

Abstract
Transmembrane P1B-type ATPase pumps catalyze the extrusion of transition metal ions across cellular lipid membranes to maintain essential cellular metal homeostasis and detoxify toxic metals. Zn(ii)-pumps of the P1B-2-type subclass, in addition to Zn2+, select diverse metals (Pb2+, Cd2+ and Hg2+) at their transmembrane binding site and feature promiscuous metal-dependent ATP hydrolysis in the presence of these metals. Yet, a comprehensive understanding of the transport of these metals, their relative translocation rates, and transport mechanism remain elusive. We developed a platform for the characterization of primary-active Zn(ii)-pumps in proteoliposomes to study metal selectivity, translocation events and transport mechanism in real-time, employing a “multi-probe” approach with fluorescent sensors responsive to diverse stimuli (metals, pH and membrane potential). Together with atomic-resolution investigation of cargo selection by X-ray absorption spectroscopy (XAS), we demonstrate that Zn(ii)-pumps are electrogenic uniporters that preserve the transport mechanism with 1st-, 2nd- and 3rd-row transition metal substrates. Promiscuous coordination plasticity, guarantees diverse, yet defined, cargo selectivity coupled to their translocation.
Transmembrane P1B-2 type ATPase pumps catalyze thepromiscuous extrusion of transition metal ions across cellular membranes by plastic recognition and electrogenic translocation of diverse metal substrates.
In all kingdoms of life, transmembrane metal pumps belonging to the P1B-type ATPase family are key primary active transporters responsible for the extrusion across cellular membranes of essential metal ions (e.g. Cu+, Zn2+) when exceeding cellular quotas, as well as toxic transition and post-transition metal ions (e.g. Cd2+, Pb2+, Hg2+) upon exposure and accumulation.1–6 Zn(ii)-pumps of to the P1B-2 subclass, can transport not only Zn2+ ions, but potentially other toxic metals, by utilizing the energy generated by ATP hydrolysis to catalyze substrate translocation across the lipid bilayers against electrochemical gradients.7–16 As for all P-type ATPases,17–19 Zn2+-pumps transport substrate following the Post-Albers catalytic cycle scheme in which the transporter structure alternates between the E1 and E2 states open to different faces of the membrane, permitting substrate access, binding to the transmembrane domain, and release to opposite sides of the phospholipid bilayer, via specific ATP-dependent conformational changes (Fig. 1).2,20 Metal binding to the membrane (M) domain, cargo transmembrane occlusion, and metal release are achieved through the concerted action of the M-domain with a soluble nucleotide binding domain (N-domain), responsible for ATP binding and hydrolysis, a phosphorylation domain (P-domain, which undergoes catalytic auto-phosphorylation subsequent to ATP hydrolysis), and an actuator module (A-domain) that transduces the energy derived from ATP hydrolysis to the transmembrane region allowing conformational changes to occur, as well as contribute to dephosphorylation.17,18 Thus, metal ion(s) bind to the high-affinity transmembrane site(s) in the E1 state, an inward-open conformation towards the cytosol, and are occluded in the transmembrane domain upon ATP hydrolysis and auto-phosphorylation (E1P state) at a conserved aspartate residue of the DKTGTLT motif, characteristic of all P-type ATPases, in their cytosolic phosphorylation domain. Upon conformational changes leading to the outward-open E2P state, substrate ions are released to the opposite side of the membrane, thereby triggering dephosphorylation (E2 state), with the help of the A-domain in the E2P → E2 → E1 transition, to regenerate the E1 state. The structure and catalytic scheme permit high-thermodynamic stability in the substrate binding site necessary for metal recognition and binding, as well as high kinetic lability to guarantee metal release through the transport cycle with a high turnover rate.2,4
Fig. 1. The catalytic Post-Albers cycle. Schematic representation of the catalytic Post-Albers cycle in P-Type ATPases exemplified for a Zn(ii)-pump. The transmembrane domain featuring the transmembrane high-affinity M2+ (M2+ = Zn2+, Cd2+, Hg2+, and Pb2+) binding-site is shown in grey color. The A-, P-, and N-domains are shown in yellow, blue and red, respectively. The two N-terminal helices (TM-A and -B) characteristic of P1B-type ATPases, the soluble N-terminal metal binding domain and His-rich terminal tail are colored in blue (based on CmZntA topology). Movements of the corresponding domains based on the catalytic cycle of SERCA are indicated by colored arrows.

P1B-type ATPases transmembrane (M) domain possess a common architecture composed of 8 transmembrane helices (MA, -MB, and M1-6), featuring signature sequences involved in intramembrane substrate recognition,2,4,5,21–24 as also revealed by crystal structures and XAS studies of Cu(i)-pumps, Zn(ii)-pumps and Zn(ii)/Co(ii)-pumps.25–32 P1B-type ATPases subfamilies feature distinct cargo selectivity and exploit a common topological framework, to catalyze the selective transport of diverse 1st-, 2nd-, and 3rd-row transition and post transition metals.1 The cargo specificity patterns correlate with conserved amino acid motifs present in transmembrane helices M4, M5 and M6. These motifs provide ligands for coordination of substrate ions into high-affinity transmembrane metal binding site(s) (TM-MBS) in the M-domain. Thus, P1B-ATPases are classified into subclasses based on these conserved sequences, P1B-1 (Cu+/Ag+ pumps), P1B-2 (Zn2+/Cd2+/Pb2+), P1B-3 (Cu+/Cu2+), P1B-4 (Zn2+/Co2+/Cd2+ pumps), P1B-5 (Ni2+/Fe2+(?)) and P1B-6/-7-types (unknown selectivity).5,6,24,32
High-resolution structural studies on metal-free Zn2+-pumps in the substrate-free E2P states and biochemical investigations of P1B-2-type ATPase homologs from E. coli and S. sonnei (EcZntA and SsZntA) established the framework by which Zn-pumps perform their function.7–9,11–13,20,28,33–35 Key amino acids present on transmembrane helices M4, M5 and M6 are critical for Zn2+ ion binding and formation of the putative high affinity transmembrane binding site. In SsZntA, Cys392, Cys394 and Asp714 have been proposed to act as transmembrane ligands for metal binding, while the conserved Lys693 acts as a built-in counter-ion potentially overcoming the necessity of the involvement of counter-ions to be co-transported during the transport catalytic cycle.20 Recently, via X-ray absorption spectroscopy and biochemical characterization, we revealed that “coordination plasticity” in metal selection by the Cys and Asp residues on M4 and M6 governs the substrate recognition and binding promiscuity (Zn2+, Cd2+, Hg2+ and Pb2+) in a Zn2+-pump from P. aeruginosa (PaZntA).28 Yet, the transport of all these metals has not been demonstrated on purified systems and the molecular level understanding of the translocation mechanism for the metals that stimulate ATPase activity, acting as putative substrates, and the relative substrate translocation rates under turnover remain poorly understood. In this work, the Zn2+ P1B-2-type ATPase pump (ZntA) from Cupriavidus metallidurans, a homologue of PaZntA and SsZntA has been selected to investigate the metal cargo selection and translocation mechanism with all its substrates. C. metallidurans is a Gram-negative bacterium that is adapted to survive under heavy metal stress and overexpresses several P1B-type ATPases, including ZntA, to guarantee that toxic metals are efficiently extruded from the cell cytosol. Thus, CmZntA P1B-type ATPases are ideal candidates to study metal pumps involved in metal detoxification.36,37
In this present study we developed a platform based on a diverse set of “turn-on” fluorescence metal detector probes, encapsulated in the CmZntA proteoliposome lumen to monitor 1st-, 2nd- and 3rd-row transition and post-transition metal transport events across lipid bilayers in real-time. In parallel, we monitored putative secondary-ion translocation with pyranine (a pH sensitive probe), and we studied the development of transmembrane potential during substrate(s) translocation with Oxonol VI, a fluorescent reporter probe sensitive to membrane potential changes. By coupling the obtained information with metal-dependent ATPase activity assays and Extended X-ray Absorption Fine Structure (EXAFS) analysis to probe the metal coordination promiscuity in the transmembrane selection site, we demonstrate that, despite differences in coordination, Zn2+-pumps follow a common electrogenic uniporter transport mechanism for all substrates that are actively translocated (Zn2+, Cd2+, Hg2+, and Pb2+), and that vectorial cargo transport is not coupled to proton-counter transport. These promiscuity, coordination plasticity, and translocation mechanism differentiate Zn(ii)-pumps form other well characterized P-type ATPases (e.g., the sarcoendoplasmic Ca2+/H+ pump – SERCA), that function as antiporters.17–19,38–44
Materials and methods
CmΔZntA166–794 recombinant expression and purification
The DNA coding the sequence for CmΔZntA166–794 lacking the N-terminal soluble HMBD and including a N-terminal hexa-histidine tag was generated by polymerase chain reaction (PCR) from the cDNA coding for full-length ZntA from Cupriavidus metallidurans (Uniprot ID: Q1LEH0). The PCR product was cloned into a modified pET19b vector containing a thrombin cleavage site to cleave the poly-histidine tag. The plasmid construct was transformed in E. coli BL21 (DE3) GOLD competent cells for recombinant protein expression. The transformed cells were grown at 37 °C in Terrific Broth (TB) media supplemented with glycerol containing 50 μg mL−1 ampicillin until the OD600 reached a value of 2. Cell cultures were then cooled at 23 °C and protein expression was induced by adding isopropyl thiogalactopyranoside (IPTG) to a final concentration of 0.3 mM. The cell cultures were then incubated at 23 °C for 18 h and harvested by centrifugation (20 min, 4 °C, 14 000 g, Thermo Scientific Sorvall LYNX 6000 centrifuge). Cells were resuspended to a final cell concentration of 0.1 g cells mL−1 in lysis buffer (20 mM Tris/HCl pH = 8, 150 mM NaCl, 5 mM MgCl2, supplemented with DNase I from bovine pancreas (30 μg mL−1; Sigma-Aldrich) and an EDTA-free protease inhibitor cocktail (1 tablet per 250 mL; Roche). Cells were lysed by passing three-times through a microfluidizer (Microfluidics M-110P) at a pressure of 20 000 psi. The cell debris was removed via centrifugation (20 min, 4 °C, 20 000 g Thermo Scientific Sorvall LYNX 6000 centrifuge). The membrane fraction was then collected by ultracentrifugation (1 h, 4 °C, 205 100 g, Beckman Optima XPN80) and resuspended to a final concentration of 1 g of original cells per mL of buffer (20 mM Tris/HCl pH = 8, 500 mM NaCl, 1% (w/v) glycerol, EDTA-free protease inhibitor cocktail (1 tablet per 50 mL; Roche)). The resuspended membrane fraction was frozen in liquid N2 and stored at −80 °C until purification.
CmΔZntA166–794 was purified by Ni-NTA affinity chromatography using a 5 mL His-trap column (GE Healthcare). Typically, a 10 mL membrane suspension was diluted to 50 mL in ice-cold extraction buffer (20 mM Tris HCl pH = 8, 500 mM NaCl, 25 mM imidazole, 5 mM β-mercaptoethanol, and EDTA-free protease inhibitor cocktail (1 tablet per 50 mL Roche)). Membrane proteins were extracted by addition of 1% (w/v) 7-cyclohexyl-1-heptyl-β-d-maltoside (Cymal-7) detergent, upon stirring for 1 h at 4 °C. The remaining membranes and protein aggregates were removed by ultracentrifugation (30 min, 4 °C, 205 100 g, Beckman Optima XPN80) and the extracted proteins were collected in the supernatant.
The supernatant was subsequently loaded onto a 5 mL Ni-NTA affinity chromatographic column connected to AKTA pure FPLC system pre-equilibrated with binding buffer (20 mM Tris/HCl pH = 8, 500 mM NaCl, 25 mM imidazole, 0.05% (w/v) Cymal-7, 1 mM dithiothreitol (DTT)) and washed with the same buffer (50 CV) to remove impurities and unbound proteins. CmΔZntA166–794 was subsequently eluted in elution buffer (8 CV; 20 mM Tris/HCl, pH = 8, 500 mM NaCl, 500 mM imidazole, 0.05% (w/v) Cymal-7, 1 mM DTT). Imidazole was immediately removed by injecting the collected sample into a Hi prep 26/10 desalting column connected to the AKTA pure FPLC and equilibrated with desalting buffer (20 mM MOPS/NaOH pH = 7, 500 mM NaCl, 0.05% (w/v) Cymal-7, 1 mM DTT) for reconstitution in proteoliposomes and ATPases assays, or 20 mM Tris pH = 8, 500 mM NaCl, 0.05% (w/v) Cymal-7, 1 mM DTT, 1 mM EDTA, for poly-histidine tag removal with thrombin cleavage and XAS sample preparation. Thrombin cleavage was conducted at 4 °C for 22 h by diluting the protein to a final concentration of 0.2 mg mL−1 and adding restriction grade purified human thrombin (Novagen) to a final concentration of 0.25–0.5 U mL−1. The purified protein was concentrated to a final concentration of ∼5–10 mg mL−1 with a 100 kDa molecular weight cut-off (MWCO) membrane filters using Amicon spinning devices. Low molecular weight impurities or any protein aggregates were removed, and the monodispersed protein fraction was collected by loading the concentrated protein onto a Superdex 200 10/300 size exclusion chromatography column and eluted with argon-saturated 20 mM Tris/HCl pH = 8, 500 mM NaCl, 1 mM DTT, 0.05% (w/v) Cymal-7 or 20 mM or with MOPS/NaOH pH = 7, 100 mM NaCl, 1 mM DTT 0.05% (w/v) Cymal-7 to incorporate protein into proteoliposomes. All protein concentrations were determined by absorption spectroscopy at 280 nm with the calculated extinction coefficient ε280 = 65 430 M−1 cm−1. Samples throughout the purification steps and purity of the final protein was confirmed by SDS-PAGE gel electrophoresis (4–15% Tris-Glycine Mini-PROTEAN gels, BioRad).
CmΔZntA166–794 reconstitution in proteoliposomes (small unilamellar vesicles – SUVs)
Purified CmΔZntA166–794 in Cymal-7 micelles was reconstituted in small unilamellar vesicles (SUVs) prepared at 1 : 25 (w/w) protein-to-phospholipid ratio. Lipids selected for liposome preparation were E. coli polar lipid extract and l-α-phosphatidylcholine (from chicken egg; Avanti Polar Lipids) in 3 : 1 ratio (w/w). The lipid mixture was dried under rotation in a glass balloon flask under nitrogen stream and dried in a vacuum desiccator overnight. The following day, lipids were hydrated and re-suspended in H2O, 1 mM DTT (pre-treated with Chelex-100 resin beads (Bio-Rad)). The lipid suspension was subsequently buffered to a final concentration of 20 mM MOPS/NaOH, pH 7, 100 mM NaCl, 1 mM DTT using a 10× buffer stock, to set the final total lipid concentration at 25 mg mL−1. SUVs were generated by three freeze thaw cycles of the lipid mixture in liquid nitrogen and subsequent 11 extrusions through a series of polycarbonate membranes with decreasing pore sizes (1 μm, 400 nm, and 200 nm) using a 1 mL air-tight syringe system (Avanti, Polar Lipids, Inc). SUVs were destabilized by adding Cymal-7 to a final concentration of 0.02% (w/v), and subsequent tilting for 1 h at room temperature. For the CmΔZntA166–794 reconstitution in destabilized SUVs, a concentrated protein stock (∼5–10 mg mL−1) was added to achieve a final 1 : 25 (w/w) protein : lipid ratio. Similarly, control liposomes were prepared by adding the same volume of buffer as in purified protein stocks. Both mixtures were tilted for 1 h at 4 °C. For detergent removal, Bio-Beads (SM-2; Bio-Rad) were utilized. Bio-Beads were activated by a series of sequential washing steps with methanol, ethanol and water and finally dried by vacuum filtration. Detergent removal from SUVs samples was performed by adding the pre-activated Bio-Beads to the control liposomes and proteoliposomes by tilting at 4 °C (Bio-Bead slurry concentration: 40 mg mL−1), with fresh Bio-Bead exchanges after 1, 12, 14, and 16 h. Control liposomes and proteoliposomes were then harvested by ultracentrifugation at 160 000 × g, 45 min, 4 °C (Sorvall mX120+ Micro-Ultracentrifuge). After centrifugation, the control liposome and proteoliposome pellets were re-suspended in 20 mM MOPS/NaOH, pH = 7, 100 mM NaCl, 1 mM DTT (treated with Chelex-100) to a final lipid concentration of 25 mg mL−1. The efficiency of protein incorporation in proteoliposomes was estimated by analyzing the supernatant of ultracentrifugation and re-suspended proteoliposomes by SDS-PAGE gels. Both proteoliposomes and control liposomes were flash frozen and stored at −80 °C until use in real-time transport assays. The number of CmΔZntA166–794 molecules per proteoliposome was calculated using the protein concentration, lipid concentration, and average volume of proteoliposomes based on the diameter of SUVs measured with dynamic light scattering (DLS, see below), assuming spherical shape for the proteoliposomes. The total area of lipid heads (STOT) is the total area of the inner layer (Sin) and the outer layer (Sout). Assuming the thickness of the lipid bilayer is, 3 nm (h = 3 nm), STOT can be calculated as: STOT = Sout + Sin = 4πr2 + 4π(r − h)2.
The amount of lipid molecules per SUV (NTOT) can be determined with the average surface area of a polar head, (SL) of 0.7 nm2 per phospholipid: NTOT = STOT/SL. The MW of a SUV was calculated and thereby the proteoliposome concentration was calculated, assuming the average MW of a phospholipid of 750 g mol−1. The number of CmΔZntA166–794 molecules per SUV was calculated knowing the final CmΔZntA166–794 concentration in the proteoliposomes solutions and the distribution range calculated based on the DLS vesicle diameter standard deviation. The CmΔZntA166–794 orientation distribution cannot be controlled in the reconstitution procedure.45,46 Thus, for all CmΔZntA166–794 activity determinations in proteoliposomes a 1 : 1 orientation distribution was assumed.
SUV analysis with UV-Vis dynamic light scattering
Size distribution and polydispersity of proteoliposomes and control liposomes were determined by UV-visible dynamic light scattering (DLS) on a Zetasizer Nano ZS (Malvern Panalytical) instrument. Samples were loaded in disposable micro cuvettes and size distribution were measured at 25.0 °C using a 633 nm laser wavelength, a scattering angle of 175°, using a medium refractive index of 1.33 and material refractive index of 1.51.
X-Ray absorption spectroscopy sample preparation
CmΔZntA166–794–Zn2+, CmΔZntA166–794–Cd2+, CmΔZntA166–794–Hg2+ and CmΔZntA166–794–Pb2+ in detergent micelles solutions after thrombin treatment as described above, were generated by addition of 1 molar equiv. of ZnCl2, CdCl2, HgCl2 or Pb(CH3COO)2 to the purified CmΔZntA166–794 in 20 mM Tris–HCl pH = 8, 500 mM NaCl, 0.05% (w/v) Cymal-7 and 20% (w/v) glycerol. The samples were concentrated to 0.3–0.5 mM and loaded into custom-made polycarbonate XAS sample cells (Vantec, Canada), sealed with metal-free tape, flash frozen in liquid nitrogen and stored in liquid nitrogen until data collection.
X-Ray absorption spectroscopy data collection
X-ray absorption spectroscopy (XAS) measurements were performed at the Stanford Synchrotron Radiation Light source with the SPEAR 3 storage ring of 450–500 mA at 3.0 GeV. Zinc and cadmium K-edge data, as well as lead and mercury L3-edge data were collected at beamline 7–3 with a wiggler field of 2 Tesla and employing an Si(220) double-crystal monochromator and a vertically-collimating pre-monochromator harmonic rejection mirror. Alternatively, for cadmium K-edge data collection, harmonic removal was achieved by detuning the monochromator crystal by 50%. The incident and transmitted X-ray intensities were monitored using nitrogen-filled ionization chambers and X-ray absorption was measured by monitoring the fluorescence yield using an array of 30 germanium detectors. Copper, silver, arsenic, or selenium filters were placed between the cryostat and the germanium detector to reduce scattered X-rays not associated with Zn, Cd, Hg or Pb fluorescence, respectively. During data collection, the samples were maintained at a temperature of ∼10 K using an Oxford instruments liquid helium flow cryostat. The Zn XAS spectra were measured using 10 eV steps in the pre-edge region (9430–9640 eV), 0.35 eV steps in the edge region (9640–9690 eV) and 0.05 Å−1 increments in the EXAFS region (collected up to k = 14.2 Å−1). A total of six 40 min scans were accumulated, and the energy was calibrated by reference to the absorption of a standard Zn metal foil measured simultaneously with each scan, assuming a lowest energy inflection point of the zinc foil at 9660.7 eV. The Cd XAS spectra were measured using 10 eV steps in the pre-edge region (26 500–26680 eV), 0.5 eV steps in the edge region (26 680–26,750 eV) and 0.05 Å−1 increments in the EXAFS region (to k = 14.2 Å−1). A total of five – 40 min scans were accumulated, and the energy was calibrated by reference to the absorption of a standard Cd metal foil measured simultaneously with each scan assuming a lowest energy inflection point of the cadmium foil at 26 714.0 eV. The Hg XAS spectra were measured using 10 eV and 1 eV steps in the pre-edge region (12 150–12,240 and 12 240– 12 265 eV, respectively), 0.35 eV steps in the edge region (12 265–12,350 eV) and 0.05 Å−1 increments in the EXAFS region (to k = 14.2 Å−1). A total of twelve ∼ 40 min scans were accumulated, and the energy was calibrated by reference to the absorption of a standard HgCl2 sample measured simultaneously with each scan, assuming a lowest energy inflection point of the reference at 12 285.0 eV. The Pb XAS spectra were measured using 10 eV steps in the pre-edge region (12 810–13,020 eV), 0.35 eV steps in the edge region (13 020–13,070 eV) and 0.05 Å−1 increments in the EXAFS region (to k = 13.2 Å−1). A total of eleven 40 min scans were accumulated, and the energy was calibrated by reference to the absorption of a standard lead metal foil measured simultaneously with each scan, assuming a lowest energy inflection point of the lead foil at 13 038.0 eV.
X-Ray absorption spectroscopy data analysis
The XAS data for each metal edge (Zn, Cd, Hg, and Pb) was processed and normalized using Sixpack software package.47 Fluorescence channels from each scan were visually checked and bad channels were deleted. Energy calibration for each scan of Zn K-edge was performed by assigning the first maximum in the first derivative spectrum of the Zn-foil to 9660.7 eV. The energy calibrated XAS data for Zn is an average of six scans that was normalized and corrected by setting the K-edge energy of Zn to 9670 eV and with Rbkg of 1. A linear function was used for fitting the pre-edge range of −200 to −50 eV and quadratic polynomial functions with 7–8 spline points were used to fit the post-edge range of −50 to +785 eV relative to E0. The edge jump was normalized by setting the difference between the corrected pre-edge and post-edge baselines to 1. The EXAFS data were converted to k-space using the relationship [2me (E − E0)/ħ2]1/2, where me is the mass of electron and ħ is the Plank's constant divided by 2π. The k3-weighted EXAFS data were Fourier-transformed over the k-range 0–13.5 Å−1 using a Hanning window and fit in r-space over the range 1–2.5 Å (first shell) using an S0 value of 0.9 using the Artemis software program (see below). The r-space data shown in the figures were not corrected for phase shifts.
Energy calibration for each scan of Cd K-edge was performed by assigning the first maximum in the first derivative spectrum of the Cd-foil to 26 714 eV. The energy calibrated XAS data for Cd is an average of five scans that was normalized, and corrected by setting the K-edge energy of Cd to 26 714 eV and with Rbkg of 1. A linear function was used for fitting the pre- edge range of −250 to −150 eV and quadratic polynomial functions with 7–8 spline points were used to fit the post-edge range of +150 to +788.7 eV relative to E0. The edge jump was normalized by setting the difference between the corrected pre-edge and post-edge baselines to 1. The EXAFS data were converted to k-space using the relationship [2me (E − E0)/ħ2]1/2, where me is the mass of electron and ħ is the Plank's constant divided by 2π. The k3-weighted EXAFS data were Fourier- transformed over the k-range 0–13.2 Å−1 using a Hanning window and fit in r-space over the range 1–2.6 Å (first shell) using an S0 value of 0.9 using the Artemis software program. The r-space data shown in the figures were not corrected for phase shifts.
For Hg, energy calibration for each scan of Hg L3-edge was performed by assigning the first maximum in the first derivative spectrum of the Hg-foil to 12 285 eV. The energy calibrated XAS data for Hg is an average of twelve scans that was normalized, and background corrected by setting the K-edge energy of Hg to 12 285 eV and with Rbkg of 1. A linear function was used for fitting the pre-edge range of −200 to −50 eV and quadratic polynomial functions with 7–8 spline points were used to fit the post-edge range of +150 to +880 eV relative to E0. The edge jump was normalized by setting the difference between the corrected pre-edge and post-edge baselines to 1. The EXAFS data were converted to k-space using the relationship [2me (E − E0)/ħ2]1/2, where me is the mass of electron and ħ is the Plank's constant divided by 2π. The k3-weighted EXAFS data were Fourier-transformed over the k-range 0–13.0 Å−1 using a Hanning window and fit in r-space over the range 1–2.5 Å (first shell) using an S0 value of 0.9 Artemis software program (see below). The r-space data shown in the figures were not corrected for phase shifts.
For Pb, energy calibration for each scan of Pb L3-edge was performed by assigning the first maximum in the first derivative spectrum of the Pb-foil to 13 040 eV. The energy calibrated XAS data for Pb is an average of eleven scans that was normalized, and corrected by setting the L3-edge energy of Pb to 13 040 eV and with Rbkg of 1. A linear function was used for fitting the pre-edge range of −200 to 150 eV and quadratic polynomial functions with 7–8 spline points were used to fit the post-edge range of −50 to +677.5 eV relative to E0. The edge jump was normalized by setting the difference between the corrected pre-edge and post-edge baselines to 1. The EXAFS data were converted to k-space using the relationship [2me (E − E0)/ħ2]1/2, where me is the mass of electron and ħ is the Plank's constant divided by 2π. The k3-weighted EXAFS data were Fourier-transformed over the k-range 0–12.5 Å−1 using a Hanning window and fit in r-space over the range 1–2.8 Å (first shell) using an S0 value of 0.9 using the Artemis software program (see below). The r-space data shown in the figures were not corrected for phase shifts. For all samples, no radiation damage was observed, and confirmed by no changes in the XANES spectra between the first and last data collection scans for each metal.
The Artemis software program with FEFF6 and IFEFFIT algorithm was used to generate and fit single scattering paths for each data set.48 Single – scattering fits were generated over an r-space of 1–2.5 Å (or 2.6 for Cd and 2.8 Å for Pb). The EXAFS fitting equation used was:
 |
1 |
where f(k) is the scattering amplitude, δ(k) is the phase-shift, N is the number of neighboring atoms, r is the distance to the neighboring atoms, and σ2 is a Debye–Wallace factor reflecting the mean square deviation in the distance to the nearest neighbor (thermal and static disorder).
To compare the different models fit to the data set, IFEFFIT utilizes three goodness of fit parameters: χ2, reduced χ2 and the R-factor. χ2 is given by eqn (2), where Nidp is the number of independent data points, N2 is the number of uncertainties to minimize, Re(f) is the real part of EXAFS function and Im(fi) is the imaginary part of the EXAFS fitting function.
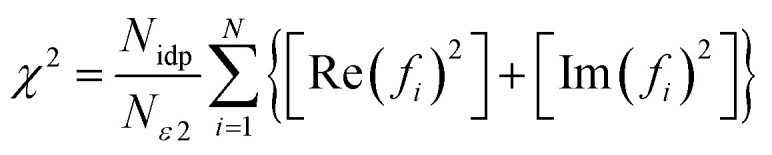 |
2 |
Reduced χ2 is given by eqn (3) where Nidp is the number of refining parameters and Nvar is the number of adjustable parameters. Additionally, IFEFFIT calculates the R-factor for the fit, which is given by eqn (4), and is scaled to the magnitude of the data making it proportional to χ2.
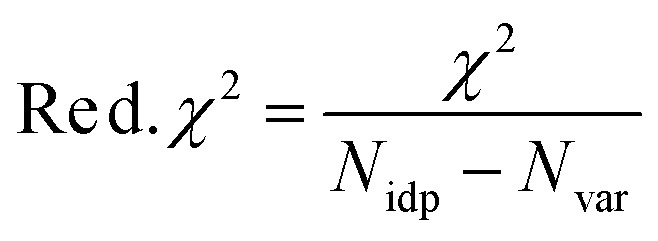 |
3 |
 |
4 |
In comparing different models, minimizing the R-factor and reduced χ2 parameter, and reasonable values of σ2 were used to determine the model that was the best fit for the data. The R-factor will generally improve with increasing number of adjustable parameters, while reduced χ2 will go through a minimum and then increase, indicating that the model is overfitting the data. The resolution of the data for each metal edge was determined by eqn (5).
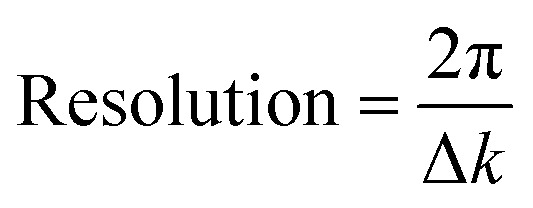 |
5 |
Determination of metal-stimulated ATPase activity of detergent-solubilized CmΔZntA166–794
MilliQ water and buffers used for all the ATPase activity assays were Chelex-treated prior to enzymatic assays. First, 40 μL reaction mixtures containing 10 μM MgCl2, 100 mM cysteine and purified CmΔZntA166–794 (0.25 mg mL−1) were placed in separate wells of a 96-well plate. To test the substrate selectivity of CmZntA, 100× stocks (final concentration = 50 μM) of selected 1st-, 2nd- and 3rd-row transition and post-transition metals (Mn2+, Co2+, Ni 2+, Cu+, Zn2+, Pb2+, Cd2+, and Hg2+) were prepared in Chelex treated MilliQ water. Metal solutions were placed in separate wells of a 96-well plate containing the CmΔZntA166–794 reaction mixtures and 10 mM ATP was subsequently added to the wells to initiate the reaction. The reaction mixture was incubated at 30 °C for 30 min with shaking at 350 rpm in a thermomixer (Eppendorf). Inorganic phosphate generated in the reaction was quantified using the Malachite Green phosphate assay. The malachite green working reagent (10 μL) from a Malachite Green phosphate assay kit (MAK 307; Sigma-Aldrich) was added to the reaction mixture after reaction completion and the developed complex was determined by measuring the absorbance at 620 nm using a Tecan Spark 20 M plate reader. Control experiments were carried out in the absence of MgCl2. The inorganic phosphate (Pi) generated in reaction mixture was determined using a series of Pi calibration solutions (0–40 μM) reacted with the working reagent from the Malachite Green phosphate assay kit as described for the samples. A calibration curve was generated by linear regression and the inorganic phosphate (Pi) generated in the reaction mixture calculated and reported as nmol Pi min−1 mgCmΔZntA−1.
In the ATPase activity assays, the following were determined: (i) the Pi in control samples lacking magnesium, which is required for ATP binding and hydrolysis by CmΔZntA166–794 – i.e. background Pi from buffer, ATP stocks, and Pi generated by ATP hydrolysis in water – which are subtracted from all samples (approx. 10 nmoL ml−1); (ii) Pi in CmΔZntA166–794 samples without metal supplementation (i.e. Pi arising from potential metal contamination and ATPase activity uncoupling); (iii) Pi corresponding to the metal-dependent ATPase activity stimulation by the different metal substrates. Only a minimal metal-independent background ATPase activity by CmΔZntA166–794 was observed (<10% compared to the maximal activity, either due to uncoupling or background metal contamination), while the metal-dependent-stimulation of CmΔZntA166–794 ATPase activity could be reliably quantified (by subtracting the background in the absence of Mg2+; ESI Fig. S1†).
Michaelis–Menten kinetic parameters for each metal ion that showed metal-stimulated ATPase activity were obtained by determining the ATPase activity as a function of increasing metal concentrations, using 100× metal stocks (ZnCl2, CdCl2, Pb(CH3COOH)2 and HgCl2) prepared in Chelex-treated MiliQ water. Metal-stock aliquots were added to the reaction mixtures in the 96 well plates to obtain final metal concentrations in the range of 1–100 μM, and the ATPase assays were carried out as described above by using the Malachite Green ATPase activity assay. Data were fitted to a Michaelis–Menten-like equation to determine the KM,M2+ and Vmax for each metal: v = (Vmax × [M2+]/(KM,M2+ + [M2+]).
Determination of metal-stimulated CmΔZntA166–794 ATPase activity in proteoliposomes
The putative substrate selectivity was determined by investigating the metal-stimulated CmΔZntA166–794 ATPase activity in proteoliposomes similarly to the protocol used for the detergent-solubilized protein. The Michaelis–Menten-like kinetic parameters for substrates showing ATPase activation were determined as a function of increasing metal concentrations using 100× metal stocks (ZnCl2, CdCl2, Pb(CH3COOH)2 and HgCl2; final concentrations 1–100 μM) in Chelex-treated MilliQ water. The proteoliposomes and control liposomes were extruded prior to the ATPase assays by sequential extrusion through 1 μm, 0.4 μm and 0.2 μm filters using a 1 mL gas-tight syringe system and diluted to obtain a protein concentration of 0.25 mg mL−1. The reaction mixtures were prepared in Eppendorf tubes as described for the detergent-solubilized protein, and the samples incubated at 30 °C with shaking at 350 rpm for 30 min (Eppendorf). The samples were subsequently centrifuged (Thermo Scientific Sorvall Legend Micro 17R) at 12 000 × g for 3 min to remove any aggregated lipids. The supernatants from the reaction mixtures were then transferred into a 96-well plate and the Malachite green working reagent from the Malachite-green phosphate assay kit was added. The developed complex was determined by measuring the absorbance at 620 nm using a Tecan Spark 20 M plate reader. Control experiments were carried out in the absence of MgCl2.
Real-time substrate transport assays in CmΔZntA166–794 proteoliposomes (Zn2+, Cd2+, Pb2+ and Hg2+)
To monitor real-time translocation of metal substrates in proteoliposomes, transport assays were conducted by encapsulating a diverse set of turn-on fluorescent probes responsive to different substrate metal ions. To monitor the kinetics of Zn2+ and Cd2+ translocation, the turn-on fluorescent probe Fluozin-3 was encapsulated in the proteoliposome and control liposome lumen. Proteoliposome and control liposomes were diluted to a lipid concentration of 12.5 mg mL−1 in transport buffer (20 mM MOPS/NaOH, pH = 7, 100 mM NaCl and 1 mM DTT). Fluozin-3 was added to a final concentration of 10 μM and the probe was encapsulated in the proteoliposome lumen by 3 freeze thaw cycles followed by sequential extrusion through membrane filters of decreasing pore sizes (1 μm, 0.4 μm and 0.2 μm). Fluozin-3-encapsulated proteoliposomes and control liposomes were centrifuged to collect the liposome pellet (160 000 g, 45 min, 4 °C; Sorvall Mx 120+ micro- ultracentrifuge). The proteoliposome and control liposomes were washed by resuspending the pellet in the proteoliposome buffer (all solutions were treated with Chelex-100 to remove any metal contaminants that can interfere with the assays). Excess Fluozin-3 was removed by an additional ultracentrifugation step and proteoliposomes were re-suspended in proteoliposome buffer (20 mM MOPS/NaOH, pH = 7, 100 mM NaCl and 1 mM DTT) to a final lipid concentration of 25 mg mL−1 ([protein] = 1 mg mL−1). For the transport assays, 120 μL proteoliposomes or control liposomes were placed in a sub-micro quartz cell (Starna Cells) and equilibrated at 30 °C. MgCl2 and ATP were subsequently added to final concentrations of 10 mM and 1 mM, respectively (from 1 M and 100 mM stocks, respectively), and the reaction mixture was mixed by pipetting. The transport reactions were triggered by addition of ZnCl2 or CdCl2 to a final concentration of 40 μM from 100× concentrated stock solutions. The time-dependent fluorescence change was monitored for 750 s in 0.5 s intervals (λexc = 480 nm, slit width = 5.00 nm; λem = 515 nm, slit width = 5.00 nm). (F − F0)/F0 was calculated using the recorded fluorescence before/immediately after the addition of Zn2+/Cd2+ as F0, using a Fluormax-4 spectrofluorometer (Horiba scientific). To determine the kinetics for Pb2+ and Hg2+, the turn-on fluorescent chelators Leadmium Green and Calcium Green were respectively encapsulated, using the same protocols as described for Fluozin-3, with the exception that Leadmium Green was encapsulated in the lumen at a final concentration of 20 μM. For Pb2+ transport the time-dependent fluorescence change was monitored for 750 s in 0.5 s intervals (λexc = 490 nm, slit width = 5.00 nm; λem = 516 nm, slit width = 5.00 nm). (F − F0)/F0 was calculated using the recorded fluorescence before/immediately after the addition of Pb2+ as F0. Prior to liposomal encapsulation acetyl groups on the Leadmium Green probe were removed by hydrolysis to make it impermeable to lipids. Leadmium Green (50 μg) was dissolved in 50 μL of DMSO, and subsequently 50 μL of methanol and 25 μL of 2 M NaOH were added. The mixture was incubated at 37 °C for 15 minutes and kept at room temperature for additional 30 min. Subsequently, 2 M HCl (25 μL) was added to the mixture to neutralize the pH. For Hg2+ transport the time-dependent fluorescence change was monitored for 750 s in 0.5 s intervals (λexc = 506 nm, slit width = 5.00 nm; λem = 531 nm, slit width = 5.00 nm). (F − F0)/F0 was calculated using the recorded fluorescence before/immediately after the addition of Hg2+ as F0. For all transport assays, control experiments were performed on CmΔZntA166–794 proteoliposomes as described above but in the absence of Mg2+ or ATP. A separate set of control experiments were also performed using the same procedure with control liposomes (liposomes without protein). All kinetic traces were collected with at least 3 replicates per sample.
Calibration of fluorescent chelator probes via titration with Zn2+, Cd2+, Pb2+ and Hg2+
Zn2+, Cd2+, Pb2+ and Hg2+ stock solutions were freshly prepared prior to the measurements. Each fluorescence chelator was titrated with the respective metal ion in transport buffer (20 mM MOPS/NaOH, pH = 7, 100 mM NaCl and 1 mM DTT), in the presence of 12.5 mg mL−1 control liposomes. Metal titrations were carried out in a 1 mL sub-micro quartz fluorometer cell (Starna Cells) with a Fluoromax-4 spectrofluorometer (Horiba scientific). For Fluozin-3 (λexc = 480 nm, slit width = 1.0 nm; λem = 515 nm, slit width = 1.0 nm) and Calcium Green (λexc = 506 nm, slit width = 1.0 nm; λem = 531 nm, slit width = 1.0 nm) 10 μM solutions were used, while for Leadmium Green (λexc = 490 nm, slit width = 1.0 nm; λem = 520 nm, slit width = 1.0 nm) a final concentration of 20 μM was utilized. For each titration, a solution containing the fluorescent probe (120 μL) was placed in the quartz cell and the initial fluorescence measured for 50 s. After 50 s, the metal (M2+) solutions (1.2 μL from 100 × stocks) at increasing concentration (0–50 μM) were added and resulting fluorescence intensity change was measured. (F − F0)/F0 was calculated using the recorded fluorescence before the addition of M2+ (F0) and the fluorescence after metal addition and emission signal stabilization (F).
Determination of putative H+ counter-transport in proteoliposomes
To determine if H+ are counter-transported ions by CmΔZntA during the substrate transport cycle, proteoliposomes and control liposomes were buffered in 10 mM MOPS pH = 7, 100 mM NaCl, 1 mM DTT, with 1 mM pyranine (λexc = 450 nm; λem = 515 nm) encapsulated in the proteoliposome and control liposome lumen by freeze-thaw membrane fracture and extrusion through a gas tight extruder system, similar to the encapsulation of Fluozin-3. Pyranine-encapsulated proteoliposomes and control liposomes were collected by ultracentrifugation (160,000 g, 45 min, 4 °C; Sorvall Mx 120+ micro-ultracentrifuge). The pellets were resuspended in proteoliposome buffer (10 mM MOPS pH = 7, 100 mM NaCl and 1 mM DTT). An additional centrifugation and resuspension cycle was performed to remove any remaining extralumenal pyranine. Transport assays to test H+ counter-transport were performed as described for the metal transport assay for each metal substrate. Proteoliposomes or control liposomes (120 μL) were placed in a sub-micro quartz cell (Starna Cells). MgCl2 and ATP were added to the reaction mixtures to final concentrations of 10 mM and 1 mM, respectively, and mixed by pipetting. The reaction was initiated by addition of ZnCl2, CdCl2, Pb(CH3COOH)2, or HgCl2 to final concentrations of 40 μM from 100× concentrated stock solutions. The time-dependent fluorescence change (ΔF/F0) was recorded at 30 °C for 750 s in 0.5 s intervals (λexc = 450 nm, slit width = 1.00 nm; λem = 515 nm, slit width = 1.00 nm) on a Fluoromax-4 spectrofluorometer (Horiba scientific). Control experiments were performed following a similar protocol as with proteoliposomes in the absence of Mg2+ or ATP. A separate set of control experiments were performed on control liposomes and measurements were conducted as described for the proteoliposomes.
Measurement of transmembrane potential development during metal substrate transport events
To test whether an electrogenic potential develops during transport events, the membrane potential sensitive fluorescent probe Oxonol VI was encapsulated in the proteoliposome and control liposome lumens by freeze-thaw membrane fracture, followed by extrusion through membrane filters of decreasing pore sizes (1 μm, 0.4 μm and 0.2 μm). A similar buffer (10 mM MOPS pH = 7, 100 mM NaCl and 1 mM DTT) was used as described in the counterion transport assays. Oxonol VI-encapsulated proteoliposomes and control liposomes were centrifuged to collect the vesicles (160,000 g, 45 minutes, 4 °C; Sorvall Mx 120+ micro-ultracentrifuge). The proteoliposome and control pellets were resuspended in the same buffer (10 mM MOPS pH = 7, 100 mM NaCl and 1 mM DTT). Excess Oxonol VI was removed by an additional washing step with buffer followed by ultracentrifugation (160,000 g, 45 min, 4 °C; Sorvall Mx 120+ micro-ultracentrifuge). Membrane potential development metal transport assays were performed by placing the proteoliposomes or control liposomes (120 μL) in a sub-micro quartz cell (Starna Cells). MgCl2 and ATP were added to the reaction mixtures (10 mM and 1 mM respectively) and thoroughly mixed by pipetting. The transport reactions were triggered by adding ZnCl2, CdCl2, Pb(CH3COOH)2, or HgCl2 to a final concentration of 40 μM from 100× concentrated stock solutions. The time-dependent fluorescence change (ΔF/F0) was recorded for 400 s in 0.5 s intervals (λexc = 580 nm, slit width = 5.00 nm; λem = 660 nm, slit width = 5.00 nm). Control experiments were performed in the absence of Mg2+ or ATP.
A separate set of control experiments were performed on control liposomes and measurements were conducted as described for the proteoliposomes.
Results and discussion
Expression, purification, and reconstitution of functional CmΔZntA166–794 in proteoliposomes
As in other P1B-type ATPases, wild-type CmZntA possesses an N-terminal ferredoxin-like metal binding domain (MBD, amino acids 90–154), flanked by a His/Cys-rich sequence potentially involved in additional metal binding.49,50 These ferredoxin-like cytoplasmic MBDs appear to exhibit a regulatory function by modulating the ATPase turnover rates and are not responsible in conferring metal selectivity, as their truncation does not abolish transport.11,20,28 This regulatory function has been demonstrated in ZntAs from different organisms.11,20,28 However, MBDs can interfere with metal coordination analysis at the high-affinity transmembrane metal selection site. Thus, the N-term CmZntA MBD was deleted, and CmΔZntA166–794 (abbreviated to CmΔZntA throughout) utilized throughout the studies to guarantee metal binding exclusively to the transmembrane binding site without any interferences from the MBD, in particular for X-ray Absorption Spectroscopy (XAS) characterization. To conduct in vitro functional studies, a protocol was established for expression, purification, and subsequent incorporation of CmΔZntA in native-like small unilamellar phospholipid bilayer vesicles, also called proteoliposomes. CmΔZntA possesses the classic P1B-type ATPases topology, with a high sequence similarity to S. Sonnei, and identical signature motifs in M4/5/6 typical of Zn(ii)-pumps where the transmembrane substrate binding site is present, allowing reliable prediction of its structure via homology modeling (Fig. 2a and b).
Fig. 2. CmΔZntA homology model, purification, reconstitution in proteoliposomes, and metal-dependent ATPase activity. (a) 3D homology model of CmΔZntA166–794 predicted with Robetta Protein Structure Prediction Server (http://robetta.bakerlab.org/; M2+ = Zn2+. Cd2+, Hg2+, Pb2+) and (b) close-up view of the putative transmembrane metal binding site involved in substrate selection. Cys438, Cys440 (on M4) and Asp777 (on M6) create the putative high affinity binding site conserved in ZntAs, and Lys756 (on M5) acts as a built-in counterion. (c) Size exclusion chromatogram of purified CmΔZntA in Cymal-7 micelles. (d) Corresponding SDS-PAGE gel of CmΔZntA and SDS-PAGE analysis of CmΔZntA incorporation in proteoliposomes (PL) vs. control (Cont.) liposomes (P: proteoliposomes/liposomes pellet isolated by ultracentrifugation; S: corresponding supernatant). (e) Dynamic light scattering (DLS) analysis of control liposomes and CmΔZntA proteoliposomes characterizing the average SUV diameter (in nm) and size distribution. (f) Relative metal-stimulated CmΔZntA ATPase activity in the presence of different metals in Cymal-7 micelles (metal concentration = 50 μM). (g) Relative metal-stimulated ATPase activity in the presence of different metals for CmΔZntA reconstituted in proteoliposomes (metal concentration = 50 μM). All data are mean ± s.d. (n = 3).

Recombinantly expressed CmΔZntA was purified by a combination of immobilized metal affinity chromatography (IMAC) and size exclusion chromatography (SEC). SEC analysis of purified CmΔZntA revealed a monodispersed distribution in Cymal-7 detergent micelles without significant aggregation, as indicated by the absence of any peak at the column void volume (Fig. 2c). The protein purity (>95%) was verified by SDS-PAGE (Fig. 2d) confirming the successful expression and purification protocol in detergent micelles.
In parallel, to study CmΔZntA in a native-like environment, the detergent-solubilized purified protein was reconstituted in unilamellar proteoliposome vesicles. The incorporation efficiency of CmΔZntA into proteoliposomes was determined by SDS-PAGE band-intensity densitometric analysis, upon liposome separation by ultracentrifugation, followed by protein quantification in the soluble and proteoliposome fractions. The results revealed >95% encapsulation efficiency in the lipid bilayers (Fig. 2d). To verify the formation of small unilamellar vesicles (SUVs), the size distribution of control liposomes and CmΔZntA proteoliposomes was determined by dynamic light scattering (DLS). A monodispersed size distribution was observed for both control liposomes and proteoliposomes, with sizes of 195.10 ± 78.05 nm (PDI = 0.116) and 200.00 ± 78.05 nm (PDI = 0.243) respectively (Fig. 2e), corresponding to the reconstitution of ∼150 CmΔZntA molecules per liposome vesicle (see Material and Methods; estimated distribution range: 50–300 CmΔZntA).
Metal-dependent ATPase activity of CmΔZntA in Cymal-7 micelles was determined for 1st-, 2nd- and 3rd-row transition and post-transition metal ions to identify which metal can act as a transported substrate (Fig. 2f and ESI Fig. 1†). The results reveal that, in Cymal-7 micelles, CmΔZntA, similarly to PaZntA, is activated by Pb2+ and group 12 metals (Zn2+, Cd2+, Hg2+). Michaelis–Menten-like analysis of the metal-dependent ATPase activity in detergent micelles as a function of metal concentrations, revealed activation with KM,M2+ values in the low μM range and ATPase hydrolysis rates following the order Pb(ii) > Zn(ii) > Hg(ii) ∼ Cd(ii) (Fig. 3a–d and Table 1).
Fig. 3. M2+-dependent CmΔZntA ATPase activity in Cymal-7 micelles. (a) Pb2+-dependent, (b) Zn2+-dependent, (c) Cd2+ dependent, and (d) Hg2+-dependent specific ATPase activity, as a function of metal concentration (0–100 μM). Data were fitted with a Michaelis–Menten-like equation as described in Materials and methods. All the ATPase activity measurements were corrected for the background ATPase activities obtained with control samples without Mg2+, required as cofactor for ATP binding and hydrolysis. All data are mean ± s.d. (n = 3).

Metal-dependent Michaelis–Menten-like ATPase parameters for CmΔZntA166–794 in detergent micelles and proteoliposomes derived from data fitting in Fig. 3 and 4.
| Metal | Detergent | Proteoliposomes | ||
|---|---|---|---|---|
| K M,M2+ (μM) | V max (nmol Pi mg−1 min−1) | K M,M2+ (μM) | V max (nmol Pi mg−1 min−1) | |
| Pb(ii) | 20.3 ± 3.3 | 1.32 ± 0.07 | 9.1 ± 2.0 | 3.10 ± 0.16 |
| Zn(ii) | 10.2 ± 2.6 | 1.07 ± 0.07 | 14.3 ± 4.9 | 2.17 ± 0.22 |
| Cd(ii) | 13.9 ± 4.8 | 0.71 ± 0.08 | 14.1 ± 5.1 | 1.86 ± 0.07 |
| Hg(ii) | 10.2 ± 3.4 | 0.70 ± 0.07 | 10.8 ± 4.9 | 1.37 ± 0.17 |
To verify functional incorporation of CmΔZntA in proteoliposomes, analysis of the metal-stimulated ATPase activity and Michaelis–Menten-like fitting confirmed ATPase rates following the order Pb(ii) > Zn(ii) > Hg(ii) > Cd(ii) (Fig. 2g and 4, and Table 1).
Fig. 4. M2+-dependent CmΔZntA ATPase activity in proteoliposomes. (a) Pb2+-dependent, (b) Zn2+-dependent (c) Cd2+-dependent and (d) Hg2+-dependent specific ATPase activity as a function of metal concentrations (0–100 μM). Data were fitted with a Michaelis–Menten-like equation as described in Materials and methods. All the ATPase activity measurements were corrected for background values for control liposomes and all data are mean ± s.d. (n = 3).

Thus, the metal selectivity profiles upon CmΔZntA incorporation in proteoliposomes are comparable to the detergent solubilized protein. However, the ATP hydrolysis catalytic activity and turnover rates were increased 2-fold in proteoliposomes, suggesting a better stabilization of the protein in the lipid environment and establishing the basis to conduct transport assays in SUVs.
Determination of metal substrate coordination by XAS
In characterized ZntAs the presence of a single high-affinity metal binding site in the transmembrane domain has been demonstrated for Zn2+, Cd2+, Pb2+, and Hg2+ by ICP-MS analysis and metal titration electronic absorption spectroscopy.9,28 To study the coordination chemistry of substrate metal ions at the high affinity transmembrane binding site and confirm that coordination plasticity, resulting in promiscuity, is a common feature in Zn2+-pumps, metal-bound M2+-CmΔZntA samples were generated in Cymal-7 micelles (M2+ = Zn2+, Cd2+, Pb2+, and Hg2+).
To prevent potential interferences caused by metal binding to the N-term poly-histidine tag, the latter was removed on purified CmΔZntA by thrombin digestion followed by size exclusion chromatography. The structure and coordination environment of CmΔZntA transmembrane metal sites (M2+ = Zn2+, Cd2+, Pb2+, and Hg2+) were probed using K-edge (for Zn, and Cd) and L3-edge (for Hg and Pb) X-ray absorption spectroscopy (XAS). Extended X-ray absorption fine structure (EXAFS) data for each metal were analyzed to obtain information about the number and nature of scatterers, and corresponding bond lengths. The EXAFS data, Fourier transforms and best fits from the EXAFS analysis are shown in Fig. 5 and Table 2.
Fig. 5. X-ray absorption spectroscopic analysis (XAS) of CmΔZntA-M2+. K-edge experimental EXAFS data (black line) and the best fit (red line), with corresponding Fourier transforms, of CmΔZntA-Zn2+ (a and b) and CmΔZntA-Cd2+ (c and d); L3-edge EXAFS data and corresponding Fourier transforms for CmΔZntA-Hg2+ (e and f) and CmΔZntA-Pb2+ (g and h; for details on data collection and analysis see the Material and methods section). Parameters for the fits are reported in Table 2. (i) Models of the coordination geometries and metal–ligand bond distances for CmΔZntA–Zn2+, CmΔZntA–Cd2+, CmΔZntA–Hg2+ and CmΔZntA–Pb2+.

Selected best EXAFS fits for the Zn, Cd, Hg, Pb-site in CmΔZntA166–794a.
| Metal-site | Shelld (N) | r d (Å) | σ 2 c d (×10−3 Å−2) | R-factord (%) | Red. χ2 |
|---|---|---|---|---|---|
| Zn-site | 2N/O | 1.99(0) | 2(0) | 2.91 | 24.8 |
| 2S | 2.29(0) | 4(0) | |||
| Cd-site | 2N/O | 2.32(2) | 5(0) | 1.73 | 6.4 |
| 2S | 2.48(2) | 2(2) | |||
| Hg-site | 1N/O | 2.48(3) | 5(0)b | 1.55 | 7.5 |
| 2S | 2.33(1) | 3(0) | |||
| Pb-site | 1N/O | 2.52(2) | 1(2) | 7.34 | 17.7 |
| 2S | 2.67(2) | 9(1) |
r – Inter atomic distance (in parentheses are mean-square deviations). σ2 – Debye–Waller factor (in parentheses are mean-square deviations in DW factor).
σ 2 was fixed during fitting.
- Values reported as 0 represent small positive numbers after 3 significant figures (e.g. 0.003 with 0.002 error = 3(2); 0.0004 with 0.0001 error = 0(0)).
The mean-square deviation values in parentheses are related to fit precision, while R-factors are fitting errors. These are distinct from model accuracies, which are expected to be larger (approx. 0.02 Å for r, and approx. 20% for N and corresponding Debye–Waller factors – σ2). Typically, N and σ2 are highly correlated, and N was fixed in curve fitting to limit the correlation.
Systematic first shell (1–2.5 Å) analysis of the of K-edge EXAFS spectrum for Zn2+-bound CmZntA revealed that models with a four-, five- or six-coordinate shell with a mixed ligand set composed of N/O- and S- donors featured better fits (R-factor < 5% and reduced χ2) over shells lacking sulphur ligation. As no additional Cys/Met residues are present beyond the CPC motif on M4, fits with more than 2S were not considered. Among the best fits a four-coordinate mixed ligand set with two N/O donors and two S donors, resulted in acceptable Debye–Wallace (σ2) and good fit parameters (R-factor and reduced χ2). For five- or six-coordinate mixed-ligand (N/O and S) fits the σ-factors for N/O scatterers increased and resulted in bond lengths unresolvable within the resolution of the data (0.13 Å). Considering the composition of the proposed metal-binding site from the structure of CmZntA, featuring two cysteine residues (Cys438 and Cys440) and one aspartic acid residue (Asp777), the four-coordinate fit by two N/O (at 1.99 Å) and two S (at 2.29 Å) represents the most reliable fit (Table 2, Fig. 5a–b and i, and ESI Table S1†). The bond distances fit well with those of Zn2+ protein binding sites featuring tetrahedral coordination by mixed S and N/O ligands, and consistent with the XAS analysis on PaZntA and E. Coli ZntA, with zinc bound in a (distorted) tetrahedral coordination by two cysteines in the conserved CPC motif (in M4), and by the carboxylate group form a conserved Asp in M6 (residues fully conserved in P1B-2 ATPases), in a bidentate-fashion.28,51 In agreement, the Asp on M6, which is in spatial proximity to the CPC motif, is directly involved in all metal substrate coordinations, and its mutation abolish metal biding and metal-dependent ATPase activity.20,28,33
Similarly to Zn2+-bound CmΔZntA, first shell analysis of the K-edge EXAFS spectrum of Cd2+–CmΔZntA showed that a combination of N/O- and S-donors in a four/five coordination site resulted in good fits and σ2-factors, and satisfactory fit parameters (R-factor and reduced χ2). The best fits were obtained for a four (2N/O and 2S) or a five-coordinate (4N/O and 1S) site (ESI Table S1†). Considering the conservation of the transmembrane coordinating residues in CmΔZntA and PaΔZntA, coordination by two sulfurs (at 2.32 Å) from the conserved Cys residues (Cys438 and Cys440) in M4 and 2 oxygens from the conserved Asp in M6 (bidentate) in a tetrahedral/distorted tetrahedral fashion is likely, as observed in PaZntA,28 despite a pentacoordinate model cannot be excluded. Nevertheless, the bond lengths significantly increased by 0.3 Å indicating promiscuity to accommodate first- and second-row transition metal substrates. The ability to uptake and accommodate Zn2+ and Cd2+ in similar coordination at the TM-MBS, despite the increased bond length due to the larger ionic radius and softer nature of Cd2+, is compatible with the absence of an uptake selectivity filter and the existence of an electronegative funnel connecting the cytoplasmic-membrane interface to the intramembranous high-affinity site.2,20
For Hg2+-bound CmΔZntA, analysis of the L3-edge EXAFS spectrum showed that models containing two S-donors remarkably resulted in good fit parameters. By progressive variation of the number of N/O ligands, the best fit was obtained with a mixed ligand set of two S-donors (at 2.33 Å) and one weakly interacting N/O (at 2.48 Å) (Table 2, Fig. 5e–f, and ESI Table S1†). Addition of further N/O scatterers still resulted in reasonable fits (R-factor < 5%) but worse overall metrics (the Hg-site model in Fig. 5i also includes a possible second N/O scatterer, in gray). The conserved Asp on M6, in spatial proximity to the CPC motif, is directly involved in metal binding, and its mutation abolish metal binding and metal-dependent ATPase activity, supporting that a linear 2S coordination is unlikely. Consistently, X-ray absorption near edge structure (XANES) features in CmΔZntA–Hg2+ (ESI Fig. S2†) are consistent with a non-linear Hg2+ complex, with an absorption maximum below 12 330 eV (which in linear bis-l-cysteinate Hg(Cys)2 is above 12 330 eV), no pronounced 2p3/2 → 6s/5d transition at 12 280 eV, and no shoulder at 12 295 eV.52,53 Overall, these results are consistent with Hg2+ binding to the transmembrane selection site in an irregular trigonal/tetrahedral geometry by two Cys sulfurs and additional weakly bonded N/O interaction(s), resulting in distortion form an ideal linear digonal coordination.28 This suggests that deviations from a linear 2S Hg2+ geometry would contribute to reduce the binding affinity and increase the kinetic lability, allowing metal release in the catalytic cycle and preservation of a relatively high translocation turnover.
As observed for the other metal substrates, the first shell L3-edge EXAFS analysis of Pb2+–CmΔZntA revealed that fits with two- S-donors at 2.67 Å resulted in satisfactory σ2 and R-factors (<5%). The addition of one N/O-donor resulted in the best model, while attempts to resolve the two S-donors resulted in fits with failed radial distances and beyond the resolution of the data (0.14 Å). Considering that the conserved Asp on M6 has been demonstrated to be a Pb(ii)-binding ligand in ZntAs, and that trigonal over digonal coordination is favored in Pb(ii)-complexes, the fit with two S-donors at 2.67 Å (2 Cys) and a single N/O donor at 2.52 Å (Asp) is considered the best fit in Pb(ii)–CmΔZntA (Table 2, Fig. 5g–h, and ESI Table S1†). This is fully consistent with the trigonal pyramidal Pb(ii)S2O/N site, with a lone pair occupying the apical position (hemidirected) observed in PaZntA.28 Moreover, the XANES spectra for CmΔZntA bound to each of the substrates are highly similar in shape and intensity to that obtained for PaZntA (ESI Fig. S2†), further corroborating the nature of the metal binding sites determined by EXAFS analysis.28
Overall, the XAS analysis and comparison with PaZntA indicate that coordination promiscuity is a conserved feature in P1B-2-type ATPases.28 This plasticity in metal recognition guarantees substrate promiscuity, via substrate binding with different coordination numbers and bond distances while common coordination ligands, controlling the promiscuous recognition of substrates that are postulated to be translocated across the lipid bilayer. While changes in bond lengths (>0.3 Å for S ligands and > 0.5 Å for N/O ligands) are significant from the coordination chemistry point of view (which we here define as coordination “plasticity”), they can be accommodated by backbone and/or side chain flexibility. The observed ability to bind, select, and translocate remarkably diverse metals (in terms of size, polarizability, coordination number and geometry) is notable compared to other transmembrane cation transporters and ion channels. In several ion channels, selectivity filters have been demonstrated to be not promiscuous and “rigid” allowing for strict selection of metal substrates over others possessing very similar coordination properties but different ionic radius (e.g. K+ channels, KcsA).54 On the other hand, in other metal transporter families (e.g. Zn(ii) transporters of the CDF family – ZnTs) strict selectivity or limited promiscuity has been observed for Zn(ii)/Cd(ii), but not for other 3rd-row transition and post transition metals.55–59 Thus, the coordination plasticity observed in ZntAs appears to be an important feature that evolved for their function. This chemistry underlies a crucial role in cellular metal detoxification imparting the ability to select both essential metals (when exceeding the metal “quotas”) as well non-essential toxic counterparts. Diverse substrate selection via formation of complexes with distinct coordination numbers (N), geometry and bond distances (Zn2+ and Cd2+N = 4, tetrahedral; Pb2+N = 3, trigonal pyramidal; Hg2+N = 2, distorted digonal with 1–2 additional weakly coordinating ligands), while preserving similar coordination environments, is optimized for promiscuous metal detoxification. In addition, divergence from ideal ligands sets and coordination geometries, potentially resulting in very high binding affinities and preventing metal release under translocation turnover, appear to underly the ability of P1B-2 type ATPases to simultaneously achieve efficient and promiscuous translocation.
Real-time detection of Zn2+, Cd2+, Pb2+ and Hg2+ transport in CmΔZntA proteoliposomes
P1B-2-type Zn(ii) ATPases appear to possess a wider promiscuity toward substrates compared to other P1B-type ATPase metal pumps by plastic selection of Zn2+, Cd2+, Pb2+, and Hg2+ in the transmembrane domain. Based on the metal-dependent stimulation of ATPase activity and XAS analysis, our investigations reveal that CmΔZntA also possess a conserved cargo promiscuity. However, despite the identification of selectivity in P1B-2-type Zn(ii) ATPases that has been inferred based on metals that stimulate ATP hydrolysis, direct translocation of these cargo, their relative transport rates, and transport mechanisms (uniport vs. cotransport, and electrogenicity) remain elusive due to the lack of tools for in vitro analysis of purified proteins in lipid bilayers that would allow study of diverse 1st-, 2nd- and 3rd-row transition and post-transition metal translocation events in real-time in a native-like environment. To reveal the breadth of CmΔZntA ion substrates, a fluorescence-based multi-probe approach was developed to monitor metal-substrate transport in proteoliposome supramolecular assemblies (Fig. 6). In search of metal-dependent, turn-on, and membrane impermeable fluorescence chelators to be encapsulated in the proteoliposome lumen for real-time metal translocation detection, a series of fluorescent probes were screened as turn-on sensors for Zn2+, Cd2+, Pb2+ and Hg2+. We selected Fluozin-3 as fluorescent sensor for Zn2+ and Cd2+, Leadmium Green for Pb2+, and Calcium Green for Hg2+. Metal titration of these probes in the presence of control liposomes revealed a linear fluorescence turn-on response for increasing metal concentrations, with break points at 1 : 1 metal/probe ratios (ESI Fig. S3†). The fluorescence response for each metal-probe pair could be normalized to compare the relative metal transport rates for different substrates. Complete metal binding to the probes is fast and occurs within the manual mixing time (approx. 5 s), thus allowing real-time detection of the translocation events by CmΔZntA for all its substrates (ESI Fig. S4†). To monitor ATP-dependent metal ion translocation across the phospholipid bilayer by CmΔZntA, the probes were encapsulated in the proteoliposome/liposome lumen by freeze-thaw membrane fracture and subsequent extrusion through 200 nm polycarbonate filters. Subsequently, proteoliposomes were supplemented with ATP and Mg2+, and substrate translocation was triggered by addition of the corresponding metal ion (40 μM, a concentration above the KM as determined by metal-stimulation ATPase assays), and the fluorescence response was measured as a function of time (Fig. 7).
Fig. 6. Real-time transport of Zn2+, Cd2+, Pb2+ and Hg2+ in proteoliposomes. Schematic representation of multi-probe experimental approach to test real-time metal translocation events by CmΔZntA reconstituted in proteoliposomes. The membrane impermeable, turn-on fluorescence probes Leadmium Green, Fluozin-3, and Calcium Green were selected for Pb2+, Zn2+/Cd2+, and Hg2+ respectively. The Leadmium Green chemical structure is not reported as it is protected intellectual property of the manufacturer.

Fig. 7. Real-time metal transport in CmΔZntA proteoliposomes monitored with fluorescent probes encapsulated in the proteoliposome lumen. (a) Leadmium Green (20 μM) reporting Pb2+ transport (λexc = 490 nm; λem = 520 nm), (b) Fluozin-3 (10 μM) reporting Zn2+ transport (λexc = 480 nm; λem = 515 nm), (c) Fluozin-3 (10 μM) reporting Cd2+ transport (λexc = 480 nm; λem = 515 nm), and (d) Calcium Green (10 μM) reporting Hg2+ transport (λexc = 506 nm; λem = 531 nm). Red lines correspond to M2+ transport traces with standard deviations shaded in red. Black – proteoliposomes supplemented with M2+ and ATP but no Mg2+. Blue – proteoliposomes supplemented with M2+ and Mg2+ but no ATP. Signals were normalized for the relative turn-on fluorescence response of different probes, and corrected for background signals of control liposomes. All data are mean ± s.d. (n = 3).

After supplementing the proteoliposomes with metals, kinetic traces revealed a time-dependent increase in fluorescence signal (ΔF/F0) as a function of time, with plateaus reached in approx. 10 min. The net fluorescence increase response ΔF/F0 was corrected for background metal leakage by performing similar recordings on control liposomes. Overall background metal permeations for all substrates were negligible. As a further control, identical experiments were conducted excluding either ATP or Mg2+ (required as cofactor for ATP binding and hydrolysis), resulting in no significant fluorescence signal increase over time. Thus, these analyses demonstrated ATP-dependent CmΔZntA-mediated metal translocation into the proteoliposome lumen, establishing that ZntA acts as a promiscuous primary active pump for diverse 1st-, 2nd- and 3rd-row transition and post-transition metals (Zn2+, Cd2+, Hg2+ and Pb2+). To compare the relative rates of translocation for different cargos, each fluorescence response was normalized with a conversion factor accounting the relative fluorescence response of the different sensor probes. For each substrate, the normalized transport traces were fitted with an exponential function and the apparent initial velocities were extrapolated from the first derivative of the fit. The results reveal that the apparent normalized initial velocities (δF/δt), when the metal concentration gradient is steepest across the phospholipid bilayer, follow the order: Pb2+ (δF/δt = (12.46 ± 0.26) × 10−3 s−1) > Zn2+(δF/δt = (4.20 ± 0.57) × 10−3 s−1) > Hg2+(δF/δt = (2.68 ± 0.65) × 10−3 s−1) ≈ Cd2+ (δF/δt = (1.48 ± 0.19) × 10−3 s−1). These data demonstrate for the first time that ZntAs can translocate a diverse set of metal ions that stimulate ATP hydrolysis across the lipid bilayers. The correspondence between the relative initial velocities and relative metal-stimulated ATPase activities for the different cargos further confirms the coupling between ATP hydrolysis and substrate translocation, as expected form the Post-Albers cycle, even for non-essential toxic metals as Pb2+, Cd2+, and Hg2+.
Assessing proton (counter)-transport and electrogenicity in real-time with pyranine and Oxonol VI
For SsZntA, it has been suggested that Zn2+ translocation occurs without proton counter-transport.20 This mechanism is supported by the SsZntA structure as determined in its phosphoenzyme ground state (E2P), which displays a wide extracellular release pathway from the high-affinity site.20 This release conduit closes in the E2.Pi state, in which the metal binding residue (D714) interacts with a conserved residue (K693), acting as a potential built-in counter ion to stimulate Zn2+ release (Fig. 1). However, structural features of the release pathway in ZntA also resemble P2-type ATPases such as the sarcoplasmic/endoplasmic reticulum Ca2+/H+-ATPase (SERCA).2,18,20,39,42 While in SsZntA the built-in counter-ion can potentially stabilize an occluded E2.Pi form preventing proton counter-flux, in Zn(ii) pumps protons could be involved in cargo release with substrates other than Zn2+.20 Indeed, the preservation of translocation mechanism with a diverse range of substrates resulting in an electrogenic transport modality remains unknown. Moreover, the substrate release mechanism in Zn(ii)-pumps appears different in Cu(i)-translocating P1B-1-type ATPases. In CopA (Cu(i) P1B-1-type ATPase pumps) the built-in counterion residues are not conserved, and a different mechanism prevents H+ back transport. CopA possesses a narrow release pathway with the conduit remaining largely unchanged from the E2P to E2.Pi state (which represent a transition state of dephosphorylation) transition, suggesting that Cu+ release is mediated by consecutive ligand exchange reactions.25,26,29,46 Thus, unique modalities of Cu+ and Zn2+ transport in P1B-type ATPase pumps have been proposed, suggesting that substrate release mechanism can vary depending on the metal translocated and/or the subfamily considered. We thus expanded our “multi-channel” strategy with probes responsive to diverse stimuli (fluorescent metal turn-on probes, pH probes, and membrane potential probes)45,46 to investigate the mechanism of Zn(ii)-pump translocation with all metal substrates that stimulate ATP hydrolysis (Zn2+, Cd2+, Pb2+, and Hg2+). Considering the different coordination modes of these substrates possessing different ligand nuclearities (e.g., tetrahedral vs. trigonal pyramidal), different modalities during translocation could be envisaged.
To determine whether substrate translocation is coupled to proton (counter)-transport or not (as in the Ca2+/H+-pump SERCA and Cu(i) P-type ATPases, respectively), transport experiments in the presence of metal substrates and ATP-Mg2+ were performed upon encapsulating the fluorescent pH indicator pyranine in the proteoliposome lumen. Pyranine(8-hydroxy-1,3,6-pyrenetrisulfonate) is a membrane-impermeable pH indicator that can be encapsulated in proteoliposomes and whose fluorescence emission intensity (at 515 nm) changes as a function of protonation of its 8-hydroxyl group (pKa ≃ 7.2).60 During substrate transport turnover, a pyranine time-dependent fluorescence change allow for the detection of a proton flux (if any) upon metal cargo translocation and its directionality (inwards vs. outward).
Pyranine was encapsulated in the CmΔZntA proteoliposome lumen by freeze-thaw membrane fracture followed by extrusion though a series of membrane polycarbonate filters. The SUVs were subsequently supplemented with ATP and Mg2+, and metal transport triggered by the addition of M2+ metal ions (M2+ = Zn2+, Cd2+, Pb2+, and Hg2+).
Time-dependent kinetic traces monitored at the pyranine emission maxima revealed no detectable pyranine fluorescence change in CmΔZntA proteoliposomes for any of the tested metal substrates (Fig. 8a). Thus, while the binding geometry/ligand distances are different for each metal substrate recognized at the high-affinity binding site in the transmembrane domain, those differences in coordination do not alter the overall mechanism of cargo translocation. Thus Zn2+-pumps are not Zn2+/H+ antiporters, a key mechanistic difference compared to other well characterized P-type ATPases like SERCA, the plasma membrane calcium ATPases PMCA, and the bacterial Ca2+/H+ pump LMCA.38,41,44 In the absence of proton-flux coupling and considering that Zn2+ pumps do not require other cations (e.g. Na+ or K+) to catalyze cargo translocation,20 we speculated that ZntAs must follow a primary active electrogenic uniporter transport mechanism.
Fig. 8. Investigation of H+ counter-transport and development of transmembrane potential in CmΔZntA proteoliposomes. (a) Determination of H+ counter-ion transport via monitoring luminal pH changes as a function of time with the pH-indicator pyranine (1 mM) under conditions utilized in metal substrate transport assays (M2+ = 40 μM). Measurements were corrected for background signals of control liposomes. All data are mean ± s.d. (n = 3). (b) Real-time measurements of transmembrane potential generation in CmΔZntA proteoliposomes under conditions utilized for metal transport (M2+ = 40 μM), determined upon encapsulating the membrane potential probe Oxonol VI in the proteoliposome lumen as described in Materials and methods. Control experiment in the absence of Mg2+ are reported and labeled (no Mg2+). Signals were corrected for background values for control liposomes. Data are mean ± s.d. (n = 3).

To verify this hypothesis, we encapsulated the transmembrane potential probe Oxonol VI in the CmΔZntA proteoliposome lumen and conducted metal transport assays. Oxonol VI (bis-(3-propyl-5-oxoisoxazol-4-yl)pentamidine oxonol) acts as a slow responsive fluorescent transmembrane-potential (Ψ) sensor, by a voltage-dependent partitioning between the aqueous phase and lipid bilayers.61,62 Changes in relative fluorescence intensity can report time-dependent transmembrane potential changes and their directionality.63
Upon encapsulation in the lumen, CmΔZntA proteoliposomes and control liposomes were supplemented with ATP and Mg2+, and substrate translocation was triggered by the rapid addition of a metal substrate (M2+ = 40 μM; M2+ = Zn2+, Cd2+, Hg2+, and Pb2+). After mixing, the fluorescence traces revealed a time-dependent fluorescence increase of the Oxonol-VI emission (λexc = 580 nm; λem = 660 nm) (Fig. 8b) for all tested metal substrates (M2+ = Zn2+, Cd2+, Pb2+ and Hg2+), confirming the development of a net positive-inside membrane potential in the lumen upon translocation. Moreover, the initial velocity of fluorescence traces followed a qualitatively similar ranking as determined in the metal transport assays and ATPase activity assays (Pb2+ ∼ Zn2+ > Cd2+ ∼ Hg2+), further confirming that, in CmΔZntA, metal translocation, transmembrane potential development, and ATP hydrolysis are coupled. Thus, the combined analyses reveal that Zn(ii)-pumps are promiscuous electrogenic pumps.
Conclusions
By developing a proteoliposome-based multi-probe platform with fluorescent reporters responsive to diverse stimuli, we reveal that Zn(ii) pumps are promiscuous, primary active, and electrogenic uniporters that catalyze the translocation of 1st-, 2nd-, and 3rd-row transition and post-transition metals across biological membranes. Considering the highly conserved subclass-specific amino acid residues in the transmembrane helices of all Zn2+-pumps, the transport mechanism is expected to be preserved throughout the P1B-2-type ATPases subfamily members. In addition, the XAS analysis established that substrate selectivity in Zn2+-pumps is primarily governed by coordination chemistry properties in the transmembrane domain, and that coordination plasticity at the high affinity transmembrane binding site allows for promiscuous substrate recognition and selection for translocation via formation of complexes with diverse coordination numbers and bond distances, while maintaining overall compatible coordination properties. The observed differences in the transport rates for the four substrates corroborate that the different substrate coordination revealed by XAS might underly the different metal association/dissociation rates and labilities necessary for substrate release and contribute to the different relative rates of translocation. Differences in metal charge density, polarizability, coordination number and geometry are expected to result in different binding and release kinetics to the transmembrane metal binding site. Despite these differences, the coupling between ATP hydrolysis and translocation is preserved. The conclusion that the ATPase activity and metal transport rates are “coupled” is supported by multiple line of evidence: (i) CmΔZntA ATP hydrolysis occurs and is significantly stimulated only in the presence of the transported metal substrates; (ii) no metal translocation and transmembrane potential generation is observed in the presence of ATP without Mg2+ (which is strictly required as a cofactor for ATP binding and hydrolysis by all P-type ATPases; Fig. 7), demonstrating that ATP binding and hydrolysis is required for substrate translocation; (iii) the relative order of ATPase activity and translocation “efficiency” observed for metal-stimulated ATPase hydrolysis (Vmax/KM,M2+) and for metal translocation ([δF/δt]/KM) follow the same trend (ESI Table S2†). Overall, plasticity and promiscuity allows for the adaptation of a conserved topological framework, characteristic for all P1B-type ATPases, to the generation of pumps with a characteristic selectivity pattern that, despite the diversity of selected substrates, can preserve the same translocation mechanism, as observed herein for Zn(ii)-pumps that act as primary active electrogenic uniporters. The cellular needs for exporter pumps capable of recognizing and translocating both 1st-row essential metals (Zn2+; toxic when exceeding necessary cellular “quotas”) and non-essential toxic substrates such as diverse 2nd- and 3rd-row transition and post-transition metals (Cd2+, Hg2+, and Pb2+), is met via fine tuning of the coordination chemistry and mechanistic properties of the pumps. The developed methodology also provides new tools to investigate metal translocation events in real-time with a wide variety of metal cargos, providing novel possibilities for dissecting the transport mechanism of other poorly characterized promiscuous transmembrane metal transporters.
Data availability
Data for this paper are available upon request to the corresponding author.
Author contributions
Sameera S. Abeyrathna: investigation, formal analysis, methodology, writing – original draft; Nisansala S. Abeyrathna, Priyanka Basak, Gordon W. Irvine, Limei Zhang: investigation, formal analysis, methodology, writing – review & editing; Gabriele Meloni: conceptualization, supervision, investigation, formal analysis, methodology, writing – original draft, resources.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
The work was supported by National Institute of General Medical Sciences (NIH, R35GM128704 to G. M.), the National Science Foundation (CHE-2045984 to G. M. and CHE-1846908 to L. Z.), and the Robert A. Welch Foundation (AT-1935-20170325 and AT-2073-20210327 to G. M.). G. W. I. was supported by an NSERC postdoctoral fellowship (Research Council of Canada). We thank the staff at SSRL Beamline 7-3/9-3 for support during XAS data collection. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00347g
References
- Smith A. T. Smith K. P. Rosenzweig A. C. Diversity of the metal-transporting P1B-type ATPases. J. Biol. Inorg. Chem. 2014;19(6):947–960. doi: 10.1007/s00775-014-1129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsel O. Gronberg C. Autzen H. E. Wang K. Meloni G. Nissen P. Gourdon P. Structure and Function of Cu(I)- and Zn(II)-ATPases. Biochemistry. 2015;54(37):5673–5683. doi: 10.1021/acs.biochem.5b00512. [DOI] [PubMed] [Google Scholar]
- Raimunda D. Long J. E. Padilla-Benavides T. Sassetti C. M. Arguello J. M. Differential roles for the Co2+/Ni2+ transporting ATPases, CtpD and CtpJ, in Mycobacterium tuberculosis virulence. Mol. Microbiol. 2014;91(1):185–197. doi: 10.1111/mmi.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello J. M. Raimunda D. Gonzalez-Guerrero M. Metal transport across biomembranes: emerging models for a distinct chemistry. J. Biol. Chem. 2012;287(17):13510–13517. doi: 10.1074/jbc.R111.319343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig A. C. Arguello J. M. Toward a molecular understanding of metal transport by P(1B)-type ATPases. Curr. Top. Membr. 2012;69:113–136. doi: 10.1016/B978-0-12-394390-3.00005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello J. M. Gonzalez-Guerrero M. Raimunda D. Bacterial transition metal P1B-ATPases: transport mechanism and roles in virulence. Biochemistry. 2011;50(46):9940–9949. doi: 10.1021/bi201418k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyrolles O. Wolschendorf F. Mitra A. Niederweis M. Mycobacteria, metals, and the macrophage. Immunol. Rev. 2015;264(1):249–263. doi: 10.1111/imr.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S. J. Liu J. Stemmler A. J. Mitra B. Conservative and nonconservative mutations of the transmembrane CPC motif in ZntA: effect on metal selectivity and activity. Biochemistry. 2007;46(12):3692–3703. doi: 10.1021/bi0616394. [DOI] [PubMed] [Google Scholar]
- Liu J. Dutta S. J. Stemmler A. J. Mitra B. Metal-binding affinity of the transmembrane site in ZntA: implications for metal selectivity. Biochemistry. 2006;45(3):763–772. doi: 10.1021/bi051836n. [DOI] [PubMed] [Google Scholar]
- Hou Z. Mitra B. The metal specificity and selectivity of ZntA from Escherichia coli using the acylphosphate intermediate. J. Biol. Chem. 2003;278(31):28455–28461. doi: 10.1074/jbc.M301415200. [DOI] [PubMed] [Google Scholar]
- Mitra B. Sharma R. The cysteine-rich amino-terminal domain of ZntA, a Pb(II)/Zn(II)/Cd(II)-translocating ATPase from Escherichia coli, is not essential for its function. Biochemistry. 2001;40(25):7694–7699. doi: 10.1021/bi010576g. [DOI] [PubMed] [Google Scholar]
- Sharma R. Rensing C. Rosen B. P. Mitra B. The ATP hydrolytic activity of purified ZntA, a Pb(II)/Cd(II)/Zn(II)-translocating ATPase from Escherichia coli. J. Biol. Chem. 2000;275(6):3873–3878. doi: 10.1074/jbc.275.6.3873. [DOI] [PubMed] [Google Scholar]
- Rensing C. Mitra B. Rosen B. P. The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc. Natl. Acad. Sci. U. S. A. 1997;94(26):14326–14331. doi: 10.1073/pnas.94.26.14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz V. Schmidt-Vogler C. Strohmeyer P. Weber S. Kleemann D. Nies D. H. Herzberg M. Behind the shield of Czc: ZntR controls expression of the gene for the zinc-exporting P-type ATPase ZntA in Cupriavidus metallidurans. J. Bacteriol. 2021;203(11):e00052-21. doi: 10.1128/JB.00052-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer J. Nies D. H. CzcP is a novel efflux system contributing to transition metal resistance in Cupriavidus metallidurans CH34. Mol. Microbiol. 2009;73(4):601–621. doi: 10.1111/j.1365-2958.2009.06792.x. [DOI] [PubMed] [Google Scholar]
- Roberts C. S. Muralidharan S. Ni F. Mitra B. Structural Role of the First Four Transmembrane Helices in ZntA, a P1B-Type ATPase from Escherichia coli. Biochemistry. 2020;59(47):4488–4498. doi: 10.1021/acs.biochem.0c00770. [DOI] [PubMed] [Google Scholar]
- Bublitz M. Morth J. P. Nissen P. P-type ATPases at a glance. J. Cell Sci. 2011;124(Pt 15):2515–2519. doi: 10.1242/jcs.088716. [DOI] [PubMed] [Google Scholar]
- Dyla M. Kjaergaard M. Poulsen H. Nissen P. Structure and Mechanism of P-Type ATPase Ion Pumps. Annu. Rev. Biochem. 2019;89:583–603. doi: 10.1146/annurev-biochem-010611-112801. [DOI] [PubMed] [Google Scholar]
- Toyoshima C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta. 2009;1793(6):941–946. doi: 10.1016/j.bbamcr.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Wang K. Sitsel O. Meloni G. Autzen H. E. Andersson M. Klymchuk T. Nielsen A. M. Rees D. C. Nissen P. Gourdon P. Structure and mechanism of Zn2+-transporting P-type ATPases. Nature. 2014;514(7523):518–522. doi: 10.1038/nature13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa-Aponte L. Ramirez D. Arguello J. M. The interplay of the metallosensor CueR with two distinct CopZ chaperones defines copper homeostasis in Pseudomonas aeruginosa. J. Biol. Chem. 2019;294(13):4934–4945. doi: 10.1074/jbc.RA118.006316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielazinski E. L. Gonzalez-Guerrero M. Subramanian P. Stemmler T. L. Arguello J. M. Rosenzweig A. C. Sinorhizobium meliloti Nia is a P1B-5-ATPase expressed in the nodule during plant symbiosis and is involved in Ni and Fe transport. Metallomics. 2013;5(12):1614–1623. doi: 10.1039/c3mt00195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimunda D. Long J. E. Sassetti C. M. Arguello J. M. Role in metal homeostasis of CtpD, a Co2+ transporting P1B4-ATPase of Mycobacterium smegmatis. Mol. Microbiol. 2012;84(6):1139–1149. doi: 10.1111/j.1365-2958.2012.08082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello J. M. Eren E. Gonzalez-Guerrero M. The structure and function of heavy metal transport P1B-ATPases. BioMetals. 2007;20(3–4):233–248. doi: 10.1007/s10534-006-9055-6. [DOI] [PubMed] [Google Scholar]
- Gourdon P. Liu X. Y. Skjorringe T. Morth J. P. Moller L. B. Pedersen B. P. Nissen P. Crystal structure of a copper-transporting PIB-type ATPase. Nature. 2011;475(7354):59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- Andersson M. Mattle D. Sitsel O. Klymchuk T. Nielsen A. M. Moller L. B. White S. H. Nissen P. Gourdon P. Copper-transporting P-type ATPases use a unique ion-release pathway. Nat. Struct. Mol. Biol. 2014;21(1):43–48. doi: 10.1038/nsmb.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronberg C. Hu Q. Mahato D. R. Longhin E. Salustros N. Duelli A. Lyu P. Bagenholm V. Eriksson J. Rao K. U. Henderson D. I. Meloni G. Andersson M. Croll T. Godaly G. Wang K. Gourdon P. Structure and ion-release mechanism of PIB-4-type ATPases. Elife. 2021;10:e73124. doi: 10.7554/eLife.73124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallenito M. J. Irvine G. W. Zhang L. Meloni G. Coordination promiscuity guarantees metal substrate selection in transmembrane primary-active Zn2+ pumps. Chem. Commun. 2019;55(73):10844–10847. doi: 10.1039/C9CC05936A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattle D. Zhang L. Sitsel O. Pedersen L. T. Moncelli M. R. Tadini-Buoninsegni F. Gourdon P. Rees D. C. Nissen P. Meloni G. A sulfur-based transport pathway in Cu+-ATPases. EMBO Rep. 2015;16(6):728–740. doi: 10.15252/embr.201439927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni G. Zhang L. Rees D. C. Transmembrane type-2-like Cu2+ site in the P1B-3-type ATPase CopB: implications for metal selectivity. ACS Chem. Biol. 2014;9(1):116–121. doi: 10.1021/cb400603t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. T. Ross M. O. Hoffman B. M. Rosenzweig A. C. Metal Selectivity of a Cd-, Co-, and Zn-Transporting P1B-type ATPase. Biochemistry. 2017;56(1):85–95. doi: 10.1021/acs.biochem.6b01022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. T. Barupala D. Stemmler T. L. Rosenzweig A. C. A new metal binding domain involved in cadmium, cobalt and zinc transport. Nat. Chem. Biol. 2015;11(9):678–684. doi: 10.1038/nchembio.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S. J. Liu J. Hou Z. Mitra B. Conserved aspartic acid 714 in transmembrane segment 8 of the ZntA subgroup of P1B-type ATPases is a metal-binding residue. Biochemistry. 2006;45(18):5923–5931. doi: 10.1021/bi0523456. [DOI] [PubMed] [Google Scholar]
- Dutta S. J. Liu J. Mitra B. Kinetic analysis of metal binding to the amino-terminal domain of ZntA by monitoring metal-thiolate charge-transfer complexes. Biochemistry. 2005;44(43):14268–14274. doi: 10.1021/bi050761k. [DOI] [PubMed] [Google Scholar]
- Hou Z. J. Narindrasorasak S. Bhushan B. Sarkar B. Mitra B. Functional analysis of chimeric proteins of the Wilson Cu(I)-ATPase (ATP7B) and ZntA, a Pb(II)/Zn(II)/Cd(II)-ATPase from Escherichia coli. J. Biol. Chem. 2001;276(44):40858–40863. doi: 10.1074/jbc.M107455200. [DOI] [PubMed] [Google Scholar]
- Janssen P. J. Van Houdt R. Moors H. Monsieurs P. Morin N. Michaux A. Benotmane M. A. Leys N. Vallaeys T. Lapidus A. Monchy S. Medigue C. Taghavi S. McCorkle S. Dunn J. van der Lelie D. Mergeay M. The complete genome sequence of Cupriavidus metallidurans strain CH34, a master survivalist in harsh and anthropogenic environments. PLoS One. 2010;5(5):e10433. doi: 10.1371/journal.pone.0010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies D. H. The biological chemistry of the transition metal "transportome" of Cupriavidus metallidurans. Metallomics. 2016;8(5):481–507. doi: 10.1039/C5MT00320B. [DOI] [PubMed] [Google Scholar]
- Inesi G. Tadini-Buoninsegni F. Ca2+/H+ exchange, lumenal Ca2+ release and Ca2+/ATP coupling ratios in the sarcoplasmic reticulum ATPase. J. Cell Commun. Signaling. 2014;8(1):5–11. doi: 10.1007/s12079-013-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen C. Picard M. Winther A. M. Gyrup C. Morth J. P. Oxvig C. Moller J. V. Nissen P. The structural basis of calcium transport by the calcium pump. Nature. 2007;450(7172):1036–1042. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- Bublitz M. Musgaard M. Poulsen H. Thogersen L. Olesen C. Schiott B. Morth J. P. Moller J. V. Nissen P. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 2013;288(15):10759–10765. doi: 10.1074/jbc.R112.436550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faxen K. Andersen J. L. Gourdon P. Fedosova N. Morth J. P. Nissen P. Moller J. V. Characterization of a Listeria monocytogenes Ca(2+) pump: a SERCA-type ATPase with only one Ca(2+)-binding site. J. Biol. Chem. 2011;286(2):1609–1617. doi: 10.1074/jbc.M110.176784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller J. V. Olesen C. Winther A. M. Nissen P. The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 2010;43(4):501–566. doi: 10.1017/S003358351000017X. [DOI] [PubMed] [Google Scholar]
- Abe K. Irie K. Nakanishi H. Suzuki H. Fujiyoshi Y. Crystal structures of the gastric proton pump. Nature. 2018;556(7700):214–218. doi: 10.1038/s41586-018-0003-8. [DOI] [PubMed] [Google Scholar]
- Gong D. Chi X. Ren K. Huang G. Zhou G. Yan N. Lei J. Zhou Q. Structure of the human plasma membrane Ca2+-ATPase 1 in complex with its obligatory subunit neuroplastin. Nat. Commun. 2018;9(1):3623. doi: 10.1038/s41467-018-06075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeyrathna S. S. Abeyrathna N. S. Thai N. K. Sarkar P. D'Arcy S. Meloni G. IroT/MavN Is a Legionella Transmembrane Fe(II) Transporter: Metal Selectivity and Translocation Kinetics Revealed by in Vitro Real-Time Transport. Biochemistry. 2019;58(43):4337–4342. doi: 10.1021/acs.biochem.9b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeyrathna N. Abeyrathna S. Morgan M. T. Fahrni C. J. Meloni G. Transmembrane Cu(I) P-type ATPase pumps are electrogenic uniporters. Dalton Trans. 2020;49(45):16082–16094. doi: 10.1039/D0DT01380C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S. M. Phys. Scr. 2005;2005:1011–1014. doi: 10.1238/Physica.Topical.115a01011. [DOI] [Google Scholar]
- Ravel B. Newville M. Athena A. HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- Banci L. Bertini I. Ciofi-Baffoni S. Su X. C. Miras R. Bal N. Mintz E. Catty P. Shokes J. E. Scott R. A. Structural basis for metal binding specificity: the N-terminal cadmium binding domain of the P1-type ATPase CadA. J. Mol. Biol. 2006;356(3):638–650. doi: 10.1016/j.jmb.2005.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L. Bertini I. Ciofi-Baffoni S. Finney L. A. Outten C. E. O'Halloran T. V. A new zinc-protein coordination site in intracellular metal trafficking: solution structure of the Apo and Zn(II) forms of ZntA(46-118) J. Mol. Biol. 2002;323(5):883–897. doi: 10.1016/S0022-2836(02)01007-0. [DOI] [PubMed] [Google Scholar]
- Raimunda D. Subramanian P. Stemmler T. Arguello J. M. A tetrahedral coordination of Zinc during transmembrane transport by P-type Zn2+-ATPases. Biochim. Biophys. Acta. 2012;1818(5):1374–1377. doi: 10.1016/j.bbamem.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalilehvand F. Leung B. O. Izadifard M. Damian E. Mercury(II) cysteine complexes in alkaline aqueous solution. Inorg. Chem. 2006;45(1):66–73. doi: 10.1021/ic0508932. [DOI] [PubMed] [Google Scholar]
- Manceau A. Lemouchi C. Rovezzi M. Lanson M. Glatzel P. Nagy K. L. Gautier-Luneau I. Joly Y. Enescu M. Structure, Bonding, and Stability of Mercury Complexes with Thiolate and Thioether Ligands from High-Resolution XANES Spectroscopy and First-Principles Calculations. Inorg. Chem. 2015;54(24):11776–11791. doi: 10.1021/acs.inorgchem.5b01932. [DOI] [PubMed] [Google Scholar]
- Gouaux E. Mackinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310(5753):1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- Wei Y. Fu D. Binding and transport of metal ions at the dimer interface of the Escherichia coli metal transporter YiiP. J. Biol. Chem. 2006;281(33):23492–23502. doi: 10.1074/jbc.M602254200. [DOI] [PubMed] [Google Scholar]
- Lu M. Fu D. Structure of the zinc transporter YiiP. Science. 2007;317(5845):1746–1748. doi: 10.1126/science.1143748. [DOI] [PubMed] [Google Scholar]
- Kolaj-Robin O. Russell D. Hayes K. A. Pembroke J. T. Soulimane T. Cation Diffusion Facilitator family: Structure and function. FEBS Lett. 2015;589(12):1283–1295. doi: 10.1016/j.febslet.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Xue J. Xie T. Zeng W. Jiang Y. Bai X. C. Cryo-EM structures of human ZnT8 in both outward- and inward-facing conformations. Elife. 2020;9:e58823. doi: 10.7554/eLife.58823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch E. Lin W. Chai J. Hershfinkel M. Fu D. Sekler I. Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity. Proc. Natl. Acad. Sci. U. S. A. 2012;109(19):7202–7207. doi: 10.1073/pnas.1200362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement N. R. Gould J. M. Pyranine (8-hydroxy-1,3,6-pyrenetrisulfonate) as a probe of internal aqueous hydrogen ion concentration in phospholipid vesicles. Biochemistry. 1981;20(6):1534–1538. doi: 10.1021/bi00509a019. [DOI] [PubMed] [Google Scholar]
- Clarke R. J. Apell H. J. A stopped-flow kinetic study of the interaction of potential-sensitive oxonol dyes with lipid vesicles. Biophys. Chem. 1989;34(3):225–237. doi: 10.1016/0301-4622(89)80061-4. [DOI] [PubMed] [Google Scholar]
- Apell H. J. Bersch B. Oxonol VI as an optical indicator for membrane potentials in lipid vesicles. Biochim. Biophys. Acta. 1987;903(3):480–494. doi: 10.1016/0005-2736(87)90055-1. [DOI] [PubMed] [Google Scholar]
- Holoubek A. Vecer J. Opekarova M. Sigler K. Ratiometric fluorescence measurements of membrane potential generated by yeast plasma membrane H+-ATPase reconstituted into vesicles. Biochim. Biophys. Acta. 2003;1609(1):71–79. doi: 10.1016/S0005-2736(02)00656-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for this paper are available upon request to the corresponding author.