

Abstract
The synthesis of a 17α-linked C2-symmetric testosterone dimer and its dihydrotestosterone analog is reported. The dimers were synthesized using a short five-step reaction sequence with 28% and 38% overall yield for the testosterone and dihydrotestosterone dimer, respectively. The dimerization reaction was achieved by an olefin metathesis reaction with 2nd generation Hoveyda-Grubbs catalyst. The dimers and their corresponding 17α-allyl precursors were tested for the antiproliferative activity on androgen–dependent (LNCaP) and androgen–independent (PC3) prostate cancer cell lines. The effects on cells were compared with that of the antiandrogen cyproterone acetate (CPA). The results showed that the dimers were active on both cell lines, with an increased activity towards androgen–dependent LNCaP cells. However, the testosterone dimer (11) was fivefold more active than the dihydrotestosterone dimer (15), with an IC50 of 11.7 μM vs. 60.9 μM against LNCaP cells, respectively, and more than threefold more active than the reference drug CPA (IC50 of 40.7 μM). Likewise, studies on the interaction of new compounds with drug-metabolizing cytochrome P450 3A4 (CYP3A4) showed that 11 was a fourfold stronger inhibitor than 15 (IC50 of 3 μM and 12 μM, respectively). This suggests that changes in the chemical structure of sterol moieties and the manner of their linkage could largely affect both the antiproliferative activity of androgen dimers and their crossreactivity with CYP3A4.
Keywords: Prostate cancer, Testosterone, Dihydrotestosterone, Androgen dimers, CYP3A4
1. Introduction
There is a need to develop original anticancer agents for prostate cancer (PCa). Indeed, recent Canadian cancer statistics indicate that 2 in 5 Canadians are expected to be diagnosed with cancer in their lifetime. Through the year 2022, approximately 1 in 4 Canadians is expected to die from the disease [1]. The three most widespread cancers are projected to be lung (30000 new cases), breast in females (28600 new cases) and prostate in males (24600 new cases) [1]. For PCa alone, 4600 deaths are anticipated in 2022, which would make it the fifth most deadly cancer with 22.6% mortality rate. Considering these statistics, the aging world population and our keen interest in developing new strategies for the treatment of PCa, we designed and investigated several types of dimeric testosterone (TS) molecules as prospective “natural antiandrogens”. In fact, the design of dimeric steroids is a subject of intense research [2]. Those compounds are designed to dock on two androgen receptor units to hinder their homodimerization, which would normally occur upon interaction with free androgens, TS or dihydrotestosterone (DHT). The correct conformation of this complex is crucial for phosphorylation, translocation into the cell nucleus and, ultimately, triggering cell growth [3]. In earlier investigations, we synthesized a variety of androgen dimers linked at 7α position of the steroid nucleus [4–7]. The goal of these studies was to utilize the natural hormone TS (1) for construction of prospective antiandrogens for the treatment of PCa. These dimers were linked either by a but-2-ene (cis-2 and trans-2) chain [4,5] or, more recently, by a hexatriene chain (3) [6] (Fig. 1). A diester chain linked another series of dimers (4a-d) that were synthesized to study the effect of the length of the spacer between the two TS units [7] (Fig. 1). In all these compounds, the tether chains were purposely linked to the 7α position suitably located in the middle of the steroid nucleus, far from the key binding sites of TS (3-C=O and 17β-OH) required for interaction with the androgen receptor (AR) [8]. This manuscript presents our recent effort in this promising research field. For the first time, we used the 17α instead of 7α position for linking the TS units. Furthermore, using this approach, we prepared the corresponding DHT dimer for biological comparison and for evaluation of crossreactivity with CYP3A4, a cytochrome P450 monooxygenase responsible for the oxidation of TS and other endogenous and xenobiotic substances [5,9]. As indicated earlier, TS and DHT interact with the AR via two main functional groups the 3-C=O and 17β-OH, a key step to trigger their biological response [8]. Nevertheless, it is important to indicate that the 17α position is known to accommodate some structural modifications leading to important biologically active steroid molecules such as the drug ethynylestradiol (5), a well-known contraceptive agent [10]. Other examples are etonogestrel (6) and levonorgestrel (7), used as a mean of birth control in women [11] (Fig. 2). Even though it can be risky to functionalize a steroid near the sites where it binds to its receptor, here we demonstrate that the 17α position can be harnessed for the development of unique antiandrogenic drugs.
Fig. 1.

TS (1) with diverse examples of dimers studied in our laboratory. They are linked at 7α by a but-2-ene chain (cis-2 or trans-2), a hexatriene chain (3) or a diester chain (4a-d).
Fig. 2.

Structure of important birth control steroids: ethynylestradiol (5), Etonogestrel (6) and Levonorgestrel (7).
2. Chemistry
The synthetic path for the preparation of the dimers is described in Scheme 1. TS was reacted with ethylene glycol under acidic condition to give the crude acetal 8 quantitatively. The same reaction gave the DHT acetal 12 with 98% yield, after purification. The conjugated acetal 8 was unstable upon purification by flash chromatography on silica. Indeed, only 43% of acetal 8 was recovered upon purification. So, it was decided to use the crude material 8 for the next synthetic step. Therefore, the crude acetal 8 and the pure acetal 9 were oxidized with pyridinium chlorochromate on alumina in dichloromethane [12]. Then, the TS derivative 10 was purified by flash chromatography, with 65% overall yield from TS. Compound 13 was obtained with 93% yield and used as such for the next step. Stereospecific addition of allylmagnesium bromide on the 17-keto function, followed by the deprotection of the acetal in acidic media, gave the momomeric allyl precursors of the final dimers, compounds 10 and 14, with 74% and 84% yield, respectively. The synthesis of TS dimer 11 and DHT dimer 15 was performed using a metathesis reaction condition and the 2nd generation Hoveday-Grubbs catalyst with 58% and 49% yield, respectively [13,14]. All intermediates and the final dimers were fully characterized by infrared (IR) and nuclear magnetic resonance (1H and 13C NMR) spectroscopy and by high-resolution mass spectrometry (HRMS) analyses.
Scheme 1. Reagents and conditions:

a) HOCH2CH2OH, TsOH, toluene, reflux, 15 h, (TS, 100%/DHT, 98%); b) PCC, alumine, CH2Cl2, 22 °C 3 h (65%/93%); c) 1. CH2=CH–CH2MgBr, Et2O, CH2Cl2, −78 °C, 30 min; 2. HCl 10% aq., THF, 22 °C, 15 h, (74%/84%); d) 5% Hoveyda-Grubbs cat. 2nd gen., CH2Cl2, 22 °C, 15 h (58%/49%).
3. Results and discussion
3.1. Antiproliferative activity of new dimers and precursors on prostate cancer cell lines
The antiproliferative activity of TS dimer (11), DHT dimer (15) and their respective precursors (10, 14) was measured using the MTT assay on two PCa cell lines: androgen-dependent LNCaP (AR+) and androgen-independent PC3 (AR−) [15,16]. The antiandrogen drug cyproterone acetate (CPA) was used as a positive control. The results from the antiproliferative activity assay are shown in Table 1, where IC50 is the compound concentration inhibiting cell growth by 50%. Firstly, it is noteworthy that all compounds were more active towards the androgen-dependent LNCaP cells. This demonstrates that the activity of the novel steroids is linked to their interaction with the AR, conferring a certain selectivity to the biological action. When the precursor 10 is dimerized into dimer 11, the IC50 decreases from 34.0 μM to 11.7 μM against LNCaP cells. A threefold improvement in the inhibitory potency indicates that, in this case, the dimerization is clearly beneficial for enhancing the biological activity. The inhibitory activity is weaker for PC3 cells but the same trend is observed: precursor 10 exhibits a nearly twofold higher IC50 relative to that of 11 (77.9 μM vs. 43.4 μM, respectively). This indicates that in LNCaP (AR+) cancer cells the TS dimer 11 most probably interacts with the active site of the AR in a way that prevents cell growth. Importantly, in LNCaP cells, compound 11 is ~3.5 times more potent than the reference drug CPA (IC50 of 40.7 μM), highlighting the relevance of the concept of dimeric steroids.
Table 1.
Antiproliferative activity of TS dimer 11, DHT dimer 15, their respective precursors (10 and 14) and CPA on androgen-sensitive (LNCaP) and androgen-insensitive (PC3) human prostate adenocarcinoma cell lines.
| Compound | IC50 (μM)a | |
|---|---|---|
| LNCaP (AR+) | PC3 (AR−) | |
| 10 (17α-allyl-TS) | 34.0 | 77.9 |
| 11 (17α-TS2) | 11.7 | 43.4 |
| 14 (17α-allyl-DHT) | 52.2 | 91.3 |
| 15 (17α-DHT2) | 60.9 | 105.3 |
| CPAb | 40.7 | 84.3 |
Inhibitory concentration (IC50) is concentration of drug inhibiting cell growth by 50%.
Cyproterone acetate.
We envisioned that the DHT analogs would show greater activities, since DHT binds more strongly to the active site of the AR. However, the antiproliferative activities of the DHT-based compounds were lower for both cell lines (Table 1). The precursor 14 displayed an IC50 of 91.3 μM against PC3 cells and 52.2 μM against LNCaP cells. The respective potency of the DHT dimer 15 was even lower: IC50 of 105.3 μM and 60.9 μM, respectively. This rather surprising result, considering the higher affinity of DHT towards the AR, is unlikely linked to the pharmacodynamic action of the DHT analogs but could be due to their lower solubility in the cell culture media and decreased concentration at the receptor site. Nevertheless, the results clearly demonstrate that the new compounds, especially the TS dimer 11, are capable of modulating conformational changes in the active site leading to the AR inhibition and, thus, they are suitable for probing the androgen-dependent growth of PCa cell lines.
3.2. Interaction of new compounds with CYP3A4
CYP3A4 is the major and most clinically relevant drug-metabolizing enzyme that also largely contributes to biotransformation of several endogenous compounds, such as TS, androstenedione and DHT [17,18]. CYP3A4 has a large and malleable active site and can accommodate two or more sterol molecules, exhibiting positive binding cooperativity [9]. Moreover, our prior studies demonstrated that CYP3A4 can form complexes with sterol dimers as well. We found that the 7α-linked TS dimers can bind to the active site of CYP3A4 and inhibit its activity [5,6]. This crossreactivity is undesired, because it could affect clearance of drugs predominantly metabolized by CYP3A4 and contribute to drug-drug interactions. Further, changes in the catalytic activity of CYP3A4 could alter systemic androgen levels and, subsequently, androgen receptor signaling, prostate growth and cancer pathogenesis. Therefore, it is important to assess the inhibitory potential of new sterol dimers towards CYP3A4.
As previously, the dissociation constant (Kd; a measure of the binding affinity) for new compounds was determined based on spectral titrations of recombinant CYP3A4. Upon association, all compounds induced a low-to-high spin transition in CYP3A4 (blue shift in the Soret band, Fig. 3A–D) but to a different extent. The binding of precursors was positively cooperative, as evident from sigmoidal titration plots derived for 10 and 14 (right insets in Fig. 3A, C) and the Hill coefficients (nH) higher than unity (Table 2). The high-spin content and the precursor concentration at half-saturation (S50; equivalent to Kd) were similar: ~70% and 17–22 μM, respectively (Table 2). In contrast, the binding of dimers to CYP3A4 was non-cooperative, with notable differences in Kd and the extent of the high-spin transition. Compared to the DHT-based 15, the TS-based 11 had a 40-fold higher affinity and induced a near complete rather than partial high-spin shift (Fig. 3B, D; Table 2).
Fig. 3.
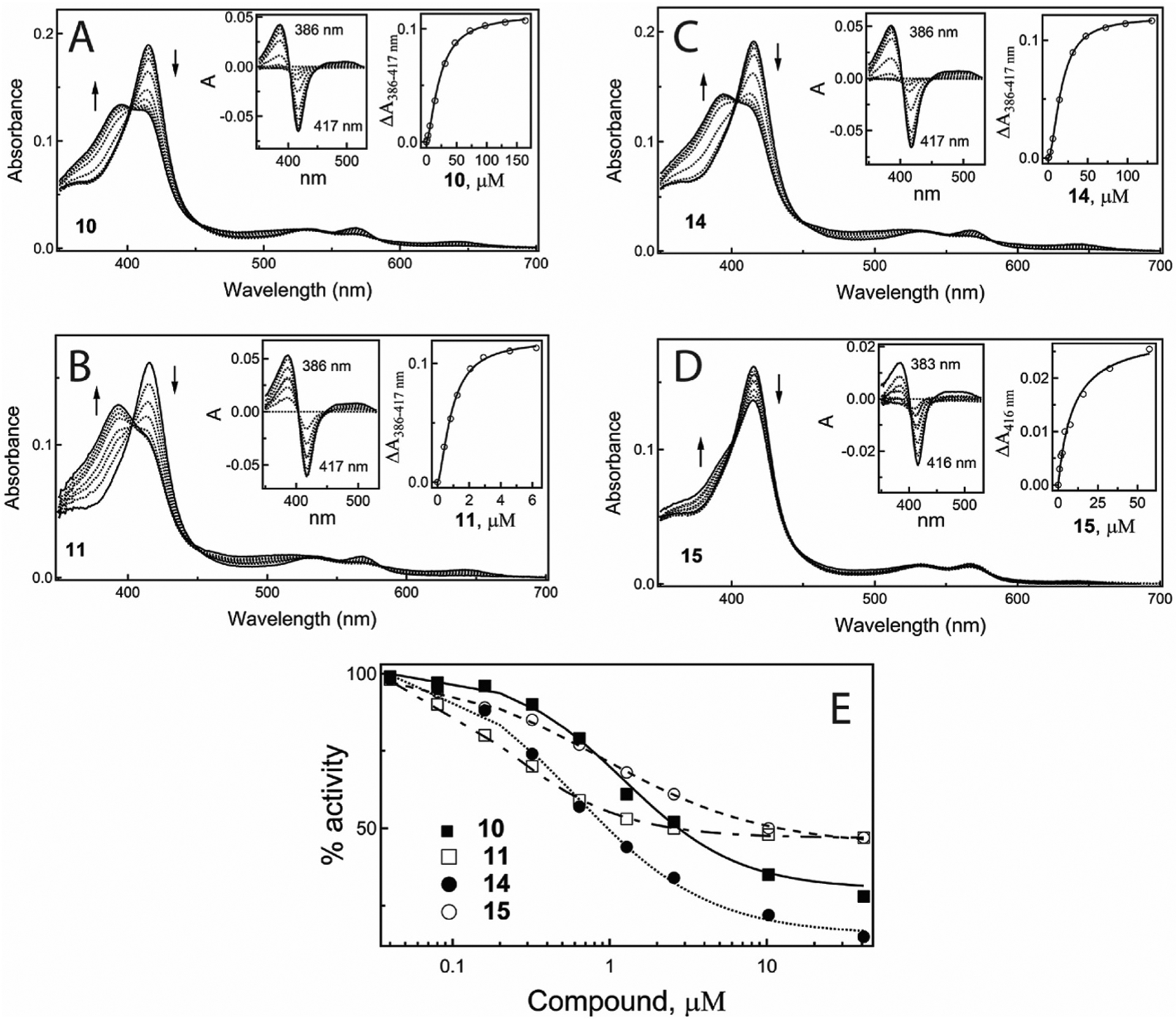
A, B - Spectral changes in CYP3A4 induced by the TS-based compounds 10 and 11, respectively. C, D - Spectral changes in CYP3A4 induced by the DHT-based compounds 14 and 15, respectively. Absorbance spectra of 1.5–2 μM CYP3A4 at different ligand concentrations were recorded in 0.1 M phosphate buffer, pH 7.4, at room temperature. Left insets are the difference spectra; right insets are titration plots with sigmoidal (A and C) or hyperbolic fittings (B and D). The derived Kd, S50 and nH values are given in Table 2.
Table 2.
Interaction of new TS and DHT dimers (11 and 15) and their precursors (10 and 14) with recombinant CYP3A4.
| Compound | Kd (μM) a | S50 (μM) b | nH c | high spin (%)d | IC50 (μM) e |
|---|---|---|---|---|---|
| 10 | 22 ± 4 | 1.52 ± 0.02 | 70 ± 2 | 2.8 ± 0.3 | |
| 11 | 0.24 ± 0.02 | 92 ± 4 | 3.0 ± 0.2 | ||
| 14 | 17 ± 2 | 1.76 ± 0.06 | 71 ± 3 | 1.0 ± 0.1 | |
| 15 | 9.6 ± 0.6 | 33 ± 3 | 12 ± 2 |
All values represent an average of three measurements ± standard deviation.
Spectral dissociation constant determined from hyperbolic fittings to titration plots shown in right insets in Fig. 3B, D.
Ligand concentration at half-saturation (equivalent to Kd) and Hill coefficient, respectively. Both parameters were determined from sigmoidal fittings to titration plots shown in right insets in Fig. 3A, C.
Ligand concentration at half-saturation (equivalent to Kd) and Hill coefficient, respectively. Both parameters were determined from sigmoidal fittings to titration plots shown in right insets in Fig. 3A, C.
The high-spin content estimated at the end of equilibrium titrations relative to azamulin-bound CYP3A4 (100% high-spin conversion).
Half-inhibitory concentration for the BFC debenzylase activity of CYP3A4.
The inhibitory potency of new compounds was assessed for the 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) debenzylase activity of CYP3A4 in a soluble reconstituted system. The IC50 values for the TS-based 10 and 11 were virtually similar (2.8 μM and 3.0 μM, respectively), whereas those for the DHT-based compounds differed by an order of magnitude: 1.0 μM for 14 and 12 μM for 15 (Table 2). Thus, compared to 11, 15 was a fourfold weaker inhibitor of CYP3A4. It should be noted that neither dimer could fully inhibit CYP3A4, with the maximal inhibition level limited to ~46% (Fig. 3E). Interestingly, at sub-micromolar concentrations, 11 inhibited CYP3A4 more potently than its precursor 10, while 15 was less potent than 14 within the entire concentration range. Such distinct properties of dimeric compounds were surprising, as TS and DHT differ only in one bond saturation and have similar lipophilicity (logP of 3.3 and 3.4, respectively). One possible explanation is that elimination of the double bond in 15 affects the dimer conformation in aqueous solutions and/or leads to formation of higher order oligomers that are less favorable for the interaction with CYP3A4 and, possibly, the AR. It should be emphasized though that the PCa cell lines used in this study do not appreciably express CYP3A4 and, hence, the compounds’ inhibitory activity towards CYP3A4 should not interfere with the cell proliferation assays (Table 1).
4. Conclusion
In this study, we designed, synthesized and characterized a 17α-TS dimer and its DHT analog as potential “natural antiandrogens” for the treatment of PCa. The new dimers were tested on both AR+ and AR− PCa cells. The TS dimer 11 was ~3.7 times more potent on AR + LNCaP than AR− PC3 cells, with IC50 of 11.7 μM and 43.4 μM, respectively, and ~3.5 times more active than the reference drug CPA towards AR + PCa cells. Contrary to our expectations, the DHT dimer 15 was less active than 11, possibly due to its lower solubility that could lead to a differential conformation or aggregation in cell culture medium. In the future, AR binding experiments could confirm those conclusions. The same trend was observed for the inhibitory activity towards CYP3A4, where 15 displayed a fourfold lower potency than 11. Thus, alterations in the chemical structure and the manner of conjugation of sterol nuclei could largely affect both the antiproliferative activity of androgen dimers and their crossreactivity with CYP3A4.
5. Experimental protocols
5.1. Biological methods
The antiproliferative activity of all compounds was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay [15,16]. LNCaP androgen-sensitive human prostate adenocarcinoma and PC3 androgen-insensitive human prostate adenocarcinoma cells were obtained from the American Type Culture Collection (Manassas, VA). LNCap and PC3 cells were cultured in RPMI medium (Hyclone, Logan, UT) supplemented with 10% of calf serum and Penicillin-Streptomycin-Glutamine (complete RPMI media). The cells were maintained at 37 °C in a moisture saturated atmosphere containing 5% CO2.
5.1.1. Antiproliferative activity assay
Cell viability of compounds 10, 11, 14, 15 and CPA was determined by the MTT assay [15,16]. All compounds were initially dissolved in 100% dimethyl sulfoxide (DMSO; Sigma Chemical Company, Oakville, Canada) to prepare stock solutions at 2.5 mM, 5 mM, 10 mM, 20 mM, 40 mM, 80 mM, and 160 mM. These stock solutions were 500-fold diluted in complete RPMI media before cell treatment. Then, a solution of 0.2% DMSO in complete RPMI media was used as vehicle in control groups. Briefly, 8×103 cells/well were seeded into 96-well plate and incubated overnight. After culturing for 48 h in fresh medium containing various concentrations of compounds, the cells were stained with MTT solution (0.5 mg/mL; Sigma-Aldrich) for 3 h at cell culture condition followed by dissolving with 10% Triton-X 100 in acidic isopropanol (0.1 N HCl). The cell growth was determined by measuring optical density (OD) value at 590 nm using the Synergy HT Microplate Reader (from Bio-Tek). Readings obtained from treated cells were compared with measurements of control cells plates fixed on the treatment day, and the percentage of cell growth inhibition was then calculated for each compound. Each condition included three replicate wells with three independent repeats.
5.2. In vitro studies on CYP3A4
Codon-optimized full-length human CYP3A4 was produced as reported previously [19]. Equilibrium ligand binding to CYP3A4 was monitored in a Cary 300 spectrophotometer at ambient temperature in 0.1 M phosphate buffer, pH 7.4, supplemented with 20% glycerol and 1 mM dithiothreitol. Sterol dimers and precursors were dissolved in DMSO and added to a 1.5–2 μM protein solution in small aliquots, with the final solvent concentration <2%. The Kd values were determined from hyperbolic or sigmoidal fits to the titration plots that were built based on the difference absorbance spectra. Inhibitory potency of sterol dimers and precursors for the BFC debenzylation activity of CYP3A4 was assessed in a soluble reconstituted system at 37 °C as described in detail elsewhere [20].
5.3. Chemistry
All reactions were performed under an inert atmosphere of nitrogen and with anhydrous solvents. The starting material, reactant and solvents were obtained commercially and used as such or purified and dried by usual methods [21]. The organic solutions were dried over magnesium sulfate (MgSO4), filtered and evaporated on a rotary evaporator under reduced pressure. All reactions were checked by UV fluorescence. Commercial TLC plates were Sigma T 6145 (polyester silica gel 60 Å, 0.25 mm). Flash column chromatography was performed according to the method of Still et al. on Merck grade 60 silica gel, 230–400 mesh [22]. All solvents used in flash chromatography were distilled before use.
The infrared spectra were taken on a Nicolet Impact 420 FT-IR spectrophotometer. The samples were analyzed as powders. Mass spectral assays were obtained using a MS model 6210, Agilent technology instrument. The high resolution mass spectra (HRMS) were obtained by TOF (time of flight) using ESI (electrospray ionization) using the positive mode (ESI+) (Université du Québec à Montréal). Nuclear magnetic resonance (NMR) spectra were recorded either on a Varian 200 MHz or on a Bruker 400 MHz NMR apparatus. Samples were dissolved in deuterated chloroform (CDCl3) for data acquisition using the residual solvent signal as internal standard (chloroform δ 7.26 ppm for 1H NMR and δ 77.23 ppm for 13C NMR). Chemical shifts (δ) are expressed in parts per million (ppm), the coupling constants (J) are expressed in hertz (Hz). Multiplicities are described by the following abbreviations: s for singlet, d for doublet, t for triplet and m for multiplet, and bs for broad singlet.
Note: The nomenclature of the androgenic molecules described here was based on the androgen skeleton (4-androsten-17β-ol-3-one) for clarity to the readers.
5.3.1. Synthesis of the 17-α-linked TS dimer and its dihydrotestosterone analog
5.3.1.1. Preparation of androst-4-en-17β-ol spiro-(1,3)dioxolan (8).
Ethylene glycol (1.8 mL, 32.2 mmol) and p-toluenesulphonic acid (136.0 mg, 0.79 mmol) were added to a solution of TS (1) (3.80 g, 13.2 mmol) dissolved in benzene (40 mL). A Dean-Stark apparatus was set up and the solution heated at reflux for 12 h. After cooling to room temperature, the reaction mixture was diluted with ether (40 mL), transferred into a separatory funnel and washed three times with water (3 × 30 mL). The organic phase was dried with anhydrous magnesium sulfate, filtered and evaporated under reduced pressure to give the crude product with 100% yield. It is noteworthy that this compound is quite sensitive to the acidic condition of silica, so the crude material is used at the next step. Yet, a portion of the acetal was purified by flash chromatography (hexane/acetone, 90/10) to give 8 used for the following spectral analysis. Mp: 156–157 °C; IR ν (cm−1): 3224 (O–H), 1252 (C–O), 1098 (C–O); 1H NMR (200 MHz, CDCl3, δ ppm): 5.36–5.33, (1H, m, 4-CH), 3.94 (4H, s, O–CH2–CH2–O), 3.64 (1H, t, J = 8.6 Hz, 17-CH), 2.61–2.53 (1H, m, 2-CH2 ax.), 1.04 (3H, s, 19-CH3), 0.76 (3H, s, 18-CH3); 13C NMR (200 MHz, CDCl3, δ ppm): 140.2 (C-4), 121.9 (C-5), 109.4 (C-3), 81.9 (C-17), 64.4, 64.2 (O–CH2–CH2–O), 51.3, 49.8, 42.7, 41.8, 36.7, 36.6, 36.3, 32.0, 31.3, 31.0, 30.5, 23.4, 20.6, 18.9, 11.0; HRMS (ESI+): (M + H)+ calculated for C21H32O3 = 332.2351; found = 332.2353.
5.3.1.2. Preparation of androst-4-en-17-one spiro-(1,3)dioxolan (9).
Pyridinium chlorochromate (1.86 g, 8.6 mmol) and alumina (5.79 g, 56.8 mmol) were added to a solution of the crude acetal (8) (1.19 g, 3.6 mmol) dissolved in methylene chloride (15 mL). The mixture was stirred at room temperature for 3 h. Afterwards, the reaction mixture was filtered on silica gel with methylene chloride as the eluent (130 mL). The organic phase was then dried, filtered, evaporated and purified by flash chromatography (hexane/acetone, 90/10) to give 773.2 mg (65% from TS 1) of the oxidized product 9. Mp: 181–183 °C; IR ν (cm−1): 1736 (C=O), 1092 (C–O). 1H NMR (200 MHz, CDCl3, δ ppm): 5.28–5.38, (1H, m, 4-CH), 3.93 (4H, s, O–CH2–CH2–O), 2.61–2.38 (2H, m, 2-CH2 ax., 16-CH2 pseudo-ax.), 1.04 (3H, s, 19-CH3), 0.87 (3H, s, 18-CH3).; 13C NMR (200 MHz, CDCl3, δ ppm): 221.1 (C-17), 140.4 (C-4), 121.4 (C-5), 109.3 (C-3), 64.4, 64.2 (O–CH2–CH2–O), 51.7, 49.8, 47.5, 41.8, 36.7, 36.2, 35.8, 31.5, 31.4, 31.0, 30.6, 21.9, 20.3, 18.9, 13.5; HRMS (ESI+): (M + H)+ calculated for C21H30O3 = 330.2195; found = 330.2194.
5.3.1.3. Preparation of 17α-allyl-androst-4-en-3-one-17β-ol (10).
The oxidized steroid (9) (425.6 mg, 1.24 mmol) was dissolved in freshly distilled tetrahydrofuran (4 mL) under an inert atmosphere and the reaction vessel was then placed in an ice bath. In the meantime, magnesium turnings (109.5 mg, 4.51 mmol) were immersed in dry diethyl ether (3.5 mL) in a two-neck round bottom flask surmounted by a condenser and the setup purged with nitrogen. Then, allyl bromide (0.39 mL, 4.51 mmol) was added to the flask and the reaction mixture was stirred vigorously. After approximately 2 min of stirring, the solution starts to boil. Once the reflux was over and the turnings disappeared, the mixture was transferred into the flask containing the steroid solution via a syringe. The mixture was stirred at 0 °C for 30 min and then at room temperature for 1 h. Afterwards, the reaction mixture was diluted with diethyl ether (30 mL), transferred into a separatory funnel and washed once with an HCl 10% solution (20 mL) and three times with water (3 × 20 mL). The organic phase was dried with anhydrous magnesium sulfate, filtered and evaporated under reduced pressure. The crude material was then dissolved in a mixture of THF (4 mL) and 10% aqueous solution of HCl (2 mL) in order to remove the acetal protective group. The solution was agitated for 16 h before being diluted with ether (30 mL), transferred into a separatory funnel and washed three times with water (3 × 20 mL). The organic phase was dried with anhydrous magnesium sulfate, filtered and evaporated under reduced pressure. The crude material was purified by flash chromatography (hexane/acetone, 90/10) to give 312.0 mg (74%) of the desired material. Mp: 53–55 °C; IR ν (cm−1): 3440 (O–H), 1662 (C=O enone), 1614 (C=C), 1231 (C=O); 1H NMR (200 MHz, CDCl3, δ ppm): 6.04–5.90 (1H, m, CH2–CH=CH2), 5.72 (1H, s, 4-CH), 5.22–5.10 (2H, m, CH2–CH=CH2), 1.20 (3H, s, 19-CH3), 0.93 (3H, s, 18-CH3); 13C NMR (200 MHz, CDCl3, δ ppm): 199.5 (C-3), 171.2 (C-5), 134.6 (CH2–CH=CH2), 123.9 (C-4), 119.4 (CH2–CH=CH2), 82.2 (C-17), 53.7, 50.0, 46.0, 41.7, 38.7, 36.4, 35.8, 34.9, 34.0, 32.8, 31.7, 31.6, 23.7, 20.7, 17.4, 14.3; HRMS (ESI+): (M + H)+ calculated for C22H32O2 = 328.2402; found = 328.2404.
5.3.1.4. Preparation of ((E)-17,17’-(but-2-ene-1,4-diyl))bis(androst-4-en-3-one-17β-ol) (11).
The 17α-allyl-androst-4-en-3-one-17β-ol (10) (108.6 mg, 0.33 mmol) was dissolved in methylene chloride (3.0 mL). Under an inert atmosphere of nitrogen, a solution of 2nd generation Hoveyda-Grubbs catalyst (27.6 mg, 0.03 mmol) in methylene chloride (1.0 mL) was added to the reaction mixture. The solution was stirred for 48 h in the dark. The nitrogen atmosphere was constantly purged during the first 2 h of the reaction, and the loss of the solvent was compensated if evaporation occurs. Likewise, during the remaining course of the reaction, the reaction flask was regularly purged with nitrogen. Afterwards, the solvent was evaporated and the crude material immediately purified by flash chromatography (hexane/acetone, 85/15) to give 60.4 mg (58%) of the dimer. Mp: Decomposition begins at 232 °C; IR ν (cm−1): 3440 (O–H), 1661 (C=O enone), 1615 (C=C), 1231 (C–O); 1H NMR (200 MHz, CDCl3, δ ppm): 5.79–5.48 (4H, s broad, 4-CH, 4′-CH and CH2–CH=CH–CH2), 1.20 (3H, s, 19-CH3), 0.92 (3H, s, 18-CH3); 13C NMR (200 MHz, CDCl3, δ ppm): 199.6 (C-3), 171.2 (C-5), 130.7 (CH2–CH=CH–CH2), 123.9 (C-4), 82.4 (C-17), 53.8, 50.1, 46.0, 40.5, 38.7, 36.4, 35.7, 35.0, 34.0, 32.8, 31.7, 31.6, 23.7, 20.7, 17.4, 14.4; HRMS (ESI+): (M + H)+ calculated for C42H60O4 = 628.4492; found = 628.4491.
5.3.1.5. Preparation of androst-17β-ol spiro-(1,3)dioxolan (12).
The experimental procedure was the same as for the synthesis of the androst-5-en-17β-ol spiro-(1,3)dioxolan (8). The crude material was purified by flash chromatography (hexane/acetone, 90/10) to give 98% of the desired product 12. Of note, contrary to acetal 8, this acetal was stable during silica gel purification. Mp: 158–159 °C; IR ν (cm−1): 3383 (O–H), 1258 (C–O), 1100 (C–O); 1H NMR (400 MHz, CDCl3, δ ppm): 3.93 (4H, s, O–CH2–CH2–O), 3.63 (1H, t, J = 8,6 Hz, 17-CH), 2.13–2.02 (1H, m, 2-CH2 ax.), 0.82 (3H, s, 19-CH3), 0.73 (3H, s, 18-CH3); 13C NMR (400 MHz, CDCl3, δ ppm): 109.4 (C-3), 82.0 (C-17), 64.2 (O–CH2–CH2–O), 54.2, 51.0, 43.8, 43.0, 38.0, 36.8, 36.1, 35.6, 35.5, 31.5, 31.2, 30.6, 28.5, 23.4, 20.8, 11.4, 11.2; HRMS (ESI+): (M + H)+ calculated for C21H34O3 = 334.2508; found = 334.2493.
5.3.1.6. Preparation of androst-17-one spiro-(1,3)dioxolan (13).
The experimental procedure was essentially the same as for the synthesis of the androst-5-en-17-one spiro-(1,3)dioxolan (9), but the crude material was clean and did not require purification. The oxidized product 7 was obtained with 93% yield. Mp: 151–152 °C; IR ν (cm−1): 1740 (C=O), 1096 (C–O); 1H NMR (400 MHz, CDCl3, δ ppm): 3.92 (4H, s, O–CH2–CH2–O), 2.45–2.38 (1H, dd, J = 19.2 Hz and 8.12 Hz, 16-CH2 pseudo-ax.), 2.09–2.00 (1H, m, 4-CH2 ax.), 0.84 (3H, s, 19-CH3), 0.82 (3H, s, 18-CH3); 13C NMR (400 MHz, CDCl3, δ ppm): 221.4 (C-17), 109.2 (C-3), 64.2 (O–CH2–CH2–O), 54.2, 51.4, 47.8, 43.7, 38.0, 36.0, 35.9, 35.7, 35.1, 31.6, 31.1, 30.8, 28.3, 21.8, 20.5, 13.8, 11.4; HRMS (ESI+): (M + H)+ calculated for C23H33O2 = 332.2351; found = 332.2338.
5.3.1.7. Preparation of 17α-allyl-androst-3-one-17β-ol (14).
The experimental procedure was the same as for the synthesis of the 17α-allyl-androst-4-en-3-one-17β-ol (10). The crude material was purified by flash chromatography (hexane/acetone, 90/10) to give 84% of the desired product 14. This product was already synthesized by a different synthetic path [23]. Mp: 117–118 °C; IR ν (cm−1): 3446 (O–H), 1701 (C=O ketone), 1645 (C=C), 1253 (C–O); 1H NMR (400 MHz, CDCl3, δ ppm): 6.04–5.90 (1H, m, CH2–CH=CH2), 5.21–5.13 (2H, m, CH2–CH=CH2), 1.03 (3H, s, 19-CH3), 0.91 (3H, s, 18-CH3); 13C NMR (400 MHz, CDCl3, δ ppm): 212.0 (C-3), 134.8 (CH2–CH=CH2), 119.4 (CH2–CH=CH2), 82.4 (C-17), 53.8, 50.4, 46.8, 46.3, 44.7, 41.8, 38.6, 38.2, 36.2, 35.8, 35.0, 31.8, 31.5, 28.9, 23.8, 21.1, 14.5, 11.5; HRMS (ESI+): (M + H)+ calculated for C22H34O2 = 330.2559; found = 330.2545.
5.3.1.8. Preparation of ((E)-17,17’-(but-2-ene-1,4-diyl))bis(androst-3-one-17β-ol) (15).
The experimental procedure was the same as for the synthesis of the ((E)-17,17’-(but-2-ene-1,4-diyl))bis(androst-4-en-3-one-17β-ol) (11). The crude material was purified by flash chromatography (hexane/acetone, 85/15) to give 49% of the desired dimer 15. Mp: Decomposition begins at 243 °C; IR ν (cm−1): 3480 (O–H), 1709 (C=O ketone), 1607 (C=C), 1221 (C–O); 1H NMR (400 MHz, CDCl3, δ ppm): 5.70 (2H, s, CH2–CH=CH–CH2), 1.03 (6H, s, 19,19′-CH3), 0.89 (6H, s, 18,18′-CH3); 13C NMR (400 MHz, CDCl3, δ ppm): 212.0 (C-3,3′), 130.8 (CH2–CH=CH–CH2), 82.6 (C-17,17′), 53.8, 50.4, 46.8, 46.2, 44.7, 40.6, 38.6, 38.2, 36.2, 35.8, 35.1, 31.8, 31.5, 28.9, 23.8, 21.1, 14.5, 11.5; HRMS (ESI+): (M + H)+ calculated for C42H64O4 = 632.4805; found = 632.4796.
Supplementary Material
Acknowledgments
This work was sponsored by a grant from the “Ministère de l’Économie et de l’Innovation”, Québec government to C. Reyes-Moreno and G. Bérubé, and by the National Institutes of Health Grant ES025767 to I. F. Sevrioukova.
Abbreviations:
- BFC
7-benzyloxy-4-(trifluoromethyl)coumarin
- CYP3A4
cytochrome P450 3A4
- CPA
cyproterone acetate
- DHT
dihydrotestosterone
- PCa
prostate cancer
- TS
testosterone
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2023.115222.
Data availability
No data was used for the research described in the article.
References
- [1].Brenner DR, Poirier A, Woods RR, Ellison LF, Billette J-M, Demers AA, Zhang SX, Yao C, Finley C, Fitzgerald N, Saint-Jacques N, Shack L, Turner D, Holmes E, For the Canadian cancer statistics advisory committee. Projected estimates of cancer in Canada in 2022, CMAJ (Can. Med. Assoc. J.) 194 (17) (2022) E601–E607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Naha L, Sarker SD, Steroid Dimers: Chemistry and Applications in Drug Design and Delivery, first ed., 2012, p. 440. West Sussex, United Kingdom. [Google Scholar]
- [3].Paquin A, Reyes-Moreno C, Bérubé G, Recent advances in the use of the dimerization strategy as a means to increase the biological potential of natural or synthetic molecules, Molecules 26 (8) (2021) 2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bastien D, Leblanc V, Asselin E, Bérubé G, First synthesis of separable isomeric testosterone dimers showing differential activities on prostate cancer cells, Bioorg. Med. Chem. Lett 20 (2010) 2078–2081. [DOI] [PubMed] [Google Scholar]
- [5].Denisov IG, Mak PJ, Grinkova YV, Bastien D, Bérubé G, Sligar SG, Kincaid JR, The use of isomeric testosterone dimers to explore allosteric effects in substrate binding to cytochrome P450 CYP3A4, J. Inorg. Biochem 158 (2016) 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Paquin A, Oufqir Y, Sevrioukova IF, Reyes-Moreno C, Bérubé G, Innovative C2-symmetric testosterone and androstenedione dimers: design, synthesis, biological evaluation on prostate cancer cell lines and binding to recombinant CYP3A4, Eur. J. Med. Chem 220 (2021), 1134965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vesper A-R, Lacroix J, Gaudreault RC, Tajmir-Rihai H-A, Bérubé G, Synthesis of novel C2-symmetric testosterone dimers and evaluation of antiproliferative activity on androgen-dependent and –independent prostate cancer cell lines, Steroids 115 (2016) 98–104. [DOI] [PubMed] [Google Scholar]
- [8].Nadal M, Prekovic S, Gallastegui N, Helsen C, Abella M, Zielinska K, Gay M, Vilaseca M, Taulès M, Houtsmuller AB, van Royen ME, Claessens F, Fuentes-Prior P, Estébanez-Perpiñá E, Structure of the homodimeric androgen receptor ligand-binding domain, Nat. Commun 8 (2017), 14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Denisov IG, Baas BJ, Grinkova YV, Sligar SG, Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation, J. Biol. Chem 282 (2007) 7066–7076. [DOI] [PubMed] [Google Scholar]
- [10].Sojo-Aranda I, Cortes-Gallegos V, Ethynylestradiol from contraceptive formulations and the ovarian response: an estrogen dose-dependency on natural estradiol concentrations, Gynecol. Endocrinol 9 (1995) 63–66. [DOI] [PubMed] [Google Scholar]
- [11].McNicholas C, Swor E, Wan LJF, Peipert, Prolonged use of the etonogestrel implant and levonorgestrel intrauterine device: 2 years beyond Food and Drug Administration-approved duration, Am. J. Obstet. Gynecol 216 (6) (2017) 586 e1–e586 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cheng Y-S, Liu W-L, Chen S, Pyridinium chlorochromate adsorbed on alumina as a selective oxidant for primary and secondary alcohols, Synthesis 3 (1980) 223–224. [Google Scholar]
- [13].Connon SJ, Blechert S, Recent developments in olefin cross-metathesis, Angew. Chem., Int. Ed 42 (2003) 1900–1923. [DOI] [PubMed] [Google Scholar]
- [14].Yee NK, Farina V, Efficient large-scale synthesis of BILN 2061, a potent HCV protease inhibitor, by a convergent approach based on ring-closing metathesis, J. Org. Chem 71 (2006) 7133–7145. [DOI] [PubMed] [Google Scholar]
- [15].Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB, Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing, Cancer Res. 47 (1987) 936–942. [PubMed] [Google Scholar]
- [16].Ford CHJ, Richardson VJ, Tsaltas G, Comparison of tetrazolium colorimetric and [3H]-uridine assays for in vitro chemosensitivity testing, Cancer Chemother, Pharmacology 24 (1989) 295–301. [DOI] [PubMed] [Google Scholar]
- [17].Waxman DJ, Attisano C, Guengerich FP, Lapenson DP, Cytochrome P-450 steroid hormone metabolism catalyzed by human liver microsomes, Arch. Biochem. Biophys 263 (1988) 424–436. [DOI] [PubMed] [Google Scholar]
- [18].Cheng Q, Sohl CD, Yoshimoto FK, Guengerich FP, Oxidation of dihydrotestosterone by human cytochromes P450 19A1 and 3A4, J. Biol. Chem 287 (2012) 29554–29567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sevrioukova IF, High-level production and properties of the cysteine-depleted cytochrome P450 3A4, Biochemistry 56 (2017) 3058–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Samuels ER, Sevrioukova IF, Rational design of CYP3A4 inhibitors: a one-atom linker elongation in ritonavir-like compounds leads to a marked improvement in the binding strength, Int. J. Mol. Sci 22 (2021) 8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Armarego WLF, Purification of Laboratory Chemicals, Eight Edition, Butterworth-Heinemann, Oxford, 2017, p. 1198, 9780128054574. [Google Scholar]
- [22].Still WC, Kahn M, Mitra A, Rapid chromatographic technique for preparative separations with moderate resolution, J. Org. Chem 43 (1978) 2923–2925. [Google Scholar]
- [23].Ray S, Salman M, Ruiz AA, Stotter PL, Chamness GC, Androgen receptor affinity chromatography: synthesis and properties of 17α-epoxypropyl-dihydrotestosterone sepharose, J. Steroid Biochem 24 (1986) 1111–1115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.