

ABSTRACT
Dysbiosis of gut microbial community is associated with the pathogenesis of CD and may serve as a promising noninvasive diagnostic tool. We aimed to compare the performances of the microbial markers of different biological levels by conducting a multidimensional analysis on the microbial metagenomes of CD. We collected fecal metagenomic datasets generated from eight cohorts that altogether include 870 CD patients and 548 healthy controls. Microbial alterations in CD patients were assessed at multidimensional levels including species, gene, and SNV level, and then diagnostic models were constructed using artificial intelligence algorithm. A total of 227 species, 1047 microbial genes, and 21,877 microbial SNVs were identified that differed between CD and controls. The species, gene, and SNV models achieved an average AUC of 0.97, 0.95, and 0.77, respectively. Notably, the gene model exhibited superior diagnostic capability, achieving an average AUC of 0.89 and 0.91 for internal and external validations, respectively. Moreover, the gene model was specific for CD against other microbiome-related diseases. Furthermore, we found that phosphotransferase system (PTS) contributed substantially to the diagnostic capability of the gene model. The outstanding performance of PTS was mainly explained by genes celB and manY, which demonstrated high predictabilities for CD with metagenomic datasets and was validated in an independent cohort by qRT-PCR analysis. Our global metagenomic analysis unravels the multidimensional alterations of the microbial communities in CD and identifies microbial genes as robust diagnostic biomarkers across geographically and culturally distinct cohorts.
KEYWORDS: Crohn’s disease, microbiome biomarkers, noninvasive diagnosis, artificial intelligence, phosphotransferase system
Introduction
Crohn’s disease (CD), one of the two main forms of inflammatory bowel disease (IBD), is characterized by skip lesions and transmural inflammation of the gastrointestinal tract. The incidence of CD has risen globally in the past two decades, causing substantial economic burdens for patients and society.1,2 Currently, diagnosis of CD is mainly based on the combined evaluation of endoscopic, radiographic, and pathological findings.3,4 However, the diagnostic power of endoscopy is often limited by patient compliance, bowel preparation quality, and other uncontrollable factors.5 Therefore, a sensitive, specific, and convenient noninvasive diagnostic tool for CD is urgently needed.
Serological and fecal biomarkers, such as C-reactive protein and fecal calprotectin, have been used as indicators to evaluate inflammatory activity in IBD.6,7 However, the accuracy and specificity of these biomarkers are not satisfactory. Recently, the diagnostic potential of microbial signatures has emerged as potential diagnostic markers for IBD.8–12 For instance, Pascal et al. constructed a diagnostic model using microbial species abundance and achieved a sensitivity of 81.8% for CD.11 Similarly, Franzosa et al. reported a model that achieved an area under the ROC curve (AUC) of 0.92.12 Along this line, future efforts are needed to conduct similar analyses that incorporate multiple cohorts of distinct cultural and geographical backgrounds to identify markers of universal value.
Notably, species abundance may not be an accurate representation of the microbial functions as reflected by the fact that the nomenclatures of many gut microbial species are currently and constantly being adjusted. In this regard, the diagnostic value of microbial genes and their polymorphisms has become a popular subject of investigation13–16 (Figure 1b). For example, microbial functional genes outperformed microbial species in distinguishing CRC from controls14. Similarly, a recent study demonstrated high accuracy of microbial SNVs for diagnosing CD17. Currently, an integrated investigation on multidimensional signatures of CD at species, gene, and SNV levels is lacking and seems to be warranted in the clinic.
Figure 1.
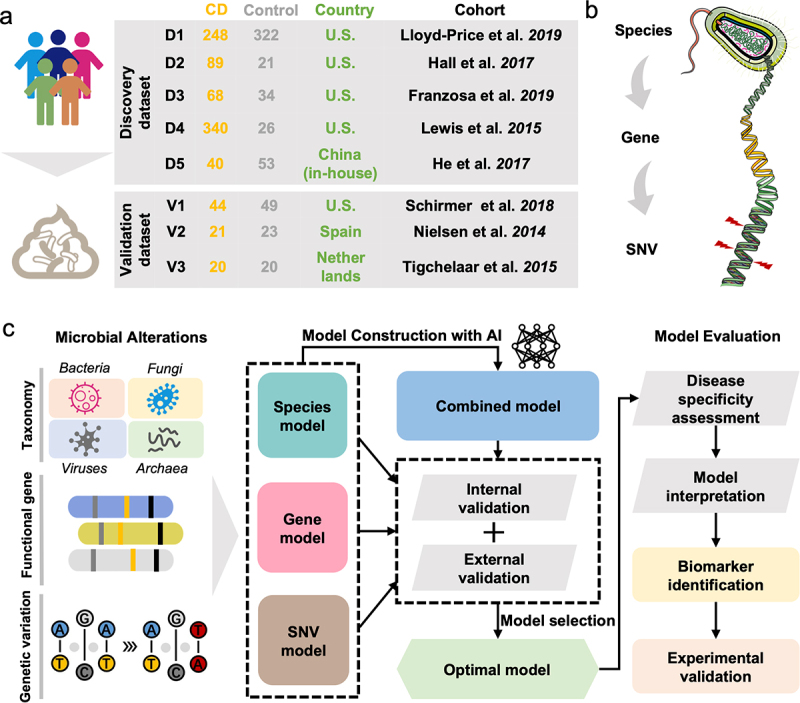
Overview of the fecal samples included in this study and the analysis protocol. (a) We collected a total of 1418 samples from eight cohorts with fecal shotgun metagenomic data. The discovery dataset included PRJNA398089 (D1), PRJNA385949 (D2), PRJNA400072 (PRISM) (D3), SRP057027 (D4) and PRJEB15371 (D5). The validation dataset included PRJNA389280 (V1), PRJEB1220 (V2) and PRJNA400072 (LifeLines DEEP and NLIBD) (V3). (b) Three levels of analysis were conducted in this study: species, gene, and microbial SNV levels. (c) the overall workflow of the study: Firstly, the microbial alterations were identified to retrieve the differential multidimensional signatures of gut microbiome. Subsequently, diagnostic models were constructed and the optimal model was selected according to the performances of the models in internal and external validations. Finally, disease specificity of model was evaluated and model interpretation was conducted for final determination of the microbial biomarker, and then biomarkers were validated by qRT-PCR analysis.
In this study, using large numbers of whole-metagenome sequencing (WMS) samples from multiple cohorts, we constructed diagnostic models for CD and systematically assessed the predictabilities of multidimensional signatures. Candidate biomarkers for CD diagnosis were identified and further validated by qRT-PCR with an independent cohort. Collectively, these results uncover multidimensional alterations of microbial communities in CD patients and provide universal and robust biomarkers for CD diagnosis.
Results
Characterization of multicohort WMS data and study design
In this study, we collected eight fecal shotgun metagenomics datasets from published studies to characterize the gut microbiome in CD patients compared to that in healthy controls (Figure 1a). Patients with a non-inflamed status and those treated with antibiotics were excluded. In total, we included 785 samples from CD patients and 456 healthy control samples across geographically distinct regions from the U.S.A. and China as the discovery dataset. In addition, 85 CD samples and 92 controls from three independent cohorts from the U.S.A., Spain, and the Netherlands were included as the validation dataset. The overall protocol for this study (Figure 1c) was based on the workflow of a previous study18 with modifications.
Multidimensional alterations in gut microbial profiles in CD patients
First, the effects of major confounders were assessed to be not significant (Figure S1), and differential signatures were identified at the species, gene, and SNV levels after adjusting for batch effects using the MMUPHin approach. At the species level, we found that alpha and beta diversities were significantly different between CD patients and controls (Figure 2a-b). A total of 80 bacterial species were identified with significantly different abundances between CD and control, such as Escherichia coli, Flavonifractor plautii, Klebsiella pneumoniae, and Bacteroides intestinalis. (Figure 2c ; Supplementary Table S5). Besides, 147 non-bacterial species including 70 fungi, 42 viruses, and 35 archaea exhibited differential abundances between CD and controls, such as Aspergillus rambellii, Capronia epimyces, Bacteroides phage B124–14, Klebsiella virus KpV80, and DPANN group archaeon LC1Nh (Figure S2 and Supplementary Table S5). Further, we investigated the differences in microbial interactions between CD and controls by performing co-abundance analysis via SparCC. Interestingly, interactions among intra-kingdom species were more frequently observed in the network of CD, compared to the network of controls (Figure S3), indicating large-scale alterations in the structure and function of the gut microbiome in CD.
Figure 2.
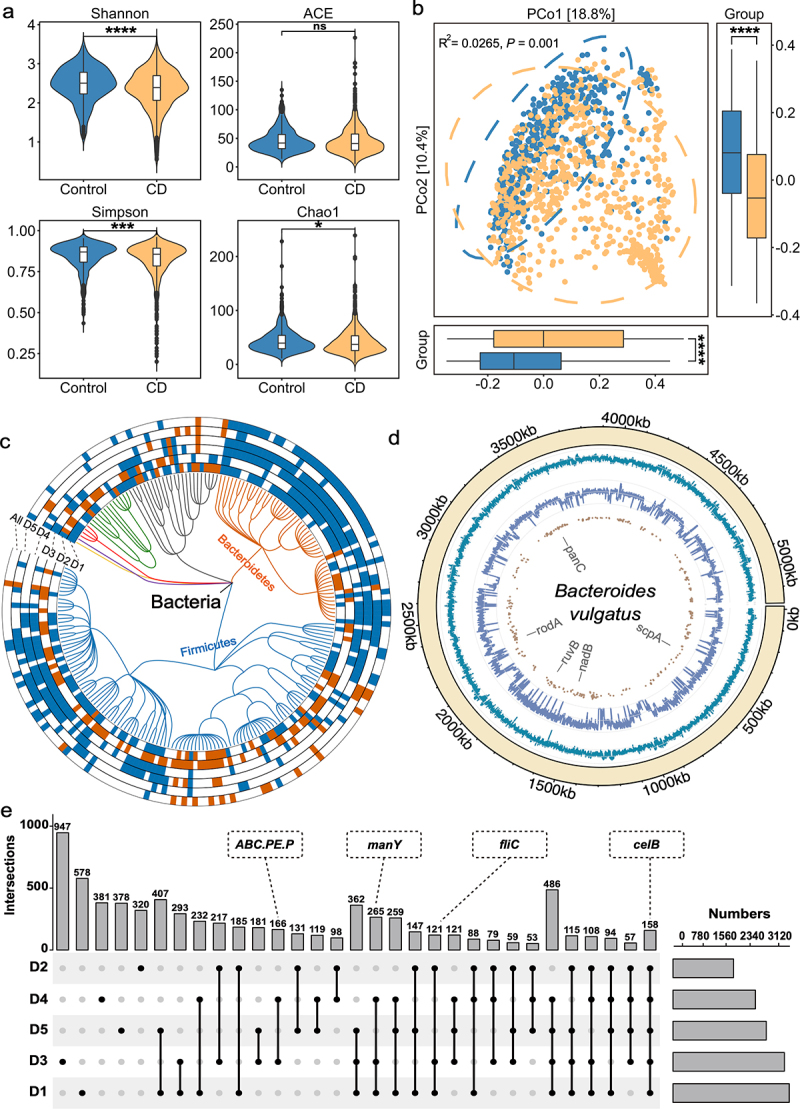
Multidimensional alterations in the gut microbiome of CD patients at species-, gene- and SNV-levels. (a) Alpha diversity measured by Shannon, ACE, Simpson and Chao1 index of patients with CD (orange, n = 785) and control individuals (blue, n = 456); *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. (b) Principal coordinate analysis (PCoA) of samples from all five cohorts based on Bray–Curtis distance, which shows that microbial compositions were different between groups (R2 = 0.0265, P = 0.001). P values were calculated by 999 permutations (two-sided test). (c) Phylogenetic tree showing the differential bacteria species, grouped by the phyla. The differential species in each dataset are shown in each circle ‘D1-D5’ (P < 0.05, two-sided test); the meta-analysis results in integrated dataset were marked by ‘All’. Increased and decreased abundances are indicated by red and blue, respectively. (d) the chord diagram shows the distributions of annotated SNVs in Bacteroides vulgatus genome. The outer circle represents the genome of B. vulgatus; the inner circles represent the GC-content (cyan indigo lines), sequencing depth (purple lines) and sites of differential SNVs (brown points) in the genome, respectively. (e) UpSet plot showing the number of differential KO genes identified via MaAsLin2 in each dataset and those shared by the datasets. The number above each column represents the intersection size of differential KO genes. The connected dots represent the common differential genes across connected cohorts. The set size on the right represents the number of differential genes in each cohort.
Next, we assessed the microbial alterations at the KEGG Orthology (KO) gene level and identified 497 genes with increased abundance and 1043 genes with decreased abundance in CD patients, such as the genes encoding cellobiose PTS system EIIC component (celB), mannose PTS system EIIC component (manY), flagellin (fliC) and peptide/nickel transport system permease protein (ABC.PE.P) (Figure 2e; Supplementary Table S6). For better understanding of these differential KO genes, we performed gene set enrichment analysis (GSEA). As a result, 59 enriched pathways were identified, including 18 pathways with increased abundances and 41 with decreased abundances in CD patients (Figure S4a and Supplementary Table S7). In detail, propanoate metabolism, quorum sensing, phosphotransferase system (PTS), and purine metabolism exhibited increased abundances in CD, while biosynthesis of secondary metabolites, pantothenate, and CoA biosynthesis exhibited decreased abundances in CD.
For microbial SNV-level analysis, a total of seven commonly observed species that have sufficient coverage were annotated (see in Method section), with the number of SNVs ranging from 74 with Bacteroides rodentium to 99,305 with Bacteroides vulgatus (Figure S4b and Figure S5). In total, 21,877 differential SNVs were identified in the seven annotated species (Figure S4c). For instance, Bacteroides vulgatus, belonging to the most commonly encountered Bacteroides species in the human colon, had 11,134 significantly differential SNVs that were located on genes, such as panC, rodA, and ruvB (Figure 2d ; Supplementary Table S8). These differential SNVs are potential candidates of risk factors mediating abnormal gene functions. Collectively, we systemically assessed the multidimensional microbial alterations in CD patients compared to controls, and identified differential signatures for further diagnostic model construction.
Diagnostic models for CD based on microbial multidimensional signatures
Based on all differential signatures at species, gene, and microbial SNV levels, we constructed models using feedforward neural network (FNN) algorithm. At the species level, we first evaluated the capability of single-kingdom species for distinguishing CD from controls. The average AUCs of the cross-validation based on fungal, viral, and archaeal signatures were 0.89, 0.81, and 0.76, respectively. Compared to non-bacterial species, bacterial species demonstrated a better performance in ten-fold cross-validation (average AUC = 0.94) (Figure S6a-d). Furthermore, we merged single-kingdom signatures together, and found that the species model based on multi-kingdom signatures had higher diagnostic accuracy with an average AUC of 0.97 (Figure 3a ; Figure S6e). Interestingly, we noticed that several fungal species, including A. rambellii and A. ochraceoroseus, were top-ranking features of the model with high SHAP values, suggesting their largely alterations in CD patients and may be associated with CD pathogenesis (Figure S7a and Supplementary Table S9).
Figure 3.
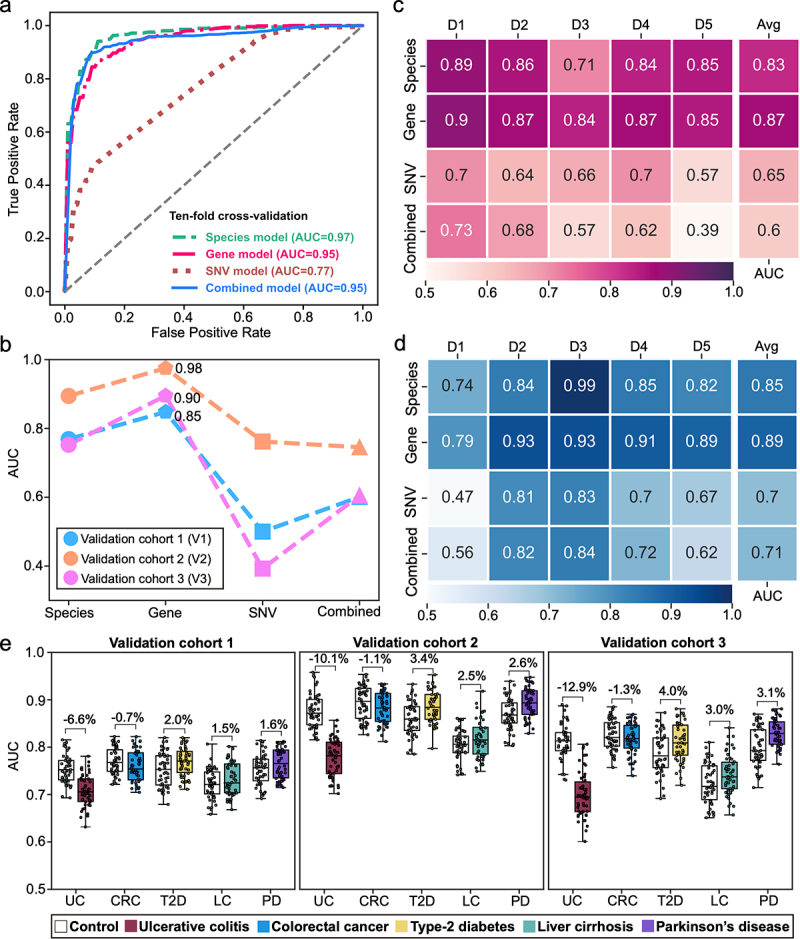
The performance of diagnostic models constructed with multidimensional signatures. (a) the ROC curves from ten-fold cross-validation of species, gene, SNV, and combined diagnostic models. (b) the AUCs of species, gene, SNV, and combined diagnostic models in external validation dataset. (c) the AUCs of each model in cohort-to-cohort validation. Each number represents the average AUC of validation with the cohort specified by its column tag as the training cohort, and all other cohorts as the validation cohorts. (d) the AUC of each model in LOCO validation. Each number represents the resulting AUC of validation with the cohort specified by its column tag as the validation cohort while the other cohorts combined as training cohort. (e) Prediction performances as AUC values on the validation cohorts when adding an external set of control and case samples from non-CD disease cohorts (ulcerative colitis (UC), colorectal cancer (CRC), type-2 diabetes (T2D), liver cirrhosis (LC) and Parkinson’s disease (PD)). Gray and colored bars are the AUCs after adding control and case samples from the non-CD disease cohorts, respectively.
Subsequently, we constructed a diagnostic model using all of the 1047 differential KO genes. The gene model achieved an average AUC of 0.95 in 10-fold cross-validation, slightly lower than that of the multi-kingdom species model (Figure 3a). From feature importance evaluation, we found that CDP-abequose synthase (rfbJ), type VI secretion system protein ImpB (impB), nitrite reductase (NO-forming) (nirK), and celB were the most important KO genes with SHAP values ranged from 0.006 to 0.008 (Supplementary Table S10). Notably, the KO gene celB was found to be significantly increased in CD patients of each dataset (Figure S7b), suggesting an outstanding contribution of celB gene to the diagnostic power of the model.
Furthermore, we explored the diagnostic potentials of microbial SNVs. The SNV model achieved an average AUC of 0.77 in cross-validation (Figure 3a). The most important SNVs were mainly from Bacteroides species including B. ovatus, B. vulgatus, and B. uniformis (Figure S8a and Supplementary Table S11). As the most widely colonized microbes in the gut, Bacteroides species contributes to the major diagnostic power of the SNV model in our results.
Finally, we constructed a model with the combination of species, gene, and SNV signatures (Figure S9d). The combined model achieved an average AUC of 0.95. Interestingly, the performance of combined model was not significantly improved compared to species and gene models, and most of the top-ranking features were from KO genes (Figure 3a ; Figure S8b). These results suggest that the gene signatures are the most powerful biomarkers for CD. All the evaluation metrics of our diagnostic models in model training and validation are provided in Supplementary Table S12 and Figure S10.
Gene model achieves superior robustness and generalization
To assess the robustness and generalization of species, gene, SNV, and combined models, we performed internal and external validations. With the internal validation cohorts, the gene model achieved the highest average AUCs of 0.87 and 0.89 in cohort-to-cohort transfer and leave-one-cohort-out (LOCO) validation, respectively (Figure 3c-d), compared to other diagnostic models at species and SNV levels (Figure S11a-g). In external validation, the gene model also exhibited the best performance, with an average AUC of 0.91 in three independent cohorts (Figure 3b). Taken together, the gene model demonstrated superior diagnostic capability compared to the species, SNV models and even the combined model.
Gene model is highly specific for CD
To ascertain the discriminative power of the gene model, that is, the model is specific for CD but not other microbiome-related diseases, we chose five microbiome-related diseases including UC, CRC, PD, T2D, and LC to evaluate the disease specificity of the gene model. Adding UC samples to three independent validation cohorts decreased the AUC by 6.6%, 10.1%, and 12.9%, respectively (Figure 3e). These changes were not significant considering the baseline values of the altered AUCs when adding CD samples to the validation dataset (decreased AUCs by 10.7%, 17.5%, and 20.5%, respectively, Figure S11h). With CRC cohort, slight and insignificant changes of AUCs in validation (decreased by 0.7%, 1.1%, and 1.3%, respectively) were observed. Similarly, slight and insignificant changes of AUC were observed in validations with T2D (increased by 2.0%, 3.4%, and 4.0%, respectively), liver cirrhosis (increased by 1.5%, 2.5%, and 3.0%, respectively), and PD (increased by 1.6%, 2.6%, and 3.1%, respectively). Altogether, the slight changes in AUCs suggest limited effects of the samples with non-CD diseases on the CD model, indicating that our diagnostic model is specific for CD.
Outstanding contributions of phosphotransferase system to the diagnostic capability of the gene model
To evaluate the respective contributions of each gene set and of key gene feature in the gene model, the KO gene features were grouped by gene set, and the importance of each gene set was evaluated as described in Methods section. Relative to the baseline AUC of 0.91, the abundance disturbance of the gene sets including quorum sensing, PTS, and ABC transporters caused the greatest decrease of AUC in the predictive model by 1.09% to 1.70% (Figure 4a). Further, we performed recursive feature elimination using gene sets and reconstructed diagnostic models. We found that the AUC of cross-validation did not decrease significantly until the glycerolipid metabolism gene set was eliminated, which confirmed the important contribution of quorum sensing, PTS, ABC transporters, fructose and mannose metabolism, and glycerolipid metabolism to the diagnostic model (Figure S12a). To further strengthen these results, we constructed a sub-model with genes of these five gene sets, which achieved an AUC of 0.89 in cross-validation (Figure S12b). The sub-model displayed decent robustness in internal validations and achieved an average AUC of 0.81 in external validation (Figure S12c). Notably, we found that celB was the most important feature of the sub-model (Supplementary Table S13). These results suggest that the above-identified gene sets are key contributors to the diagnostic capabilities of the gene model.
Figure 4.
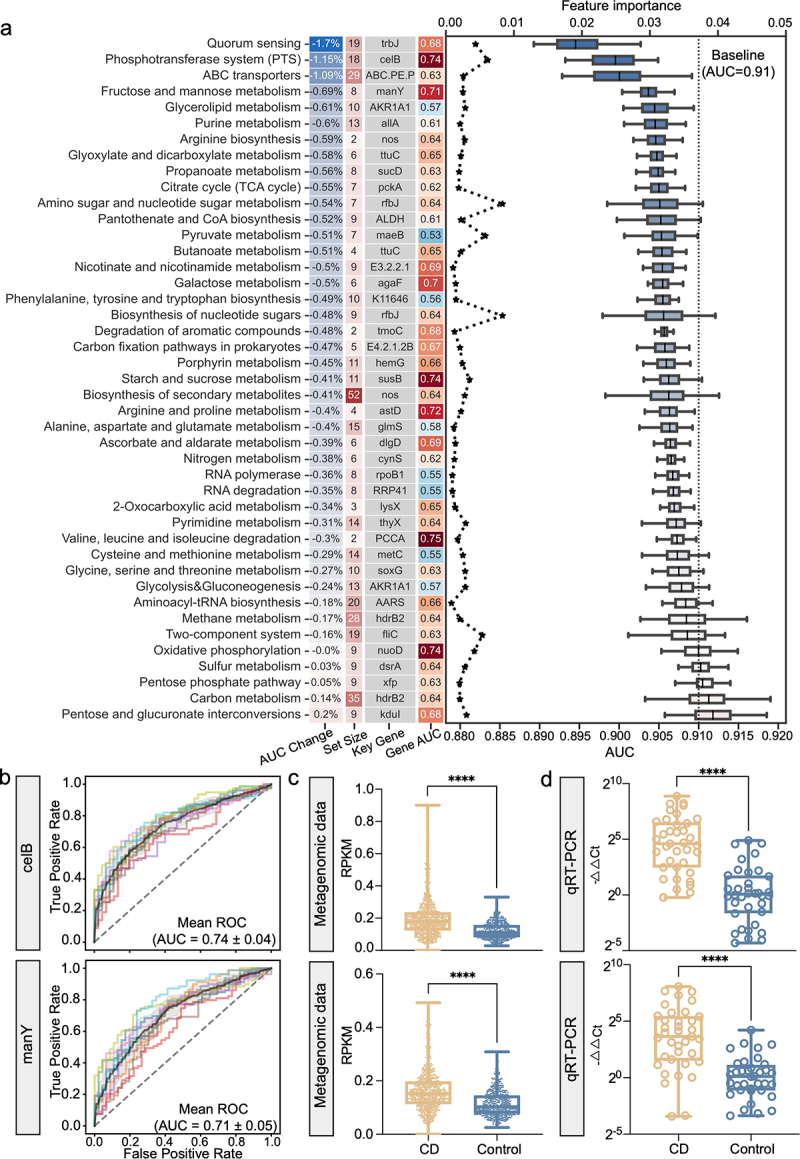
The model interpretation of the gene model. (a) the left column lists the average percent change of AUC after shuffling the abundance values of the genes in each gene set in validation dataset with the background color indicating the degrees of AUC change; the center left column lists the number of KO genes in each gene set with the background color indicating the set size; the center right column is the representative signature of each gene set; and the right column lists the cross-validation AUC of the representative microbial gene with the background color indicating an increased (red) or decreased (blue) AUC. The line plot shows the values of feature importance of the representative signatures (upper horizontal axis); the box plot shows the AUCs of each gene set in validation dataset with the dotted line representing the baseline AUC of 0.91 (lower horizontal axis). (b) The ROC curve shows the diagnostic performance of microbial genes celB and manY, respectively. (c-d) The box plot shows the abundances of celB (upper) and manY (lower) in metagenomic data (c) and qRT-PCR data (d) (N = 37, CD; N = 36, control), respectively. Data are presented as mean ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Next, we assessed the prediction power of representative KO genes of each gene set (Supplementary Table S14). Notably, celB and manY displayed excellent diagnostic capabilities with AUCs of 0.74 and 0.71, respectively (Figure 4b). Since celB and manY (also a member of fructose and mannose metabolism) are both members of the PTS, the above-mentioned results indicated that the PTS gene set mediated the most significant functional alterations of the gut microbiome in CD patients. Finally, we validated the abundances of celB and manY in an independent cohort of CD patients and controls using qRT-PCR. Consistent with the metagenomic data (Figure 4c), both celB and manY were significantly more abundant in CD patients (Figure 4d). Additionally, we validated the abundances of those genes that belong to important pathways and with high feature importance using qRT-PCR (Figure S13). These results revealed the respective contributions of individual gene feature to the diagnostic capability of the gene model and identified celB and manY as the individual biomarkers with the highest predictive power for diagnosing CD.
Altered interactions within and between each level of microbial signatures in CD
For a global understanding of the interactions among all microbial signatures in CD, we investigated the associations among all the microbial signatures via HALLA (Figure 5a-b). In both CD and control networks, considerable associations were observed between KO genes and species, but few were observed between SNVs and the other two levels (|correlation| > 0.4) (Figure 5b, Figure S14a, d). More associations were observed in the network of CD (206 associations) (Figure S14c) than in the network of controls (163 associations) (Figure S14f ; Supplementary Table S15–S16). Interestingly, there were more negative associations between the gene- and the species-signatures in the control network than that in the CD network. For example, D-nopaline dehydrogenase (nos), type IV secretion system protein TrbJ (trbJ) genes were negatively associated with R. hominis, R. bassiana, and C. aerofaciens. Notably, we found that KO genes had a stronger degree centrality than the species in the CD network (Figure S14b). Moreover, compared with the control network, these KO genes in CD tended to form isolated clusters, as exemplified by the independent module consisting celB and manY in the CD network (Figure 5a)
Figure 5.
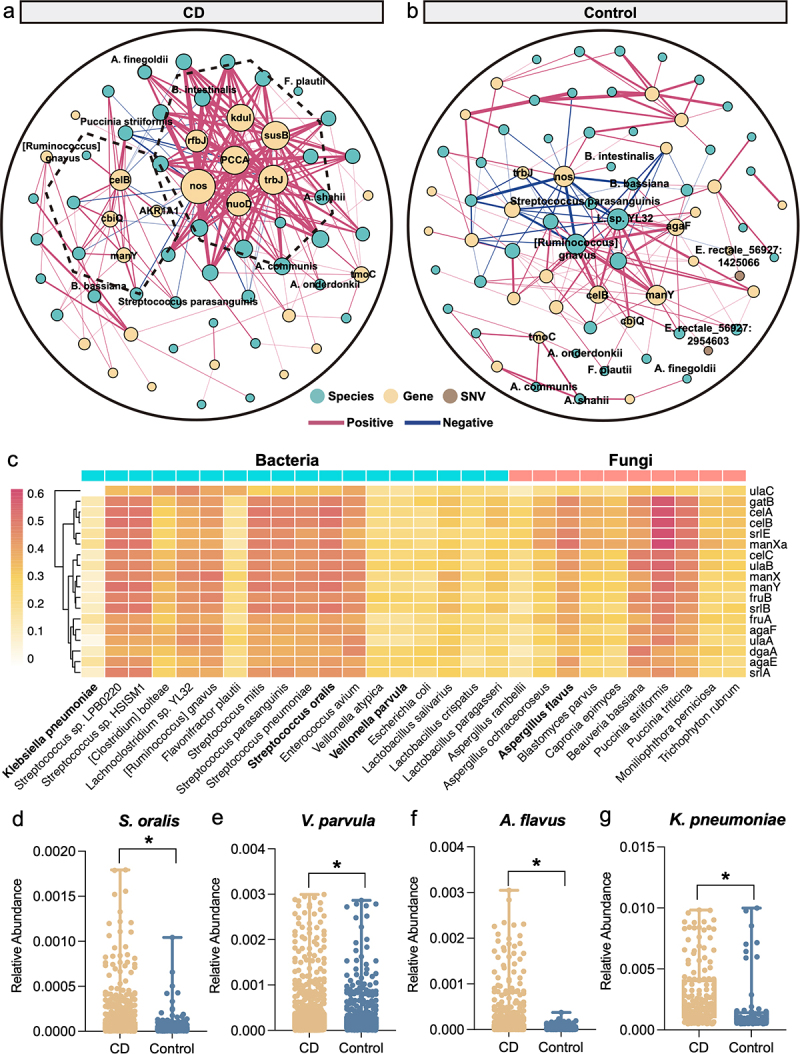
The cross-talk among multidimensional signatures. (a-b) Correlations among species-, gene and SNV signatures in the control (a) and the CD (b) networks. Node color indicates different levels of signatures: species (green), KO genes (yellow), and SNVs (brown). Red line indicates positive interaction; and blue line indicates negative interaction (|correlation|>0.4, FDR < 0.05). (c) The correlations between genes in PTS and microbial species (bacterial species, blue columns; and fungal species, orange column). The color bar represents the ranges of correlation coefficients. (d-g) the relative abundance of S. oralis (d), V. parvula (e), A. rambellii (f) and K. pneumoniae (g) in CD and controls.
In addition, we found that the oral microorganisms Streptococcus and Veillonella spp. were positively correlated with PTS-related genes (Figure 5c). In detail, Streptococcus oralis (R = 0.5, P < 2.2e-16) and Veillonella parvula (R = 0.2, P = 5e-13) were increased in CD patients and were positively correlated with the PTS-related genes (Figure 5d-e, Figure S15a-b). Besides, Aspergillus rambellii (R = 0.42, P < 2.2e-16) and K. pneumoniae (R = 0.13, P < 6.6e-06) with increased abundance also showed a positive correlation with gene celB (Figure 5f-g, Supplementary Figure S15c-d). Several studies have reported relevant evidence of the impact of Aspergillus rambellii and K. pneumoniae infection on gut ecology.19–21
Discussion
In this study, multidimensional microbial signatures of CD were systematically analyzed with multiple cohorts of distinct cultural and geographical backgrounds. By comparing the diagnostic capabilities of the microbial signatures including differential species, genes, and SNVs, the gene model achieved superior accuracy and robustness in distinguishing CD from controls, and the gene model was specific for CD against other microbiome-related diseases. Finally, the major contributing genes in the gene model were identified and validated, and their pathogenic characteristics in CD are highlighted.
Multidimensional alterations of the gut microbiome in CD patients contain massive amounts of information that can predict the disease state. Therefore, we employed a deep learning method to fit the underlying characteristics of the gut microbiome in patients with CD. With the microbial species models, while bacterial species achieved the best performance among single-kingdom models, multi-kingdom models with both bacterial and non-bacterial species achieved better accuracy than the single-kingdom models, which is similar to our observations with the microbial models for colorectal cancer.14
Comparing models of the three different types, the gene model demonstrated the best generalization and robustness in model evaluations compared to the species-, SNV- and combined models. This is reasonable, considering that homologous genes of different microorganisms may contribute to the same abnormalities in the gut microbiome in connection to specific pathological processes22.
By examining the contributions of individual gene set and gene to the diagnostic capabilities of the gene model, we found that genes that belong to the PTS gene set had a great impact on the model accuracy in abundance disturbance analysis. The importance of the PTS gene set in the diagnosis model was also demonstrated in recursive feature elimination analysis and in cross-validation of the sub-model. In gut bacteria, PTS is known as a system that catalyzes sugar transport as well as sugar phosphorylation23,24. In addition, PTS regulates a wide variety of transport, metabolic processes, biofilm formation, and virulence;25 thus, it is considered a comprehensive regulation and coordination system. We observed that the CD patients exhibited increased abundance in PTS and that the KO genes in PTS were associated with the differential species in CD (Figure 5c).
Furthermore, numerous studies have suggested colonization of the gut by oral commensals, such as Streptococcus, Prevotella, Veillonella, Haemophilus, and Bifidobacterium, in inflammatory bowel diseases26,27. In our results, the increased abundances of S. oralis and V. parvula and their positive correlations with the PTS-related genes indicated the essential role of these species in CD pathogenesis. Consistently, several studies reported that S. oralis26,28 and V. parvula29 were enriched in IBD patients and closely associated with IBD pathogenesis. Therefore, we hypothesize that the nutrient environment alteration related to PTS biological process under gut dysbiosis may be responsible for the ectopical colonization of the oral microorganisms.
More importantly, the KO gene celB, which encodes the enzyme IIC component (EIIC) of cellobiose PTS, exhibited the highest predictability among all gene markers and an increased abundance in CD patients. These observations support the outstanding potential for the microbial gene celB of PTS to be used as a biomarker for noninvasive CD diagnosis. Moreover, celB was associated with K. pneumoniae, which is in line with the roles of celB component of PTS in biofilm formation and virulence of K. pneumoniae,30 and the roles of K. pneumoniae in the initiation and perpetuation of the pathological damage of CD were also demonstrated21. We also observed a significant increase of K. pneumoniae in CD patients (Figure 5g). Therefore, it is reasonable to hypothesize that the interaction between celB and K. pneumoniae contributes to the development of CD. Overall, these findings reveal the crucial microbes involved in the pathogenesis of CD, which still requires further investigation.
Another microbial gene manY was also identified as a biomarker for CD diagnosis. manY encodes the EIIC component of mannose PTS system (man-PTS) that is a part of the PTS regulatory network. The hairpin tips of IIC in man-PTS are coordinated with mannose and mediate mannose transport31. Interestingly, previous studies found that man-PTS and cellobiose-PTS were upregulated in gut microbes by changing from a low-fat diet to a high-fat, high-sugar diet31,32, suggesting that the PTS of gut microbes is sensitive to the nutritional environment of mucosal surfaces. Thus, the upregulation of celB and manY in CD likely indicates upregulation of the biological activities of cellobiose-PTS and man-PTS in association with CD pathology. That is, manY may also be involved in the pathogenesis of CD. However, the cause of these alterations in CD remains unclear and requires further investigation.
Moreover, we noticed a trend toward more isolated clusters of microbial genes in the CD network (Figure 5a). The concept of “guild” may be a possible explanation of such phenomenon. In a “guild”, or “functional groups”, the microbial members perform similar functions and tend to exhibit co-abundance patterns by thriving or declining together within a community33,34. “Guilds” may exist in both healthy and CD microbial communities. However, when the gut ecosystems were disturbed, “Guilds” may response differently. In our study, the independent module consisting celB and manY in the CD network (Figure 5a-b), which may represent a concentrated function of the “guild” in the phosphotransferase system (PTS)-related niches and nutrients, where genes are with increased abundance and become a major “guild” in CD microbial community. Considering that they occupy the same metabolic and spatial niches by exploiting the same class of environmental resources in a similar way,35,36 the competitions among guild members could be intense.
Collectively, our study systematically analyzed multimodal microbial signatures including species, genes, and SNVs in CD patients. In addition, we identified universal biomarkers across distinct cultural and geographical backgrounds by integrating multiple cohorts, and improved discrimination power for CD by taking advantage of the excellent adaptability and learning ability of AI. Moreover, one of the strengths of our study comes from the significance of the identified microbial gene markers. The altered abundances in these genes suggest a possibly pathogenic origin for CD pathogenesis. However, this study did not provide an in-depth investigation on the microbial features of distinct inflammatory statuses and locations in CD patients for lack of relevant information. Besides, the discrepancy of several genes between metagenomics and qRT-PCR was likely due to the relatively smaller sample size in qRT-PCR validation and relatively lower resolution of the qRT-PCR method in microbiome analysis.
Conclusions
Our global metagenomic analysis unravels the multidimensional alterations of the microbial communities in CD and identifies microbial genes as robust diagnostic biomarkers across cohorts. These genes are functionally related to the pathogenesis of CD. Future studies on these genes may lead to the development of an effective, non-invasive diagnostic tool for CD.
Materials and methods
Study inclusion and data acquisition
For the discovery dataset, we used PubMed to search for studies that published fecal shotgun metagenomic data from CD patients and controls. Raw FASTQ files of 1241 fecal samples from four studies were downloaded from the European Nucleotide Archive (ENA) using the following identifiers: PRJNA398089 (D1) for Lloyd-Price et al.9, PRJNA385949 (D2) for Hall et al.37, PRJNA400072 (PRISM) (D3) for Franzosa et al.12, and SRP057027 (D4) for Lewis et al.38 (Figure 1a).
For the validation dataset, the raw data of 177 samples from three studies were collected from the ENA using the following identifiers: PRJNA389280 (V1) for Schirmer et al.39, PRJEB1220 (V2) for Nielsen et al.40 and PRJNA400072 (LifeLines DEEP and NLIBD) (V3) for Tigchelaar et al.12 (Figure 1a). The clinical characteristics of patients are presented in Supplementary Table S1. We excluded CD patients in remission and those treated with antibiotics within 1 month of sample collection in both discovery and validation datasets.
To evaluate whether the prediction model is specific for CD rather than non-CD diseases, we further collected five cohorts of non-CD diseases including ulcerative colitis (UC) from PRJNA400072 PRISM12, colorectal cancer (CRC) from PRJEB2792813, type-2 diabetes (T2D) from PRJEB178641, liver cirrhosis (LC) from PRJEB633742, and Parkinson’s disease (PD) from PRJEB1778443.
Patient recruitment and sample collection of Chinese cohorts
The Chinese cohort consisted of 40 CD and 53 control samples that were sequenced and published in He et al.44 (Supplementary Table S2). The CD patients and controls were enrolled at the Sixth Affiliated Hospital of the Sun Yat-sen University, Guangdong Province, China. The raw metagenomic sequencing data were available from the ENA Database (Accession No. PRJEB15371).
For qRT-PCR validation, we enrolled CD patients and controls at the Shanghai Tenth People’s Hospital. Patients diagnosed with CD were included in the study. Potential participants were excluded if they were pregnant, diagnosed with indeterminate colitis, had an acute gastrointestinal infection, or had received antibiotic therapy within 3 months. In total, we collected 73 fecal samples (N = 37 for CD and N = 36 for control, Supplementary Table S3) that were then stored at −80°C before DNA extraction. The study was approved by the Institutional Review Board at the Shanghai Tenth People’s Hospital, Tongji University, Shanghai (No. 20KT863), and informed consent was obtained from each participant.
Quality control of WMS sequencing data
For preprocessing the WMS sequencing data, quality control was performed using KneadData V0.6.0. Subsequently, reads with length lower than 50 bp, or with low-quality bases were filtered out by Trimmomatic software (V0.32). Furthermore, reads that mapped to the mammalian genome, bacterial plasmids, UNiVec sequences, and chimeric sequences were removed.
Annotation and abundance estimation of microbial taxa, genes, and SNVs
For multi-kingdom species level analysis, a customized reference database was constructed with 18,756 bacterial, 359 archaeal, and 9346 viral reference genomes from the NCBI RefSeq database (accessed in January 2020), and 1094 fungal reference genomes from the NCBI RefSeq database, FungiDB (http://fungidb.org) and Ensemble (http://fungi.ensembl.org) (all accessed in January 2020). Quality-filtered reads were aligned and quantified using Kraken2 and Bracken software, respectively.
For microbial gene-level analysis, quality-filtered metagenomes were assembled into contigs with Megahit (v1.2.9) using ‘meta-sensitive’ parameters. Contigs shorter than 500-bp were excluded for further analysis. Prodigal (v2.6.3) software was used to predict genes at the metagenome mode (−p meta). A non-redundant microbial gene reference was constructed with CD-HIT using a sequence identity cutoff of 0.95, and a minimum coverage cutoff of 0.9 for the shorter sequences. The reference was annotated with the EggNOG mapper (v2.0.1) based on EggNOG orthology data. Subsequently, CoverM (V4.0) was used to estimate gene abundance by mapping reads to the non-redundant reference and to calculate the coverage of genes in the original contigs. Finally, the abundance of KEGG Orthology (KO) groups were calculated by summing the expression of genes annotated to the same KOs. KO is a collection of orthologous genes in organisms based on sequence similarity45, which represents similar molecular functions of genes/proteins. Thus, KO abundance can provide an overall profile of gene functions in gut microbes.
For SNV-level analysis, MIDAS (V1.3.2) was used to perform microbial SNV annotation. A customized reference genome database was constructed to include seven species with sufficient coverage (>3×) in at least 20% of all samples. The WMS reads were then mapped to the reference database for SNV calling. Subsequently, the SNV profiles of all samples were merged, with only bi-allelic positions chosen. The other parameters were identical to those of the preset option ‘—core_snps’ (merge_midas.py snps – core_snps).
All processed metagenomic data in this study has been uploaded in the National Omics Data Encyclopedia under accession no. OEP003761.
Diagnostic model construction and evaluation
Model construction
Artificial intelligence (AI) algorithm called feedforward neural network (FNN) was employed to construct the diagnostic model. In detail, the hidden layers were activated by a rectified linear unit (ReLU) activation function, and the output layer was activated by a sigmoid function. Subsequently, we performed stratified ten-fold cross-validation to avoid overfitting issues and model estimation using Scikit-learn 1.1.0. Finally, we trained the diagnostic model with well-optimized hyperparameter combinations using TensorFlow 2.8.0. In order to comprehensively evaluate the performances of our models and facilitate the comparisons between similar diagnostic studies, we assessed the model performance with various metrics, especially the normalized Matthews correlation coefficient (MCC)46 that can produce a more accurate and informative score in evaluating binary classifications. The feature importance was evaluated with SHapley Additive exPlanations (SHAP)47 to explain the output of machine learning model.
Model interpretation
To better interpret the compositions and corresponding contributions of features in model, we grouped KO genes by gene sets based on the priori knowledge of the KEGG database. Subsequently, we randomly shuffled the abundance values of KO genes of a gene set in validation dataset, and performed predictions using the constructed diagnostic model. The decrease of AUC was considered to indicate the importance of gene set to the diagnostic model. The above procedure was repeated for 50 times.
Evaluation of the model’s robustness and generalization
To test the robustness and generalization of the selected optimal model among distinct cohorts, we performed cohort-to-cohort transfer and leave-one-cohort-out (LOCO) validation as described in our previous studies18,48. For cohort-to-cohort transfer, diagnostic models were trained on one single cohort and validated on each of the remaining cohorts. For LOCO validation, one single cohort was set as the validation dataset, while all other cohorts were pooled together as the discovery dataset.
Disease specificity assessment of prediction model
Using non-CD diseases samples of UC, CRC, T2D, LC, and PD, we evaluated the disease specificity of the predictive model for CD, following the method described by Thomas et al.49 In detail, we randomly selected 10 control samples and 10 case samples from non-CD external data and added them into the control group in the validation dataset. If the model is specific for CD, the model would not perform worse with the addition of a case relative to the addition of the controls, because the model does not cover the characteristics of non-CD diseases. This procedure was repeated 50 times.
Validation of microbial genes by qRT-PCR
gDNA was extracted using the TIANamp Stool DNA Kit (Cat# 4992205, TIANGEN) according to the manufacturer’s instructions. The primers used for validation are listed in Supplementary Table S4. To perform the qRT-PCR analysis, the reaction mixture contained the primer pair with concentrations diluted to 0.2 μM and 10 ng gDNA in a 10 μl final volume with the SYBR Green qPCR Mix (Thermo Fisher Scientific). The cycling program was as follows: pre-denaturation at 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 s and annealing at 60°C for 60 s, followed by melting curve analysis. The qRT-PCR results were quantitated by calculating −ΔΔCt values between candidate genes and the 16S gene. The significance of the comparison between CD and control samples was tested by a two-sided Wilcoxon rank-sum test (P < 0.05).
Statistical analysis
Confounder analysis
Permutational multivariate analysis of variance analysis (PERMANOVA) was performed to quantify the effects of potential confounding factors. The total variance of a given signature was compared to the variance explained by group and the variance by confounding factors (age, BMI, sex, and disease location) in a linear model. Variance calculations were performed using the same procedure as in our previous study.48
Differential signature identification
A formal meta-analysis approach called MMUPHin50 was performed to identify CD-related differential microbial signatures, which enabled the batch effect correction and combination of multiple microbial community studies. The MMUPHin approach can fit environmental exposures, phenotypes, and population structures across microbial community studies via a combat-like extended linear regression method50. When fitting our microbiome data using the multivariate linear modeling framework in MMUPHin, the cohort factor was treated as the main “batch” and the other confounders, including age, BMI, sex, and disease location, were treated as covariates of biological interest. Overall, MMUPHin provides a meta-analysis by aggregating individual study results with established fixed effect models to identify consistent overall effects.
Alpha and beta diversity analysis
Alpha diversity of taxonomic profiles including Shannon, ACE, Simpson, and Chao1 index were calculated using R (V4.0.5) “vegan” (V2.5.7) package. Beta diversity between groups was calculated based on Bray-Curtis distance using PERMANOVA called adonis test, and significance was evaluated with 999 permutations.
Co-abundance analysis
First, we generated species abundance profiles of CD and controls, respectively. We then employed SparCC to perform a co-abundance analysis of differential multi-kingdom species. Correlations between differential multi-kingdom species were determined by estimating the observations of Dirichlet distribution for 50 times. Then, SparCC resampled the original dataset through a bootstrap method to obtain random datasets. Later, pseudo-p-values are calculated from these random data sets to assess the significance of the initial observation scores. The network was visualized with Gephi (V0.9.5).
Multidimensional signatures association analysis
To further explore the potential associations between multidimensional signatures, Hierarchical All-against-All association testing (HALLA, V 0.8.20) was performed. We generated species, gene, and SNV profiles of CD patients and controls, respectively. Subsequently, the associations between the species, gene, and SNV signatures were calculated in pairs using HALLA. After that, we merged the output correlation matrices. Correlations with |cor| > 0.4 and P-values <0.05 were used to construct the network and visualized with Gephi (V0.9.5).
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82170542 to RZ, 92251307 to RZ, 32200529 to DW, 82000536 to NJ), the National Key Research and Development Program of China (2021YFF0703700/2021YFF0703702 to RZ), Guangdong Province “Pearl River Talent Plan” Innovation and Entrepreneurship Team Project (2019ZT08Y464 to LZ), the program of Guangdong Provincial Clinical Research Center for Digestive Diseases (2020B1111170004), and National Key Clinical Discipline. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Funding Statement
The work was supported by the National Natural Science Foundation of China [82170542, 92251307, 32200529, 82000536]; National Key Research and Development Program of China [2021YFF0703700/2021YFF0703702]; Guangdong Province “Pearl River Talent Plan” Innovation and Entrepreneurship Team Project [2019ZT08Y464]; Program of Guangdong Provincial Clinical Research Center for Digestive Diseases [2020B1111170004], and National Key Clinical Discipline.
Abbreviations
ABC.PE.P: peptide/nickel transport system permease protein; agaF: N-acetylgalactosamine PTS system EIIA component; AKR1A1: alcohol dehydrogenase (NADP+); ALDH: aldehyde dehydrogenase (NAD+); allA: ureidoglycolate lyase; AUC: area under the ROC curve; CD: Crohn’s disease; celB: cellobiose PTS system EIIC component; CRC: colorectal cancer; EIIC: enzyme IIC component; ENA: European Nucleotide Archive; fliC: flagellin; FNN: Feedforward neural network; GSEA: gene set enrichment analysis; IBD: inflammatory bowel disease; impB: type VI secretion system protein ImpB; KO: KEGG Orthology; LC: liver cirrhosis; LOCO: leave-one-cohort-out; maeB: malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+); manY: mannose PTS system EIIC component; nirK: nitrite reductase (NO-forming); pckA: phosphoenolpyruvate carboxykinase (GTP); PD: Parkinson’s disease; PTS: phosphotransferase system; ReLU: rectified linear unit; rfbJ: CDP-abequose synthase; ROC: receiver operating characteristic; SHAP: SHapley Additive exPlanations; SNVs: single nucleotide variants; sucD: succinyl-CoA synthetase alpha subunit; T2D: type-2 diabetes; tcPp: toxin coregulated pilus biosynthesis protein P; tmoC: toluene monooxygenase system ferredoxin subunit; trbJ: type IV secretion system protein TrbJ; ttuC: tartrate dehydrogenase/decarboxylase/D-malate dehydrogenase; UC: ulcerative colitis; WMS: whole-metagenome sequencing
Authors’ contributions
LZ, ZL, QH, and RZ conceived and designed the project. SG performed the public data collection, microbiome analysis, AI modeling, and bioinformatics analysis. XG recruited the participants, collected the fecal samples, and performed the qRT-PCR analysis. SG and XG drafted the manuscript. RZ, DW, ZF, NJ, RS, WG, QH, ZL, and LZ revised the manuscript. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All of the processed data in this study has been uploaded in the National Omics Data Encyclopedia under accession no. OEP003761. The raw metagenomic data are available in the European Nucleotide Archive (https://www.ebi.ac.uk/ena/) under accession Nos. PRJNA398089, PRJNA385949, PRJNA400072, SRP057027, PRJEB15371, PRJNA389280, PRJEB1220, PRJEB27928, PRJEB17784, PRJEB1786, and PRJEB6337. The data relevant to the study are included in the article or uploaded as supplementary information. The code and scripts are available on GitHub (https://github.com/tjcadd2020/Diagnosis-for-CD).
Ethics approval and consent
All participants provided written informed consent prior to data collection. The study was approved by the Institutional Review Board at the Shanghai Tenth People’s Hospital, Tongji University, Shanghai (No. 20KT863).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2221428
References
- 1.Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–18. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 2.Alatab S, Sepanlou SG, Ikuta K, Vahedi H, Bisignano C, Safiri S, Sadeghi A, Nixon MR, Abdoli A, Abolhassani H.. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(1):17–30. doi: 10.1016/S2468-1253(19)30333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feuerstein JD, Cheifetz AS. Crohn disease: epidemiology, diagnosis, and management. Mayo Clin Proc. 2017;92(7):1088–1103. doi: 10.1016/j.mayocp.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Maaser C, Sturm A, Vavricka SR, Kucharzik T, Fiorino G, Annese V, Calabrese E, Baumgart DC, Bettenworth D, Borralho Nunes P, et al. ECCO-ESGAR guideline for diagnostic assessment in IBD Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13(2):144–164. doi: 10.1093/ecco-jcc/jjy113. [DOI] [PubMed] [Google Scholar]
- 5.Feld LD, Kirk K, Feld AD. A high quality approach to addressing complications of endoscopy and optimizing risk management strategies. Tech Innovations In Gastrointestinal Endoscopy. 2022;24(4):390–395. doi: 10.1016/j.tige.2022.03.006. [DOI] [Google Scholar]
- 6.Lewis JD. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology. 2011;140(6):1817–1826.e2. doi: 10.1053/j.gastro.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosli MH, Zou G, Garg SK, Feagan SG, MacDonald JK, Chande N, Sandborn WJ, Feagan BG. C-Reactive protein, fecal calprotectin, and stool lactoferrin for detection of endoscopic activity in symptomatic inflammatory bowel disease patients: a systematic review and meta-analysis. American Journal Of Gastroenterology. 2015;110:802–819. quiz 20. doi: 10.1038/ajg.2015.120. [DOI] [PubMed] [Google Scholar]
- 8.Dubinsky M, Braun J. Diagnostic and prognostic microbial biomarkers in inflammatory bowel diseases. Gastroenterology. 2015;149(5):1265–74.e3. doi: 10.1053/j.gastro.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Q, Zhang X, Hu Z. Application of artificial intelligence modeling technology based on multi-omics in noninvasive diagnosis of inflammatory bowel disease. J Inflamm Res. 2021;14:1933–1943. doi: 10.2147/JIR.S306816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, et al. A microbial signature for Crohn’s disease. Gut. 2017;66:813–822. doi: 10.1136/gutjnl-2016-313235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4(2):293–305. doi: 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tierney BT, Tan Y, Kostic AD, Patel CJ. Gene-level metagenomic architectures across diseases yield high-resolution microbiome diagnostic indicators. Nat Commun. 2021;12(1):2907. doi: 10.1038/s41467-021-23029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu NN, Jiao N, Tan JC, Wang Z, Wu D, Wang AJ, Chen J, Tao L, Zhou C, Fang W, et al. Multi-kingdom microbiota analyses identify bacterial–fungal interactions and biomarkers of colorectal cancer across cohorts. Nat Microbiol. 2022;7(2):238–250. doi: 10.1038/s41564-021-01030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Y, Nguyen LH, Franzosa EA, Huttenhower C. Strain-level epidemiology of microbial communities and the human microbiome. Genome Med. 2020;12(1):71. doi: 10.1186/s13073-020-00765-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C, Chen K, Wang Y, Cen C, Zhai Q, Zhang J. Establishing a novel colorectal cancer predictive model based on unique gut microbial single nucleotide variant markers. Gut Microbes. 2021;13(1):1–6. doi: 10.1080/19490976.2020.1869505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang S, Chen D, Ma C, Liu H, Huang S, Zhang J. Establishing a novel inflammatory bowel disease prediction model based on gene markers identified from single nucleotide variants of the intestinal microbiota. iMeta. 2022;1(3):1. doi: 10.1002/imt2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao W, Chen W, Yin W, Zhu X, Gao S, Liu L, Wu D, Zhu R, Jiao N. Identification and validation of microbial biomarkers from cross-cohort datasets using xMarkerfinder. Protocol Exchange. 2022. [DOI] [PubMed] [Google Scholar]
- 19.Coker OO, Liu C, Wu WKK, Wong SH, Jia W, Sung JJY, Yu J. Altered gut metabolites and microbiota interactions are implicated in colorectal carcinogenesis and can be non-invasive diagnostic biomarkers. Microbiome. 2022;10(1):35. doi: 10.1186/s40168-021-01208-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallen-Adams HE, Suhr MJ. Fungi in the healthy human gastrointestinal tract. Virulence. 2017;8(3):352–358. doi: 10.1080/21505594.2016.1247140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rashid T, Ebringer A, Wilson C. The role of Klebsiella in Crohn’s disease with a potential for the use of antimicrobial measures. Int J Rheumatol. 2013;2013:610393. doi: 10.1155/2013/610393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schirmer M, Garner A, Vlamakis H, Xavier RJ. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. 2019;17(8):497–511. doi: 10.1038/s41579-019-0213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lengeler JW, Jahreis K. Bacterial PEP-dependent carbohydrate: phosphotransferase systems couple sensing and global control mechanisms. Contrib Microbiol. 2009;16:65–87. [DOI] [PubMed] [Google Scholar]
- 24.Västermark A, Saier MH Jr.. The involvement of transport proteins in transcriptional and metabolic regulation. Curr Opin Microbiol. 2014;18:8–15. doi: 10.1016/j.mib.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saier MH Jr. The bacterial phosphotransferase system: new frontiers 50 years after its discovery. J Mol Microbiol Biotechnol. 2015;25(2–3):73–78. doi: 10.1159/000381215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han Y, Wang B, Gao H, He C, Hua R, Liang C, Xin S, Wang Y, Xu J. Insight into the relationship between Oral Microbiota and the Inflammatory Bowel Disease. Microorganisms. 2022;10(9):10. doi: 10.3390/microorganisms10091868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdelbary MMH, Hatting M, Bott A, Dahlhausen A, Keller D, Trautwein C, Conrads G. The oral-gut axis: salivary and fecal microbiome dysbiosis in patients with inflammatory bowel disease. Front Cell Infect Microbiol. 2022;12:1010853. doi: 10.3389/fcimb.2022.1010853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu S, Mok J, Gowans M, Ong DEH, Hartono JL, Lee JWJ. Oral Microbiome of Crohn’s disease patients with and without Oral Manifestations. J Crohns Colitis. 2022;16(10):1628–1636. doi: 10.1093/ecco-jcc/jjac063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rojas-Tapias DF, Brown EM, Temple ER, Onyekaba MA, Mohamed AMT, Duncan K, Schirmer M, Walker RL, Mayassi T, Pierce KA, et al. Inflammation-associated nitrate facilitates ectopic colonization of oral bacterium Veillonella parvula in the intestine. Nat Microbiol. 2022;7(10):1673–1685. doi: 10.1038/s41564-022-01224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu M-C, Chen Y-C, Lin T-L, Hsieh P-F, Wang J-T, Camilli A, Camilli A. Cellobiose-specific phosphotransferase system of Klebsiella pneumoniae and its importance in biofilm formation and virulence. Infect Immun. 2012;80(7):2464–2472. doi: 10.1128/IAI.06247-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeckelmann JM, Erni B. The mannose phosphotransferase system (Man-PTS) - Mannose transporter and receptor for bacteriocins and bacteriophages. Biochimica et Biophysica Acta (BBA) - Biomembr. 2020;1862(11):183412. doi: 10.1016/j.bbamem.2020.183412. [DOI] [PubMed] [Google Scholar]
- 32.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, Fu H, Xue X, Lu C, Ma J, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359(6380):1151–1156. doi: 10.1126/science.aao5774. [DOI] [PubMed] [Google Scholar]
- 34.Wu G, Zhao N, Zhang C, Lam YY, Zhao L. Guild-based analysis for understanding gut microbiome in human health and diseases. Genome Med. 2021;13(1):22. doi: 10.1186/s13073-021-00840-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Y, Kitamoto S, Kamada N. Microbial adaptation to the healthy and inflamed gut environments. Gut Microbes. 2020;12(1):1857505. doi: 10.1080/19490976.2020.1857505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, Lagoudas GK, Vatanen T, Fornelos N, Wilson R, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9(1):103. doi: 10.1186/s13073-017-0490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman E, Hoffmann C, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe. 2015;18(4):489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, Ananthakrishnan AN, Andrews E, Barron G, Lake K, et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol. 2018;3(3):337–346. doi: 10.1038/s41564-017-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32(8):822–828. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 41.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Krogh Pedersen H, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528(7581):262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513(7516):59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 43.Bedarf JR, Hildebrand F, Coelho LP, Sunagawa S, Bahram M, Goeser F, Bork P, Wüllner U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017;9(1):39. doi: 10.1186/s13073-017-0428-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He Q, Gao Y, Jie Z, Yu X, Laursen JM, Xiao L, et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. Gigascience. 2017;6:1–11. doi: 10.1093/gigascience/gix050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chicco D, Jurman G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. Bmc Genomics. 2020;21(1):6. doi: 10.1186/s12864-019-6413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lundberg SM, Lee S-I. A unified approach to interpreting model predictions. NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems; Long Beach California USA. 2017. p. 4768–4777. [Google Scholar]
- 48.Wu Y, Jiao N, Zhu R, Zhang Y, Wu D, Wang AJ, Fang S, Tao L, Li Y, Cheng S, et al. Identification of microbial markers across populations in early detection of colorectal cancer. Nat Commun. 2021;12(1):3063. doi: 10.1038/s41467-021-23265-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25(4):667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma S, Shungin D, Mallick H, Schirmer M, Nguyen LH, Kolde R, Franzosa E, Vlamakis H, Xavier R, Huttenhower C, et al. Population structure discovery in meta-analyzed microbial communities and inflammatory bowel disease using MMUPHin. Genome Biol. 2022;23(1):208. doi: 10.1186/s13059-022-02753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the processed data in this study has been uploaded in the National Omics Data Encyclopedia under accession no. OEP003761. The raw metagenomic data are available in the European Nucleotide Archive (https://www.ebi.ac.uk/ena/) under accession Nos. PRJNA398089, PRJNA385949, PRJNA400072, SRP057027, PRJEB15371, PRJNA389280, PRJEB1220, PRJEB27928, PRJEB17784, PRJEB1786, and PRJEB6337. The data relevant to the study are included in the article or uploaded as supplementary information. The code and scripts are available on GitHub (https://github.com/tjcadd2020/Diagnosis-for-CD).