

Abstract
The diverse structures and profound biological activities of lignan natural products have enticed significant effort in the exploration of new methodologies for their total synthesis. We have prepared γ-butyrolactone oximes from readily available δ-nitro alcohols via Boc2O mediated cyclization. The mild conditions are compatible with a wide range of functional groups, and this methodology has been applied to the total synthesis of five lignan natural products.
Graphical Abstract
Lignan natural products, abundantly found in nature, possess a diverse range of biological activities (Scheme 1a). Hinokinin and cubebin are lignan natural products first isolated from the seeds of the Piper cubeba flower in 19381 but have since been found in a number of various plant species. Hinokinin has shown strong anti-inflammatory activity2 but is most notably recognized for its antitrypanosomal activity,3 with its emerging potential as a Chagas’ disease therapeutic being heavily studied.4 Bicubebin, isolated from the Aristolochia pubescens tubercula, features a unique dilignan carbon skeleton.5 An elegant total synthesis of bicubebin B was previously accomplished in 2017 by Barker et. al through an acid catalyzed dimerization that furnished (−)-bicubebin B in less than 1% overall yield among isomers (−)-bicubebin A and (+)-bicubebin C (Scheme 1b).6 In addition, Aryltetralin lignans such as podophyllotoxin and isodeoxypodophyllotoxin7 are important natural products with diverse biological activities, including anticancer, antiviral, insecticidal, anti-inflammatory, and immunosuppressive properties.8 The natural abundance of aryltetralin derivatives such as podophyllotoxin and its congeners in Podophyllum peltatum is high; thus, nearly all the clinically used podophyllotoxin is obtained by extraction from the natural source. However, if medicinal chemists intend to have a thorough structure–activity relationship study, a de novo synthesis of aryltetraline derivatives is an obvious prerequisite.
Scheme 1.

γ-Butyrolactone Oxime Carbonates for Lignan Natural Product Synthesis
Given the importance of lignan products, particularly the signature motif of polysubstituted butyrolactones, it comes with no surprise that synthetic chemists have invested substantial effort in the development of asymmetric techniques.9 Recent developments in α,β-disubstituted γ-butyrolactone synthesis have focused on carbene, transition metal, and organo-catalyzed reactions to achieve stereo-selective γ-butyrolactone cyclization methods.9a In 2015, Johnson et al. employed chiral N-heterocyclic carbene (NHC) catalysts with racemic α,β-unsaturated aldehydes and βhalo-α-ketoesters to form γ-butyrolactones featuring three stereocenters.10 In 2017, Bhat and colleagues developed an enantioselective method to achieve γ-butyrolactones through a three-stage Knoevenagl condensation/Michael addition/decarboxylative lactonization mechanism using a quinine based amine organocatalyst.11 Their employment of an organocatalyzed Michael addition to achieve high enantioselectivity was inspiring in the development of our methodology. In 2019, Singh et al. developed a one-pot methodology using multi-catalytic steps to form α,β-disubstituted γ-butyrolactones, although it required longer reaction times to achieve moderate yields.12
A relatively unexplored pathway to α,β-disubstituted γ-butyrolactone natural products is through the oxime functional group.13 Due to the two-fold nucleophilic reactivity of their nitrogen and oxygen sites, oximes are valuable building blocks in organic synthesis and versatile scaffolds in the formation of heterocyclic functional groups and derivatizations.14 The variability of the oxygen substituent can be used to selectively attune the reactivity of oximes by forming oxime ethers, esters, and carbonates alike. Previously, Naito et al. formed butyrolactone ester oximes from enaminones by a sulfanyl radical addition reaction.15 The sulfanyl radical initiator successfully allowed them to achieve α,β-substituted oxime scaffolds, previously reserved for carbohydrate oximes, but mechanistically limited their enantiocontrol, leading to a mixture of cis and trans isomers.
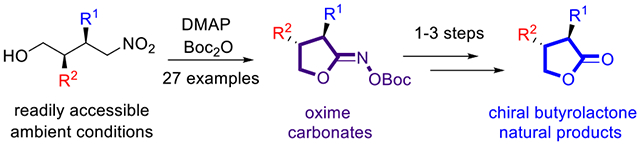
Therefore, we developed a methodology to selectively prepare trans-α,β-substituted butyrolactone O-(Boc) oxime carbonates via activation of δ-nitro alcohol precursors under mild conditions, using cost-effective reagents: 4-dimethylamino pyridine (DMAP) and di-tert-butyl carbonate (Boc2O). In this approach, δ-nitro alcohols can be conveniently obtained by Michael addition of carbonyl α-carbons to nitroalkenes. Through organocatalysis,16 and under ambient conditions, a variety of substituents can be readily incorporated into the nitro alcohols. The methodology was utilized in the concise preparation of α,β-substituted γ-butyrolactones,9d important structural motifs of several lignan natural products including (−)-hinokinin, (−)-bicubebin B, and (−)-isodeoxypodophyl-lotoxin (Scheme 1).
Optimization was performed on δ-nitro alcohol 1a upon which various activators were screened as well as solvents and base additives. Acetic anhydride (Ac2O),17 and methanesulfonyl chloride (MsCl) were ineffective under the tested conditions (Table 1, entries 1 and 2). Switching the activator to ethyl chloroformate (ClCO2C2H5) provided promising results, and the desired product was obtained in 60% yield in 3 h (entry 3). Further optimization led to the identification of Boc2O as a superior activator, which promoted formation of the desired product in 83% isolated yield, with complete consumption of the starting material in 1 h (entry 4). Basic additives, such as triethylamine, had a detrimental impact on the reaction yield and a quick survey of other solvents, such as dichloromethane and tetrahydrofuran, failed to improve the yield as compared with using acetonitrile as the optimal solvent (entries 5–7). We found that the combination of Boc2O and DMAP in MeCN best promoted formation of the desired product.
Table 1.
Optimization Tablea

| ||||
|---|---|---|---|---|
| entry | solvent | activator | time (h) | yield (%)b |
| 1 | MeCN | Ac2O | 24 | N.R. |
| 2 | MeCN | MsCl | 3 | N.R. |
| 3 | MeCN | ClCO2C2H5 | 3 | 60 |
| 4 | MeCN | Boc2O | 1 | 83c |
| 5d | MeCN | Boc2O | 3 | 42 |
| 6 | DCM | Boc2O | 3 | 43 |
| 7 | THF | Boc2O | 2 | 15 |
General procedure unless otherwise stated: substrate 1a (0.2 mmol), DMAP (0.04 mmol), activation group (0.5 mmol), and solvent (1.0 mL) were stirred under nitrogen at rt.
NMR yield detected by using bromo-1,2-methylene dioxybenzene as an internal standard.
Isolated yield.
Et3N additive (0.5 mmol).
With the optimized reaction conditions in hand, we proceeded to interrogate the generality of the methodology (Scheme 2). The mildness of the Boc2O/DMAP combination allowed for broad functional group tolerance for both aldehyde and nitroalkene substrate precursors. To maximize the synthetic potential of the methodology, our cyclization precursors can be used as a crude mixture obtained from a preceding conjugate addition reaction18 without an observable diminution in yields as compared to using purified materials. Aliphatic aldehydes (shown in red) with saturated alkyl groups and those bearing a pendant neutral or electron rich aromatic motif are good substrates for the Michael addition and the subsequent lactone oxime tert-butyl carbonate formation (2a–2c, 2h–2t). Aldehydes with a TBS protecting group also provided the desired lactone oxime carbonate in good yields (2u–2y). A wide variety of nitroalkenes (shown in blue) are also compatible with current cyclization conditions. Phenyl, alkoxyphenyl, halophenyl, heterocycles, and naphthyl are all proper substituents for nitroalkenes. In addition, nitrobutadiene is an excellent Michael acceptor and cyclization precursor, which further provides a vinyl bromide functionality to the butyrolactone oxime carbonates (2o, 2t, 2x, 2aa). It is worth noting that β-nitroenamine is a valid acceptor for conjugate addition19 and oxime carbonate formation, resulting in amino butyrolactone derivatives that could be used for unnatural amino acid synthesis (2p). Butyrolactone oxime carbonate derived from N-toluenesulfonyl indole (2j) demonstrated high yields even on a 1 mmol scale. Single crystal X-ray crystallography analysis of highly crystalline compound 2j also allowed us to unambiguously determine the oxime carbonate geometry to be Z.20
Scheme 2.

Substrate Scopea
aGeneral procedure unless otherwise stated: nitroalkene (0.2 mmol), aldehyde (0.4–0.6 mmol), (R/S)-3 cat. (0.04 mmol), p-nitrophenol (0.04 mmol) in toluene (1 mL) under nitrogen at 0 °C to rt for 2–4 h before addition of MeOH (1 mL) and NaBH4 (1.0 mmol) at 0 °C. Crude 1 (0.2 mmol), DMAP (0.04 mmol), and Boc2O (0.5 mmol) were stirred in MeCN (1 mL) at rt for 1–2 h.
With our butyrolactone oxime carbonates scope established, we briefly investigated the derivatization of this important chemical scaffold (Scheme 3). Complete removal of the oxime moiety from 2a was achieved in a single operation by treatment of HCl in dioxane resulting in butyrolactone 4 (±)-aspergilfuranone A, a cytotoxic lignan first isolated from the Aspergillus fungus in 2021.21 This cleanly demonstrates our methodologies power as a γ-butyrolactone lignan building block. Next, the broader derivatization potential of γ-butyrolactone oximes was explored. Aspergilfuranone A 4 was subjected to Grignard addition and Ru-catalyzed oxidation to afford compound 5 in 63% yield.22 Upon treatment of trifluoroacetic acid in DCM, 2a was neatly converted to lactone oxime 6 in excellent yield. Oxime 6 was oxidized by tert-butyl hypochlorite to generate a nitroso intermediate, which, when reacted with butadiene, furnished protected hetero Diels–Alder adduct 7 in 52% yield, successfully displaying the potential of our substrates in nitroso reactions compared to traditional carbohydrate auxiliaries.23 In addition, lactone oxime derived from 2j underwent cycloaddition with dimethyl acetylene dicarboxylate (DMAD) followed by isomerization to produce compound 8 in 81% yield.24
Scheme 3.

Derivatizations of Lactone Oxime Carbonates
Following the successful derivatization procedures, we turned our attention to the total synthesis of lignan natural products; hinokinin, cubebin, bicubebin, and isodeoxypodo-phyllotoxin, featuring γ-butyrolactone carbon skeletons. For the synthesis of (−)-hinokinin, (−)-cubebin, and (−)-bicubebin B, we started from aldehyde 9, which was prepared from piperonal using a previously established procedure (Scheme 4).25 Henry reaction of 9 under basic conditions, formed nitro alcohol S4 in 91% yield, which was readily dehydrated in the presence of mesylate chloride and triethylamine affording nitroalkene 10 in 91% yield. Asymmetric Michael addition between prepared 11 (derived from cinnamic acid S5) and nitroalkene 10 using catalyst (S)-3 provided optically enriched δ-nitro alcohol 12 in 10:1 dr, >99% ee, and 81% yield. When our lactone oxime carbonate conditions were applied, δ-nitro alcohol 12 was readily converted to oxime carbonate 13 in 64% yield at 0 °C. The reaction was carried out at a lower temperature to avoid interaction of the electron rich aromatics with the activated nitro group. Removal of the Boc oxime carbonyl was accomplished in a single step by treatment with HCl in dioxane to furnish natural product (−)-hinokinin 14 in 87% yield. Hinokinin 14 was directly reduced to (−)-cubebin 15 in nearly quantitative yield in the presence of DIBALH in toluene at −78 °C.
Scheme 4.

Synthesis of (−)-Hinokinin, (−)-Cubebin, and (−)-Bicubebin
To access dilignan natural product (−)-bicubebin B, we sought to apply a glycosylation approach using anomeric acetate 16 as a glycosyl donor and 15 as a glycosyl acceptor. Indeed, the glycosylation proceeded cleanly to afford (−)-bicubebin B 17 in 61% yield using Lewis acid SnCl4 at −78 °C. To our great pleasure, the major stereoisomer was 17 and only trace amounts of other isomers were observed. The major byproduct was the C-aryl glycoside isocubebinic ether 18 generated via intramolecular cyclization in 20% yield.6
The synthesis of (−)-isodeoxypodophyllotoxin26 commenced in a similar fashion with an enantioselective Michael addition of aldehyde 11 to trimethoxy nitroalkene 19 (derived from S8) under organocatalysis (Scheme 5). The resulting aldehyde was reduced by sodium borohydride to afford δ-nitro alcohol 20 in 67% yield, 9:1 dr and 90% ee. Alcohol 20 was then subjected to our cyclization conditions, and oxime carbonate 21 was obtained in 57% yield. To avoid the undesired reactivity from highly nucleophilic aromatics, the removal of the oxime carbonate group was achieved in two steps in excellent yield. The final oxidative cyclization was accomplished by using DDQQ in trifluoroacetic acid27 to isolate (−)-isodeoxypodophyllotoxin 23 in 56% yield.
Scheme 5.

Synthesis of (−)-Isodeoxypodophyllotoxin
In summary, we have developed a practical approach to access trans-α,β-substituted butyrolactone oximes from readily available δ-nitro alcohols via Boc2O mediated cyclization under mild conditions. The mildness of the method and ease of access to various δ-nitro alcohols allow expedient access to a wide variety of polysubstituted butyrolactones, an important motif in many bioactive natural products. By applying the methodology, we successful prepared a series of natural lignans including (±)-aspergilfuranone A, (−)-hinokinin, (−)-cubebin, (−)-bicubebin B, and (−)-isodeoxypodophyllotoxin. We expect our Michael addition–lactone oxime formation will find broader applications in natural product synthesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institute of General Medical Sciences (1R15GM12068501) and the University of Central Florida for their financial support. We thank Dr. David Richardson for NMR assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c03727.
General information, crystal data of 2j, optimization details, procedures, characterization data, 1H and 13C NMR spectra (PDF)
Accession Codes
CCDC 2209687 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223 336033 The Supporting Information is available free of charge at.
The authors declare no competing financial interest.
Contributor Information
Katelyn B. Bobek, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States
Nameer S. Ezzat, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States; Department of Chemistry, University of Mosul, Mosul 41002, Iraq
Brandon S. Jones, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States
Yujia Bian, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States.
Thomas E. Shaw, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States
Titel Jurca, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States.
Hongya Li, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States; College of Life Sciences, Hebei Agricultural University, Baoding, Hebei 071000, P.R. China.
Yu Yuan, Department of Chemistry, University of Central Florida, Orlando, Florida 32816, United States.
Data Availability Statement
The data underlying this study is available in the published article and its online Supporting Information.
REFERENCES
- (1).Haworth RD; Woodcock D The constituents of natural phenolic resins. Part xiii. The synthesis of dl-, d-, and l-hinokinin. J. Chem. Soc 1938, 1985–1989. [Google Scholar]
- (2).(a) da Silva R; de Souza GHB; da Silva AA; de Souza VA, ; Pereira AC; Royo VD; Silva M; Donate PM; Araujo A; Carvalho JCT; Bastos JK Synthesis and biological activity evaluation of lignan lactones derived from (−)-cubebin. Bioorg. Med. Chem. Lett 2005, 15, 1033–1037. [DOI] [PubMed] [Google Scholar]; (b) Lima TC; Lucarini R; Volpe AC; de Andrade CQJ; Souza AMP; Pauletti PM; Januario AH; Simaro GV; Bastos JK; Cunha WR; Borges A; Laurentiz RD; Conforti A; Parreira RLT; Borges CHG; Caramori GF; Andriani KF; Silva M In vivo and in silico anti-inflammatory mechanism of action of the semisynthetic (−)-cubebin derivatives (−)-hinokinin and (−)-o-benzylcubebin. Bioorg. Med. Chem. Lett 2017, 27, 176–179. [DOI] [PubMed] [Google Scholar]
- (3).(a) de Souza VA; da Silva R; Pereira AC; Royo VD; Saraiva J; Montanheiro M; de Souza GHB; da Silva AA; Grando MD; Donate PM; Bastos JK; Albuquerque S; Silva M Trypanocidal activity of (−)-cubebin derivatives against free amastigote forms of trypanosoma cruzi. Bioorg. Med. Chem. Lett 2005, 15, 303–307. [DOI] [PubMed] [Google Scholar]; (b) Saraiva J; Vega C; Rolon M; da Silva R; Silva M; Donate PM; Bastos JK; Gomez-Barrio A; de Albuquerque S In vitro and in vivo activity of lignan lactones derivatives against trypanosoma cruzi. Parasitol. Res 2007, 100, 791–795. [DOI] [PubMed] [Google Scholar]; (c) Jackson Y; Wyssa B; Chappuis F Tolerance to nifurtimox and benznidazole in adult patients with chronic chagas’ disease. J. Antimicrob. Chemother 2020, 75, 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Marcotullio MC; Pelosi A; Curini M Hinokinin, an emerging bioactive lignan. Molecules 2014, 19, 14862–14878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).de Pascoli IC; Nascimento IR; Lopes LMX Configurational analysis of cubebins and bicubebin from aristolochia lagesiana and aristolochia pubescens. Phytochemistry 2006, 67, 735–742. [DOI] [PubMed] [Google Scholar]
- (6).Davidson SJ; Pearce AN; Copp BR; Barker D Total synthesis of (−)-bicubebin a, b, (+)-bicubebin c and structural reassignment of (−)-cis-cubebin. Org. Lett 2017, 19, 5368–5371. [DOI] [PubMed] [Google Scholar]
- (7).Novelo M; Cruz JG; Hernandez L; Peredmiranda R; Chai HY; Mar W; Pezzuto JM Cytotoxic constituents from hyptisverticillata. 6. Chemical studies on mexican hyptis species and. 2. Biologically-active natural-products from mexican medicinal-plants. J. Nat. Prod 1993, 56, 1728–1736.8277312 [Google Scholar]
- (8).(a) Ardalani H; Avan A; Ghayour-Mobarhan M Podophyllotoxin: A novel potential natural anticancer agent. Avicenna J. Phytomed 2017, 7, 285–294. [PMC free article] [PubMed] [Google Scholar]; (b) Yu X; Che ZP; Xu H Recent advances in the chemistry and biology of podophyllotoxins. Chem. -Eur. J 2017, 23, 4467–4526. [DOI] [PubMed] [Google Scholar]
- (9).(a) Hur J; Jang J; Sim J A review of the pharmacological activities and recent synthetic advances of gamma-butyrolactones. Int. J. Mol. Sci 2021, 22, 2769. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mao B; Fananas-Mastral M; Feringa BL Catalytic asymmetric synthesis of butenolides and butyrolactones. Chem. Rev 2017, 117, 10502–10566. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Murauski KJR; Jaworski AA; Scheidt KA A continuing challenge: N-heterocyclic carbene-catalyzed syntheses of gamma-butyrolactones. Chem. Soc. Rev 2018, 47, 1773–1782. [DOI] [PubMed] [Google Scholar]; (d) Seitz M; Reiser O Synthetic approaches towards structurally diverse gamma-butyrolactone natural-product-like compounds. Curr. Opin. Chem. Biol 2005, 9, 285–292. [DOI] [PubMed] [Google Scholar]
- (10).Goodman CG; Walker MM; Johnson JS Enantioconvergent synthesis of functionalized gamma-butyrolactones via (3 + 2)-annulation. J. Am. Chem. Soc 2015, 137, 122–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Khopade TM; Sonawane AD; Arora JS; Bhat RG Direct organocatalytic multicomponent synthesis of enantiopure γ-butyrolactones via tandem knoevenagel-michael-lactonization sequence. Advanced Synthesis & Catalysis 2017, 359, 3905–3910. [Google Scholar]
- (12).Mahto P; Rana NK; Shukla K; Das BG; Joshi H; Singh VK Asymmetric multifunctional modular organocatalysis: One-pot direct strategy to enantiopure alpha,beta-disubstituted gamma-butyrolactones. Org. Lett 2019, 21, 5962–5966. [DOI] [PubMed] [Google Scholar]
- (13).(a) Degnan AP; Meyers AI Enantioselective synthesis of rigid 2-aminotetralins. Utility of silicon as an oxygen and nitrogen surrogate in the tandem addition reaction. J. Org. Chem 2000, 65, 3503–3512. [DOI] [PubMed] [Google Scholar]; (b) For the cyclization of enol see: Ansell GB; Moore DW; Nielsen AT Formation and crystal structure of 3-(4-bromophenyl)-2-hydroxyimino-6,7-dihydro-4(5h)-benzofuranone - michael addition of cyclohexane-1,3-dione to 4-bromo-omega-nitrostyrene. J. Chem. Soc. D 1970, 0, 1602–1603. [Google Scholar]; (c) Dominianni SJ; Chaney MO; Jones ND Base catalyzed condensation of dimedone with β-nitrostyrene. Tetrahedron Lett. 1970, 11, 4735–4736. [Google Scholar]; (d) Ansell GB; Moore DW; Nielsen AT Intramolecular reactions of nitro-olef in-cyclohexane-1.3-dione michael adducts. Crystal structure of 3- (4- b romophenyl)-6,7-di hydro-2- hyd roxyiminobenzofuran-4(5h)-one. J. Chem. Soc. B 1971, 0, 2376–2382. [Google Scholar]; (e) Gomezsanchez A; Galan J; Rico M; Bellanato J Studies on sugar nitro-olefins. Part 6.’ Synthesis of (3/?)-3,5,6,7-tetrahydro2-hydroxyimino-3-(penta-o-acetylpentitol-i -yl)benzofuran-4(2h)-ones from 3,4,5,6,7-penta-o-acetyl-l,2-dideoxy-l -nitrohept-i -enitols and cyclohexane1,3-diones. J. Chem. Soc., Perkin Trans 1 1985, 2695–2700. [Google Scholar]
- (14).Rykaczewski KA; Wearing ER; Blackmun DE; Schindler CS Reactivity of oximes for diverse methodologies and synthetic applications. Nature Synthesis 2022, 1, 24–36. [Google Scholar]
- (15).Miyata O; Nishiguchi A; Ninomiya I; Aoe K; Okamura K; Naito T Radical cyclization in heterocycle synthesis. 11. A novel synthesis of alpha,beta-disubstituted gamma-lactones via sulfanyl radical addition-cyclization using hydroximates as a tether. J. Org. Chem 2000, 65, 6922–6931. [DOI] [PubMed] [Google Scholar]
- (16).Phillips AMF Organocatalytic asymmetric nitro-michael reactions. Curr. Org. Synth 2016, 13, 687–725. [Google Scholar]
- (17).Berrocal MV; Gil MV; Roman E; Serrano JA Reactions of hydroxylated sodium nitronates with acetic anhydride/pyridine. Tetrahedron 2002, 58, 5327–5333. [Google Scholar]
- (18).Hayashi Y; Gotoh H; Hayashi T; Shoji M Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric michael reaction of aldehydes and nitroalkenes. Angew. Chem., Int. Ed 2005, 44, 4212–4215. [DOI] [PubMed] [Google Scholar]
- (19).Zhu SL; Yu SY; Wang Y; Ma DW Organocatalytic michael addition of aldehydes to protected 2-amino-1-nitroethenes: The practical syntheses of oseltamivir (tamiflu) and substituted 3-aminopyrrolidines. Angew. Chem., Int. Ed 2010, 49, 4656–4660. [DOI] [PubMed] [Google Scholar]
- (20).X-ray crystallography data are deposited at CCDC 2209687.
- (21).Wang G-K; Li Y; Liu H-T; Su J-L; Luo L; Zhu J-Y; Wang K; Liu J-S Aspergilfuranones a-d, four norlignanolides from the peucedanum praeruptorum endophytic fungus aspergillus udagawae. Tetrahedron 2021, 82, 131951. [Google Scholar]
- (22).Linz G; Weetman J; Abdelhady AF; Helmchen G Asymmetric diels-alder reactions - epc-synthesis of a stable sarkomycin precursor (cyclosarkomycin). Tetrahedron Lett. 1989, 30, 5599–5602. [Google Scholar]
- (23).Defoin A; Joubert M; Heuchel JM; Strehler C; Streith J Enantioselective diels-alder reaction with an alpha-chloronitroso dienophile derived from 5-o-acetyl-2,3-isopropylidenedioxy-d-ribose. Synthesis 2000, 2000, 1719–1726. [Google Scholar]
- (24).Yokoyama M; Yamada N Synthesis of spiro and bicyclic nucleosides from ribose nitrile oxide with dimethyl acetylenedicarboxylate. Tetrahedron Lett. 1989, 30, 3675–3676. [Google Scholar]
- (25).(a) Sasai H; Suzuki T; Arai S; Arai T; Shibasaki M Basic character of rare-earth-metal alkoxides - utilization in catalytic c-c bond-forming reactions and catalytic asymmetric nitroaldol reactions. J. Am. Chem. Soc 1992, 114, 4418–4420. [Google Scholar]; (b) Davies SG; Goddard DC; Roberts PM; Russell AJ; Smith AD; Thomson JE; Withey JM Strategies for the construction of morphinan alkaloid ab-rings: Regioselective friedel-crafts-type cyclisations of gamma-aryl-beta-benzoylamido acids with asymmetrically substituted gamma-aryl rings. Tetrahedron: Asymmetry 2016, 27, 274–284. [Google Scholar]
- (26).Brown E; Daugan A An easy preparation of (−) and (+)-beta-piperonyl-gamma-butyrolactones, key-intermediates for the synthesis of optically-active lignans. Tetrahedron Lett. 1985, 26, 3997–3998. [Google Scholar]
- (27).(a) Planchenault D; Dhal R; Robin JP Synthesis of nonphenolic bisbenzocyclooctadiene lignan lactones and aporphinic alkaloids, by oxidative coupling with new agents in fluoro acid-medium. 4. Tetrahedron 1993, 49, 5823–5830. [Google Scholar]; (b) Venkateswarlu R; Kamakshi C; Moinuddin SGA; Subhash PV; Ward RS; Pelter A; Hursthouse MB; Light ME Transformations of lignans, part v. Reactions of ddq with a gmelinol hydrogenolysis product and its derivatives. Tetrahedron 1999, 55, 13087–13108. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study is available in the published article and its online Supporting Information.