was assessed in a single session, occurring between 9.5 and 12.5 months of age. In an A-not-B task, infants searched, after performance-adjusted delays, for an object hidden in a new location. Overall, the cocaine-exposed (CE) infants did not differ from non-CE controls recruited from the same at-risk population. However, comparison of heavier-CE (n = 9) to the combined group of lighter-CE (n = 10) and non-CE (n = 32) infants revealed significant differences on A-not-B performance, as well as on global tests of mental and motor development. Covariates investigated included socioeconomic status, marital status, race, maternal age, years of education, weeks of gestation, birth weight, as well as severity of prenatal marijuana, alcohol, and tobacco exposure. The relationship of heavier-CE status to motor development was mediated by length of gestation, and the relationship of heavier-CE status to mental development was confounded with maternal gestational use of cigarettes. The relationship of heavier-CE status to A-not-B performance remained significant after controlling for potentially confounded variables and mediators, but was not statistically significant after controlling for the variance associated with global mental development.
Children exposed prenatally to cocaine are at increased risk for neurological dysfunction secondary to in utero hypoxia (Wood, Plessinger, & Clark, 1987). There may also be direct effects of cocaine, which readily crosses the placental barrier and reaches the fetus (Volpe, 1992). In this investigation of executive function (EF) in cocaine-exposed (CE) infants, a delay tolerance version of the A-not-B task was employed.
EF tasks are generally sensitive to developmental dysfunction (Pennington, 1991) and have been recommended for neurobehavioral toxicology investigations (Krasgnor et al., 1994). EF is a constellation of abilities, the composition of which is a topic of active debate, but often-cited candidates include working memory, inhibitory control, and planning (Fuster, 1989). The A-not-B task requires all three and fits well with the construct of EF because it requires goal-directed action in the context of response competition.
The critical trials of the A-not-B task are those in which a toy is hidden in a new location (B) and the infant must resist searching for it in the location (A) where they found it on previous trials. When the infant searches at the A location, despite seeing it hidden in the B location, they commit the A-not-B error that gives the task its name. The delay between when the toy is hidden and when the infant is allowed to reach for it is critical to success on the A-not-B task (Harris, 1975). The length of the delay directly affects whether the infant finds the object at its new location or perseverates to the old location (see Marcovitch & Zelazo, 1999; Wellman, Cross, & Bartsch, 1986, for reviews). Further, the ability to tolerate delay increases with age (Bell & Adams, 1999; Diamond, 1985; Matthews, Ellis, & Nelson, 1996). For example, Diamond (1985) found that, at 8 months of age, infants only consistently succeeded if the delay was less than 1 sec, but at 12 months of age the same infants consistently succeeded at a 6-sec delay.
The A-not-B task was selected for the current investigation because of the clinical and experimental evidence suggesting that performance may be sensitive to disruptions of the frontal cortical–subcortical system and the supply of the neurotransmitter dopamine (see Noland, 2001, for a review). Like human infants, adult monkeys with experimental prefrontal lesions commit the A-not-B error (Diamond, 1990; Diamond & Goldman-Rakic, 1989). Human infants with early-treated phenylketonuria, whose dopamine production is disrupted, are different from control groups, in that they commit the A-not-B error at shorter delays (Diamond, Prevor, Callender, & Druin, 1997). There is also evidence that individual differences in frontal functioning predict performance on the A-not-B task. Individual infants with more-developed frontal lobe functioning, as assessed by EEG measures, show greater delay tolerance in A-not-B performance (Bell & Adams, 1999; Bell & Fox, 1992).
Experimental work with animals suggests that direct fetal CE causes persistent alterations in dopamine-reliant neurons (Friedman, Yardin, & Wang, 1996; Levitt, Harvey, Friedman, Simansky, & Murphy, 1997; Minabe, Ashby, Heyser, Spear, & Wang, 1992; Spear, Kirstein, & Frambes, 1989). A separate line of animal studies suggests that functioning of the prefrontal cortex is dependent on the availability of dopamine (Sawaguchi & Goldman-Rakic, 1991; Taylor, Elsworth, Roth, Sladek, & Redmond, 1990) and that CE affects the level of dopamine metabolite in human infants (Needleman, Zuckerman, Anderson, Mirochnick, & Cohen, 1993). Thus, prenatal CE that disrupts the dopaminergic system is likely to negatively affect the cognitive functions supported by the prefrontal cortex (see Mayes, 1999, for a review of this hypothesis).
One previous study has investigated A-not-B performance in CE children. Espy, Kaufmann, and Glisky (1999) found that CE children were less successful on an A-not-B task, but not different from controls on general tests of mental and motor development. Further, the CE children differed on A-not-B performance, even when mental abilities as assessed by the Bayley Scales of Infant Development (BSID-II; Bayley, 1993) were controlled for. Espy et al. (1999) was presented as a preliminary study and was important in suggesting that A-not-B performance of CE children does not match that of nonexposed children. However, there are several design issues that limit the conclusions that can be drawn from this study. The test administrators knew the drug-exposure status of the children in advance of the assessment. Further, the controls differed from the CE group in socioeconomic status (SES) and were recruited as a “normal” sample against which the CE group could be compared. Given that toddler cognitive abilities are positively correlated with parental education (Roberts, Bornstein, Slater, & Barrett, 1999), it is reasonable to suggest that performance on the A-not-B task may have varied by SES.
Espy et al. (1999) also acknowledged, but did not attempt to control for, the greater prenatal marijuana and alcohol use by the cocaine-using mothers. Recent studies suggest sensitivity of EF to prenatal marijuana exposure (see Fried & Smith, 2001, for a review), prenatal alcohol exposure (Kodituwakku, Handmaker, Cutler, Weathersby, & Handmaker, 1995; Mattson, Goodman, Caine, Delis, & Riley, 1999; Noland, et al., 2003) and a correlation between maternal responsivity and A-not-B performance (Ayoun, 1998). Finally, the children in the CE group were younger, and the degree to which this disadvantaged them on the A-not-B task was not addressed.
Building on the Espy et al. (1999) finding, we administered a delay tolerance version of the A-not-B task, as well as the BSID–II, to CE infants and nonexposed, SES-similar infants. Infants, CE and non-CE alike, were recruited prospectively from a population of infants at risk for prenatal drug exposure. The non-CE infants were recruited into two groups: a control group with no drug or alcohol exposure and a noncocaine drug (alcohol, tobacco, marijuana) exposed (NCDE) comparison group. Based on previous research reporting threshold effects of prenatal CE on cognitive (Jacobson, Jacobson, Sokol, Martier, & Chiodo, 1996) and behavioral (Singer, Arendt, Minnes, Farkas, & Salvator, 2000) assessments, infants were also divided into heavier-CE, lighter-CE, and non-CE groups. In addition to maternal substance-use variables, several environmental and gestational growth variables were assessed, and those that differed between groups and were related to the outcome measure were controlled for statistically in the data analysis.
METHOD
Participants
Fifty-one infants, ages 9.5 to 12.5 months of age, were recruited from a cohort who had been enrolled in an investigation of cardiac effects of prenatal drug-exposure (Mehta et al., 2001, 2002). The hospital staff at the county hospital for Cleveland, OH, identified mother–infant dyads at risk for prenatal drug exposure. This sample was the source for all the infants in the study. In a procedure described in the next section, drug-exposure status was determined by a combination of biological assay and maternal interview before hospital discharge. Infants were excluded from the cardiac study if they or their mothers had medical problems requiring pharmacological or surgical treatment, or if they had birth weights less than 1500 g, 5-min Apgar less than 6, or gestational age (GA) less than 34 weeks. Birth size was recorded from hospital charts, as was GA.
Caregivers who had indicated willingness to participate in the behavioral study, and whose infants were in the appropriate age range, were contacted by phone and the behavioral assessment scheduled. The 51 infants were brought in to the research lab, where they were tested by examiners blind to exposure status. Three additional infants were recruited and came to the lab for the testing session. Their data were not included, because they were tested in the piloting phase of the experiment (n = 2), or because they were unwilling or unable to retrieve the object on any two trials during the warm-up phase of the A-not-B test (n = 1). Compensation was given in an amount established in previous studies: $50 for time and $5 for transportation. The procedures were reviewed and approved by the institutional review board of the participating hospitals, and informed consent was obtained from the caregiver before participation in this test session.
Drug-screening and group assignment.
Based on the design described in previous publications (Mehta et al., 2001; Mehta et al., 2002), the original three groupings included CE, NCDE, and no-drug-exposure (ND) groups. Assignment was based on maternal report, as well as meconium, infant urine, and maternal urine testing. These procedures were developed for a previous study, and the advantages associated with combining self-report and meconium information, for the purposes of assigning exposure groups, are described in detail in Arendt, Singer, Minnes, and Salvator (1999). Meconium is the content of the bowels, which begins forming early in the second trimester and is excreted by the neonate in the first postpartum days.
For inclusion in the CE group, only evidence of alcohol, marijuana, nicotine, and cocaine was acceptable, and infants whose mothers used other illicit drugs were excluded. For the NCDE group, alcohol, marijuana, and tobacco exposure were acceptable, but not cocaine or other illicit drugs. For the ND group, no drug or alcohol use was acceptable. Maternal urine was screened for cocaine metabolites, cannabinoids, opiates, PCP, and amphetamines. Infant urine was screened for all of these substances, if maternal urine was positive for any of them. Regardless of urine toxicology, meconium was collected from multiple diapers and analyzed for cocaine and its metabolites (benzoylecgonine, metahydroxybenzoylecgonine, cocaethylene), cannabinoids (THC), opiates, phencyclidine, amphetamines, and benzodiazepines.
Maternal self-report of drug-use was assessed with a version of Streissguth et al.’s Maternal Postpartum Questionnaire (Singer et al., 1997; Streissguth, 1986). The mothers were asked about amount per occasion and frequency of substance use in four time periods: the month prior to pregnancy, and during each trimester of pregnancy. The severity scores for the four periods were then averaged.
Based on the averaged cocaine severity score and the meconium analysis of cocaine metabolites, infants in the CE group were divided into heavier- and lighter-CE groups, based on cutoffs established in a previous cohort of mother–infant dyads recruited from the same hospital (Singer et al., 1999, 2001). Infants were classified as heavier-exposed when CE, as indicated by maternal report, was above the 70th percentile or concentration of meconium cocaine metabolites was above the 75th percentile for the previous cohort. For the level of CE analysis, the NCDE and ND groups combined into a single no-cocaine (NC) group.
Materials
A-not-B apparatus.
Three hiding wells were recessed into a foam core platform (see Figure 1 for dimensions), which was placed on a 62-cm-high table. The adult holding the infant was seated in a swivel chair (allowing for a 360° turn), 54 cm off the ground. The toys (small animal figurines) were too large to swallow, but small enough to fit completely in the hiding well. The covers for the wells were identical white washcloths (24 × 25 cm rectangles).
FIGURE 1.
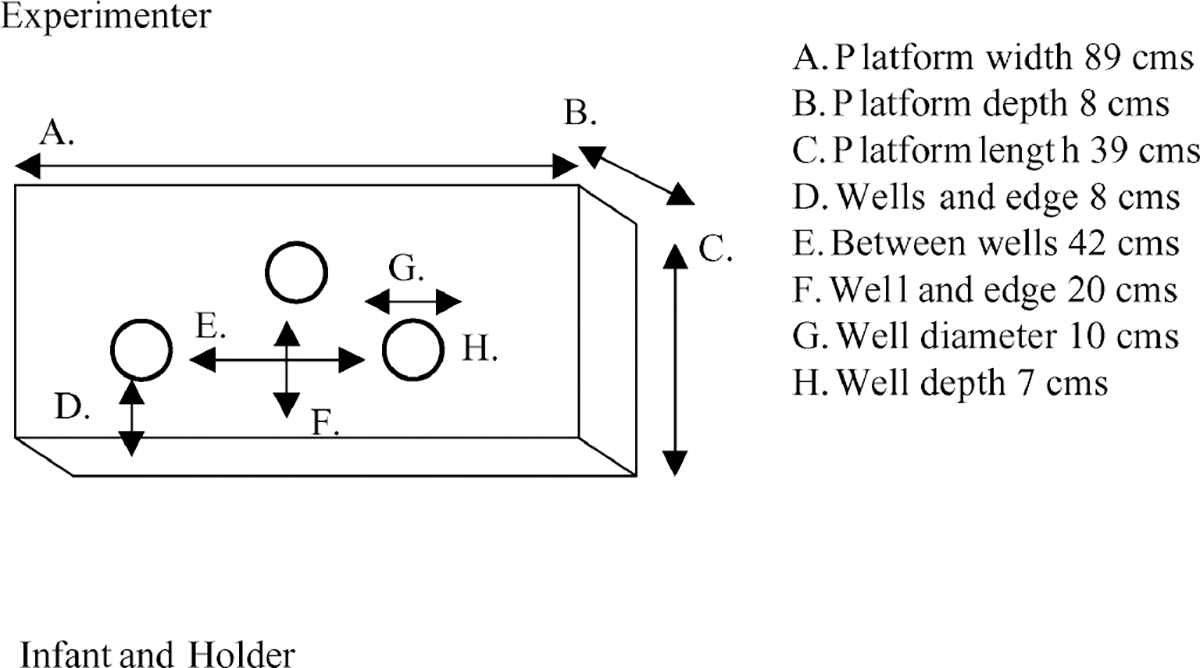
Top view of A-not-B apparatus, platform with three identical hiding wells.
Design and Procedure
A-not-B task design.
As with the delay tolerance A-not-B task employed by Bell and colleagues (Bell & Fox, 1992; Bell & Adams, 1999) and Diamond and colleagues (Diamond, 1985; Diamond et al., 1997), we aimed to identify how long a delay between hiding and search an infant could tolerate and still succeed on a B (reversal) trial. A set of three AB sequences was administered at a given delay, to determine if the infant would consistently fail the B trials at that delay. The criterion for consistent failure was two of three B trials failed, which is an adaptation of Diamond’s (1985) A-not-B error criterion. As with the Bell version, we have extended the scale downward to rate the performance of infants whose object search abilities are not yet well-enough developed to consistently produce the A-not-B error.
As the infant watched, the experimenter dropped the toy into the hiding well, then covered both test wells with the cloths. The experimenter then indicated that the adult holding the infant should turn away and wait, while the experimenter counted the passage of the delay out loud. For the 0-sec delay trials, the holder did not turn away from the test apparatus. Instead, the experimenter sought to draw the infant’s gaze away from the wells, and retrieval was permitted as soon as the gaze was broken. Search continued until the infant uncovered a hiding well or turned their attention away from the hiding apparatus.
Each AB began with A trials (toy hidden in location A) which continued until either one of two things happened: (a) the infant earned a B trial by succeeding on two consecutive A trials or (b) the infant failed out of the A trials, with 5 consecutive or 10 total failed A trials in an AB sequence. If the infant succeeded at two consecutive A trials, the next trial was a B trial, and the toy was hidden in the other test well (location B).
One of the two lateral wells served as the warm-up-only well. The lateral well, which served as the warm-up well, alternated between infants. There was no delay on the warm-up trials, and only the warm-up well was covered. To pass the warm-up criteria, an infant was required to succeed on two consecutive trials. Up to 10 warm-up trials were administered.
Complementary to the designation of the warm-up well, only one of the two lateral wells was used as a test trial hiding well for each infant. The lateral test well was the A location on the first AB sequence, for all infants, and the center well was always the B location for the first AB sequence. For the subsequent AB sequences, the B hiding well in the preceding AB sequence became the first A-trial hiding well for the next sequence. The criterion for success on the test trials required the infant to uncover only the correct well.
If an infant failed two of the three B trials at a given delay, then the test session was ended. If the infant passed two B trials at a given delay, then a new set of AB sequences was begun with a 2-sec-longer delay. If an infant failed on all three B trials or was unable to earn a B trial at a given delay, then a new set was begun with a 2-sec-shorter delay.
Testing continued until one of the following occurred: the infant failed on two of three reversal trials at a given delay, the infant performed below this criterion at the 0 sec delay, or the infant could no longer be induced to attend to the hiding event.
The delay used in the initial set of three AB sequences was based on chronological age. For 10-month-old infants (range = 9 months, 14 days–10 months, 7 days), the initial delay was 2 sec. For the 11-month-old infants (range = 10 months, 8 days–11 months, 23 days), the initial delay was 4 sec; and for the 12-month-old infants (range = 11 months, 24 days–12 months, 8 days), the delay was 6 sec.
Delay tolerance A-not-B rating scale.
Delay tolerance on the A-not-B task was assessed on a scale similar to that developed by Bell and colleagues (Bell & Adams, 1999; Bell & Fox, 1992). The scale ranged from a 0 (performance never merited a single reversal trial) to 7 (failed A-not-B criteria at an 8-sec delay). This scale is presented in detail in Table 1.
TABLE 1.
A-not-B Rating Scale
| 0 | Finds completely hidden object during warm-up |
| 1 | Finds object in one of two hiding wells, but on no consecutive trials |
| 2 | Merits at least one B trial, but never successful on a B trial |
| 3 | At 0 sec delay, fails two of three B trials |
| 4 | At 2 sec delay, fails two of three B trials |
| 5 | At 4 sec delay, fails two of three B trials |
| 6 | At 6 sec delay, fails two of three B trials |
| 7 | At 8 sec delay, fails two of three B trials |
Other infant assessments.
The BSID–II, a general test of infant mental and motor development (Bayley, 1993), was administered after the A-not-B task. There is an adjustment for GA prescribed by the BSID–II manual. For parity with the A-not-B rating scale, this adjustment was not used, and the chronological age Mental Development Index (MDI) and Psychological Developmental Index (PDI) scores are reported. The BSID–II Behavioral Rating Scale (BRS) is based on the examiner’s rating of the infant’s behavior during the test session.
Data Analysis
All self-report and drug variables were positively skewed and, for parametric statistics, were normalized by a log × +1 transformation. To eliminate the noise associated with the wide range of postnatal age-at-test, A-not-B rating was adjusted for number of postnatal days, in every comparison. This also increased the similarity of the A-not-B rating to the BSID–II index scores, which are adjusted for age-at-test. Main effects of CE grouping and level-of-CE grouping (one-way analysis of variance, General Linear Model) on outcome variables were examined first. Next, we conducted the following planned comparisons of outcome variables, through t tests for BSID–II measures and regression models for A-not-B rating: CE/NCDE versus ND; CE versus NC; heavier-CE versus Lighter-CE/NC. Significant relationships between cocaine and outcome variables were explored with a series of stepwise regression analyses, controlling for relevant covariates. Qualified potential confounders and mediators were forced into the first steps of a stepwise regression model, after which the CE variable was entered, to determine if it had any incremental predictive validity.
Entry into regression analysis was limited to variables that were related ( p < .10) to both the target CE grouping (χ2, t tests) and the target outcome variables (Pearson correlational analysis, multiple regression). Thus, both the potential confounders and the potential mediators qualified for entry in the models, if they were simultaneously related to the outcome variable and different between the CE groups. Race, gender, parity, number of prenatal care visits, maternal age, years of education, marital status, per person household income, and severity of exposure variables for noncocaine variables were evaluated as potentially confounded variables. Weeks of gestation and birth weight were evaluated as potential mediators of cocaine effects, because previous research suggests both birth weight and GA at birth are related to general mental development and are effected by CE.
Only potential confounders were included in the first model. If the CE grouping variable was significant in this first model, then the second model would include both potential confounders and mediators. The specificity of any significant effect of CE on A-not-B rating was further investigated in a third model. Following Espy et al. (1999), MDI was added to the model with the potential confounders and mediators.
RESULTS
Birth Mother Characteristics by Group
The CE, NCDE, and ND groups were similar on SES, but differed on several maternal (Table 2), including number of prenatal visits, percent born preterm, birth size, and use or nonuse of alcohol and tobacco. The cocaine-using women were older and had more previous pregnancies (parity) than the ND women. The CE women were older and reported more alcohol and tobacco use than the NCDE women (Table 2). All of these group differences have been reported in other studies of CE with samples from urban hospitals (see Jacobson & Jacobson, 2001, for a review).
TABLE 2.
Maternal Characteristics by Drug-Exposure Group
|
CE
a
|
NCDE
b
|
ND
c
|
χ 2 | F/t | p <d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | SD | % | M | SD | % | M | SD | % | ||||
|
| ||||||||||||
| Race (non-White) | 74 | 67 | 82 | 1.0 | .6 | |||||||
| Low SES | 89 | 87 | 76 | 1.2 | .6 | |||||||
| Married | 16 | 27 | 24 | 0.6 | .8 | |||||||
| Alcohol, any use | 79 | 40 | 5.4 | .04 | ||||||||
| Marijuana, any use | 37 | 13 | 2.4 | .2 | ||||||||
| Tobacco, prenatal use | 100 | 80 | 4.2 | .08 | ||||||||
| Years of education | 11.2 | 1.2 | 11.1 | 1.6 | 12.1 | 1.3 | 2.3 | .104 | ||||
| Age (years) | 28.7 | 6.3 | 23 | 5.9 | 23.1 | 3.6 | 6.5 | .003e,f | ||||
| Parity (previous births) | 3.6 | 2.2 | 2.5 | 1.5 | 2.2 | .6 | 3.4 | .03e | ||||
| No. prenatal visitsg | 6.3 | 3.7 | 9 | 3 | 8.5 | 3.4 | 3.3 | .05 | ||||
| Amount alcoholh,i | 5.4 | 5.8 | .2 | .3 | 18.1 | .001 | ||||||
| Amount marijuanah,i | .5 | 1 | .5 | 1.4 | 0.0 | .97 | ||||||
| Amount tobaccoi, j | 8.8 | 5.8 | 4.8 | 6.2 | 18.6 | .001 | ||||||
Note. CE = cocaine exposure; NCDE = noncocaine drug exposure; ND = no drug exposure; SES = socioeconomic status.
n = 19.
n = 15.
n = 17.
Probability estimates based on Fishers Exact Test, no pairwise comparisons conducted. NCDE vs. ND, α = .05.
CE vs. ND, α = .05.
CE vs. NCDE, α = .05.
Adjusted for gestational age.
Maternal self-report, averaged drinks or joints per day × number days per week.
Significance tests used log + 1 transformed data.
Maternal self-report, averaged cigarette per day.
According to self-report, the median number of rocks of cocaine used per week, averaged across pregnancy, was 7 by the women in the heavier-CE group and .6 by the women in the lighter-CE group. Correspondingly, the self-report cocaine severity scores of heavier-CE group women are higher than those of the lighter-CE women (Table 3). When divided into these levels of exposure groups, there were no differences in the socioeconomic variables, but the groups differed on several other maternal variables, including number of prenatal visits, and use or nonuse of alcohol, tobacco, and marijuana. The heavier-CE woman were older, had higher parity, and used more alcohol and tobacco than the NC controls. The lighter-CE group women were older and used more alcohol than the NC controls.
TABLE 3.
Maternal Characteristics by Level of Cocaine Exposure Groups
|
Heavier CE
a
|
Lighter CE
b
|
No CE
c
|
χ 2 | F/t | p <d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | SD | % | M | SD | % | M | SD | % | ||||
|
| ||||||||||||
| Race (non-White) | 89 | 60 | 75 | 2.1 | .4 | |||||||
| Low SES | 89 | 90 | 81 | 0.6 | 1 | |||||||
| Married | 11 | 20 | 25 | 0.8 | .9 | |||||||
| Alcohol, any use | 89 | 70 | 19 | 18.5 | .001 | |||||||
| Marijuana, any use | 22 | 50 | 6 | 10.2 | .004 | |||||||
| Tobacco, any use | 100 | 100 | 38 | 19.5 | .001 | |||||||
| Years of education | 11.2 | 1.6 | 11.1 | 1.7 | 11.7 | 1.5 | 0.6 | .5 | ||||
| Age (years) | 29 | 4.9 | 28.4 | 7.7 | 23.1 | 4.7 | 6.5 | .004e,f | ||||
| Parity (previous births) | 4.4 | 1.7 | 2.9 | 2.4 | 2.3 | 1.1 | 6.4 | .003f | ||||
| No. prenatal visitsg | 6.4 | 4.6 | 6.1 | 2.8 | 8.8 | 3.1 | 3.2 | .05 | ||||
| Amount alcoholh,i | 6.7 | 6.3 | 4.3 | .4 | < .1 | 32.1 | .001e,f | |||||
| Amount marijuanah,i | .18 | .5 | .73 | 1.2 | .21 | .96 | 2.0 | .15 | ||||
| Amount tobaccoi, j | 9.2 | 6.7 | 8.4 | 5.2 | 2.3 | 4.7 | 18.6 | .001e,f | ||||
| Amount cocaineh,i | 48.7 | 102 | 1.1 | 1.5 | 3.5 | .005 | ||||||
Note. CE = cocaine exposure; SES = socioeconomic status.
n = 9.
n = 10.
n = 30.
Probability estimates based on Fishers Exact Test, no pairwise comparisons conducted.
Lighter CE vs. NC, α = 0.05.
Heavier CE vs. NC, α = 0.05.
Adjusted for gestational age.
Maternal self-report, averaged across pregnancy drinks, rocks, or joints per day × days per week.
Significance tests used log + 1 transformed data.
Maternal self-report, averaged across pregnancy cigarettes per day.
Birth Outcomes by Group
The CE, NCDE and ND groups were different on the percent born preterm and birth size (Table 4). The heavier-CE group infants were born earlier and smaller than the NC infants and were born earlier and shorter than the lighter-CE group infants (Table 4).
TABLE 4.
Infant Birth Characteristics by Drug-Exposure Group and by Level of Cocaine Exposure by Drug Exposure Group
|
Cocaine
a
|
NCDE
b
|
ND
c
|
χ 2 | F | p<d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | SD | % | M | SD | % | M | SD | % | ||||
|
| ||||||||||||
| Born <38 weeks | 31 | 7 | 0 | 8.5 | .003 | |||||||
| Male | 47 | 60 | 56 | 0.5 | .8 | |||||||
| Gestational age (weeks) | 38.5 | 2.1 | 39 | 1.3 | 39.2 | 1.0 | 1.1 | .3 | ||||
| Birth weight (kgs) | 2.8 | .53 | 3.1 | .32 | 3.1 | .44 | 3.2 | .05 | ||||
| Birth length (cms) | 47.7 | 2.7 | 49.7 | 2.3 | 49.7 | 2.4 | 3.9 | .03 | ||||
| Head circumference (cms) | 33.3 | 1.6 | 33.8 | 1.2 | 34 | 1.4 | 1.1 | .4 | ||||
| Age at test (days) | 338 | 20 | 332 | 19 | 330 | 14 | 1.1 | .4 | ||||
|
By Level of CE Groups
|
||||||||||||
|
Heavier CE
e
|
Lighter CE
f
|
No CE
g
|
χ 2 | F | p <h | |||||||
| M | SD | % | M | SD | % | M | SD | % | ||||
|
| ||||||||||||
| Born < 38 weeks | 56 | 10 | 3 | 16.5 | .001h | |||||||
| Male | 67 | 30 | 56 | 2.9 | .21h | |||||||
| Gestational age (weeks) | 37.6 | 2.3 | 39.3 | 1.6 | 39.1 | 1.3 | 4.3 | .02i,j | ||||
| Birth weight (kgs) | 2.64 | .59 | 2.94 | .46 | 3.13 | .38 | 4.5 | .017i | ||||
| Birth length (cms) | 45.9 | 1.3 | 49.3 | 2.7 | 49.7 | 2.3 | 10 | .001h,i,j | ||||
| Head circumstance (cms) | 33.0 | 1.4 | 33.6 | 1.9 | 33.9 | 1.3 | 1.4 | .26 | ||||
| Age at test (days) | 335 | 22 | 341 | 19 | 331 | 16 | 1.3 | .28 | ||||
Note. NCDE = noncocaine drug-exposure; ND = no drug exposure; CE = cocaine exposure.
n = 19.
n = 15.
n = 17.
Probability estimates based on Fishers Exact Test, no pairwise comparisons conducted.
n = 9.
n = 10.
n = 32.
CE vs. ND, pairwise comparison significant, α = .05.
Heavier CE vs. NC, pairwise comparison significant, α = .05.
Lighter CE vs. heavier CE, pairwise comparison significant, α = .05.
Outcome by Drug Exposure Grouping
There were no main effects of drug exposure status (CE, NCDE, ND) on A-not-B rating, F(2, 48) = 0.2, p < .81; BSID–II MDI, F(2, 48) = 0.1, p < .94; BSID–II PDI, F(2, 49) = 0.2, p < .79; or BRS total, F(2, 48) = 1.24, p < .3. Nor were there effects of drug exposure (CE/NCDE vs. ND) or cocaine exposure (CE vs. NC) in planned comparisons.
Level of CE Grouping
There was a main effect of level of cocaine exposure grouping (heavier-CE, lighter-CE, NC) on A-not-B rating, and trends for MDI and PDI (see Table 5). There was no effect on the BRS scores. In planned comparisons of heavier-CE group and the combined group of all other infants, there were significant relationships between heavier-CE status and all three outcome variables.
TABLE 5.
Performance by Level of Cocaine Exposure Groups
|
Heavier CE
a
|
Lighter CE
b
|
No CE
c
|
χ 2 | F | p <d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | SD | % | M | SD | % | M | SD | % | ||||
|
| ||||||||||||
| A-not-B Rating > 4 | 33 | 80 | 56 | 4.2 | .12 | |||||||
| A-not-B Ratinge | 2.6 | 1.5 | 4.8 | 1.5 | 3.6 | 1.4 | 5.7 | .006f,g | ||||
| MDIh | 88.6 | 12 | 96.3 | 11 | 96.6 | 9 | 2.36 | .106f | ||||
| PDIh | 86.6 | 11 | 97.6 | 10 | 94.8 | 11 | 2.93 | .063f | ||||
| Percentile on BRS | 51.8 | 30 | 58.8 | 31 | 67.7 | 27 | 1.2 | .29 | ||||
Note. CE = cocaine-exposure; MDI = Mental Development Index; PDI = Psychological Development Index; BRS = Behavioral Rating Scale.
n = 9.
n = 10.
n = 32.
Probabilty based on Fisher’s Exact test, pairwise comparison not conducted.
Means and regression analysis adjusted for age at test (days), estimated test statistic given.
Heavier CE vs. lighter CE/NC, p < .05.
Heavier CE vs. lighter CE, pairwise comparison, p < .05.
Based on chronological age.
Confounders and mediators.
The significant relationships, which led to the identification of potential confounders and mediators, are reported in Table 6. GA at birth was identified as a potential mediator of the relationship of heavier-CE and A-not-B performance. However, heavier-CE remained a significant predictor of A-not-B performance, when it was entered last in a regression that included GA at birth (Table 7). Birth weight, which might be a more accurate gauge of gestation in a population late to start prenatal care, was substituted for GA and the model was re-run, with the same outcome, heavier-CE, β −1.33 (SE .58) t = −2.21, p < .03, F(47, 3) = 3.18, p < .032, R2 = .17. Heavier CE was no longer a significant predictor of MDI score, when maternal report of severity of gestational cigarette use was included in the model (Table 8). Parity was identified as a potential confounder, and GA at birth was identified as a potential mediator of relationship of heavier CE to motor development (Table 6). In the regression model of PDI, including parity and heavier CE, the latter was, at a level corresponding to statistical trend, still related to PDI, (Table 9). However, this trend of heavier-CE status to predict motor development was eliminated when GA was included in the final model, heavier-CE, β 6.3 (SE 4.5) t = −1.4, p < .16, F(47, 3) = 2.2, p < .10, R2 = 0.12.
TABLE 6.
Identification of Variables Related (p < .10) to Outcome Measures, and Differing (p < .10) Between Groups for Heavier CE vs. Lighter CE/No Cocaine Model
|
Group Differences Heavier CE vs. All Others
|
Significant Correlations With Outcome Variables
|
|||||||
|---|---|---|---|---|---|---|---|---|
|
A-not-B Rating
a
|
PDF
b
|
MDP
b
|
||||||
| t | p < | R 2 | p < | r 2 | p < | r 2 | p < | |
|
| ||||||||
| Parity (previous births) | −3.4 | .001 | .23 | .105 | ||||
| Maternal age (years) | −2.2 | .03 | ||||||
| Cigarettes (severity score)c | −3.04 | .004 | −.29 | .04 | ||||
| Alcohol (severity score)c | −4.85 | .001 | ||||||
| Gestational age (weeks) | 2.0 | .07 | .05 | .098 | .25 | .07 | .29 | .04 |
| Birth weight (grams) | 2.15 | .06 | .28 | .04 | .30 | .04 | ||
| Length for gestationald | −3.1 | .003 | ||||||
| age (cms) | ||||||||
Note. CE = cocaine-exposure; PDI = Psychological Development Index; MDI = Mental Development Index.
Analysis adjusted for age at test (days), reported as squared semipartial correlation.
Based on chronological age.
log + 1 transformed data.
t-estimate for CE status as predictor of birth length, after gestational age entered in regression model.
TABLE 7.
Effects of Heavier Cocaine Exposure on A-not-B Rating
| Predictor | β | SE | Standardized β | t | p | Change in R2 for Final Step |
|---|---|---|---|---|---|---|
|
| ||||||
| Age at test | .02 | .01 | .25 | 1.9 | .06 | |
| Gestational age | .12 | .14 | .12 | .81 | .42 | |
| Heavier CE | −1.2 | .59 | −.30 | −2.1 | .046 | .07 |
Note. CE = cocaine-exposure. F(47, 3) = 3.4, p < .025, R2 = 0.18.
TABLE 8.
Effects of Heavier Cocaine Exposure on Chronological Age MDI
| Predictor | β | SE | Standardized β | t | p | Change in R2 for Final Step |
|---|---|---|---|---|---|---|
|
| ||||||
| Cigarettes, severity | −1.9 | 1.4 | −.20 | −1.3 | .17 | |
| Heavier CE | 5.8 | 3.9 | −.22 | −1.5 | .14 | .04 |
Note. MDI = Mental Development Index; CE = cocaine-exposure. F(48, 2) = 3.4, p < .04, R2 = 0.12.
TABLE 9.
Effects of Heavier Cocaine Exposure on Chronological Age PDI
| Predictor | β | SE | Standardized β | t | p | Change in R2 for Final Step |
|---|---|---|---|---|---|---|
|
| ||||||
| Parity | −.71 | 97 | −.11 | −.74 | .46 | |
| Heavier CE | −7.5 | 4.3 | −.27 | −1.8 | .09 | .06 |
Note. PDI = Psychological Development Index; CE = cocaine-exposure. F(48, 2) = 2.9, p < .06, R2 = 0.11.
Specificity of Cocaine’s Effect on EF
As originally planned, chronological age MDI was added to age-at-test and gestational weeks in the regression model predicting A-not-B rating from heavier-CE versus all others. Heavier-CE status was no longer a significant predictor of A-not-B rating, heavier CE, β −.94 (SE .58) t = −1.62, p < .11, F(46, 4) = 3.98, p < .007, R2 = 0.26, the change in R2 associated with the addition of MDI was nonsignficant, partial R2 = .04. On a reviewer’s suggestion, this model was re-run without GA and the results were similar. With age-at-test and MDI in the model, heavier-CE status predicted worse A-not-B rating at the level of a statistical trend, F(3, 47) = 5.35, p < . 003, R2 = 0.25, the change in R2 associated with the addition of heavier CE, F(1) = 3.51, p < .067, partial R2 = .056.
Severity of Cocaine Use
There was no relationship of maternal report of average amount of cocaine used during gestation (log + 1 transformed severity score) and any of the outcome variables.
DISCUSSION
When the mother–infant dyads were compared across the original recruitment groups (CE, NCDE, and non-drug-exposed), there were no group differences in SES, but these were group differences in birth size, obstetric history, number of prenatal visits, and maternal age. Despite these differences, there were no group differences on any of the cognitive, motor, or behavioral assessments.
An effect of CE on EF emerged when the infants were divided into heavier-CE, lighter-CE, and non-CE groups. Infants in the heavier prenatal CE group scored lower on the delay tolerance A-not-B rating scale than the combined group of lighter-CE and non-CE group infant. The heavier-cocaine-using women differed from the other women in age, parity, and use of cigarettes, alcohol; and the heavier-CE group infants were born earlier, lighter, and shorter for GA. GA at birth was the only of these variables related to A-not-B performance, qualifying it as a potential mediator. With the variance associated with GA controlled for, there was still a significant effect of heavier-CE status on A-not-B rating. The results support the suggestion that heavier prenatal cocaine exposure is associated with decreased EF in infancy, independent of cocaine’s effect on the length of gestation.
Espy et al. (1999) previously reported that children exposed to cocaine perform worse than control children on A-not-B task. However, there are several design issues that severely limit the degree to which that difference could be attributed to direct effects of cocaine exposure on the developing fetus. First, in the Espy study, the testers knew the drug-exposure status of the children during test, raising the possibility of experimenter bias. In this study, the experimenters were blind to drug-exposure status. Second, in the Espy study, there were unaddressed age and socioeconomic differences between CE and control group, either of which may have accounted for the performance differences. In this study, the controls and CE families had the same socioeconomic background, and age-at-test-associated variance was controlled for statistically.
In both these and the Espy samples, prenatal alcohol use was correlated with cocaine use. According to growing consensus in behavioral teratology, this mandates investigating the potentially confounding role of prenatal alcohol exposure (Jacobson & Jacobson, 2001). The potential for alcohol effects to be spuriously attributed to cocaine is especially high, when the target behavior is EF, which has, along with its neurological underpinnings, demonstrated vulnerability to prenatal alcohol exposure in humans (Kodituwakku et al., 1995; Mattson et al., 1999; Noland et al., 2003; Wass, Persutte, Hobbins, 2001). Although, in this study, severity of alcohol exposure was not related to A-not-B performance, investigating the relationship was a crucial, previously neglected step in exploring direct effects of prenatal cocaine exposure on EF.
The current effect of cocaine on A-not-B performance was only apparent at the heavier levels of exposure, suggesting that there may be a threshold of exposure that must be exceeded before performance is impaired. Espy et al. (1999) did not report amount of cocaine used by the mothers in their study; therefore, it is not possible to directly compare level of exposure across the two studies, nor is it feasible to speculate on the level of CE in the Espy study. The women had been in treatment (which might imply heavier cocaine use), but they also had abstained for 12 months and had retained custody of their children, both of which imply less-severe cocaine addiction.
The classification of infants into CE groups in this study was based on a combination of maternal report and analysis of meconium. In a previous sample, this combination has been demonstrated to be more effective for classification than either method alone (Arendt et al., 1999). One limitation of this methodology is that meconium is not formed in the first trimester, therefore, CE before that time would not be detectable by biological assay. The noise introduced by these false negatives would work against the hypothesis of the study and, consequently, does not limit the interpretation of the results.
However, these findings also suggest that a decreased GA is associated with decreased A-not-B performance, as has been previously reported in preterm infants (Ross, Tesman, Auld, & Nass, 1992). However this finding is in contrast to findings of the only other study, which, like this study, excluded infants born before 34 gestational weeks. In that study, preterm infants out-performed their chronological age-matched controls on an A-not-B task (Matthews et al., 1996). However, Matthews et al. (1996) study had fewer at-risk infants, in that they were not substance-exposed or born to impoverished women. Subtle brain damage may have been inflicted on this sample and left the offspring vulnerable to the effects of early birth.
We also explored the effect of heavy CE on general mental and psychomotor development. When the levels of exposure groups were compared, there was a trend toward group differences on the motor development, independent of potential confounders. However, differences in GA related to heavier CE, seem to mediate this trend. A shortened gestation has been found in other studies of CE infants (Eisen et al., 1991; Mayes, Granger, Frank, Schottenfeld, & Bornstein, 1993; Singer et al., 2001) and has been identified as a potential mediator of cocaine effects on development (Lester, Freier, & LaGasse, 1995). Global mental development index scores were lower in the heavier-CE group, but this effect is confounded with reported severity of prenatal cigarette exposure.
To investigate the possibility of CE effects on EF over and above differences in general cognitive functioning, we employed the same procedure that Espy et al. (1999) had adapted from Welsh, Pennington, Ozonoff, Rouse, and McCabe (1990). When we controlled for the variance associated with MDI in the model of heavier-CE on A-not-B performance, there was no longer a significant cocaine effect. This is in contrast to the Espy et al. (1999) report of group differences in A-not-B performance, which persisted, even after the variance associated with MDI was controlled for. Age differences in the children possibly accounts for this contrast. In the Espy study, the offspring were between 17 and 21 months of age, compared with the 10- to 12-month-olds in this study. EF may be less differentiated from general mental functioning at younger ages. This would accord with the finding from adults that effects of another teratogen (prenatal alcohol) on EF are only independent from effects on global intelligence at higher levels of intellectual functioning (Connor, Sampson, Bookstein, Barr, & Streissguth, 2001). Another possibility is that the trend for heavier-CE to predict A-not-B performance, independent, of mental development, would have, given a larger sample, reached the level of statistical significance.
Currently, any inquiry into EF capacity in infancy will be limited by the paucity of measurement instruments available for infants. The single measure of EF employed in this study may have only tapped a portion of the cognitive abilities that make up EF. Some components of EF may be more vulnerable to teratogenic effects than others (Fried & Smith, 2001; Kodituwakku et al., 1995; Noland et al., in press). Welsh, Pennington, and Grossier (1991) demonstrated that there are distinct developmental patterns for different subcomponents of EF and several EF abilities do not peak until late childhood.
We did not find relationships either of severity of prenatal marijuana exposure or of alcohol exposure on A-not-B performance, despite evidence that both impair EF in children (Fried & Smith, 2001; Kodituwakku et al., 1995; Noland et al., 2003). This may be a power issue. Because of the small sample size, only large effects on behavior could have been detected. It may also result from the restricted breadth of the current assessment. As discussed earlier, there are subcomponents of EF not tapped by the A-not-B task.
This study provides some evidence that EF is impacted by heavier prenatal cocaine exposure, and that confirms the sensitivity of the A-not-B task in detecting dysfunction early in development. An important step in future studies is to better isolate effects of prenatal cocaine from caregiver differences that have been shown to be related to A-not-B performance, such as maternal responsiveness (Ayoun, 1998).
ACKNOWLEDGMENTS
Supported by grants NIDA RO1-DA09049-01A3 and NIDA F32-05904 from the National Institutes on Drug Abuse and a grant from the Schubert Center on Child Development, Case Western Reserve University. Research presented at the International Society for Infant Studies in July 2000, at Brighton, England.
With thanks to L. Ellison, P. Gallito, P. Manning, and B. Hong for assistance in data collection, as well as to T. Lotz-Ganley, H. Lockett, and P. Weishampel for manuscript preparation.
REFERENCES
- Arendt RE, Singer LT, Minnes S, & Salvator A (1999). Accuracy in detecting prenatal drug exposure. Journal Drug Issues, 29, 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoun C (1998). Maternal responsiveness and search for hidden object and contingency learning by infants. Early Development and Parenting, 7(2), 61–72. [Google Scholar]
- Bayley N (1993). Bayley Scales of Infant Development (2nd ed.). San Antonio, TX: Psychological Corporation. [Google Scholar]
- Bell MA, & Adams SE (1999). Comparable performance on looking and reaching versions of the A-not-B task at 8 months of age. Infant Behavior and Development, 22(2), 221–235. [Google Scholar]
- Bell MA, & Fox NA (1992). The relations between frontal brain electrical activity and cognitive development during infancy. Child Development, 63, 1142–1163. [PubMed] [Google Scholar]
- Connor PD, Sampson PD, Bookstein FL, Barr HM, & Streissguth AP (2001). Direct and indirect effects of prenatal alcohol damage on executive function. Developmental Neuropsychology, 18, 331–354. [DOI] [PubMed] [Google Scholar]
- Diamond A (1985). Development of the ability to use recall to guide action, as indicated by infants’ performance on A not B. Child Development, 56, 868–883. [PubMed] [Google Scholar]
- Diamond A (1990). The development and neural bases of memory functions as indexed by the A not B and delayed response tasks, in human infants and infant monkeys. In Diamond A (Ed.), The development and neural bases of higher cognitive functions (pp. 276–317). New York: New York Academy of Sciences. [DOI] [PubMed] [Google Scholar]
- Diamond A, & Goldman-Rakic PS (1989). Comparison of human infants and rhesus monkeys on Piaget’s A-not-B task. Evidence for dependence on dorsolateral prefrontal cortex. Experimental Brain Research, 74, 24–40. [DOI] [PubMed] [Google Scholar]
- Diamond A, Prevor MB, Callender G, & Druin DP (1997). Prefrontal cortex cognitive deficits in children treated early and continuously for PKU. Monographs of the Society for Research in Child Development, 62(4), 1–205. [PubMed] [Google Scholar]
- Eisen LN, Field TM, Bandsta ES, Roberts JP, Morrow C, Larson SK, et al. (1991). Perinatal cocaine effects on neonatal stress behavior and performance on the Brazelton scale. Pediatrics, 88, 477–480. [PubMed] [Google Scholar]
- Espy KA, Kaufmann PM, & Glisky ML (1999). Neuropsychologic function in toddlers exposed to cocaine in utero: a preliminary study. Developmental Neuropsychology, 15, 447–460. [Google Scholar]
- Fried PA, & Smith AM (2001). A literature review of the consequences of prenatal marihuana exposure: An emerging theme of a deficiency in aspects of executive function. Neurotoxicology & Teratology, 23, 1–11. [DOI] [PubMed] [Google Scholar]
- Friedman E, Yardin E, & Wang HY (1996). Effect of prenatal cocaine on dopamine receptor-g protein coupling in mesocortical regions of the rabbit brain. Neuroscience, 70(2), 739–747. [DOI] [PubMed] [Google Scholar]
- Fuster JM (1989). The prefrontal cortex (2nd ed.). New York: Raven. [Google Scholar]
- Harris PL (1975). Development of search and object permanence during infancy. Psychological Bulletin, 82, 332–344. [PubMed] [Google Scholar]
- Jacobson SW, Jacobson JL, Sokol RJ, Martier SS, & Chiodo LM (1996). New evidence of neurobehavioral effects of in utero cocaine exposure. The Journal of Pediatrics, 129, 581–588. [DOI] [PubMed] [Google Scholar]
- Jacobson SW, & Jacobson JL (2001). Alcohol and drug-related effects on development: A new emphasis on contextual factors. Infant Mental Health Journal, 22(3), 416–430. [Google Scholar]
- Kodituwakku PW, Handmaker NS, Culter SK, Weathersby EK, & Handmaker SD (1995). Specific impairments in self-regulation in children exposed to alcohol prenatally. Alcoholism: Clinical and Experimental Research, 19, 1556–1564. [DOI] [PubMed] [Google Scholar]
- Krasnegor NA, Otto DA, Bernstein JH, Burke R, Chappell W, & Eckerman DA (1994). Neurobehavioral test strategies for environmental exposures in pediatric populations. Neurotoxicology and Teratology, 16, 499–509. [DOI] [PubMed] [Google Scholar]
- Lester BM, Freier K, & LaGasse L (1995). Prenatal cocaine exposure and child outcome: What do we really know? In Lewis M & Bendersky M, Mothers, babies and cocaine: The role of toxins in development (pp. 19–39). Hillsdale, NJ: Lawrence Erlbaum Associates, Inc. [Google Scholar]
- Levitt P, Harvey JA, Friedman E, Simansky K, & Murphy EH (1997). New evidence of neurotransmitter influences on brain development. Trends in Neuroscience, 20(6), 269–275. [DOI] [PubMed] [Google Scholar]
- Marcovitch S, & Zelazo PD (1999). The A-not-B error: results from a logistic meta-analysis. Child Development, 70, 1297–1313. [Google Scholar]
- Matthews A, Ellis AE, & Nelson CA (1996). Development of preterm and full-term infant ability of AB, recall memory, transparent barrier detour, and means-end tasks. Child Development, 67, 2658–2676. [PubMed] [Google Scholar]
- Mattson SN, Goodman AM, Caine C, Delis DC, & Riley EP (1999). Executive functioning in children with heavy prenatal alcohol exposure. Alcoholism: Clinical and Experimental Research, 23, 1808–1815. [PubMed] [Google Scholar]
- Mayes LC (1999). Developing brain and in utero cocaine exposure: Effects on neural ontogeny. Developmental and Psychopathology, 11, 685–714. [DOI] [PubMed] [Google Scholar]
- Mayes LC, Granger RH, Frank MA, Schottenfeld R, & Bornstein MH (1993). Neurobehavioral profiles of neonates exposed to cocaine prenatally. Pediatrics, 91, 778–783. [PubMed] [Google Scholar]
- Mehta SK, Super D, Salvator A, Singer LT, Connuck D, Goetz-Fradley LG, et al. (2001). Heart rate variability in cocaine-exposed newborn infants. American Heart Journal, 142, 828–832. [DOI] [PubMed] [Google Scholar]
- Mehta SK, Super D, Salvator A, Singer LT, Connuck D, Thomas JD, et al. (2002). Diagnostic filling abnormalities by color kinesis in newborns exposed to intrauterine cocaine. Journal of American Society of Echocardiography, 15, 447–453. [DOI] [PubMed] [Google Scholar]
- Minabe Y, Ashby CR Jr., Heyser C, Spear LP, & Wang RY (1992). The effects of prenatal cocaine exposure on spontaneously active midbrain dopamine neurons in adult male offspring: an electrophysiological study. Brain Research, 561(1), 152–156. [DOI] [PubMed] [Google Scholar]
- Needleman R, Zuckerman B, Anderson GM, Mirochnick M, & Cohen DJ (1993). Cerebrospinal fluid monoamine precursors and metabolites in human neonates following in utero cocaine exposure. Pediatrics, 92(1), 55–60. [PubMed] [Google Scholar]
- Noland JS (2001). The A-not-B task. In Singer LT and Zeskind PS (Eds.), Biobehavioral Assessment of the Infant (pp. 312–322). New York: Guilford. [Google Scholar]
- Noland JS, Singer LT, Arendt RE, Minnes S, Short EJ, & Bearer CF (2003). Executive functioning is preschool-age children prenatally exposed to alcohol, cocaine, and marijuana. Alcoholism: Clinical and Experimental Research, 27(4), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington BF (1991). Diagnosing learning disorders: a neuropsychological framework. New York: Guildford. [Google Scholar]
- Roberts E, Bornstein MH, Slater AM, & Barrett J (1999). Early cognitive development and parental education. Infant & Child Development, 8(1), 49–62. [Google Scholar]
- Ross G, Tesman J, Auld PA, & Nass R (1992). Effects of subependymal and mild intraventricular lesions on visual attention and memory in premature infants. Developmental Psychology, 28, 1067–1074. [Google Scholar]
- Sawaguchi T, & Goldman-Rakic PS (1991). D1 Dopamine receptors in prefrontal cortex: Involvement in working memory. Science, 251, 947–950. [DOI] [PubMed] [Google Scholar]
- Singer LT, Arendt RE, Fagan J, Minnes S, Bolek T, & Becker M (1999). Neonatal visual information processing in cocaine-exposed and non-exposed infants. Infant Behavior and Development, 22(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer LT, Ardendt RE, Farkas K, Minnes S, Huang J, & Yamashita T (1997). Relationship of prenatal cocaine exposure and maternal postpartum psychological distress to child developmental outcome. Developmental Psychopathology, 35, 473–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer LT, Arendt RE, Minnes S, Farkas K, & Salvator A (2000). Neurobehavioral outcomes of cocaine-exposed infants. Neurotoxicology and Teratology, 22, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer LT, Salvator A, Arendt RE, Minnes S, Farkas K, & Kliegman R (2001). Effects of cocaine/polydrug exposure and maternal psychological distress on infant birth outcomes. Neurotoxicology and Teratology, 23, 1–9. [DOI] [PubMed] [Google Scholar]
- Spear LP, Kirstein CL, & Frambes NA (1989). Cocaine effects on the developing central nervous system: behavioral, psychopharmacological and neurochemical studies of function. Annals of the New York Academy of Science, 562, 230–307. [DOI] [PubMed] [Google Scholar]
- Streissguth AP (1986). The behavioral teratology of alcohol: Performance, behavioral, and intellectual deficits in prenatally exposed children. In West JR (Ed.), Alcohol, Brain and Development (pp. 3–44). New York: Oxford University Press. [Google Scholar]
- Taylor JR, Elsworth JD, Roth RH, Sladek JR, & Redmond DE (1990). Cognitive and motor deficits in the acquisition of an object retrieval/detour task in MPTP-treated monkeys. Brain, 113, 617–637. [DOI] [PubMed] [Google Scholar]
- Volpe J (1992). Effects of cocaine on the fetus. New England Journal of Medicine, 327, 135–142. [DOI] [PubMed] [Google Scholar]
- Wass TS, Persutte WH, & Hobbins JC (2001). The impact of prenatal alcohol exposure on frontal cortex development in utero. American Journal of Obstetrics and Gynecology, 185, 737–742. [DOI] [PubMed] [Google Scholar]
- Wellman HM, Cross D, & Bartsch K (1986). Infant search and object permanence: a meta-analysis of the A-not-B error. Monographs on the Society for Research in Child Development, 51(3), 62–67. [PubMed] [Google Scholar]
- Welsh MC, Pennington BF, & Grossier DB (1991). A normative-developmental study on executive function: A window on prefrontal function in children. Developmental Neuropsychology, 7, 131–149. [Google Scholar]
- Welsh MC, Pennington BF, Ozonoff S, Rouse B, & McCabe ERB (1990). Neuropsychology of early-treated phenylketonuria: specific executive function deficits. Child Development, 61, 1697–1713. [PubMed] [Google Scholar]
- Wood J, Plessinger M, & Clark K (1987). Effects of cocaine on uterine blood flow and fetal oxygenation. Journal of the American Medical Association, 257, 957–961. [PubMed] [Google Scholar]