

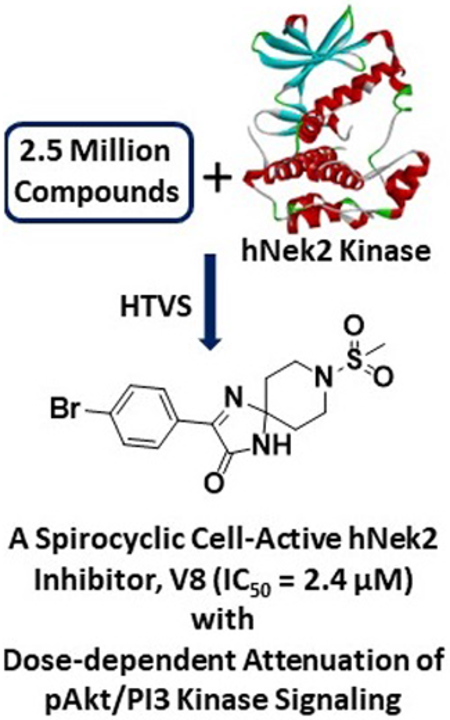
Abstract
NIMA Related Kinase 2 (Nek2) kinase is an attractive target for the development of therapeutic agents for several types of highly invasive cancers. Despite this, no small molecule inhibitor has advanced to the late clinical stages thus far. In this work, we have identified a novel spirocyclic inhibitor (V8) of Nek2 kinase, utilizing a high-throughput virtual screening (HTVS) approach. Using recombinant Nek2 enzyme assays, we show that V8 can inhibit Nek2 kinase activity (IC50 = 2.4 ± 0.2 μM) by binding to the enzyme’s ATP pocket. The inhibition is selective, reversible and is not time dependent. To understand the key chemotype features responsible for Nek2 inhibition, a detailed structure-activity relationships (SAR) was performed. Using molecular models of the energy-minimized structures of Nek2-inhibitory complexes, we identify key hydrogen-bonding interactions, including two from the hinge-binding region, likely responsible for the observed affinity. Finally using cell-based studies, we show that V8 attenuates (a) pAkt/PI3 Kinase signaling in a dose-dependent manner, and (b) proliferative and migratory phenotypes of highly aggressive human MDA-MB-231 breast and A549 lung cancer cell lines. Thus, V8 is an important novel lead compound for the development of highly potent and selective Nek2 inhibitory agents.
Keywords: Nek2 kinase, Nek2 inhibitor, Nek2 hinge pharmacophore, Nek2 HTVS, Cytotoxicity of Nek2 inhibitor, Nek2 and Akt/PI3K Signaling
Graphical Abstract
Mitosis is a fundamental cellular process that is carefully orchestrated by an exquisite group of kinases working to coordinate a sequence of complex events in a highly organized manner.1, 2 A controlled mitotic process is essential for maintaining cellular homeostasis during normal survival and growth processes. Aberrant mitosis as a result of dysregulated kinase signaling can be catastrophic, resulting in multinucleated daughter cells with abnormal chromosomal contents and more than two centrosomes. If cellular apoptotic machinery is nonfunctional in cells containing mitotic errors, cells will divide ‘asymmetrically’ in the next round of cell cycle, creating aneuploidic cells. A body of strong evidence suggest that aneuploidy and abnormal number of centrosomes can promote cancer development.3-5 One of the prominent mitotic kinases is Never in mitosis (NIMA)-related kinase 2 (Nek2) that plays critical roles in several mitotic events, including centrosomal disjunction, microtubule anchoring and stabilization, and spindle assembly checkpoint signaling.6 Overexpression of Nek2 kinase stimulates centrosome amplification (CA) and chromosomal instability (CIN), promoting the onset of tumorigenesis.7-9
Several recent studies further indicate that higher levels of Nek2 kinase can promote drug resistance, and accelerate tumor growth.8, 10-13 Results published from our laboratory further established that overabundant Nek2 can activate PI3K/Akt signaling and promote metastasis in vivo.14 The active role of Nek2 kinase in metastasis has been corroborated by other subsequent studies.12, 15 This finding is highly important since at least two thirds of deaths from solid tissue cancers are due to migration of primary tumor cells to secondary sites.16, 17 Furthermore, expression analysis in cancer cells from various origins, including the ones from highly invasive tumors, revealed overexpression of Nek2 kinase.18-22 This kinase is thus considered an attractive therapeutic target for anti-cancer drug development.23 Indeed, inhibitory perturbation of Nek2 kinase signaling by RNAi-based agents, active-site targeted small molecules, and protein-protein interaction (PPI) inhibitory agents of Nek2-Hec1 protein interface has been shown to exert strong anti-tumor effects and reverse drug resistance (Figure 1).13, 24-28 Despite this, no small molecule Nek2 inhibitors have advanced to late stage clinical trials, primarily due to lack of cellular activity, low potency, and kinase nonspecificity. Novel chemotypes are thus needed to develop them into potent, selective and cell-permeable Nek2 inhibitory agents as potential anti-cancer agents. In this work, we have implemented a High-Throughput Virtual Screening (HTVS) protocol on a library of 2.5 million compounds, and identified a novel spirocyclic compound, V8, that inhibits Nek2 kinase activity in-vitro. The binding of V8 is selective, fast and reversible, and ATP-site directed. Additionally, to provide further support that a spirocyclic motif is well-tolerated within the Nek2 active site, we explore structure-activity relationships (SAR) using a commercially available V8-analog compound library. To further probe that a 3-ring system, as in V8, was essential for affinity, a set of 3 bicylic derivatives was synthesized and biochemically evaluated. We also utilized the energy-minimized inhibitory complexes of Nek2, created by computational modeling, to investigate the key structural features responsible for Nek2 inhibition at the molecular level. Finally, using western-blot analysis, we showed that V8 is cell-active and inhibits pAkt/PI3K signaling in live human A549 lung cancer cells in a dose-dependent manner. Consistently, it also reduces cell-proliferative and migratory phenotypes in both human MDA-MB-231 breast and A549 cancer cell lines.
Figure 1.

The chemotypes utilized in the development of the reported small molecule inhibitors of Nek2 kinase activity.14, 29-31, 34, 41-45
To acquire a novel inhibitory chemotype for Nek2 kinase, we performed a HTVS of a 2.5 million compound library (ChemDiv Inc., USA and Hit2Lead Inc., USA) that was readily available for experimental evaluation. Schrodinger Maestro version 12 was used for the docking protocols that included sequential implementation of HTVS, SP, and XP modes for enhanced computational efficiency (See Supporting Information, Section I). Because each of the three docking modes follows a unique algorithm, identifying an appropriate Nek2 crystal structure from the existing PDB database was essential for accurate docking prediction. This was accomplished by screening the twenty-eight available Nek2 structures in the PDB database for each docking mode and identifying the one where strongest linear negative correlation existed between the docking score and the reported IC50 values. These efforts led to the selection of three crystal structures: 2XKF29 (for HTVS mode), 4AFE30 (for SP mode) and 2JAV31 (for XP Mode). The HTVS procedures were then implemented as depicted in Figure 2. A total of 1600 chemical structures were obtained after the XP mode of docking. A general physicochemical evaluation of the structures in combination with visual inspection, based on their drug-likeness, resulted in a set of 63 compounds. These were then subjected to further evaluation, based on novelty of the chemotypes, especially in the context of reported kinase inhibitors.
Figure 2.

HTVS workflow implemented in the discovery of spirocyclic inhibitory Nek2 kinase chemotype, V8.
We finalized a list of eight compounds, obtained them from commercial vendors (ChemDiv Inc., USA and Hit2Lead Inc., USA) and evaluated them for biochemical inhibition against the active recombinant hNek2 kinase (SignalChem Inc., Canada). A previously utilized, commercially available ADP-Glo Kinase assay for small molecule inhibitory screen was used (for ADP-Glo assay protocol, see the Supporting Information, Section II).32, 33 This initial screen identified five Nek2 inhibitory compounds. Interestingly, three of these compounds activated Nek2 kinase, although to a different degree; this may warrant further biochemical investigation and was not currently pursued. The lead spirocyclic inhibitory compound, V8, was then subjected to a concentration-dependent inhibitory experiment. The data was plotted and fitted to a standard concentration-response curve using KaleidaGraph software (Version 4.5), yielding the IC50 value of 2.4 ± 0.2 μM (Figure 3A and Table 1). To confirm that V8 was binding in the ATP-binding pocket of Nek2 kinase, [ATP] concentration was increased by 3-fold to 150 μM in the assays. As expected, the concentration-response curve shifted to the right, with the new IC50 at 17.5 ± 0.2 μM (Figure 3A and Table 1), thereby indicating that V8 was a competitive inhibitor for ATP binding to the Nek2 active site pocket. We next evaluated the selectivity of inhibition. Given that obtaining selectivity for Nek2 inhibitors against other cell cycle kinases have proven challenging, we assessed V8’s activity against CDK1, Plk1, and Aurora A kinases.31, 34 Furthermore, we included Nek7, another member of NIMA family of kinase with functional overlap with Nek2 kinase in mitotic processes involving separation of centrosomes and appropriate arrangement of the microtubule-based mitotic spindle.35 We also included EGFR enzyme, a receptor tyrosine kinase that has been shown to share inhibitor preferences with Nek2 kinase in the ATP binding site.14 A concentration-response curve was thus generated for each enzyme and the data fitted to obtain the IC50 values (Figure 3B and Table 1). Analysis of the data revealed that V8 was at least 9- to 13-fold selective towards Nek2 kinase over the other tested kinases. This suggested that V8 is a preferred Nek2 inhibitory scaffold. To futher corroborate our finding that (a) V8 was indeed a valid lead inhibitory compound, and (b) a phenyl-substituted spirocyclic scaffold was generally well tolerated, we evaluated the inhibitory efficacies of fourteen structurally homologous compounds (Table 2; V8-1 to V8-14). This study was anticipated to provide important information for the future during the scaffold optimization process. The inhibitory assessments of V8 analogs revealed that potency was only modestly affected, IC50 values increasing from 1.3-fold to 6-fold when compared to original hit V8, with substitutions at both the aromatic ring and methylsulfone group (see Table 2). To rationalize comparable potency of V8-1 to V8-14 analogs to that of V8, we performed XP-Glide docking using Nek2 crystal structure (PDB ID: 2JAV).31 The XP-Glide scores for these analogs were in the range of −8.4 to −6.5 kcal/mol (see Table 2); this is consistent with the inhibition profile observed for V8-1 to V8-14 compounds. While replacement of the 4-bromine atom with a bulky 4-tert-butyl moiety on the phenyl ring decreased the inhibitory potency nearly 4-fold, smaller aliphatic substitutions, such as the 4-methyl group in V8-2 and the 3,4-dimethyl group in V8-3, were less detrimental. In addition, cyclic substitutions on the sulfonyl group as in V8-4 and V8-9 and V8-11 to V8-14 were also somewhat well tolerated. Unsubstituted arylsulfone derivatives (V8-5 to V8-9) generally showed comparable Nek2 inhibitory potency. Isosteric replacement of the sulfone group in V8-6 with a carbonyl group led to V8-10 with retention of potency. The V8-6, V8-7, and V8-8 analogs showed a preference for strong electron-withdrawing group, fluoro, compared to the moderate electron-withdrawing chlorine and bromine atoms. Aliphatic substituents on the para-position of the phenylsulfone moiety (V8-12) are generally tolerated compared to 3,4-disubstituted (V8-14) or 2,4,6-trisubstituted (V8-13) analogs. To understand the importance of bromophenyl ring on the planar imidazolinone ring in Nek2 inhibition, we synthesized three compounds, V0-1, V0-2, and V0-3, and assessed their inhibition profile against Nek2 kinase (see Table 2). V0-1 that lacks the bromophenyl ring showed >13-fold loss of activity, compared to V8. A similar loss in activity was also noted for V0-2 that lacked both bromophenyl ring and the methylsulfone group. The methylated derivative of the spiro-piperidine ring -NH- as in V0-3 also led to a significant loss of potency. Substantial loss of Nek2 inhibitory activity by truncated analogs, V0-1, V0-2, and V0-3, are further corroborated with poor Glide scores (−6.2 to −3.8 kcal/mol). The SAR thus observed with this series of compounds indicates that phenyl-substituted spirocyclic V8 scaffold may well be suited for further optimization to obtain potent and selective Nek2 inhibitors using structure-based design approach.
Figure 3.

Acitivity-based dose-response curves of V8 against Nek2 kinase [A], and other related mitotic kinases [B], Plk1, Nek7, CDK1, and Aurora A. The points are experimental and the line joining them are the fit to obtain the IC50 values.
Table 1.
Experimental IC50 values of V8 against Nek2 kinase and other related mitotic kinases.
| Enzymes | IC50 (μM) [ATP = 50 μM] |
|---|---|
| Nek2 | 2.4 ± 0.2 |
| * 17.1 ± 0.2 | |
| Aurora A | 21.3 ± 0.8 |
| CDK1 | 29.3 ± 0.2 |
| Nek7 | 28.8 ± 0.1 |
| Plk1 | 20.4 ± 0.2 |
At [ATP] = 150 μM
Table 2.
Experimental IC50 values and XP-Glide scores for V8 and V8-analogue compounds against hNek2 kinase.
| Compound I.D. | Chemical Structure | IC50 (μM) |
XP-Glide Score (kcal/mol) |
|---|---|---|---|
| V8 |
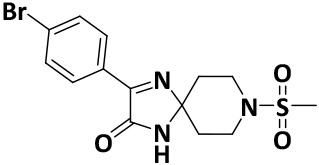
|
2.4 ± 0.2 | −7.9 |
| V8-1 |
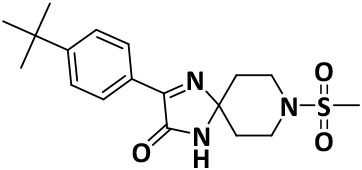
|
8.7 ± 0.2 | −7.8 |
| V8-2 |
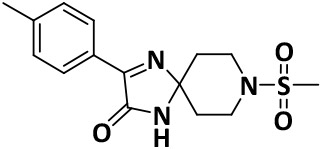
|
4.9 ± 0.6 | −7.9 |
| V8-3 |
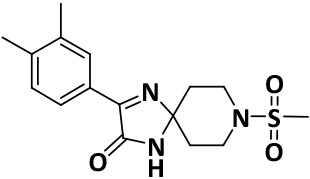
|
3.2 ± 0.1 | −8.1 |
| V8-4 |
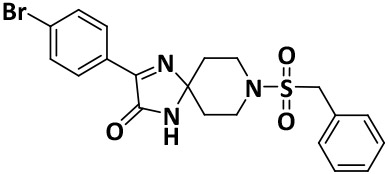
|
6.8 ± 0.1 | −8.0 |
| V8-5 |
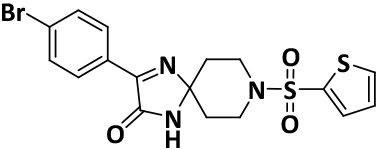
|
5.6 ± 0.1 | −7.7 |
| V8-6 |
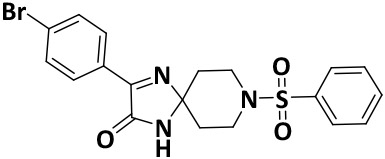
|
9.3 ± 0.1 | −7.9 |
| V8-7 |
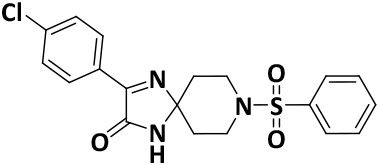
|
6.0 ± 0.1 | −8.0 |
| V8-8 |
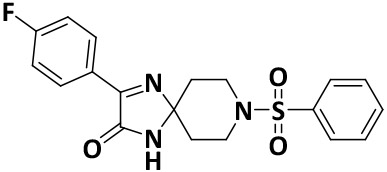
|
4.5 ± 1.0 | −8.4 |
| V8-9 |
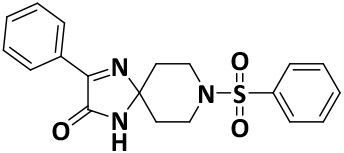
|
8.1 ± 1.0 | −7.9 |
| V8-10 |
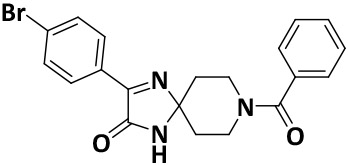
|
9.2 ± 0.7 | −6.5 |
| V8-11 |
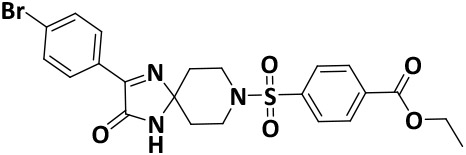
|
14.6 ± 1.0 | −8.0 |
| V8-12 |
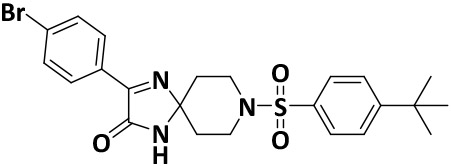
|
5.0 ± 1.0 | −7.3 |
| V8-13 |
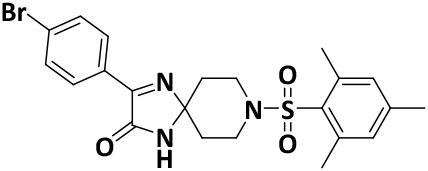
|
9.8 ± 1.3 | −6.6 |
| V8-14 |
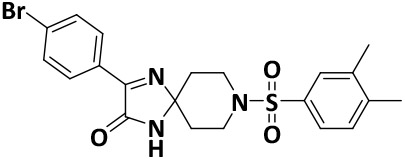
|
12.3 ± 1.2 | −7.2 |
| V0-1 |
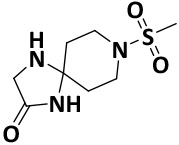
|
32.1 ± 0.6 | −5.4 |
| V0-2 |
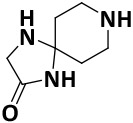
|
32.5 ± 0.7 | −6.2 |
| V0-3 |
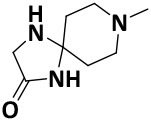
|
58.1 ± 1.0 | −3.8 |
To understand the key interactions observed in the most potent analog V8, the moderately active V8-14, and the least active V0-3 with the active site residues of Nek2 enzyme, the docked and energy minimized inhibitory complexes were analyzed at the molecular levels (Figure 4A-C). This analysis revealed that V8 retained two key hinge-binding interactions: (i) The backbone -NH- from Cys89 formed a hydrogen bonding interaction with the carbonyl oxygen (C=O) of V8, and (ii) The backbone carbonyl oxygen (C=O) from Glu87 formed a hydrogen bond interaction with -NH- group of V8. Hinge-binding interactions are critically important and are present in majority of ATP-binding site directed Type I protein kinase inhibitors.36 Other notable interaction exists where the sulfonyl oxygen (S=O) in V8 forms an electrostatic interaction with the backbone -NH- of Asp159. Furthermore, the bromophenyl ring forms aromatic-aromatic and hydrophobic interactions respectively with the side chains of Tyr88 and Ile14. Finally, the methyl of the methanesulfonyl group snugly fits in a pocket lined by the T-helix residues such as Leu162, Ala163, and Leu166 (Figure 4A). These non-covalent interactions seen in the energy-minimized inhibitory complex of V8-Nek2 kinase support the experimentally observed inhibitory potency. Although moderately active V8-14 analog preserved two hydrogen bonds with the hinge region and electrostatic interaction of the sulfone group with the backbone -NH- of Asp159 residue, its 3,4-dimethyl substituted phenyl ring formed unfavorable steric clash with the side chains of T-helix residues, Leu162, Ala163, and Leu166 (Figure 4B), thus explaining its moderate activity. Similarly, although the least active V0-3 showed two hydrogen bonds with the hinge residues in addition to the cation-π interaction between the piperidine ring -N- and Phe148, it lacked the key sulfone group and a para-substituted phenyl ring and could not fit into the T-helix to form favorable interactions with the side chain residues of the helix (Figure 4C). Collectively, it is thus inferred that a halogen substituted phenyl ring on imidazolinone scaffold and a spiropiperidine ring with methanesulfonyl moiety are key structural features responsible for Nek2 inhibition.
Figure 4.

Comparison of molecular interactions of compounds V8 (IC50 = 2.4 ± 0.2 μM), V8-14 (IC50 = 12.3 ± 1.2 μM) and V0-3 (IC50 = 58.1 ± 1.0 μM) with the Nek2 active site. [A] Active-site view of the energy minimized complex of inhibitor V8 docked into the ATP-binding pocket of Nek2 kinase (left) and its 2D-interaction map (right). Two key hinge-binding interactions are evident: The carbonyl oxygen (C=O) of V8 formed a hydrogen-bonding interaction with amide backbone NH from Cys89, and NH of V8 formed a hydrogen-bonding interaction with the backbone carbonyl oxygen (C=O) of Glu87 residue. In addition, the sulfonyl oxygen (S=O) of V8 formed an electrostatic interaction with the backbone NH from Asp159 residue. In 3D-interaction maps (left): Dashed red lines indicate hydrogen bonds, the dashed purple line indicates an electrostatic interaction, and the dashed green line indicates a cation-π interaction; ligands are shown as ball and stick model whereas active site residues are shown in stick model. [B] 3D- (left) and 2D- (right) interaction map of the V8-14 with the active site of Nek2 kinase [C] 3D- (left) and 2D- (right) interaction map of the V0-3 with the active site of Nek2 kinase. [D] Legends for 2D-interaction maps.
We have shown that V8 can bind Nek2 kinase in-silico and inhibit its activity in in-vitro kinase assay. We next determined the efficacy of V8 in a biological context in living cells. Our hypothesis was that if V8 was a cell-active inhibitor, it is anticipated to downregulate Nek2-mediated signaling pathways. Using Drosophila melanogaster as an overexpression model system, we previously reported that Nek2 overabundance enhanced cell proliferative signaling via Akt/PI3 kinase pathway in-vivo.14 This finding was also corroborated by Zhou et al.10 We therefore first investigated if treatment of human A549 lung cancer cell line with V8 would mitigate Akt/PI3 kinase signaling. Since phosphorylation of Akt at Ser-473 activates Akt/PI3 kinase signaling pathway, we investigated the effect of V8 on the cellular levels of pAkt (Ser-473).37 Western-blot analysis of V8-treated A549 cells (24 h) resulted in reduced levels of pAkt (Ser-473) in a dose-dependent manner (Figure 5). DMSO, and rac-cct 250863 - a commercially available potent inhibitor of Nek2 (IC50 = 73 nM) served as −ve and +ve controls respectively.30 Since V8 successfully downregulated pAkt/PI3 kinase signaling, we thus anticipated that cellular inhibition of Nek2 kinase by V8 would also lead to enhanced cytotoxicity and reduced cell migration.8, 12, 14 To demonstrate this, highly aggressive human MDA-MB-231 breast cancer and A549 cell lines were chosen.25 To assess cytotoxicity, cell viability was determined (see Supporting Information, Section III) using MTT assay.38 V8-treated cells exhibited reduced cell viability in both human MDA-MB-231 breast cancer (IC50 = 18.1 ± 0.2 μM) and human A549 lung cancer (IC50 = 22.7 ± 0.2 μM) cell lines (Figure 6A). Further, to assess the effect of V8 on cell’s migratory behavior, wound-healing assay (see Supporting Information, Section IV) was performed on human A549 lung cancer cell line.38 V8-treated A549 cells migrated slower than the vehicle control (Figure 6B); this is consistent with Nek2’s role in promoting cell migration.39, 40
Figure 5.

Cellular effect of Nek2 inhibitor, V8, on pAkt levels in the human lung A549 cancer cell line. Western-blot analysis of A549 cells treated with V8. DMSO and a highly potent commercially available Nek2 inhibitor, rac-cct 250863, served as −ve and +ve control respectively. A dose-dependent level of pAkt is observed upon treatment of cells with V8, compared to the control, DMSO. GAPDH served as a loading control for protein normalization.
Figure 6.

[A] Dose-dependent Effect of Nek2 inhibitor, V8, on cell viability in human MDA-MB-231 breast cancer (EC50: 18.1 ± 0.2 μM) and human A549 lung cancer (EC50: 22.7 ± 0.2 μM) cell lines. [B] Assessment of migratory behavior of human A549 lung cancer cells treated with V8. The pair of red lines represents the visual boundary at the initial scratch time (t = 0) while the pair of white lines represent the visual boundary at 24 hr post-treatment. Dose-dependent inhibiton of cell migration is observed in V8-treated A549 cells.
In summary, using HTVS protocol we have identified a novel spirocyclic lead compound, V8, that inhibits the activity of human Nek2 (IC50 = 2.4 ± 0.2 μM). The mode of inhibition is competitive with respect to ATP, reversible, and selective. A modest SAR using structurally-similar V8 analog library validates the inhibitory profile of V8 and suggests that it may be readily amenable to the development of highly potent, selective, and cell-active Nek2 inhibitors. The synthesized truncated 2-ring analogs of V8 (V0-1, V0-2, and V0-3) exhibit significantly lower affinity for Nek2 enzyme, further corroborating the observation that the 3-ring spirocyclic system in V8 is required for effective inhibition. Molecular modeling studies involving the energy-minimized structure of Nek2 inhibitory complexes provide support for the observed inhibition profile. Finally, we show that V8 inhibits pAkt/PI3K signaling pathway and has favorable anti-proliferative and anti-migratory phenotypes in two highly aggressive human MDA-MB-231 breast and human A549 lung cancer cell lines. Future studies involving an expanded and systematic SAR should identify Nek2 inhibitors that are highly potent and selective with desirable cellular profile.
Supplementary Material
Acknowledgement
Research reported in this publication was financially supported by the National Institutes of Health (NIH) under award number 1R15CA243109 to SKP. JYC was supported by NIH SC2 GM130470. SKP also gratefully acknowledges the support of Chemistry and Biochemistry Department and the Office of Dean of Math and Natural Sciences (MNS) of Queens College for their continued support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
T.T.T. is a co-founder of Hysplex, Inc. with interests in PARP inhibitor development. Other authors declare no known competing financial interests or personal relationships.
Appendix A. Supplementary data
Supplementary data to this article can be found online.
References and Notes:
- 1.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2(1): 21–32. [DOI] [PubMed] [Google Scholar]
- 2.Welburn JPI, Jeyaprakash AA. Mechanisms of Mitotic Kinase Regulation: A Structural Perspective. Front Cell Dev Biol. 2018;6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat Rev Genet. 2020;21(1): 44–62. [DOI] [PubMed] [Google Scholar]
- 4.Clyde D. Aneuploidy in the driving seat. Nat Rev Genet. 2021;22(10): 624–625. [DOI] [PubMed] [Google Scholar]
- 5.Gonczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. 2015;15(11): 639–652. [DOI] [PubMed] [Google Scholar]
- 6.Fry AM, Bayliss R, Roig J. Mitotic Regulation by NEK Kinase Networks. Front Cell Dev Biol. 2017;5: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230(1): 6–19. [DOI] [PubMed] [Google Scholar]
- 8.Rivera-Rivera Y, Marina M, Jusino S, et al. The Nek2 centrosome-mitotic kinase contributes to the mesenchymal state, cell invasion, and migration of triple-negative breast cancer cells. Sci Rep. 2021;11(1): 9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayward DG, Fry AM. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006;237(2): 155–166. [DOI] [PubMed] [Google Scholar]
- 10.Zhou W, Yang Y, Xia J, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23(1): 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Zhou W, Xia J, et al. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget. 2014;5(23): 11986–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Chen L, Feng L, et al. NEK2 promotes proliferation, migration and tumor growth of gastric cancer cells via regulating KDM5B/H3K4me3. Am J Cancer Res. 2019;9(11): 2364–2378. [PMC free article] [PubMed] [Google Scholar]
- 13.Meng L, Carpenter K, Mollard A, et al. Inhibition of Nek2 by small molecules affects proteasome activity. Biomed Res Int. 2014;2014: 273180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das TK, Dana D, Paroly SS, et al. Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis. 2013;2: e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang YY, Yen CJ, Chan SH, et al. NEK2 Promotes Hepatoma Metastasis and Serves as Biomarker for High Recurrence Risk after Hepatic Resection. Ann Hepatol. 2018;17(5): 843–856. [DOI] [PubMed] [Google Scholar]
- 16.Dillekas H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019;8(12): 5574–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024): 1559–1564. [DOI] [PubMed] [Google Scholar]
- 18.Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64(20): 7370–7376. [DOI] [PubMed] [Google Scholar]
- 19.Liu H, Liu B, Hou X, et al. Overexpression of NIMA-related kinase 2 is associated with poor prognoses in malignant glioma. J Neurooncol. 2017;132(3): 409–417. [DOI] [PubMed] [Google Scholar]
- 20.Andreasson U, Dictor M, Jerkeman M, et al. Identification of molecular targets associated with transformed diffuse large B cell lymphoma using highly purified tumor cells. Am J Hematol. 2009;84(12): 803–808. [DOI] [PubMed] [Google Scholar]
- 21.Zeng X, Shaikh FY, Harrison MK, et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. 2010;29(36): 5103–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X, Gao Y, Lu Y, Zhang J, Li L, Yin F. Upregulation of NEK2 is associated with drug resistance in ovarian cancer. Oncol Rep. 2014;31(2): 745–754. [DOI] [PubMed] [Google Scholar]
- 23.Fang Y, Zhang X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle. 2016;15(7): 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Gollahon L. Nek2-targeted ASO or siRNA pretreatment enhances anticancer drug sensitivity in triplenegative breast cancer cells. Int J Oncol. 2013;42(3): 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsunoda N, Kokuryo T, Oda K, et al. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100(1): 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki K, Kokuryo T, Senga T, Yokoyama Y, Nagino M, Hamaguchi M. Novel combination treatment for colorectal cancer using Nek2 siRNA and cisplatin. Cancer Sci. 2010;101(5): 1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayward DG, Newbatt Y, Pickard L, et al. Identification by high-throughput screening of viridin analogs as biochemical and cell-based inhibitors of the cell cycle-regulated nek2 kinase. J Biomol Screen. 2010;15(8): 918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuang SH, Lee YE, Huang LYL, et al. Discovery of T-1101 tosylate as a first-in-class clinical candidate for Hec1/Nek2 inhibition in cancer therapy. Eur J Med Chem. 2020;191: 112118. [DOI] [PubMed] [Google Scholar]
- 29.Whelligan DK, Solanki S, Taylor D, et al. Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J Med Chem. 2010;53(21): 7682–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Innocenti P, Cheung KM, Solanki S, et al. Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: structure-activity relationship, structural biology, and cellular activity. J Med Chem. 2012;55(7): 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rellos P, Ivins FJ, Baxter JE, et al. Structure and regulation of the human Nek2 centrosomal kinase. J Biol Chem. 2007;282(9): 6833–6842. [DOI] [PubMed] [Google Scholar]
- 32.Patel PR, Sun H, Li SQ, et al. Identification of potent Yes1 kinase inhibitors using a library screening approach. Bioorg Med Chem Lett. 2013;23(15): 4398–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balzano D, Santaguida S, Musacchio A, Villa F. A general framework for inhibitor resistance in protein kinases. Chem Biol. 2011;18(8): 966–975. [DOI] [PubMed] [Google Scholar]
- 34.Henise JC, Taunton J. Irreversible Nek2 kinase inhibitors with cellular activity. J Med Chem. 2011;54(12): 4133–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Upadhya P, Birkenmeier EH, Birkenmeier CS, Barker JE. Mutations in a NIMA-related kinase gene, Nek1, cause pleiotropic effects including a progressive polycystic kidney disease in mice. Proc Natl Acad Sci U S A. 2000;97(1): 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu Rev Biochem. 2011;80: 769–795. [DOI] [PubMed] [Google Scholar]
- 37.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15(23): 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 38.Pijuan J, Barcelo C, Moreno DF, et al. In vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front Cell Dev Biol. 2019;7: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Wang W, Wang Y, et al. NEK2 promotes hepatocellular carcinoma migration and invasion through modulation of the epithelial-mesenchymal transition. Oncol Rep. 2018;39(3): 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li G, Zhong Y, Shen Q, et al. NEK2 serves as a prognostic biomarker for hepatocellular carcinoma. Int J Oncol. 2017;50(2): 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solanki S, Innocenti P, Mas-Droux C, et al. Benzimidazole inhibitors induce a DFG-out conformation of never in mitosis gene A-related kinase 2 (Nek2) without binding to the back pocket and reveal a nonlinear structure-activity relationship. J Med Chem. 2011;54(6): 1626–1639. [DOI] [PubMed] [Google Scholar]
- 42.Fang Y, Kong Y, Xi J, et al. Preclinical activity of MBM-5 in gastrointestinal cancer by inhibiting NEK2 kinase activity. Oncotarget. 2016;7(48): 79327–79341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Cheng P, Pavlyukov MS, et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J Clin Invest. 2017;127(8): 3075–3089. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Coxon CR, Wong C, Bayliss R, et al. Structure-guided design of purine-based probes for selective Nek2 inhibition. Oncotarget. 2017;8(12): 19089–19124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matheson CJ, Coxon CR, Bayliss R, et al. 2-Arylamino-6-ethynylpurines are cysteine-targeting irreversible inhibitors of Nek2 kinase. RSC Med Chem. 2020;11(6): 707–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.