

Abstract
Here, we prepared UiO-66-NH2 and employed a post-synthesis modification method for its functionalization with a β-cyclodextrin (β-CD) organic compound. The resulting composite was employed as a support for the heterogenization of the Pd NPs. Various techniques, including FT-IR, XRD, SEM, TEM, EDS, and elemental mapping, were used to characterize UiO-66-NH2@β-CD/PdNPs, indicating its successful preparation. Three C–C coupling reactions, including the Suzuki, Heck, and Sonogashira coupling reactions, were promoted using the produced catalyst. As a result of the PSM, the proposed catalyst displays improved catalytic performance. In addition, the suggested catalyst was highly recyclable up to 6 times.
Here, we prepared UiO-66-NH2 and employed a post-synthesis modification method for its functionalization with a β-cyclodextrin (β-CD) organic compound.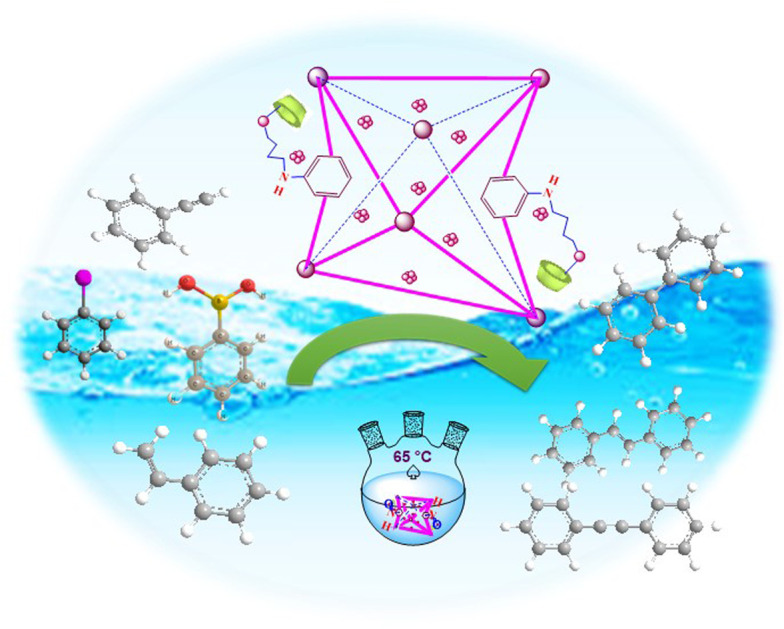
Introduction
The generation of a C–C bond is the consequence of a cross-coupling reaction, defined as the substitution of a heteroatomic nucleophile for an alkyl, aryl, or vinyl halide.1,2 Usually, a transition-metal complex is applied as a catalyst for promoting such reactions.3,4 The electrophilic counterparts in this reaction are normally halides, diazonium salts, or alcohols, while organometallic substances are used as nucleophilic counterparts.5,6 C–C cross-coupling reactions catalyzed by palladium are among the most essential techniques in contemporary organic synthesis.7,8 Forty percent of the C–C bond-forming events in pharmaceutical synthesis are catalyzed by Pd cross-coupling reactions because of their controllable reactivity and selectivity. Multiple aryl derivatives may be made in one go with high selectivity and yield without the need for intermediate product isolation.9,10 Examples include the Heck reaction, which transforms olefins with different substituents into their aryl derivatives. Non-symmetric biphenyls may be synthesized using the Suzuki method, while diaryl-alkyne compounds can be made using the Sonogashira coupling.5,11
The role of β-CDNs can be explained on the base of its capability to form inclusion complex with substrates and bringing them in the vicinity of the catalytic active sites.12,13 Also β-cyclodextrin can activate variety of organic compounds like aldehydes, ketones, anhydride, oximes, amines, nitriles and increase the rate of reactions.14,15 Here, novel β-cyclodextrin (β-CD)@1,4-dibromo butane@functionalized UiO-66-NH2/Pd-NPs composite was in situ prepared using β-CD functionalized MOF (Zr) and Pd-NPs, and C–C coupling reactions to promote formation coupling desired was developed based on the composite. A series of investigations on the β-cyclodextrin (β-CD)@1,4-dibromo butane@functionalized UiO-66-NH2/Pd-NPs composite material were conducted. The β-CD functionalized 1,4-dibromobutane has an excellent and stable dispersion effect.16 The composite was further combined with metal–organic frameworks (MOFs) to overcome the disadvantages of a single material, displaying excellent efficacy and a synergistic catalytic effect, so C–C coupling reaction is developed.17–19
The tendency of the metal nanoparticle for aggregation in solvents makes it urgent to use heterogeneous supports for their cost-effective, green, and reusable applications.12–14 The aggregation of metal NPs decreases their surface area, resulting in diminishing or even quenching the catalytic activities. Heterogenous supports not only can prevent aggregation of the NPs but can boost their catalytic performance or add some new features to the resulting composite.15
One of the most popular materials for the heterogenization of metal NPs is metal–organic frameworks (MOFs), a family of porous materials with tunable chemical and physical properties.16 The designable structure of these materials leads to their broad application in various branches of chemistry, including catalysis, photocatalysis, electrocatalysis, adsorption, separation, drug delivery, and organic transformation.17–20 Moreover, post-synthesis modification (PSM) allows for the extensive tuning of the chemical and physical characteristics in MOF by implanting a wide variety of organic and inorganic functionalities.21
The Zr4+ ions in the inorganic clusters of UiO-66-NH2 are responsible for the compound's remarkable stability in aqueous environments.22 The extraordinary stability of the UiO-66 structure may be attributed to the strong attraction between the negatively charged carboxylic acids and the positively charged Zr4+ ions, as well as the packed secondary building block (SBU) that prevents H2O molecules from reaching the zirconia cluster.23 UiO-66-NH2 is an excellent candidate for the PSM because of its high stability, intrinsic open metal sites (OMSs), large surface area, and presence of the amino group. Depending on the application, the amino groups of the organic linker can serve as a binding center to alter the electronic structure of UiO-66-NH2.24
Here, we modified the structure of an UiO-66-NH2 MOF with a β-cyclodextrin organic compound using a PSM approach and employed it as support for heterogenize the Pd NPs. The manufactured UiO-66-NH2@β-CD/PdNPs was employed as a catalyst for promoting a series of C–C coupling reactions. The suggested catalyst exhibits increased catalytic performance as a consequence of the PSM. The recommended catalyst was also extremely recyclable for up to six cycles.
Experimental
All the chemicals and reagents were purchased from Merck and were used without further purification.
Preparation of UiO-66-NH2
The process for the preparation of UiO-66-NH2 is as follows: 2.05 g of 2-aminoterephthalic acid (NH2-BDC) was transferred to a three-mouth flask containing dimethylformamide (DMF) (65 ml), under constant stirring. At this stage, 2.65 g of zirconium tetrachloride (ZrCl4) and 55 ml dimethylformamide to the above flask. Finally, 2.5 ml of concentrated hydrochloric acid (HCl) was added as the modulator. The reaction was performed under an inert atmosphere (argon gas atmosphere) for 24 hours at a temperature of 130 °C.
Synthesis of UiO-66-NH2@1,4-dibromobutane
The first stage of the PSM was carried out as follows: 0.1 g of UiO-66-NH2 was transferred to the balloon containing 60 ml of methanol, dispersed for 5 minutes, and stirred at RT for 20 minutes. Next, 1,4-dibromobutane (1.2 ml) was dissolved in 0.5 ml methanol and stirred for 10 minutes. The uniform solution of 1,4-dibromobutane in methanol was added to the stirring mixture of UiO-66-NH2 in methanol and kept stirring at 70 °C for 24 h. After the reaction process, the sediment was separated with a 10 000 rpm centrifuge, and it was treated twice with methanol and dried at 60 °C overnight.
Synthesis of UiO-66-NH2@β-CD
To prepare UiO-66-NH2@β-CD, 0.5 g of β-cyclodextrin was dispersed in 40 ml of dry dimethyl sulfoxide (DMSO) and stirred until completely dissolved. In a separate flask, 0.1 g of as-prepared UiO-66-NH2@1,4-dibromobutane was dispersed in 30 ml DMSO and stirred for 30 minutes. Next, the contents of these two flasks were gradually mixed and agitated for 24 h at 60 °C. Finally, the sediment was separated through a centrifuge, washed with dry DMSO, and dried at 60 °C overnight.
Synthesis of UiO-66-NH2@β-CD@PdNPs
0.04 g of palladium acetate (Pd(OAc)2) was dissolved in 50 ml acetonitrile at room temperature. Then, 0.2 g of the UiO-66-NH2@β-CD was added to the flask containing the transparent amber color solution of palladium acetate in acetonitrile and kept stirring at 40–45 °C for 8 h. Finally, the temperature was lowered to RT, and the reaction mixture was charged with 0.3 ml of freshly prepared hydrazine hydrate solution (3 drops of hydrazine hydrate in 3 ml of deionized water) and kept stirring at RT for another 24 h. Finally, the reaction mixture was separated using a 9000 rpm centrifuge, washed once with acetonitrile, and dried in an oven.
General procedure for the Suzuki reaction
Typically, 1 mmol of the appropriate aryl halide and 1.1 mmol of the aryl boronic acid were mixed in 78% ethanol solvent (3 ml) in a single-neck flask (10 ml). To this mixture, 0.025 g of the as-prepared UiO-66-NH2@β-CD/PdNPs, and 2 mmol of K2CO3 were added and agitated. The appropriate temperature and time for each reaction vary based on the applied precursor. TLC monitored the progress of the reaction with the solvent ratio (n-Hex : EtOAc/7 : 3). At the end of each reaction, the catalyst was filtered, the mixture was cooled to 25 °C, and the organic phase was extracted with diethyl acetate. Then it was dried over magnesium sulfate (MgSO4). After a while, MgSO4 was separated by filtration, the organic solvent was evaporated, and column chromatography was hired to purify the products in a solvent ratio of n-hexane : ethyl acetate/1 : 5. The physical data (melting point), FT-IR, and NMR technique were used for the identification of the products (ESI†).
General procedure for the Heck reaction
In a suitable round-bottomed flask (10 ml), 1 mmol of the desired aryl halide, 1.1 mmol of olefin, 2 mmol of potassium carbonate (K2CO3), dimethylformamide solvent (DMF) (3 ml), and 0.03 g of the newly synthesized UiO-66-NH2@β-CD/PdNPs catalyst was added at 85 °C. The mixture was centrifuged at the end of the reaction process to separate the catalyst. After cooling the resulting mixture, ethyl acetate was used to extract the products. The organic solvent was evaporated after drying with MgSO4. Column chromatography was hired to purify the products in a solvent ratio of n-hexane : ethyl acetate/1 : 5. The physical data (melting point), FT-IR, and NMR technique were used for the identification of the products (ESI†).
General procedure for the Sonogashira reaction
Typically, 1.2 mmol of acetylene and 1 mmol of the corresponding halobenzene were mixed in 0.3 ml of distilled water in a one-neck balloon. 20 mg of UiO-66-NH2@β-CD/PdNPs catalyst and 0.2 mmol of K2CO3 were added to this mixture, and the temperature was raised to 50 °C and kept stirring for an appropriate time (Table S4†). The progression of the reaction was tracked by TLC using an n-Hex : EtOAc: 8 : 1 solvent ratio. After the completion of each reaction, the catalyst was filtered, washed with ethanol, and dried at 60 °C for 12 hours. The filtered solution was cooled to ambient temperature, and its organic content was extracted with pure diethyl ether (Et2O). Column chromatography was hired to purify the products in a solvent ratio of n-hexane : ethyl acetate/1 : 4. The physical data (melting point), FT-IR, and NMR technique were used for the identification of the products (ESI†).
Results and discussion
This report introduces a highly efficient and recyclable Pd-based catalyst for promoting a series of C–C coupling reactions, including the Suzuki, Heck, and Sonogashira reactions, as a result of our prior efforts to develop simple and environmentally friendly methods for developing various organic reactions. First, 1,4-dibromobutane bridge ligands were incorporated into the UiO-66-NH2 molecule. To add β-cyclodextrin to the frameworks, a step-by-step PSM technique was used. The synthesized UiO-66-NH2@β-CD was then employed as a basis for the heterogenization of Pd NPs (Scheme 1). The produced UiO-66-NH2@β-CD/PdNPs were then used as a catalyst to promote C–C coupling processes.
Scheme 1. Schematic synthesis of gold nanoparticle immobilized on UiO-66-NH2@β-CD@PdNPs.

The FT-IR spectra of UiO-66-NH2, UiO-66-NH2@1,4-dibromobutane, and UiO-66-NH2@β-CD are represented in Fig. 1. The F-IR spectra of bare UiO-66-NH2 shows a broad peak at 1500–1600 and 3300–3500 cm−1, regarding the free and uncoordinated NH2 groups. Stretching vibrations of the C–N bonding of the H2BDC–NH2 are showing themselves at 1261 and 1338 cm−1. The peaks at 661 and 760 cm−1 are related to the Zr–O–Zr bonding of the zirconia ions in SBU. The modified MOF with 1,4-dibromobutane shows less intensity for the broad peak of the NH2 group at 3300–3500 cm−1, indicating less free and available uncoordinated NH2. The intensity of the same peak increases for the UiO-66-NH2@β-CD, which results from the presence of the uncoordinated OH groups of the β-cyclodextrin. Other characteristic peaks of various parts of the composite overlapped, and other characterization methods were used to prove the formation of the catalyst.
Fig. 1. FT-IR spectra of (a) UiO-66-NH2, (b) UiO-66-NH2@1,4-dibromobutane, (c) β-CD, (d) UiO-66-NH2@1,4-dibromobutane@β-CD.

The XRD pattern of the bare UiO-66-NH2 and the UiO-66-NH2@β-CD/PdNPs are represented in Fig. 2. The XRD pattern of the pristine MOF reveals all the characteristics of the UiO-66-NH2 with good intensity, which indicate the formation of its structure in excellent crystallinity. In the case of the Pd decorated β-dextrin modified MOF, the XRD pattern shows a little broadening of the peaks and a red-shift to lower 2θ, probably due to minor loss of crystallinity throughout the synthesis step. Apart from that, the XRD pattern of the resulting composite shows all the characteristics of the bare MOF, indicating its successful modification. Moreover, new peaks appeared at the XRD pattern of the UiO-66-NH2@β-DC/PdNPs regarding 111, 200, and 220 of the Pd NPs, proving their successful formation.
Fig. 2. The XRD pattern of the UiO-66-NH2 and UiO-66-NH2@β-CD/PdNPs.

SEM and TEM techniques investigated the morphology of the resulting catalyst. According to Fig. 3A, the SEM image shows the agglomerated nanoparticles with a diameter starting at approximately 55 nm. The rough surface of UiO-66-NH2@β-CD/PdNPs results from the PSM procedure. The TEM image shows the well-distributed Pd NPs over the surface of the modified MOF (Fig. 3B). Elemental composition and spatial elemental distribution of the UiO-66-NH2@β-CD/PdNPs are investigated by EDS and mapping techniques (Fig. 3C and D). The EDS spectra show the presence of O, N, C, Zr, and Pd in UiO-66-NH2 and UiO-66-NH2@β-CD/PdNPs with 7.33 wt% loading of palladium NPs, which indicates the successful formation of the desired composite. These spectra also reveal the presence of minor amounts of Br element, indicating some unreacted ligands. Mapping images indicate the uniform distribution of elements in the proposed catalyst, specifying the good quality of the synthesis procedure.
Fig. 3. (A) The SEM image, (B–D) the TEM images, (E) mapping, and the EDS spectra of UiO-66-NH2@β-CD/PdNPs.

The N2 adsorption–desorption experiment (BET) assigned in order to identify and calculate the surface area of the porosity of modified the UiO-66-NH2@1,4-dibromobutane@β-cyclodextrin@Pd-NPs as MOF catalyst at 77 K in Fig. 4. The adsorption–desorption isotherm UiO-66-NH2 indicates a type I isotherm, which suggests the micro-porosity of its structure, with a surface area of 430 cm3 g−1. This plot shows only one type of microporous in the MOF structure with a pore diameter of 1.32 nm. This result is a consequence of the successful PMS of MOF, in which 1,4-dibromo butane and @β-cyclodextrin@Pd-NPs filled pores of UiO-66-NH2 and reduced its surface area (Fig. 4).
Fig. 4. N2 adsorption–desorption experiment UiO-66-NH2@1,4-dibromobutane@β-cyclodextrin@Pd-NPs.

After careful characterization of our proposed catalyst, we assess the catalytic application of UiO-66-NH2@β-CD/PdNPs for various coupling reactions, including Heck, Sonogashira, and Suzuki. We begin our inquiry by assessing the catalytic efficacy of the proposed catalyst for promoting the Suzuki reaction. The reaction between iodobenzene and phenylboronic acid in the company of UiO-66-NH2@β-CD/PdNPs is selected as the template reaction. The effects of the time, temperature, solvent type, base, and amount of catalyst on the reaction rate are studied, and the outcomes are illustrated in Table 1. To determine the optimal solvent, the progress of the Suzuki-coupling reaction of the model reaction in the company of the suggested catalyst in various solvents, including water, toluene, DMSO, DMF, CH2Cl2, MeCN, a combination of ethanol and water, ethanol, tetrahydrofuran, and NMP, was monitored. This study reveals that ethanol shows the most satisficing outcome. The temperature was raised to 80 °C, and the best results were obtained at 70 °C. A further increase in the temperature did not boost the yield of the reaction. Hiring the TLC technique to track the reaction's progress, we determined that at 70 °C in H2O, the reaction achieved equilibrium after 60 minutes. The optimal amount of catalyst was determined by observing the progress of the reaction in the company of varying amounts of catalyst. The results of this study suggested that 25 mg of the proposed catalyst is adequate for the Suzuki reaction to proceed optimally. So, according to these investigations, the highest yields are achieved with 25 mg of UiO-66-NH2@β-CD/PdNPs catalyst at 70 °C in EtOH after 60 minutes.
The findings of the Suzuki coupling reaction optimization experimentsa.
| ||||||
|---|---|---|---|---|---|---|
| Entrance | Catalyst (mg) | Solvent | T (°C) | Base | Time (min) | Yield (%) |
| 1 | 5 | EtOH absolute | 25 | K2CO3 | 60 | 15 |
| 2 | 10 | EtOH absolute | 70 | K2CO3 | 60 | 40 |
| 3 | 20 | EtOH : H2O (1 : 1) | 70 | K2CO3 | 60 | 72 |
| 4 | 25 | EtOH 78% | 70 | K2CO3 | 60 | 99 |
| 5 | 25 | EtOH 78% | 70 | K2CO3 | 60 | 65 |
| 6 | 25 | EtOH 78% | 80 | K2CO3 | 60 | 92 |
| 7 | — | EtOH 78% | 70 | K2CO3 | 180 | — |
| 8 | 25 | EtOH 78% | 85 | K2CO3 | 60 | 83 |
| 9 | 25 | EtOH 78% | 70 | K2CO3 | 60 | 99 |
| 10 | 25 | EtOH 78% | 70 | KOH | 60 | 85 |
| 11 | 25 | EtOH 78% | 70 | K3PO4·3H2O | 60 | 78 |
| 12 | 25 | EtOH 78% | 70 | CH3COONa | 60 | 25 |
| 13 | 25 | EtOH 78% | 70 | Et3N | 60 | 50 |
| 14 | 25 | EtOH 78% | 70 | Piperidine | 60 | 30 |
| 15 | 25 | EtOH 78% | 70 | Na2CO3 | 60 | 68 |
| 16 | 25 | DMSO | 70 | K2CO3 | 60 | — |
| 17 | 25 | DMF | 70 | K2CO3 | 60 | 28 |
| 18 | 25 | 1,2-Dichloromethane CH2Cl2 | 40 | K2CO3 | 60 | 25 |
| 19 | 25 | MeCN | 70 | K2CO3 | 60 | 10 |
| 20 | 25 | PhCH3 | 70 | K2CO3 | 60 | 30 |
| 21 | 25 | NMP | 70 | K2CO3 | 60 | 35 |
| 22 | 25 | THF | 70 | K2CO3 | 60 | 20 |
Conditions for the chemical reaction: 1 mmol of iodobenzene and 1 mmol of phenylboronic acid.
The optimization test also was run for the Heck and Sonogashira reactions in the company of the proposed catalyst. Tables S1 and S2† summarize the result of this investigation for the Heck and Sonogashira reactions, respectively. The reaction of iodobenzene and styrene was chosen as the template reaction for the Heck reaction, and the reaction of iodobenzene and phenylacetylene was selected as the template reaction for the Sonogashira reaction. According to this study, using H2O as the solvent and the K2CO3 as the base in the company of 30 mg of UiO-66-NH2@β-CD/PdNPs as the catalyst at 80 °C after 120 min resulted in the best yield for the Heck reaction. The same study indicates that the optimum condition for the progress of the Sonogashira reaction is 20 mg of catalyst in H2O in the company of K2CO3 at 50 °C and 15 min of reaction time.
After determining the optimal conditions, we test the generalizability of our proposed technique by synthesizing several biphenyl derivatives from diverse precursors. In this context, several aryl halides and aryl boronic acids interacted in the company of the proposed catalyst and under optimal circumstances. Table 2 provides a summary of this study's findings. The data in this table shows that the reaction proceeds with high yields when aryl halides are present. This is true whether the aryl halide is located in the aromatic ring's ortho, meta, or para position. As can be seen in the table below, using an aliphatic halide still produced a high-quality harvest. As demonstrated by its ability to catalyze the Suzuki coupling process in the presence of iodo, bromo, and chloro derivatives of aromatic compounds, the suggested catalyst is highly effective and broadly applicable. The generality of our proposed method for the Heck and Sonogashira reactions also were conducted, and the outcomes are represented in Tables S3 and S4,† respectively. This study indicated that UiO-66-NH2@β-CD/PdNPs promote these reactions in good to excellent yields, albeit the yields of some products of the Sonogashira reaction were relatively lower.
Preparation of various organic compounds by Suzuki coupling reaction under optimum conditionsa.
| |||||
|---|---|---|---|---|---|
| Entry | Aryl boronic acid | Aryl halide | Product | Time (min) | Yield (%) |
| 1 |
|
|
|
60 | 92 |
| 2 |
|
|
|
60 | 95 |
| 3 |
|
|
|
60 | 97 |
| 4 |
|
|
|
60 | 99 |
| 5 |
|
|
|
60 | 77 |
| 6 |
|
|
|
92 | 92 |
| 7 |
|
|
|
60 | 95 |
| 8 |
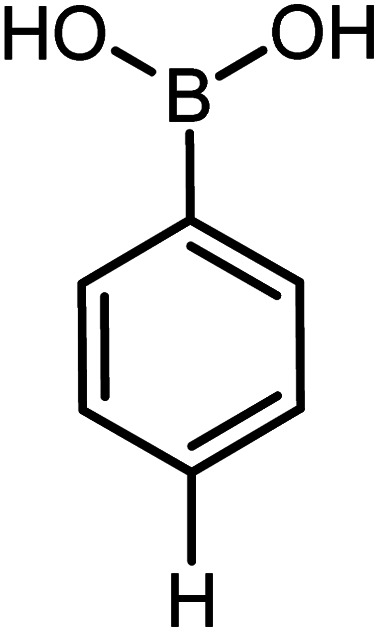
|
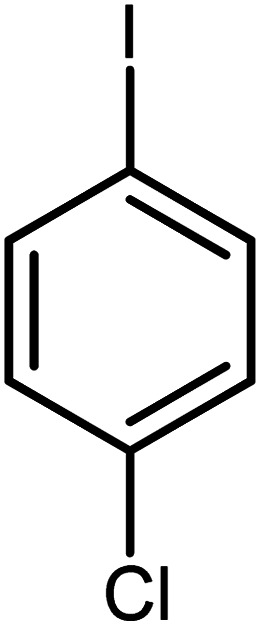
|
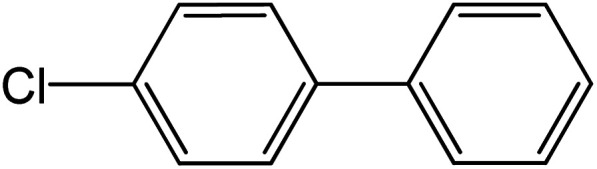
|
60 | 99 |
| 9 |
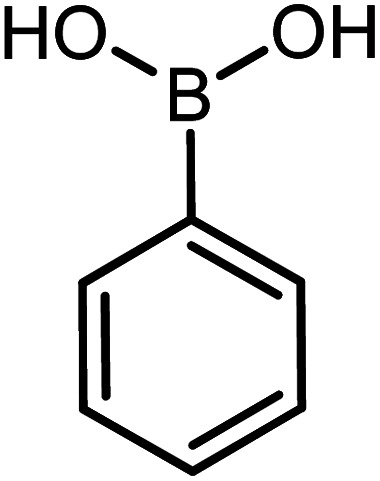
|
|
|
60 | 98 |
| 10 |
|
|
|
60 | 99 |
| 11 |
|
|
|
60 | 98 |
| 12 |
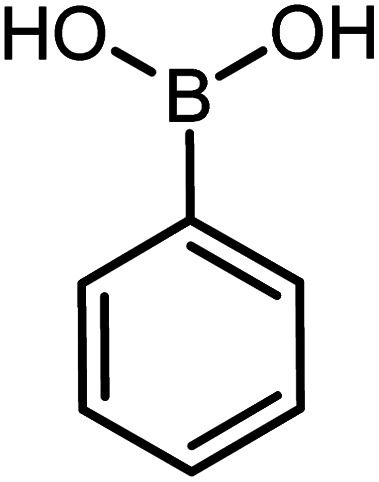
|
|
|
73 | 99 |
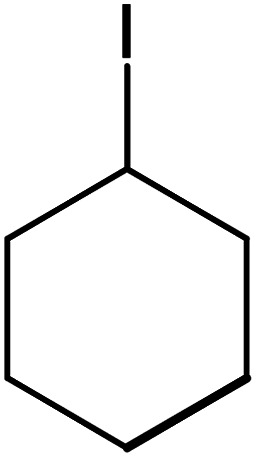
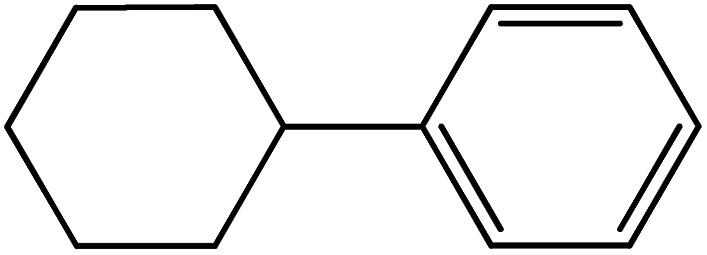
| 13 |
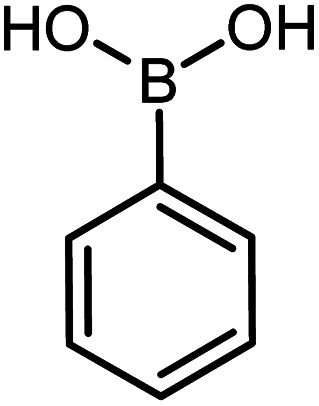
|
|
|
60 | 76 |
| 14 |
|
|
|
60 | 95 |
| 15 |
|
|
|
60 | 90 |
| 16 |
|
|
|
60 | 98 |
| 17 |
|
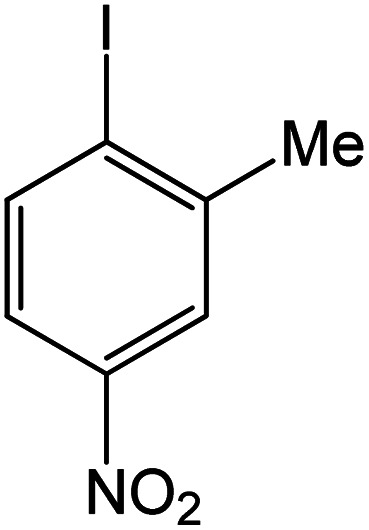
|
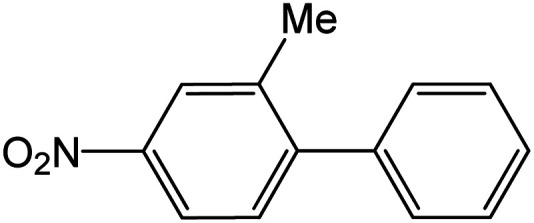
|
60 | 96 |
| 18 |
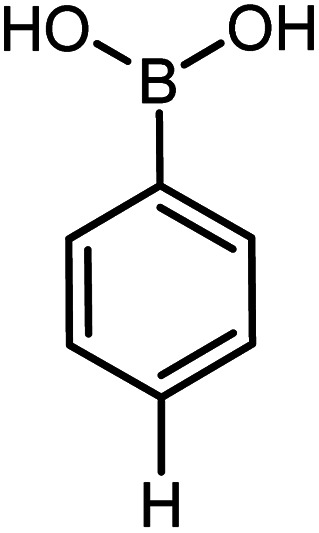
|
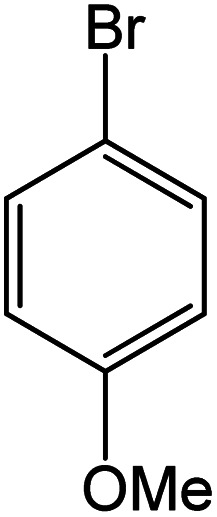
|
|
90 | 99 |
| 19 |
|
|
|
60 | 97 |
| 20 |
|
|
|
60 | 95 |
| 21 |
|
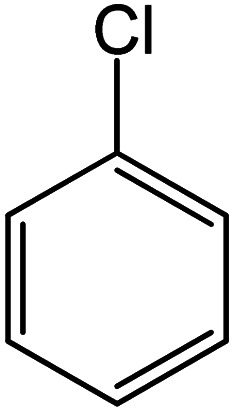
|
|
93 | 35 |
Conditions for the chemical reaction: 1 mmol of aryl halide, 1 mmol of aryl boronic acid, 70 °C, EtOH 78%, K2CO3.
The catalyst's reusability is a key feature of its scaled applications because it allows multiple runs of the same reaction. To test how well our proposed catalyst recycles, we filtered it out of the reaction liquid and washed it multiple times with ethyl acetate. We then investigate the effectiveness of the recycled catalyst for the model Suzuki coupling reaction for a total of ten cycles. In order to create the runs of the recycled catalyst, after final each reaction, the reaction mixture with the catalyst is centrifuged, to separate the catalyst on a strainer, wash with hot distilled water and hot ethanol until the product dissolves, as a result the catalyst remains on the filter to follow continue runs. As shown in Fig. 5, the catalyst's performance did not drop after three cycles; after six runs, it still performed at a level greater than 90% of its initial use. The exceptional reusability of the proposed catalyst may be attributed to the great water resistance of UiO-66-NH2 in aquatic environments. Moreover, the PSM of the UiO-66-NH2 prevents the accessibility of water molecules to the zirconium SBU by filling the pores of MOF and increasing its stability. As Fig. S1† indicates, the UiO-66-NH2@β-CD/PdNPs show excellent reusability for the Heck and Sonogashira coupling reactions up to 6 cycles.
Fig. 5. The recyclability of UiO-66-NH2@β-CD/PdNPs catalyst for the Suzuki coupling reaction.

The model reaction between iodobenzene and phenylboronic acid in the presence of UiO-66-NH2@β-CD/PdNPs is selected as the template reaction in order to perform 99% production. So, according to these investigations, the highest yields are achieved with 25 mg of UiO-66-NH2@β-CD/PdNPs catalyst at 70 °C in EtOH after 60 minutes. Also, the same test is recommended to determine the heterogeneous nature of the catalyst. The reaction conditions were similar for the two reaction vessels. Halfway through the reaction time, the catalyst was removed from one of the reaction vessels and the reaction continued without catalyst for 45 minutes. And the rate of reaction and the rate of production of products were measured. The result of the reaction for container 1, until half of the reaction of both containers 1 and 2, and the product percentage was 50% of the product after 30 minutes. However, after 60 minutes, the container containing the catalyst had 99% product and the container from which the catalyst was separated, the product percentage was the same as 50% of the product and no more product was added (Table 3).
Result of heterogenous catalyst.
| Time (min) | Container 1 | Container 2 |
|---|---|---|
| Yield (%) | Yield (%) | |
| 10 | 30 | 30 |
| 15 | 39 | 39 |
| 20 | 45 | 45 |
| 25 | 54 | 54 |
| 35 | 60 | 60 |
| 45 | 72 | 60 |
| 50 | 80 | 60 |
| 55 | 90 | 60 |
| 60 | 99 | 60 |
The Suzuki cross-coupling reaction catalytic performance of UiO-66-NH2@β-CD/PdNPs is compared to some of the published catalysts in the literature in Table 4. According to this data, our suggested catalyst has one of the highest recorded yields. This could result from meticulous post-synthesis alteration using β-cyclodextrin to alter the electrical structure of the UiO-66-NH2. The electronic structure of the UiO-66-NH2 is changed by the addition of β-cyclodextrin, which increases the palladium NPs' capacity to catalyze in the finished composite. As Table S6† indicates, the same trend happens for our proposed catalyst toward the Heck reaction.
Comparison of the catalytic performance of the proposed catalyst for the Suzuki reaction with some related reports in the literature.
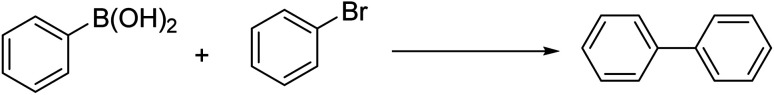
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Reaction condition | Time (min) | Yield (%) | Ref. |
| 1 | Pd–starch-2 0.1 g | Microwave 300 W, 130 °C | 2 | 99 | 25 |
| 2 | Fe3O4@CS–starch/Pd 0.1 mol% | H2O : EtOH, K2CO3, 40 °C | 15 | 98 | 26 |
| 3 | Poly(NIPAM-co-4-VP)–Pd (1) | K2CO3, H2O, 60 °C | 60 | 95 | 27 |
| 4 | GO/Fe3O4/PAMPS/Pd (0.4) | K2CO3, EtOH : H2O, 80 °C | 120 | 96 | 28 |
| 5 | MWCNTs/Pd (0.5) | K2CO3, EtOH : H2O, 60 °C | 180 | 99.2 | 29 |
| 6 | PNIPAM–HNT–Pd (1) | K2CO3, H2O, 70 °C | 90 | 97 | 30 |
| 7 | Pd NPs–HNG | K2CO3, EtOH : H2O, 60 °C | 150 | 94 | 31 |
| 8 | GO–2N–Pd(ii) (0.5) | K2CO3, H2O, 80 °C | 240 | 82 | 32 |
| 9 | G/MWCNTs/Pd (0.5) | K2CO3, EtOH : H2O, 60 °C | 60 | 97 | 33 |
| 10 | GO–NH2–Pd(ii) (1) | K2CO3, EtOH : H2O, 80 °C | 240 | 73 | 34 |
| 11 | Poly(NIPAM-4-VP-AC)–Pd (0.05) | K2CO3, H2O, Ar, 90 °C | 60 | 92 | 35 |
| 12 | GO–NHC–Pd(ii) (0.25) | K2CO3, EtOH : H2O, 60 °C | 180 | 93 | 36 |
| 13 | GO–PEG–imidazole–Pd | K2CO3, H2O, 80 °C | 5 | 93 | 37 |
| 14 | GA–FSNP@Pd (0.28 mol%) | Solvent free, 90 °C | 30 | 88 | 38 |
| 15 | GO–NHC–Pd (1 mol%) | DMF : H2O (1 : 1), 50 °C | 60 | 94 | 39 |
| 16 | Pd-NPs/P, kurdica gum | H2O, K2CO3, 60 °C | 40 | 98 | 40 |
| 17 | UiO-66-NH2@β-CD/PdNPs 0.025 g | K2CO3, EtOH 78%, 70 °C | 40 | 99 | This study |
Conclusion
We synthesized an UiO-66-NH2 and performed its functionalization with a β-cyclodextrin chemical molecule using a post-synthesis modification technique. The resultant composite was used as a support for heterogenizing Pd NPs. UiO-66-NH2@β-CD/PdNPs was characterized by XRD, SEM, TEM, EDS, and elemental mapping, demonstrating its effective preparation. Using the generated catalyst, three C–C coupling reactions, including the Suzuki, Heck, and Sonogashira coupling reactions, were catalyzed. As a consequence of the PSM, the performance of the suggested catalyst has been enhanced. Additionally, the proposed catalyst was reusable up to six times.
Conflicts of interest
The authors declare no competing interests.
Supplementary Material
Acknowledgments
The authors would like to thank the Materials and Energy Research Center (Grant No. 9911940) for the financial support of this project.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra08347g
References
- Ahadi A. Rostamnia S. Panahi P. Wilson L. D. Kong Q. An Z. Shokouhimehr M. Catalysts. 2019;9:140–150. doi: 10.3390/catal9020140. [DOI] [Google Scholar]
- Rostamnia S. Xin H. Appl. Organomet. Chem. 2013;27:348–352. doi: 10.1002/aoc.2986. [DOI] [Google Scholar]
- Devendar P. Qu R.-Y. Kang W.-M. He B. Yang G.-F. J. Agric. Food Chem. 2018;66:8914–8934. doi: 10.1021/acs.jafc.8b03792. [DOI] [PubMed] [Google Scholar]
- Rostamnia S. Rahmani T. Appl. Organomet. Chem. 2015;29:471–474. doi: 10.1002/aoc.3319. [DOI] [Google Scholar]
- Diccianni J. B. Diao T. Trends Chem. 2019;1:830–844. doi: 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]
- Alamgholiloo H. Rostamnia S. Hassankhani A. Khalafy J. Baradarani M. M. Mahmoudi G. Liu X. Appl. Organomet. Chem. 2018;32:e4539. doi: 10.1002/aoc.4539. [DOI] [Google Scholar]
- Sherwood J. Clark J. H. Fairlamb I. J. S. Slattery J. M. Green Chem. 2019;21:2164–2213. doi: 10.1039/C9GC00617F. [DOI] [Google Scholar]
- Rostamnia S. Lamei K. Pourhassan F. RSC Adv. 2014;4:59626–59631. doi: 10.1039/C4RA09157D. [DOI] [Google Scholar]
- Trzeciak A. M. Augustyniak A. W. Coord. Chem. Rev. 2019;384:1–20. doi: 10.1016/j.ccr.2019.01.008. [DOI] [Google Scholar]
- Amini M. Bagherzadeh M. Rostamnia S. Chin. Chem. Lett. 2013;24:433–436. doi: 10.1016/j.cclet.2013.03.025. [DOI] [Google Scholar]
- Taghavi R. Rostamnia S. Chem. Methodol. 2022;6:629–638. [Google Scholar]
- Yang Q. Xu Q. Jiang H. L. Chem. Soc. Rev. 2017;46:4774–4808. doi: 10.1039/C6CS00724D. [DOI] [PubMed] [Google Scholar]
- Sadeghi S. H. Yaghoobi M. Ghasemzadeh M. A. Appl. Organomet. Chem. 2022;36:e6771. doi: 10.1002/aoc.6771. [DOI] [Google Scholar]
- Sadeghi S. H. Neamani S. Moradi L. Polycyclic Aromat. Compd. 2022:1–17. [Google Scholar]
- Wu N. Nanoscale. 2018;10:2679–2696. doi: 10.1039/C7NR08487K. [DOI] [PubMed] [Google Scholar]
- Ghasempour H. Wang K.-Y. Powell J. A. ZareKarizi F. Lv X.-L. Morsali A. Zhou H.-C. Coord. Chem. Rev. 2021;426:213542. doi: 10.1016/j.ccr.2020.213542. [DOI] [Google Scholar]
- Taghavi R. Rostamnia S. Farajzadeh M. Karimi-Maleh H. Wang J. Kim D. Jang H. W. Luque R. Varma R. S. Shokouhimehr M. Inorg. Chem. 2022;61:15747–15783. doi: 10.1021/acs.inorgchem.2c01939. [DOI] [PubMed] [Google Scholar]
- Zhou H. C. J. Kitagawa S. Chem. Soc. Rev. 2014;43:5415–5418. doi: 10.1039/C4CS90059F. [DOI] [PubMed] [Google Scholar]
- Czaja A. U. Trukhan N. Müller U. Chem. Soc. Rev. 2009;38:1284–1293. doi: 10.1039/B804680H. [DOI] [PubMed] [Google Scholar]
- Safaei M. Foroughi M. M. Ebrahimpoor N. Jahani S. Omidi A. Khatami M. TrAC, Trends Anal. Chem. 2019;118:401–425. doi: 10.1016/j.trac.2019.06.007. [DOI] [Google Scholar]
- Mandal S. Natarajan S. Mani P. Pankajakshan A. Adv. Funct. Mater. 2021;31:2006291. doi: 10.1002/adfm.202006291. [DOI] [Google Scholar]
- Bai Y. Dou Y. Xie L. H. Rutledge W. Li J. R. Zhou H. C. Chem. Soc. Rev. 2016;45:2327–2367. doi: 10.1039/C5CS00837A. [DOI] [PubMed] [Google Scholar]
- Yuan S. Qin J. S. Lollar C. T. Zhou H. C. ACS Cent. Sci. 2018;4:440–450. doi: 10.1021/acscentsci.8b00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S. Qin J.-S. Lollar C. T. Zhou H.-C. ACS Cent. Sci. 2018;4:440–450. doi: 10.1021/acscentsci.8b00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budarin V. L. Clark J. H. Luque R. Macquarrie D. J. White R. J. Green Chem. 2008;10:382–387. doi: 10.1039/B715508E. [DOI] [Google Scholar]
- Veisi H. Joshani Z. Karmakar B. Tamoradi T. Heravi M. M. Gholami J. Int. J. Biol. Macromol. 2021;172:104–113. doi: 10.1016/j.ijbiomac.2021.01.040. [DOI] [PubMed] [Google Scholar]
- Lee Y. Hong M. C. Ahn H. Yu J. Rhee H. J. Organomet. Chem. 2014;769:80–93. doi: 10.1016/j.jorganchem.2014.07.013. [DOI] [Google Scholar]
- Asadi S. Sedghi R. Heravi M. M. Catal. Lett. 2017;8:2045–2056. doi: 10.1007/s10562-017-2089-2. [DOI] [Google Scholar]
- Nie R. Shi J. Du W. Hou Z. Appl. Catal., A. 2014;473:1–6. doi: 10.1016/j.apcata.2013.12.029. [DOI] [Google Scholar]
- Hong M. C. Ahn H. Choi M. C. Lee Y. Kim J. Rhee H. Appl. Organomet. Chem. 2014;3:156–161. doi: 10.1002/aoc.3103. [DOI] [Google Scholar]
- Movahed S. K. Dabiri M. Bazgir A. Appl. Catal., A. 2014;488:265–274. doi: 10.1016/j.apcata.2014.09.045. [DOI] [Google Scholar]
- Bai C. Zhao Q. Li Y. Zhang G. Zhang F. Fan X. Catal. Lett. 2014;9:1617–1623. doi: 10.1007/s10562-014-1299-0. [DOI] [Google Scholar]
- Song H. Zhu Q. Zheng X. Chen X. J. Mater. Chem. A. 2015;3:10368–10377. doi: 10.1039/C5TA00280J. [DOI] [Google Scholar]
- Shang N. Feng C. Zhang H. Gao S. Tang R. Wang C. Wang Z. Catal. Commun. 2013;40:111–115. doi: 10.1016/j.catcom.2013.06.006. [DOI] [Google Scholar]
- Zhang Y. Yang J. Zhang X. Bian F. Yu W. React. Funct. Polym. 2012;4:233–241. doi: 10.1016/j.reactfunctpolym.2012.02.010. [DOI] [Google Scholar]
- Shang N. Gao S. Feng C. Zhang H. Wang C. Wang Z. RSC Adv. 2013;3:21863–21868. doi: 10.1039/C3RA44620D. [DOI] [Google Scholar]
- Heravi M. M. Asadi S. Hoseini Chopani S. M. Jaderi E. Appl. Organomet. Chem. 2020;34:e5805. doi: 10.1002/aoc.5805. [DOI] [Google Scholar]
- Tanhaei M. Mahjoub A. Nejat R. Catal. Lett. 2018;148:1549–1561. doi: 10.1007/s10562-018-2347-y. [DOI] [Google Scholar]
- Movahed S. K. Esmatpoursalmani R. Bazgir A. RSC Adv. 2014;28:14586–14591. doi: 10.1039/C3RA46056H. [DOI] [Google Scholar]
- Veisi H. Faraji A. R. Hemmati S. Gil A. Appl. Organomet. Chem. 2015;8:517–523. doi: 10.1002/aoc.3325. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.