

Abstract
Recent developments in mass spectrometry-based single-cell proteomics (SCP) have resulted in dramatically improved sensitivity, yet the relatively low measurement throughput remains a limitation. Isobaric and isotopic labeling methods have been separately applied to SCP to increase throughput through multiplexing. Here we combined both forms of labeling to achieve multiplicative scaling for higher throughput. Two-plex stable isotope labeling of amino acids in cell culture (SILAC) and isobaric tandem mass tag (TMT) labeling enabled up to 28 single cells to be analyzed in a single liquid chromatography–mass spectrometry (LC–MS) analysis, in addition to carrier, reference, and negative control channels. A custom nested nanowell chip was used for nanoliter sample processing to minimize sample losses. Using a 145-min total LC–MS cycle time, ~280 single cells were analyzed per day. This measurement throughput could be increased to ~700 samples per day with a high-duty-cycle multicolumn LC system producing the same active gradient. The labeling efficiency and achievable proteome coverage were characterized for multiple analysis conditions.
Graphical Abstract
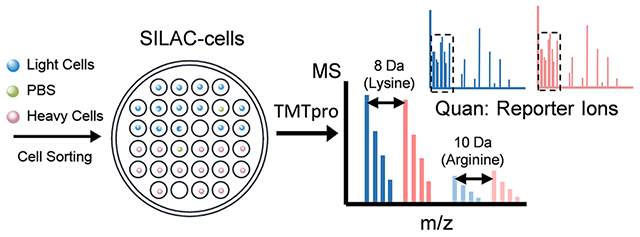
INTRODUCTION
Liquid chromatography–mass spectrometry (LC–MS) is increasingly utilized for unbiased analysis of the proteome of individual cells.1 The increased speed and sensitivity of modern mass spectrometers,2,3 along with improved sample preparation,4–10 separations,11–15 and experimental design16–19 have allowed for deeper analysis in single-cell proteomics (SCP). SCP can be broadly categorized as advancing along two tracks: label-free and label-based workflows. Label-free SCP directly analyzes protein digests from single cells, and since common peptides originating from different cells are indistinguishable, label-free SCP must analyze proteins from one cell per analysis. Label-free SCP has advanced rapidly. Unbiased proteome profiling of single mammalian cells originally required hours per sample and was limited to several hundred proteins per cell.4 With improved low-flow separations and novel MS data acquisition strategies, we can now quantify >3000 proteins from single cells in 40-min analyses, with only a modest reduction of coverage in 20-min analyses.19 Much faster LC gradients have also been explored,20,21 but these may significantly reduce proteome coverage.
Due to its early stage of development, SCP is not widely employed in biomedical research relative to, e.g., RNAseq. One of the main challenges is the throughput. Measuring more cells per unit time is needed to draw statistical conclusions for certain applications of SCP. For example, single-cell RNAseq often requires analysis of thousands of cells to identify rare cell populations, which would be prohibitively expensive for current label-free SCP approaches. Multiplexed analyses using chemical labeling have thus been explored to achieve higher throughput. For label-based SCP, peptides from each cell are tagged with unique isotopic or isobaric labels, which enables common peptides from different cells to be distinguished even when analyzed in a single LC–MS analysis. Most commonly, label-based SCP has employed isobaric tandem mass tags for multiplexed analyses as originally reported in the SCoPE-MS workflow.16 With 18-plex tandem mass tag (TMT) reagents now available, label-based SCP has proven high-throughput capabilities albeit with reduced proteome coverage relative to state-of-the-art label-free approaches. For example, Woo et al.5 reported the identification of ~1500 proteins using a nested nanoPOTS (N2) chip to simplify the pooling of different TMT-labeled cells for LC–MS analysis and improved protein recovery by reducing the sample preparation volume to <30 nL. The throughput of TMT-based SCP can be >10 times higher than that of label-free SCP given the multiplexing capability of TMT reagents.
The number of TMT reagents has increased only gradually over the years, and it is thus unlikely that the degree of multiplexing based on TMT alone will increase more than incrementally in the near term. However, combining two orthogonal forms of multiplexing, such as isotopic and isobaric labeling, enables multiplicative scaling and potentially much greater throughput. Such “hyperplexing” approaches have been explored for bulk-scale proteomics22–32 but not yet for SCP. With these strategies, isotopic labels create a mass difference between common peptides originating from different samples when measured at the MS1 stage, and isobaric tags within each isotopic set enable differentiation and quantification of proteins by their reporter ions in MS2 or MS3 spectra following fragmentation. The combination has thus far enabled two or three times the number of biological samples to be combined and analyzed in a single LC–MS analysis compared to the standard isobaric labeling. For example, Dephoure and Gygi22 reported hyperplexing that combined 3-plex stable isotope labeling of amino acids in cell culture (SILAC) and the 6-plex TMT labeling to enable 18-plex large-scale quantitative proteome studies at a time when there were far fewer TMT channels available. The isotopic and isobaric labeling strategy was then used to study protein turnover kinetics27,32 and in translational applications,24,30 taking advantage of the SILAC labeling principle and fewer missing values within a TMT-labeled set. Everley et al.23 demonstrated 54-plex targeted proteome analysis in a single LC–MS run by measuring the inhibition of protein kinase activity in MCF7 cells using 6-plex TMT, two synthesized TMT-like tags, and three mass variants of the targeted peptide. Evans and Robinson25 developed combined precursor isotopic labeling and isobaric tagging (cPILOT) and applied it to global quantification of peptides. In this workflow, low-pH stable isotope dimethylation (heavy and light) was performed first on the peptide N-terminus before high-pH TMT labeling on the lysine residues. MS3 was used to select the y1 ions to produce signal from the TMT reporter ion. This method was used to analyze the mouse liver proteome26 and peripheral proteome of Alzheimer’s disease in a mouse model.31 The cPILOT concept was then extended to a 24-plex using the dimethylation and DiLeu (N,N-dimethyl leucine) isobaric tags.28 This approach doubles the throughput, yet all quantitative information is lost from arginine-terminated tryptic peptides. With a similar strategy considering mass differences before and after fragmentation, TMT-11 and TMTpro33 reagents with different nominal masses have been combined to achieve 27-plex34 and 29-plex35 analyses.
In the present proof-of-concept work, we evaluated hyperplexing for SCP (hyperSCP) by combining 2-plex SILAC isotopic labeling with 18-plex TMTpro isobaric labeling. As many as 28 single cells were analyzed in a single LC–MS analysis, along with included carrier, negative control, and reference channels. Using a 145-min LC separation with a 60-min active elution profile, we analyzed ~280 cells per day, which could be increased to ~700 cells per day with a high-duty-cycle multicolumn nanoLC system21,36 having the same elution profile. We also explored different conditions to study the tradeoff between proteome coverage and the throughput by utilizing a carrier proteome and applying different separation gradients. To evaluate the quantitative performance of the hyperSCP workflow, 590 single cells from four different cell types were analyzed, which provided ready differentiation and an average coverage of ~800 proteins/cell.
EXPERIMENTAL SECTION
Materials.
Formic acid (FA), triethylammonium bicarbonate buffer (TEAB, 1 M, pH = 8.5), SILAC protein quantitation kit (trypsin), Pierce BCA protein assay kit, Pierce quantitative peptide assays and standards, Pierce peptide desalting spin columns, tris(2-carboxyethyl)phosphine (TCEP), chloroacetamide (CAM), TMTpro reagents, and 50% hydroxylamine were purchased from Thermo Fisher Scientific (Waltham, MA). N-Dodecyl-β-d-maltoside (DDM), sodium dodecyl sulfate, dimethyl sulfoxide (DMSO), buffered oxide etchant, (heptadecafluoro-1,1,2,2-tetrahydrodecyl)dimethylchlorosilane (PFDS), 2,2,4-trimethylpentane, and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from Sigma-Aldrich (St. Louis, MO). S-trap mini columns were purchased from ProtiFi (Farmingdale, NY). LC–MS grade water and acetonitrile used for LC mobile phase A (0.1% v/v FA in water) and B (0.1% FA in acetonitrile) were purchased from Honeywell (Charlotte, NC). All aqueous solutions were prepared using Optima LC/MS grade water from Thermo Fisher Scientific. Other unmentioned reagents were obtained from Sigma-Aldrich.
HyperSCP Chip Fabrication.
The design of the hyperSCP chip (Figures 1a and S1) was adapted from the nested nanoPOTS (N2) chip5 and fabricated using standard photolithography and wet chemical etching as described previously37 with minor modifications. Briefly, the pattern was designed using AutoCAD (Autodesk, San Rafael, CA), and a glass photomask was produced at the Integrated Microfabrication Laboratory at Brigham Young University. A microscope slide that was precoated with chromium and photoresist (75 × 25 mm, Telic, Valencia, CA) was taped to the photomask and flood exposed to UV light for 10 s using a Karl Suss photomask aligner (MA 150 CC, SUSS MicroTec, Garching, Germany). Wet etching with buffered oxide etchant was then performed to etch to a depth of ~5 μm around the designed pattern after developing and removing the chromium. The exposed glass surface was then hydrophobized with 2% PFDS (v/v) in 2,2,4-trimethylpentane. The photoresist and chromium covering the desired pattern were then removed to form hydrophilic nanowells. A 1-mm-thick PMMA frame was glued to an unpatterned microscope glass slide, which served as a cover (Figure S2) for long-term incubation.
Figure 1.
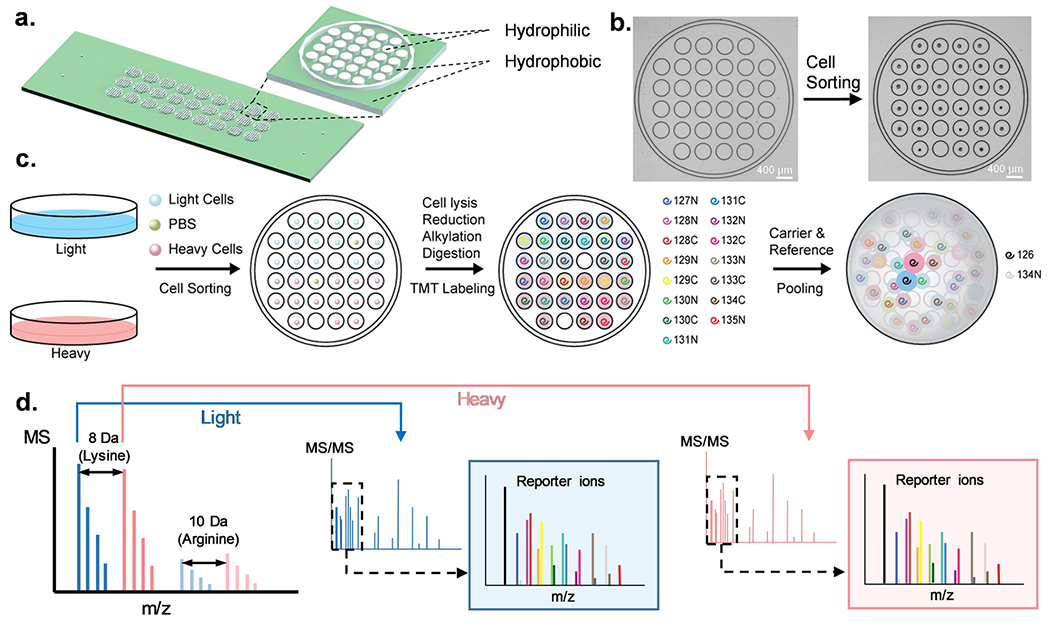
HyperSCP chip and workflow. (a) Design and structure of the hyperSCP chip. Thirty-two hydrophilic nanowells (white) are nested within a hydrophilic ring (white) that is surrounded by a hydrophobic surface (green). Twenty-seven hyperSCP sample sets can be prepared in parallel on a chip. The dimensions are shown in Figure S1. (b) Photomicrographs of a nested nanowell before and after cell sorting. Some nanowells were intentionally left empty during sorting to hold reference samples or serve as negative controls. (c) Workflow of hyperSCP sample preparation. First, the cells were cultured separately in regular media (light) or media containing 13C6 15N2 l-Lysine-2HCl and 13C6 15N4 l-Arginine-HCl (heavy). The cells and a PBS negative control sample were then sorted into each well. After cell lysis, reduction, alkylation and digestion, each single cell sample was labeled with a different TMT reagent. The carrier and reference channels were added after HA quenching, and all the peptides were pooled with 2% mobile phase B/98% mobile phase A. (d) Schematic of hyperSCP. Heavy and light precursors are identified in MS1, selected for fragmentation and analyzed by tandem MS. Reporter ions are produced for relative quantification.
Cell Culture and Harvest.
HeLa, A549, K562, and HFL1 cells were obtained from ATCC (Manassas, VA) and cultured at 37 °C with 5% CO2 using cell culture medium with 10% fetal bovine serum and 1% penicillin/streptomycin. Non-SILAC (“light”) cells were cultured in regular RPMI 1640 medium, while the SILAC-labeled (“heavy”) cells were cultured in the RPMI 1640 medium in which the lysine and arginine were replaced with 13C615N2 l-lysine-2HCl and 13C615N4 l-arginine-HCl. The cells were cultured as reported38 and split upon reaching ~70% confluency. The medium was replaced every 2–3 days. All cells were cultured in light or heavy medium for at least five doubling times for better labeling efficiency (>99%). After labeling, the cells were harvested at 70% confluency. The adherent cells (HeLa, A549, and HFL1) were first rinsed three times with iced phosphate buffered saline (PBS) before collection.
Bulk Sample Preparation for Carrier and Reference Channels.
HeLa and A549 cell digests were prepared in bulk to serve as carrier and reference channels using S-trap.39 The cells were collected using a cell scraper and lysed with 500 μL of 2% SDS buffer in a Protein LoBind tube (Eppendorf, Hamburg, Germany). DNA in the cell lysate was sheared using 18G, 22G, and 25G needles sequentially before being sonicated in ice water for 5 min. The protein concentration was determined with a BCA protein assay. 250 mM TCEP and CAM were added to reach a final concentration of 5 mM TCEP and 20 mM CAM. For reduction and alkylation, the sample mixture was then incubated at 95 °C for 10 min and sonicated for 5 min. After cooling to room temperature, the sample was acidified to pH ~1 by adding 27.5% H3PO4. The sample solution was loaded on the S-trap column with 100 mM TEAB in 90% methanol by centrifuging three times at 4000 × g for 30 s before centrifuging at 4000 × g for 1 min to remove the solvent. 125 μL of digestion buffer containing 30 μg of trypsin was then added for overnight digestion at 37 °C. The peptides were pooled by centrifuging at 4000 × g for 1 min with 50 mM TEAB, 0.2% FA, and 50% ACN sequentially, and dried using a speed vacuum concentrator. After reconstitution and aliquoting, the peptide aliquots were labeled with TMTpro-126 or TMTpro-134 N, quenched with 5% hydroxylamine, and desalted using peptide desalting spin columns. The final concentration of labeled peptides was identified using a Pierce quantitative peptide assay.
Single-Cell Sample Preparation on the HyperSCP Chip.
HeLa, A549, and HFL1 cells were collected after trypsinization, while K562 cells were collected directly. Cells were washed three times with cold (4 °C) PBS after pelletizing the cells by centrifugation at 150 × g for 5 min. The cell suspensions were filtered with a 40-μm cell strainer and kept on ice until use. Fourteen SILAC light cells, 14 SILAC heavy cells, and two PBS-containing droplets of equivalent (~300 pL) volume were sorted into nanowells within each nested well (Figure 1b,c). Both cell sorting and reagent dispensing were performed using the cellenONE platform (Cellenion, Lyon, France). The temperature of the cellenONE platform was set in the software to 1 °C below the dew point. 4 nL of 5 mM TCEP and 0.05% DDM in 100 mM HEPES (pH = 8.5) was added into each well followed by 2 nL of 45 mM CAM. A cover glass with spacer (Figure S2) was then clamped to the hyperSCP chip. The clamped chip was placed in a wet box and incubated at 70 °C for 30 min and then at 95 °C for 15 min. After being cooled to room temperature, the chip was placed back into the cellenONE platform and 5 nL of enzyme solution containing 0.25 ng of lys-c and 0.5 ng of trypsin was added into each well before incubating overnight at 37 °C for digestion. The stage temperature was adjusted to 20 °C before labeling with TMTpro reagents to prevent DMSO from freezing. To reduce evaporation, the relative humidity was set to 50% and 1 nL of DMSO was added to each well. Then 10 nL of 10 μg/μL TMTpro reagents were dispensed into the corresponding nanowells (Figure 1c) before the chip was capped with the cover glass slide and incubated at room temperature for 1 h. Unreacted TMTpro reagents were quenched with 2 nL of 5% hydroxylamine solution followed by 15 min incubation. Finally, the sample was acidified with 5 nL of 5% FA. For carrier channels, 10 ng of 126-labeled “light” HeLa and A549 digest (1:1) and 10 ng of 126-labeled “heavy” HeLa and A549 digest (1:1) were added to each nested well. Similarly, for reference channels, 0.5 ng of 134 N-labeled “light” HeLa and A549 digest (1:1) and 0.5 ng of 134 N-labeled “heavy” HeLa and A549 digest (1:1) were dispensed in each nested well. The chip was then air-dried and stored at −80 °C prior to LC–MS analysis.
NanoLC–MS/MS Analysis.
The nanoLC–MS settings were described in previous work12 with some modifications. A home-packed 100-μm-i.d. solid-phase extraction (SPE) column (Dr. Maisch C18, 1.9 μm particles) was used for desalting. A 30-cm-long, 30-μm-i.d. column packed with the same particles was used for nanoLC separation at a flowrate of ~30 nL/min. For 60-min gradient separations, the nanoLC pump (Dionex UltiMate NCP-3200RS, Thermo Fisher) was programmed to deliver a 60-min linear gradient from 8 to 25% mobile phase B, followed by a 20-min linear gradient from 25 to 45% B, column wash, and re-equilibration. The 90-min gradient increased mobile phase B from 8 to 25% over that time, and the 25–45% gradient was 30 min.
An Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific) was operated in data-dependent acquisition mode to analyze the peptides. The electrospray potential was 2000 V, and the ion transfer tube was set to 200 °C. Precursor ions of m/z 450–1600 were scanned with a resolution of 120,000 with a normalized automatic gain control (AGC) target of 300%. Peptide precursors of charge state 2–5 having intensities greater than 5E3 were selected for MS/MS analysis with a fit threshold of 50% and an isolation window of 0.7 m/z within a 1.5-s cycle time. The precursors were fragmented with 35% normalized HCD collision energy and then analyzed in the orbitrap with a resolution of 60,000, an AGC target of 200%, and a maximum injection time (IT) of 118 ms.
Database Searching and Data Analysis.
Proteome Discoverer version 2.5 (Thermo Fisher Scientific) was used to search all raw files against the UniProtKB/Swiss-Prot human database (downloaded 07/05/2020, 20,305 sequences). The precursor mass tolerance was set to 10 ppm and the fragment mass tolerance was 0.6 Da. Oxidation (+15.995 Da) on methionine residues and deamidation (+0.984 Da) on asparagine residues were selected as dynamic modifications, and TMTpro (304.207 Da) on any N-terminus and carbamidomethyl (+57.021 Da) on cysteines were set as static modifications. Two individual database searches were applied for each raw file because the software did not allow for labeling of lysine with both TMTpro and SILAC as indicated in previous work.27 For non-SILAC-labeled (light) precursors, the TMTpro (304.207 Da) on lysine was set as another static modification. For SILAC-labeled (heavy) precursors, a new modification (H25C213C13N−115N4O3, +312.221 Da) on lysine was created, which accounted for both the SILAC and TMTpro labeling. This new modification and label 13C615N4 (+10.008 Da) on arginine were set as static modifications. The intensity of reporter ions was obtained from the PSM output and processed using R-script (Figure S3) referencing code from a previous study.9 Briefly, all reporter-ion intensities were filtered for high confidence and non-contaminants before being normalized using the reference channel. The CV was calculated at the peptide level, and any single-cell data with CV > 0.3 was removed. 60% valid value was filtered at the protein level, and KNN (k = 5) imputation was performed within each cell type. For PCA and correlation studies, the common proteins quantified from all the cell lines were selected and ComBat40 was used to remove batch effects.
Data are available via ProteomeXchange with identifier PXD040455. The R-script and data description (README) file are accessible to the community41 at https://github.com/RTKlab-BYU/HyperSCP.
RESULTS AND DISCUSSION
We reasoned that higher order multiplexing combining both isotopic and isobaric labeling may be a viable approach to achieve higher throughput single-cell proteome profiling. To test this concept and identify any tradeoffs that might exist, we developed hyperSCP and evaluated its performance in terms of both proteome coverage and measurement throughput using a glass nested nanowell chip. The combination of orthogonal multiplexing approaches based on isotopic (SILAC) labeling to differentiate peptides based on their precursor masses and isobaric (TMT) labeling for differentiation based on reporter-ion masses upon fragmentation enables multiplicative scaling. While 2-plex SILAC and 18-plex TMTpro were employed in this proof-of-concept study, other combinations of isotopic and isobaric labeling might be similarly employed, including 3-plex SILAC, 18O labeling42,43 or dimethylation as isotopic labels, and iTRAQ, DiLeu,44 TMT, and TMTpro reagents as isobaric labels. Dimethylation is not preferred in the present one-pot workflow because the labeling reagents cannot be cleaned up prior to TMT labeling and because both reagents label primary amines. In the current form using a low-duty-cycle nanoLC system (one sample every 145 min with a 60-min active elution profile), the hyperSCP method achieves a measurement throughput of ~280 samples/day. By implementing a multicolumn nanoLC platform to eliminate the long wait times between active elutions,21,36 the same method would analyze nearly 700 cells per day.
Design of HyperSCP and Chips.
The hyperSCP chip design, which was an adaptation of the N2 chips,5 is shown in Figure 1a. Briefly, a 6 × 6 square array of nanowells with the four corner wells removed was designed with each nanowell having a diameter of 400 μm and a center-to-center spacing of 500 μm (Figure S1). The 32 nanowells were surrounded by a “moat” ring to form a nested well, and each nested well can hold up to 30 single-cell samples and two negative control samples. On each chip, there are 27 nested wells arranged in a 3 × 9 array such that each chip contains 864 nanowells in total. The surface of the nanowells and the surrounding moat rings were untreated hydrophilic glass, while the remaining area on the chip was hydrophobically treated. Thus, individual nanowells contained prepared single-cell samples prior to sample pooling, and then the outer moat rings confined the pooled samples for injection into the LC–MS platform (Figure 1c). With this design, each hydrophilic nanowell can hold ~25 nL of liquid, and each nested well, defined by an outer moat ring, can hold a total of 3–5 μL.
Experimental Design.
Nanoliter liquid dispensing of cells and reagents was performed using the CellenONE system, which can gently place single cells contained in ~300 pL droplets into nanowells with high positional accuracy (Figure 1b). For reagent dispensing, droplets of the desired solution are serially dispensed to reach a set volume. In hyperSCP experiments, the cells were first cultured in SILAC-heavy or SILAC-light medium. After labeling, the cells were harvested and sorted into nanowells on the hyperSCP chip. In each nested well, the top 16 nanowells were designed to prepare samples labeled with TMTpro, and the remaining 16 nanowells held the TMTpro-labeled SILAC-heavy samples (Figure 1c). However, we note that a single nanowell could contain both a light-and a heavy-labeled cell for labeling with a given TMT reagent, which could increase the number of cells processed in a chip. The TMTpro labeling is similar to the SCoPE245 or N2,5 but two sets (light and heavy) of TMTpro-labeled samples are pooled in each nested well. In each isobaric labeled sample set, a 0.5 ng reference sample in channel 134N was included for normalization and a prepared blank sample served as negative control. The use of a carrier, which consisted of a 10-ng protein digest labeled with TMTpro-126, was also evaluated. In these cases, channel 127C was not used due to isotopic contamination. When a carrier was not used, neither channel 126 nor 127C was used to match the carrier experiments (Table S1). The negative controls and the single cells were randomized in the rest of the TMTpro channels, prepared, and labeled together before pooling with carrier and reference channels (Figure 1c). The samples were then analyzed using nanoLC–MS. For a given peptide, two precursor peaks having a predictable m/z difference (SILAC light and heavy) appeared in the MS1 spectra. After fragmentation, the signals of reporter ions from both light and heavy plexes were used for relative quantification (Figure 1d).
Labeling Efficiency.
In the hyperSCP workflow, high labeling efficiency is necessary for accurate quantification. To determine the SILAC labeling efficiency, the cell samples were prepared in bulk,38 analyzed by nanoLC–MS, and database-searched using PD 2.5, with the label 13C615N2 (+8.014 Da, K) and label 13C615N4 (+10.008 Da, R) selected as dynamic modifications. The results indicated that 97% of lysines and 98% of arginines were labeled in the SILAC-heavy cells.
The labeling efficiency of TMTpro across all light cells is shown in Figure 2a. For the labeling of lysine residues and the N-termini, a dynamic modification of TMTpro was used for database searching. To evaluate possible overlabeling on histidine, serine, and threonine residues, TMTpro modifications on both lysine residues and the N-terminus were set as static modifications, and the same modifications were set as dynamic on histidine, serine, and threonine residues. Overall, >99.5% of lysine residues and >95% of N-termini were labeled with TMTpro, and ~10% of histidine, serine, and threonine residues were also labeled. This degree of overlabeling is similar to the data in bulk proteomics if the suggested amount of TMT reagent is used.46 In this study, the ratio of TMTpro reagent to proteins is even larger than what is suggested for bulk studies46 to ensure high labeling efficiency as is common practice for TMT-based SCP,5,47 but a lower amount of TMTpro may be beneficial to minimize overlabeling.
Figure 2.
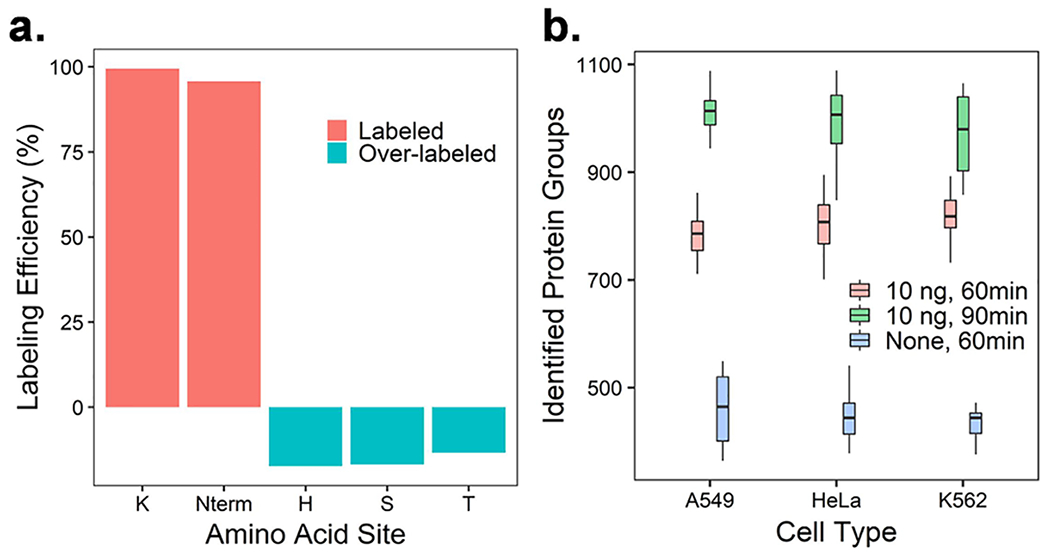
Labeling efficiency and proteome coverage for hyperSCP. (a) Overall labeling efficiency of TMTpro across four different cell lines. (b) Number of protein groups identified in single-cell channels under different gradient lengths and with and without a 10-ng carrier channel.
Proteome Coverage.
While label-free SCP can now quantify >3000 proteins per single cell,19 most TMT-based SCP provides more limited coverage of ~1000 proteins even when a carrier channel is employed. Using DDA mode, where high-abundance precursors are sampled for subsequent MS2 scans, the number of MS2 scans is limited by the long ion-accumulation time in single-cell analysis. Even longer injection times may be needed for TMT-based multiplexing methods to ensure accurate quantification.49 In addition, there may be other reasons for reduced proteome coverage including losses and inefficiencies during additional sample processing steps, changes in hydrophobicity resulting from chemical labeling, reduced fragmentation efficiency, etc., which should be studied further. Still, the tremendous gain in measurement throughput enabled by multiplexing (or in this case hyperplexing) is highly compelling for many applications, and the coverage is sufficient to, e.g., enable differentiation of cell types or treatment conditions.
As with numerous past studies achieving modest coverage for TMT SCP, we also expected reduced coverage for hyperSCP. Indeed, without a carrier channel, proteome coverage was quite modest for TMTpro labeled and hyperSCP samples as shown in Figure S4a. While hyperSCP increases measurement throughput in terms of cells profiled/day, it does not increase the number of MS2 scans/time, which become divided between heavy and light versions of each precursor. As such, some reduction in proteome coverage was expected for hyperSCP. Indeed, as shown in Figures 2b and S4a, hyperSCP doubles the throughput of TMTpro-labeled SCP but with a tradeoff of ~15% lower proteome coverage in the absence of a carrier channel. The quantifiable protein groups (≥2 unique peptides) among identified proteins are shown in Figure S4b. Compared to using TMTpro only, the percentage of quantifiable proteins in light channels in hyperSCP is slightly higher, while the heavy channels show a slightly lower percentage. A consequence of the limited MS2 scans is missing values. TMT labeling can reduce missing values within a single LC–MS analysis, but missing values are still present across different LC–MS runs due to the stochastic selection of precursors. As such, quantitative information for lower-abundance precursors might be missing across runs, or within a run due to inconsistent selection of both heavy and light versions of precursors (Figure S4c). The use of algorithms trained to select all isotopes of a given precursor, or the recently reported prioritized SCoPE approach,48 may serve to increase proteome coverage while reducing missing values.
Carrier channels containing larger samples have been used to improve the proteome coverage of single cells.16 However, the carrier proteome effect was reported to compress the signals from single cells and reduce the proportion of peptides originating from single cells in each MS2 scan.47,49,50 To reduce the carrier proteome effect, smaller carrier channel amounts or higher AGC and longer injection times are necessary. In this work, a carrier channel containing 10 ng sample was selected to increase proteome coverage while maintaining quantitative accuracy. The ratio of carrier to single-cell sample is ~50× as confirmed by SCPCompanion.49 With a 10-ng carrier, the proteome coverage increased to ~850 proteins/cell on average using the same gradient (Figure 2b). We also noticed that with a longer separation gradient (i.e., 90-min effective gradient), the protein identification can be further improved to ~1000 proteins/cell on average (Figure 2b), but the 60-min gradient was used for other experiments in this work to maintain higher throughput.
Cell Differentiation.
To evaluate the ability of the hyperSCP workflow to differentiate cells according to cell line with the observed proteome coverage, HeLa, A549, K562, and HFL1 cells were prepared and analyzed using this workflow with a 10-ng carrier channel. The CellenONE allows cell dispensing with just ~300 pL of supernatant, reducing the potential for any interfering background signals. In this work, we dispensed single droplets of PBS solution to serve as the negative control. As predicted, a lower intensity of reporter ions was observed in the negative control channel, where the remaining signals were caused by the co-isolation and crosstalk between different TMT channels or SILAC labels. The distributions of the median protein coefficients of variation (CVs) from single-cell and control channels are shown in Figure 3a. Across the 590 cells and 40 control channels analyzed in total, the single cells show a low median CV (0.17–0.3) compared to greater variation in the control samples after normalization. A filter passing cell with a median CV < 0.3 was set to remove the outliers. The number of quantifiable protein groups was ~620 depending on the cell type. Despite the relatively low proteome coverage across the different cell lines, all four cell types were readily differentiated using a PCA plot, while SILAC heavy and light cells from the same cell line clustered together nicely (Figure 3b). The correlations (Figure S5) and PCA plot (Figure S6) indicate that hyperSCP performance is sufficient to enable high-throughput analyses to explore the difference between different cell types at the single-cell level.
Figure 3.
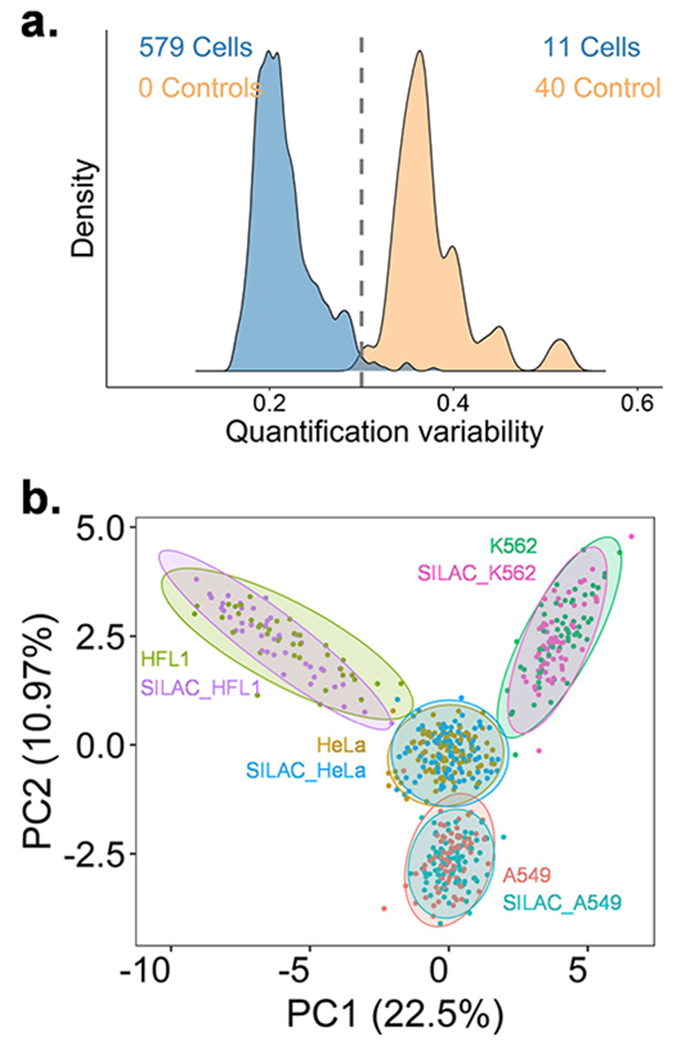
Comparison of single HeLa, A549, K562, and HFL1 cells using hyperSCP with 10 ng carrier proteome and 60-min gradient. (a) Median protein CVs from single-cell channels and negative control channels. The cutoff was set to 0.3 to remove outliers. (b) PCA plot of four cell lines. The shared proteins that have 60% valid values and were quantifiable from all cell lines were used for analysis.
Comparison of Lung Cells Using HyperSCP.
The cultured human lung adenocarcinoma cell line A549 and lung fibroblast cell line HFL1 were further studied using hyperSCP. With a 10-ng carrier, 653 proteins were found in both A549 and HFL1 cell lines across the 164 cells (Figure 4a). The individual HFL1 cells show more variability (Figure S6). A volcano plot (Figure 4b) indicates that 23 proteins were significantly underexpressed in A549 cells relative to HFL1, while 10 proteins were more highly expressed (fold change >1.5). Meanwhile, five proteins were only quantified in HFL1 cells. These proteins were annotated using the online tool DAVID (https://david.ncifcrf.gov/home.jsp). When annotated using the Gene Ontology molecular function (GOMF), about 93% of the proteins expressed more in HFL1 are related to protein binding and most of them are related to molecular binding, such as calcium ion, identical protein, cadherin, and RNA bindings. The proteins upregulated in A549 cancer cells were generally related to enzyme activities like retinal dehydrogenase, d-threo-aldose 1-dehydrogenase, and alditol:-NADP+ 1-oxidoreductase.
Figure 4.
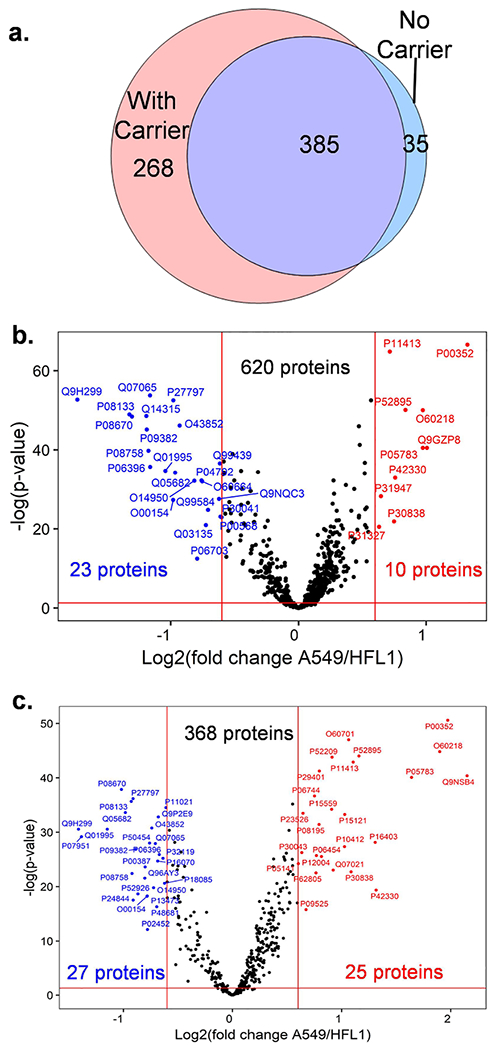
Differential protein abundances between HFL1 and A549 lung cells. (a) Venn diagram shows the quantifiable proteins found from both HFL1 and A549 cells while using a 10-ng carrier channel or no carrier channel. (b,c) Volcano plot indicates the proteins with fold change >1.5 and Benjamini–Hochberg adjusted p-value <0.05 with a 10-ng carrier channel (b) and without carrier (c).
If no carrier proteins were applied in the hyperSCP workflow, the proteome coverage was reduced but some specific proteins from the single-cell channels which are not included in the carrier proteome may be observed (Figure 4a), and their quantification may be more accurate with less interference from the carrier proteome. For example, the quantitative analysis shows some extra proteins related to protein and RNA binding in A549 cells (Figure 4c). Also, collagen was found expressed more in HFL1 cells as the main function of the fibroblast cells, while aldehyde dehydrogenase and keratin were found more in A549 cells as reported previously.51
CONCLUSIONS
A hyperSCP workflow is described and evaluated in this work, combining isotopic and isobaric labeling for higher throughput SCP. Using the 2-plex SILAC labeling and 18-plex TMTpro isobaric labeling, the hyperSCP workflow enables measurement throughput of ~280 cells/day when a long 145-min total LC cycle time is employed. The tradeoff between proteome coverage and throughput was evaluated. Compared to the TMTpro labeled SCP, the hyperSCP method doubles the throughput with a tradeoff of ~15% reduced proteome coverage in the absence of a carrier channel. Using a 10-ng carrier proteome, the proteome coverage was approximately doubled and was further increased if a longer separation gradient was employed. We explored the performance of the developed workflow for a 4-cell line study. Even with a reduced number of protein identifications, the hyperSCP is sufficiently sensitive to distinguish different cell lines and can be used for rapid concept testing. A comparison of lung cells A549 and HFL1 also shows the ability of hyperSCP to analyze cell functions. Overall, this method combines MS1 and MS2 quantitative labeling methods to improve the LC–MS throughput. Thus, it is flexible and should be adaptable to different metabolic or chemical isotopic labeling and isobaric labeling strategies. In addition, if metabolic labeling such as SILAC is used, this workflow can also be used for fast protein turnover rate studies or other biological applications.
Supplementary Material
ACKNOWLEDGMENTS
We thank Andrew Leduc and Prof. Nikolai Slavov for helpful discussions and sharing of code used for batch normalization. The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM138931. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.3c00906.
Schematic of nested nanowell dimensions (Figure S1); schematic of cover plate (Figure S2); data processing workflow (Figure S3); comparison of isobaric labeling alone vs hyperplexing (Figure S4); correlation between heavy and light labeled cells (Figure S5); and PCA plot of A549 and HFL1 lung cells (PDF)
Key showing the contents of each channel for hyperSCP and TMTpro-only experiments (Table S1) (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.3c00906
The authors declare the following competing financial interest(s): RTK has a financial interest in MicrOmics Technologies, which provided the nanoelectrospray emitters used in this study.
Contributor Information
Yiran Liang, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Thy Truong, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States.
Aubrianna J. Saxton, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Hannah Boekweg, Department of Biology, Brigham Young University, Provo, Utah 84602, United States.
Samuel H. Payne, Department of Biology, Brigham Young University, Provo, Utah 84602, United States
Pam M. Van Ry, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
Ryan T. Kelly, Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, United States
REFERENCES
- (1).Kelly RT Mol. Cell. Proteomics 2020, 19, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bekker-Jensen DB; Martínez-Val A; Steigerwald S; Rüther P; Fort KL; Arrey TN; Harder A; Makarov A; Olsen JV Mol. Cell. Proteomics 2020, 19, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Brunner A-D; Thielert M; Vasilopoulou C; Ammar C; Coscia F; Mund A; Hoerning OB; Bache N; Apalategui A; Lubeck M; Richter S; Fischer DS; Raether O; Park MA; Meier F; Theis FJ; Mann M Mol. Syst. Biol 2022, 18, No. e10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhu Y; Clair G; Chrisler WB; Shen Y; Zhao R; Shukla AK; Moore RJ; Misra RS; Pryhuber GS; Smith RD; Ansong C; Kelly RT Angew. Chem., Int. Ed 2018, 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Woo J; Williams SM; Markillie LM; Feng S; Tsai CF; Aguilera-Vazquez V; Sontag RL; Moore RJ; Hu D; Mehta HS; Cantlon-Bruce J; Liu T; Adkins JN; Smith RD; Clair GC; Pasa-Tolic L; Zhu Y Nat. Commun 2021, 12, 6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gebreyesus ST; Siyal AA; Kitata RB; Chen ESW; Enkhbayar B; Angata T; Lin KI; Chen YJ; Tu HL Nat. Commun 2022, 13, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Liang Y; Acor H; McCown MA; Nwosu AJ; Boekweg H; Axtell NB; Truong T; Cong Y; Payne SH; Kelly RT Anal. Chem 2021, 93, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ctortecka C; Hartlmayr D; Seth A; Mendjan S; Tourniaire G; Mechtler K; Biocenter V bioRxiv 2022, DOI: 10.1101/2021.04.14.439828. [DOI] [Google Scholar]
- (9).Leduc A; Huffman RG; Cantlon J; Khan S; Slavov N Genome Biol. 2022, 23, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matsumoto C; Shao X; Bogosavljevic M; Chen L; Gao Y bioRxiv 2022, 12. [Google Scholar]
- (11).Lombard-Banek C; Moody SA; Nemes P Angew. Chem., Int. Ed 2016, 55, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Anal. Chem 2020, 92, 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xiang P; Zhu Y; Yang Y; Zhao Z; Williams SM; Moore RJ; Kelly RT; Smith RD; Liu S Anal. Chem 2020, 92, 4711–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cong Y; Motamedchaboki K; Misal SA; Liang Y; Guise AJ; Truong T; Huguet R; Plowey ED; Zhu Y; Lopez-Ferrer D; Kelly RT Chem. Sci 2021, 12, 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Johnson KR; Greguš M; Ivanov AR J. Proteome Res 2022, 21, 2453–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Budnik B; Levy E; Harmange G; Slavov N Genome Biol. 2018, 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Woo J; Clair GC; Williams SM; Feng S; Tsai CF; Moore RJ; Chrisler WB; Smith RD; Kelly RT; Paša-Tolić L; Ansong C; Zhu Y Cell Syst. 2022, 13, 426–434.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Derks J; Leduc A; Wallmann G; Huffman RG; Willetts M; Khan S; Specht H; Ralser M; Demichev V; Slavov N Nat. Biotechnol 2023, 41, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Truong T; Johnston SM; Webber K; Boekweg H; Lindgren CM; Liang Y; Nydeggar A; Xie X; Payne SH; Kelly RT bioRxiv 2022, DOI: 10.1101/2022.10.18.512791. [DOI] [Google Scholar]
- (20).Williams SM; Liyu AV; Tsai C-FF; Moore RJ; Orton DJ; Chrisler WB; Gaffrey MJ; Liu T; Smith RD; Kelly RT; Pasa-Tolic L; Zhu Y Anal. Chem 2020, 92, 10588–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Webber KGI; Truong T; Johnston SM; Zapata SE; Liang Y; Davis JM; Buttars AD; Smith FB; Jones HE; Mahoney AC; Carson RH; Nwosu AJ; Heninger JL; Liyu AV; Nordin GP; Zhu Y; Kelly RT Anal. Chem 2022, 94, 6017–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dephoure N; Gygi SP Sci. Signaling 2012, 5, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Everley RA; Kunz RC; McAllister FE; Gygi SP Anal. Chem 2013, 85, 5340–5346. [DOI] [PubMed] [Google Scholar]
- (24).Klann K; Münch C Methods Mol. Biol 2022, 2428, 75–87. [DOI] [PubMed] [Google Scholar]
- (25).Evans AR; Robinson RAS Proteomics 2013, 13, 3267–3272. [DOI] [PubMed] [Google Scholar]
- (26).Evans AR; Gu L; Guerrero R; Robinson RAS Proteomics: Clin. Appl 2015, 9, 872–884. [DOI] [PubMed] [Google Scholar]
- (27).Welle KA; Zhang T; Hryhorenko JR; Shen S; Qu J; Ghaemmaghami S Mol. Cell. Proteomics 2016, 15, 3551–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Frost DC; Rust CJ; Robinson RAS; Li L Anal. Chem 2018, 90, 10664–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Aggarwal S; Talukdar NC; Yadav AK J. Proteome Res 2019, 18, 2360–2369. [DOI] [PubMed] [Google Scholar]
- (30).Klann K; Tascher G; Münch C Mol. Cell 2020, 77, 913–925.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).King CD; Robinson RAS Anal. Chem 2020, 92, 2911–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Klann K; Krause D; Münch C bioRxiv 2021, DOI: 10.1101/2021.07.06.451268. [DOI] [Google Scholar]
- (33).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA Nat. Methods 2020, 17, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang Z; Yu K; Tan H; Wu Z; Cho JH; Han X; Sun H; Beach TG; Peng J Anal. Chem 2020, 92, 7162–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sun H; Poudel S; Vanderwall D; Lee DG; Li Y; Peng J Proteomics 2022, 22, No. 2100243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Livesay EA; Tang K; Taylor BK; Buschbach MA; Hopkins DF; LaMarche BL; Zhao R; Shen Y; Orton DJ; Moore RJ; Kelly RT; Udseth HR; Smith RD Anal. Chem 2008, 80, 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhu Y; Podolak J; Zhao R; Shukla AK; Moore RJ; Thomas GV; Kelly RT Anal. Chem 2018, 90, 11756–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ong SE; Mann M Nat. Protoc 2006, 1, 2650–2660. [DOI] [PubMed] [Google Scholar]
- (39).Hailemariam M; Eguez RV; Singh H; Bekele S; Ameni G; Pieper R; Yu YJ Proteome Res. 2018, 17, 2917–2924. [DOI] [PubMed] [Google Scholar]
- (40).Johnson WE; Li C; Rabinovic A Biostatistics 2007, 8, 118–127. [DOI] [PubMed] [Google Scholar]
- (41).Gatto L; Aebersold R; Cox J; Demichev V; Derks J; Emmott E; Franks AM; Ivanov AR; Kelly RT; Khoury L; Leduc A; MacCoss MJ; Nemes P; Perlman DH; Petelski AA; Rose CM; Schoof EM; Van Eyk J; Vanderaa C; Yates JR; Slavov N Nat. Methods 2023, 20, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ye X; Luke B; Andresson T; Blonder J Briefings Funct. Genomics Proteomics 2009, 8, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bonzon-Kulichenko E; Pérez-Hernández D; Núñez E; Martínez-Acedo P; Navarro P; Trevisan-Herraz M; Del Carmen Ramos M; Sierra S; Martínez-Martínez S; Ruiz-Meana M; Miró-Casas E; García-Dorado D; Redondo JM; Burgos JS; Vázquez J Mol. Cell. Proteomics 2011, 10, No. M110.003335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Frost DC; Greer T; Li L Anal. Chem 2015, 87, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Petelski AA; Emmott E; Leduc A; Huffman RG; Specht H; Perlman DH; Slavov N Nat. Protoc 2021, 16, 5398–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zecha J; Satpathy S; Kanashova T; Avanessian SC; Kane MH; Clauser KR; Mertins P; Carr SA; Kuster B Mol. Cell. Proteomics 2019, 18, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N Genome Biol. 2021, 22, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Huffman RG; Leduc A; Wichmann C; Di Gioia M; Borriello F; Specht H; Derks J; Khan S; Khoury L; Emmott E; Petelski AA; Perlman DH; Cox J; Zanoni I; Slavov N Nat. Methods 2023, DOI: 10.1038/s41592-023-01830-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cheung TK; Lee CY; Bayer FP; McCoy A; Kuster B; Rose CM Nat. Methods 2021, 18, 76–83. [DOI] [PubMed] [Google Scholar]
- (50).Tsai CF; Zhao R; Williams SM; Moore RJ; Schultz K; Chrisler WB; Pasa-Tolic L; Rodland KD; Smith RD; Shi T; Zhu Y; Liu T Mol. Cell. Proteomics 2020, 19, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Moreb JS; Zucali JR; Ostmark B; Benson NA Cytometry, Part B 2007, 72, 281–289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.