

Abstract
Sulfur and selenium occupy a distinguished position in biology owing to their redox activities, high nucleophilicity, and acyl transfer capabilities. Thiolated/selenolated amino acids, including cysteine, selenocysteine, and their derivatives, play critical roles in regulating the conformation and function of proteins and serve as an important motif for peptide design and bioconjugation. Unfortunately, a general and concise method to attain enantiopure β-thiolated/selenolated amino acids remains an unsolved problem. Herein, we present a photoredox-catalyzed asymmetric method for the preparation of enantiopure β-thiolated/selenolated amino acids using a simple chiral auxiliary, which controls the diastereoselectivity of the key alkylation step and acts as an orthogonal protecting group in the subsequent peptide synthesis. Our protocol can be used to prepare a wide range of β-thiolated/selenolated amino acids on a gram scale, which would otherwise be difficult to obtain using conventional methods. The effect of our chemistry was further highlighted and validated through the preparation of a series of peptidyl thiol/selenol analogues, including cytochrome c oxidase subunit protein 7C and oxytocin.
Graphical Abstract
INTRODUCTION
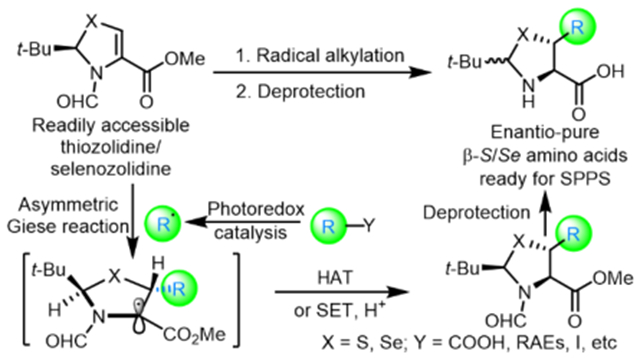
Cysteine (Cys) and its analogue selenocysteine (Sec) play irreplaceable roles in protein folding and stability,1 enzymatic activity,2 and redox regulation.3 The intricate design of free thiol and selenol is Nature’s way of realizing these critical functions in biology. The significance of cysteine and selenocysteine is arguably more conspicuously presented in the domain of selective chemical protein modification,4 the construction of native peptide bonds (native chemical ligation, NCL),5 late-stage mutagenesis (to Ala and Ser),6 disulfide bond engineering,7 and the design of peptidyl ligands.8 As a case in point, the combination of the NCL–dechalcogenation strategy,9 one of the most efficient approaches used to tether two peptidyl segments, completely relies on the specific chemoselectivities exhibited by thiolated/selenolated proteinogenic amino acids (Figure 1A). After ligation and selective dechalcogenation, peptides bearing the corresponding native residues are produced. Moreover, disulfide bridges are crucial for the stability and activity of many important therapeutic peptides and proteins. Recent studies have suggested the use of thiolated amino acids as disulfide precursors can also enhance the stability and activity of peptides.10 In addition, Cys is a fundamental element for functional peptide design, thus fine-tuning the properties of synthetic peptide ligands could be achieved with thiolated amino acid preparation.11 Nonetheless, the resultant β-thiolated/selenolated amino acids, in which the β-carbon carries the thiol/selenol, are valuable precursors to bioactive peptides and proteins.
Figure 1.
Development of a general method used for the preparation of enantiopure β-thio/selenolated amino acids. (A) NCL–dechalcogenation. (B) Representative methods for constructing thiolated/selenolated amino acids.(C) The photoredox-catalyzed asymmetric Giese reaction as a general solution to this synthetic problem.
In contrast to these powerful applications derived from thiolated/selenolated amino acids, a major challenge to their widespread utility is the limited accessibility of these prerequisite enantiopure thiolated/selenolated amino acids. Currently, the majority of methods are indirect and are confined to certain amino acid scaffolds despite significant research efforts toward the preparation of β-thiolated/selenolated amino acids reported by multiple research groups.12 Among the methods toward the synthesis of β-thiolated/selenolated amino acids, using thiol/selenol reagents and an electrophilic carbon to introduce S/Se is the most representative approach with enantiopure β-hydroxyl amino acids or Garner’s aldehyde utilized to stereoselectively construct the C–S/Se bond (Figure 1B). Unfortunately, the modification of β-hydroxyl amino acids approach is extremely impractical, as it requires expensive and rare commercially available β-hydroxyl amino acids as the precursors (e.g., $200/gram for β-hydroxy leucine) and it often leads to a diastereomeric mixture of β-thiol products. The latter tactic could achieve moderate diastereoselectivity, yet multistep manipulations are often required before incorporating β-thiol/selenol residues into solid-phase peptide synthesis.13 Therefore, a more general and concise protocol to high-value enantioselective β-thiolated/selenolated amino acids from readily accessible building blocks remains elusive.
Herein, we present a general and practical method to access enantiopure β-thiolated/selenolated amino acids via an asymmetric Giese reaction14 (Figure 1C). Our strategy starts from enantiopure selenazoline/thiazoline bearing a chiral pivalaldehyde acetal, which is easily prepared from l-Cys/Sec on a large scale. Visible-light photocatalysis generated alkyl radicals (including primary, secondary, and tertiary radicals) furnish thiazoline/selenazoline to produce the β-substituted thiolated/selenolated amino acid framework. The chiral pivalaldehyde acetal of thiazoline/selenazoline controls the diastereoectivity of the radical addition reaction and acts as a “smart” protecting group, which is orthogonal to solid-phase peptide synthesis conditions and eliminates the need for protecting group manipulations. This strategy allows the concise stereoselective synthesis of a diverse range of β-selenolated/-thiolated amino acids from a common precursor at will with high stereopurity and high yield. Products can be rapidly acquired on a multigram scale that are not easily accessible using conventional methods. Furthermore, the resulting selenazolidine/thiazolidine amino acids can be directly used in peptide synthesis after a simple operation without complex protecting-group manipulation.
RESULTS AND DISCUSSION
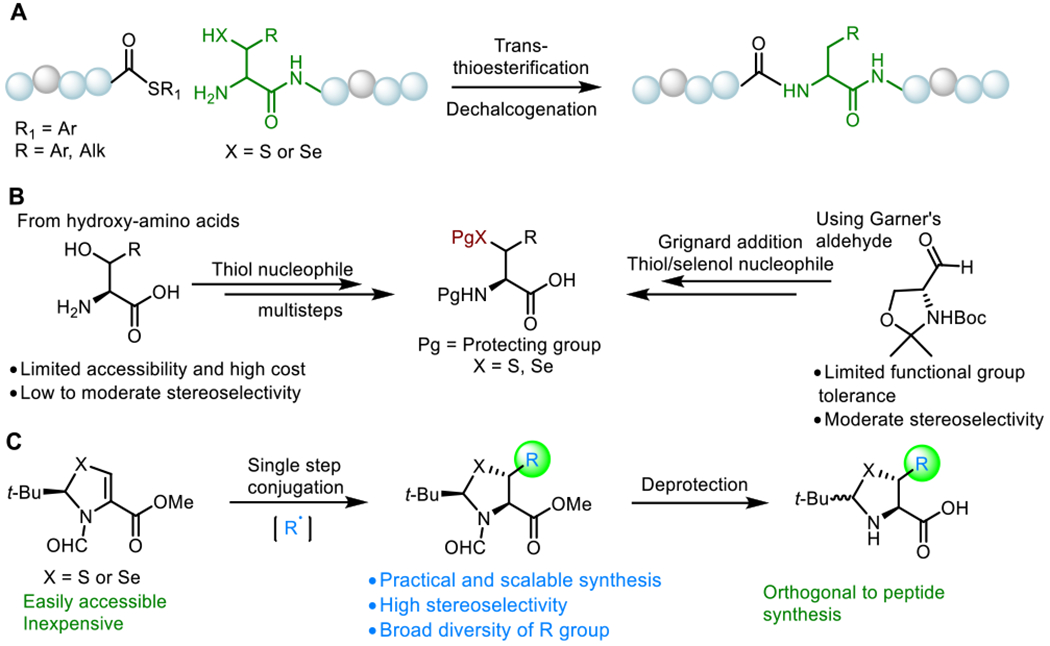
In contrast to the thiol/selenol nucleophiles used to generate the C–S/Se bond described in the existing methods, the stereoselective insertion of an alkylated group onto the β-carbon of Cys/Sec is superior. It has been reported that, using the concept of “self-regeneration of chirality centers”,15 chiral S,N-acetyl thiazoline served as a good starting point for the preparation of β-thiolated amino acids via a Michael-type alkylation.16 However, this transformation remains limited in synthesis due to its poor yield and low functional group tolerance and diversity, attributed to the severe β-elimination of the thiol group when alkyl metal reagents are employed in the reaction. The challenges notwithstanding, we envisioned that a radical 1,4-conjugate addition under mild photoredox conditions might lead to superior results. It is well-established that convenient radical generation occurs from carboxylic acids,17 redox-active esters (RAEs),18 and alkyl halides19 in the presence of a photoredox catalyst via single-electron transfer (SET). Mechanistically, production of alkyl radical via SET oxidation (carboxylic acids) or reduction (RAEs and alkyl halides) can lead to stereoselective radical addition from the reverse face of the bulky tert-butyl group in A, which generates radical intermediate B. B is reduced via H atom abstraction (HAT) or SET followed by a diastereoselective enolate protonation step to produce the trans product C, providing two new stereocenters in one step (Figure 2A). Subsequent deprotection may furnish a broad family of β-thiolated/selenolated amino acids (D), which are suitable for direct peptide coupling.
Figure 2.

Design plan and optimization of the reaction conditions used in the preparation of β-selenolated Leu. (A) Symmetric Giese reaction via photoredox catalysis. (B) Standard conditions: PC (1 mol %), 2a (1.2 equiv), HE (1.5 equiv), DIPEA (2.0 equiv), DCM, blue LED (10 W), 25 °C. Legend: (a) 18 h; (b) 48 h; (c) isolated yield; (d) diastereoselectivity determined by 1H NMR and GC analysis of the crude reaction mixture. Abbreviations: PC, photoredox catalyst; HE, Hantzsch ester; DIPEA, N,N-diisopropylethylamine; DCM, dichloromethane; DMF, N,N-dimethylformamide.
The importance of Se in biology causes selenolated amino acids to be in high demand; conversely, the sheer difficulty of handling labile Se during reactions prevents securing Se-amino acids from a large quantity. We began our exploration with the preparation of β-selenolated amino acids as more practical synthetic targets. The starting material, pivalaldehyde N,Se-acetal selenazoline 1, was quickly prepared over three steps from the diselenide derivative of l-Sec methyl ester on a 20 g scale. An N-formyl group was used to protect the nitrogen atom as well as to improve the electrophilicity of the double bond.20 Under irradiation of blue LED light (10 W), Ru(bpy)3(PF6)2 (PCA, 1 mol %) catalyzed the coupling of 1 with isopropyl N-hydroxyphthalimide ester 2a (1.2 equiv) to provide the desired product, trans-3a (β-selenolated Leu), as a single diastereoisomer in the presence of Hantzsch ester (HE, 1.5 equiv) and DIPEA (2.0 equiv). It is worth noting that 3a was obtained on a gram scale as a mixture of rotamers in near-quantitative yield (97%), as confirmed by variable-temperature 1H NMR and solvent switching (see page 10 in the Supporting Information for details) (Figure 2B). Screening of the solvent system suggested that dichloromethane (DCM) afforded the highest yield in comparison to MeCN and DMF; other photoredox catalysts such as Eosin Y (PCB) and Ir[dF(CF3)-ppy]2(dtbbpy)PF6 (PCC) could also mediate the same reaction with eroded yields. In all cases, only the single diastereoisomer trans-3a was obtained (>99:1 dr).
An ideal method is one where an inexpensive or readily accessible starting material can be converted into a diverse family of members with higher values via a single-step reaction. Subsequently, a broad scope of carboxylic acids was evaluated, including primary, secondary, and tertiary carboxylic acids with various functionalities. RAEs derived from secondary and tertiary carboxylates were initially examined (Figure 3A, Protocol A). Reaction of selenazoline 1 with a series of cyclic carboxylate N-hydroxyphthalimide esters (cyclobutyl, 2b; cyclohexyl, 2c; adamantyl, 2d) and fluorinated cyclobutyl carboxylate ester 2e yielded the desired products 3b–e in excellent yields and high diastereoselectivity on a gram scale. Notably, product 3d bearing an adamantyl substituent was obtained in >90% yield, which suggested that generating sterically bulky quaternary centers could be accomplished. The absolute configuration of 3d was confirmed via X-ray crystallography (Figure 3B).
Figure 3.
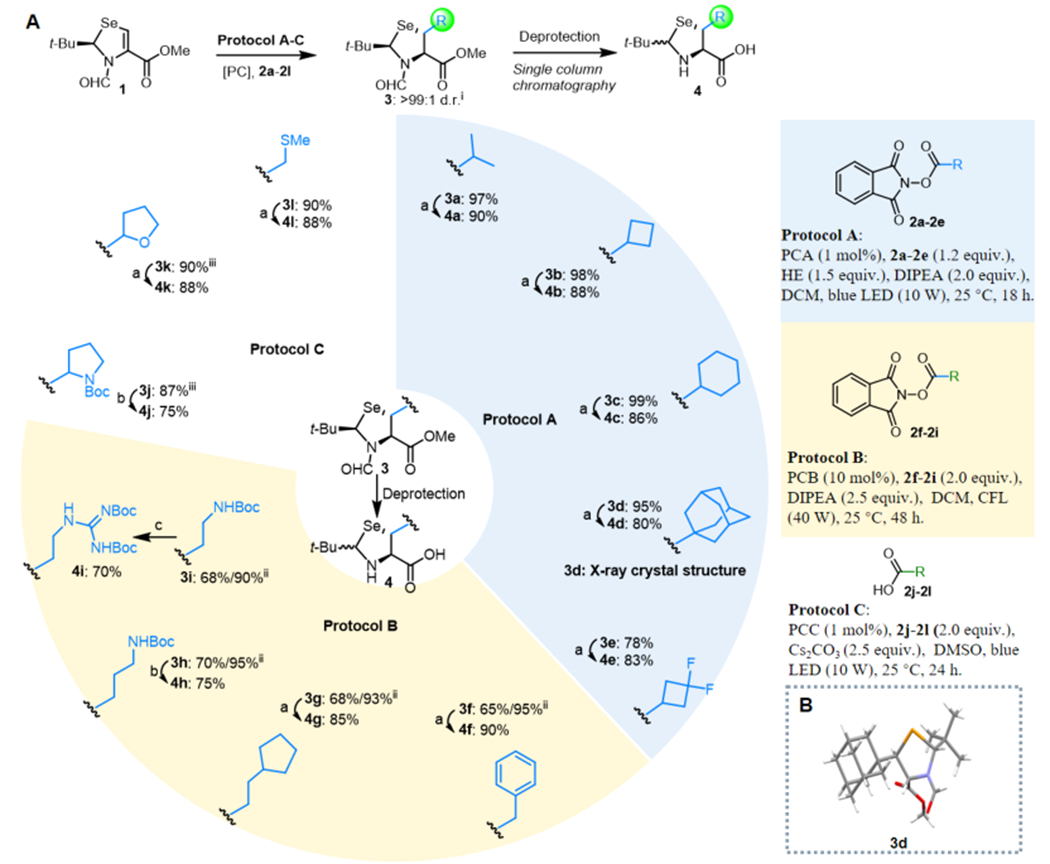
Asymmetric alkylation of selenazoline under photoredox conditions and their derivatization. (A) Scope of the β-selenolated amino acids. Deprotection conditions: (a) (1) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h, (2) 6 N HCl in dioxane, 50 °C, 8 h; (b) (1) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h, (2) 6 N HCl in dioxane, 50 °C, 8 h, (3) (Boc)2O (1.0 equiv), THF/H2O (10/1 v/v), DIPEA (2.0 equiv), 25 °C, 1 h; (c) (1) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h, (2) 6 N HCl in dioxane, 50 °C, 8 h, (3) N N′-bis-Boc-1-guanylpyrazole (1.0 equiv), DIPEA (2.0 equiv), MeOH, 25 °C, 3 h. (B) Absolute configuration of 3d as confirmed by X-ray crystallography. Legend: (i) diastereoselectivity determined by GC analysis of the crude reaction mixture; (ii) brsm, based on recovered starting material; (iii) mixture of diastereomers found.
Coupling with selenazoline 1 and primary alkyl carboxylic acids could not be accessed effectively by PCA and PCC catalysis. After extensive investigations, the optimized conditions were obtained using a 40 W household compact fluorescent light (CFL) bulb to irradiate 1 in the presence of 2.0 equiv of RAEs (2f–i) and DIPEA (2.5 equiv) in DCM using PCB (Eosin Y), which gave the desired coupling products 3f–i in good yield at room temperature (Figure 3A, Protocol B).21
Moreover, carboxylic acids bearing a stabilizing α-heteroatom (O, N, and S) were evaluated. Compounds 2j–l were added to 1 in the presence of PCC directly, giving products 3j–l in excellent yields (Figure 3A, Protocol C). In all cases, we were pleased to find that excellent diastereoselectivity was obtained under our reaction conditions.
We next sought to convert products 3 into the target amino acids 4, which might be used directly as SPPS-compatible amino acids. The desired products 4a–g,k,l can be generated from their parent compounds after saponification of methyl ester and acidic removal of the N-formyl group over two steps (Figure 3A, conditions a). Substrates 4a,l represent β-seleno Leu and Met, respectively. After acidic treatment, epimerization of the stereogenic center in the auxiliary was observed and the α,β-chiral center of the amino acids remained untouched. In the case of 3h,j, a further Boc protection step was required to form β-selenolated Lys 4h,j (Figure 3A, conditions b). Guanidinylation was performed to yield β-selenolated Arg 4i from ornithine 3i (Figure 3A, conditions c). For most cases, only a single purification was needed after these deprotection conditions, minimizing a time-consuming isolation process.
We further expanded the reaction scope to a number of β-thiolated amino acids, which are given in Figure 4. In line with the β-selenolated amino acid preparation, all of the RAEs (2a–d,f–2i,m,n) and carboxylic acids (2j–l,o) smoothly conjugated with thiazoline 5, which was readily obtained from l-cysteine methyl ester hydrochloride ($0.20/g) and pivalaldehyde ($0.25/mL) over three steps on a 100 g scale (see page 4 in the Supporting Information for details). Products 6a–d,f–o were acquired in high yields using Protocols A–C (Figure 4A). Notably, reaction of 5 with 2-iodoacetamide 2p under photoredox dehalogenation conditions (Figure 4A, Protocol A’) gave 6p in 63% yield. Synthetically valuable handles such as the sulfide of methionine (6l), pyridine (6m), acetal (6n), ketone (6o), and amide (6p) were well tolerated, and the incorporation of an adamantyl nucleus (6d) offered a ready protocol to modify peptide distribution and lipophilicity/hydrophobicity. The structure and stereochemistry of 6d were confirmed by X-ray crystallography. Pyridine-containing amino acids have significant effects on the biological properties of peptides and are often found in therapeutic peptides; therefore, substrate 6m was used to demonstrate that the pyridine motif can be smoothly introduced. Compound 6a exists as a mixture of rotamers, as confirmed by variable-temperature 1H NMR and solvent switching.
Figure 4.
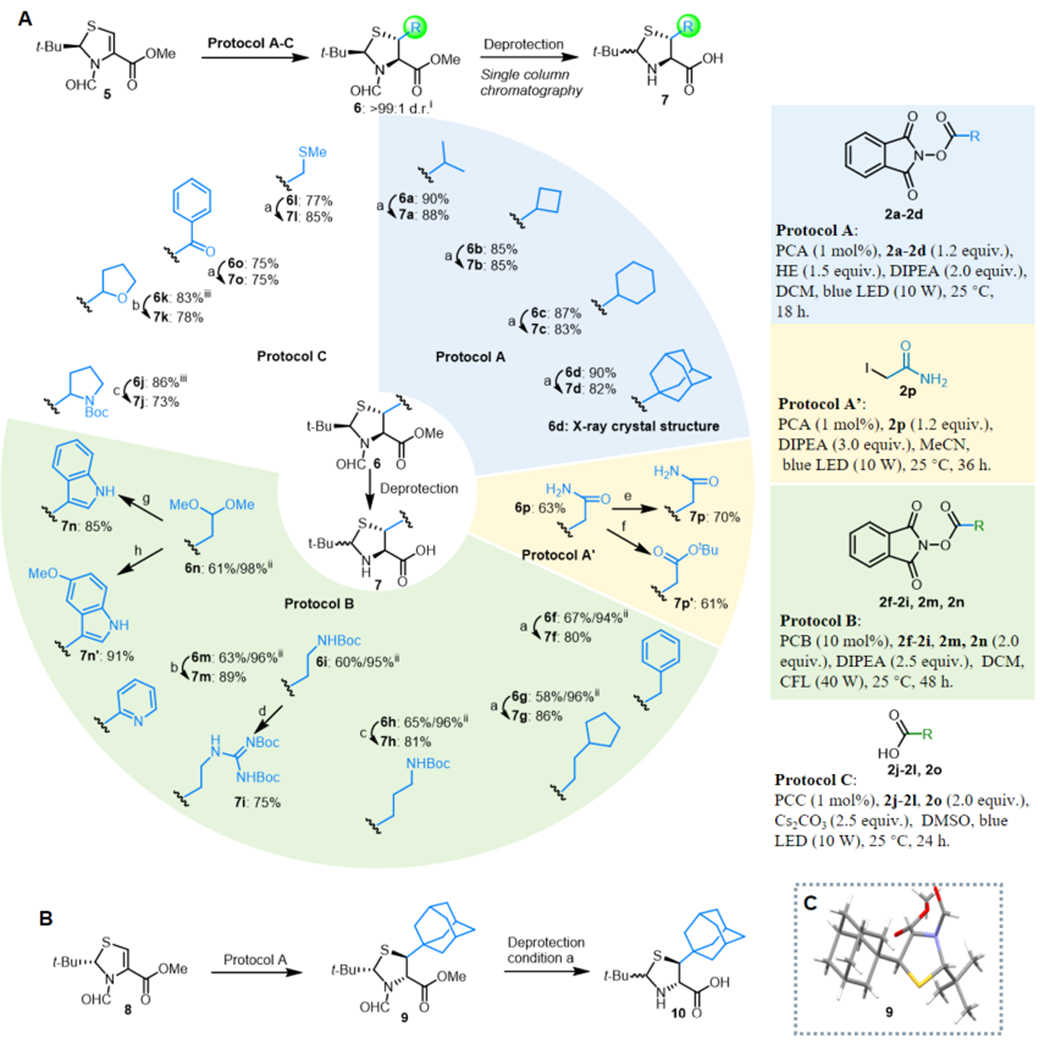
Asymmetric alkylation of thiazoline under photoredox conditions and derivatization. (A) Scope of β-thiolated amino acids. Deprotection conditions: (a) 6 N HCl, 90 °C, 8 h; (b) (1) 0.5 N HCl in MeOH, 25 °C, 12 h, (2) LiOH (10 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h; (c) (1) 6 N HCl, 90 °C, 8 h, (2) (Boc)2O (1.0 equiv), DIPEA (2.0 equiv), THF/H2O (10/1 v/v), 25 °C, 1 h; (d) (1) 6 N HCl, 90 °C, 8 h, (2) N,N′-bis-Boc-guanylpyrazole (1.0 equiv), DIPEA (2.0 equiv), MeOH, 25 °C, 3 h; (e) (1) 33% HBr in AcOH, 80 °C, 1 h, (2) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h; (f) (1) 6 N HCl, 90 °C, 30 min, (2) p-TsOH (1.0 equiv), isobutylene in DCM (50.0 equiv), 40 °C, 12 h, (3) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h; (g) (1) PhNHNH2(2.0 equiv), TFA/DCM (1/3 v/v), 25 °C, 30 min, 97%, (2) 0.5 N HCl in MeOH, rt, 12 h, (3) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h; (h) (1) p-MeO-PhNHNH2 (2.0 equiv), TFA/DCM (1/3 v/v), 97%, (2) 0.5 N HCl in MeOH, 25 °C, 12 h, (3) LiOH (10.0 equiv), H2O/MeOH (1/3 v/v), 25 °C, 6 h. (B) Synthesis of d-β-thiolated amino acids from d-cysteine. (C) Absolute configuration of 9 as confirmed by X-ray crystallography. Legend: (i) diastereoselectivity determined by GC analysis of the crude reaction mixture; (ii) brsm, based on recovered starting material; (iii) mixture of diastereomers found.
In comparison with the delicate selenolated analogues, β-thiolated amino acids are more robust in nature. Thus, methods used for the deprotection of these compounds are more straightforward. The deprotection conditions used to yield β-thiolated amino acids are summarized in Figure 4A Hydrolysis using 6 N HCl led to substrates 6a–d,f,g,o,l) directly in a single step (Figure 4A, conditions a), including β-thiolated Leu (6a) and Met (6l). Deformylation of 6k,m was carried out using 0.5 N HCl followed by saponification to give 7k,m in high yields (Figure 4A, conditions b). Similarly, acidic removal of the N-formyl group, followed by hydrolysis of the methyl ester and Boc group, gave the free amines from 6h,j, which were reprotected with Boc to give β-thiol Lys 7h,j in high yields, respectively (Figure 4A, conditions c). After acidolysis and guanidinylation, β-thiol Arg 7i was produced in three steps starting from β-thiolated Orn 6i (Figure 4A, conditions d). β-Thiol Trp analogues 7n,n′ were constructed via Fischer indole synthesis from γ-aldehyde dimethyl acetal 6n (Figure 4A, conditions g and h). β-Thiol Gln 7p and Glu 7p′ were obtained in good yields starting from 6p, respectively. Selective deformylation in the presence of the side-chain amide moiety in 6p was achieved using 33% HBr in acetic acid, and a subsequent saponification step provided β-thiol Gln 7p in 70% yield (Figure 4A, conditions e). Maintenance of the methyl ester during acidic removal of the N-formyl group together with hydrolysis of the side-chain amide moiety of 6p was possible using 6 N HCl at 90 °C for 30 min, leaving the methyl ester untouched. Functional group manipulations eventually produced β-thiol Glu 7p′ in good yield (Figure 4A, conditions f). Gram quantities of the β-thiolated amino acids could be easily prepared in the majority of these cases.
Mirror-image proteins have important therapeutic potential.22 Thus, we further explored the preparation of D-β-thiolated amino acids. With 8 (the enantiomer of 5) as the starting material, an adamantyl radical was introduced in the same manner. Compound 9 (the enantiomer of 6d) was obtained and subjected to acidolysis to produce 10 in high yield (Figure 4B). The absolute configuration of 9 was confirmed using X-ray crystallography (Figure 4C).
With these β-thiolated/selenolated amino acids in hand, we next explored their utility in peptide synthesis. Selenolated amino acids 4a,e,i,l were introduced onto a solid-supported polypeptide N-terminus. After subsequent acidic cleavage from the resin and removal of the side-chain protecting groups using TFA, peptides 11a,e,i,l were prepared, respectively (Figure 5A). In all cases, no double-coupling products were observed due to the steric bulkiness of the neighboring t-Bu group. Next, after deprotection of the auxiliary in the presence of excess MeONH2, diselenide dimer peptides 12a,e,l were obtained in good yield after HPLC purification (Figure 5A, conditions a, entries 1, 2, and 4). Unfortunately, the deprotection of the pivalaldehyde acetal of β-selenolated Arg peptide (11i) failed in the presence of MeONH2 due to the decomposition of peptide. As such, we opted for the employment of milder conditions. Interestingly, we observed that this auxiliary was stable under NCL conditions in the presence of MPAA and TCEP, yet complete acetal removal of N-terminal selenolated amino acids was achieved in the presence of 1.5 equiv of MPAA in 6 M GND·HCl/0.2 M Na2HPO4 at pH 7.0, generating the MPAA-adduct peptide 12i in good yield (Figure 5A, conditions b, entry 3). Mechanistic studies suggested that the selenazolidine ring was opened via the in situ produced disulfide of MPAA (see page 84 in the Supporting Information for details). To the best of our knowledge, the deprotection of selenoazolidine described here has some of the mildest conditions reported to date. Meanwhile, one-pot diselenide-selenoester ligation (DSL)–deselenizations23 between peptide selenoester 13 and selenolated peptides 12a,e,i,l were conducted. Peptides 14a,e,i were obtained according to a ligation and deselenization protocol in good yields (Figure 5A, conditions c). A low yield of 14l was observed during deselenization. The deselenization proceeded well when excess amounts of DTT and TCEP were used (Figure 5A, conditions d). We further examined NCL–desulfurization using β-thiolated amino acids. Peptides 15c,d,n,o were prepared via Fmoc-SPPS and TFA deprotection. Next, auxiliary removal was achieved successfully using an excess amount of MeONH2, providing thiolated peptides 16c,d,n,o in good yields (Figure 5B, conditions a, entries 1–4). Ligation of peptide 16 with peptide 13 under NCL conditions, followed by one-pot desulfurization, yielded peptides 17c,d,o smoothly (Figure 5B, conditions e). Though peptide 17n was obtained in 20% yield after desulfurization, the reaction could be improved upon treatment of a large excess of TCEP and VA-044 (Figure 5B, conditions f). Therefore, this method not only significantly expanded the utility of NCL–dechalcogenation to generate native peptides but also provided a simple strategy to access peptides containing unnatural amino acids.6a,9f,24 For example, the synthesis of fluorinated peptide 14e represents a general strategy to incorporate fluorinated amino acids into peptides, which opens up a new avenue to access fluorinated peptides.25
Figure 5.

Exploiting the utility of β-thio/selenolated amino acids. (A) DSL–deselenization using β-selenolated amino acids. (B) NCL–desulfurization using β-thiolated amino acids. (C) One-pot synthesis of cytochrome c oxidase subunit 7C (22). (D) Crude HPLC trace of the final deselenization to give 22 and ESI mass spectrum of 22. Conditions for auxiliary removal: (a) 6 M GND·HCl, 0.2 M Na2HPO4, 0.2 M MeONH2, pH 4.0; (b) 6 M GND·HCl, 0.2 M Na2HPO4, MPAA (1.5 equiv), pH 6.8. Conditions for ligation, 13 (1.2 equiv), 6 M GND·HCl, 0.2 M Na2HPO4, TCEP (2.0 equiv), pH 6.2, 25 °C, 16 h. Conditions for deselenization: (c) 6 M GND·HCl, 0.2 M Na2HPO4, DTT (9.0 equiv), TCEP (50 equiv), pH 5.2, 25 °C, 16 h; (d) 6 M GND·HCl, 0.2 M Na2HPO4, DTT (250 equiv), TCEP (50 equiv), pH 5.2, 25 °C, 16 h. Conditions for desulfurization: (e) 6 M GND·HCl, 0.2 M Na2HPO4 200 mM TCEP, 10 mM VA-044, 50 mM GSH, 37 °C, 16 h; (f) 6 M GND·HCl, 0.2 M Na2HPO4, 500 mM TCEP, 30 mM VA-044, 150 mM GSH, 37 °C, 16 h. Legend: (i) yield according to the original loading of the resin; (ii) yield of ligation and deselenization; (iii) yield of ligation and desulfurization. Abbreviations: GND, guanidine; MPAA, 4-mercaptophenylacetic acid; TCEP, tris(carboxyethyl)phosphine; VA-044, 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride; DTT, dithiothreitol; GSH, glutathione; Fmoc SPPS, Fmoc solid-phase peptide synthesis.
To further exhibit the practicality of this method, cytochrome c oxidase subunit protein 7C26 (22) was prepared by a one-pot synthesis using the selenolated Lys building block 4h as an example. As shown in Figure 5C, MPAA-thioester 18 and peptide 19 were combined under NCL conditions to yield peptide 20 via in situ production of MPAA disulfide from 18. Without separation, the crude reaction mixture was directly combined with selenoester 21, followed by an in situ chemoselective deselenization reaction using DTT and TCEP to give protein 22 in 38% isolated yield over four steps. Overall, we have successfully synthesized cytochrome c oxidase subunit 7C (22) via sequential one-pot NCL activation of the auxiliary and DSL–deselenization chemistry without any purification or solvent removal. The facile synthesis of 22 demonstrated the application of construction of β-selenolated amino acids, and the MPAA-mediated deprotection of selenazolidine could expand and enhance selenolated amino acid mediated ligations.
Disulfides have been extensively used to stabilize peptide conformations and confine structure rigidity. Current methods for disulfide bond engineering are limited to the replacement of the disulfide bond with thioether, selenoether, diselenide, and hydrocarbon bridges.27 In order to further understand the effect of introducing cysteine analogues on protein stability, a series of oxytocin (a hormone stimulating parturition and lactation drug) analogues (23a,d,h) were prepared via SPPS and subsequent oxidative folding, which contain various N-terminal β-thiolated amino acid residues (Figure 6A). The thermal stability of native oxytocin and its analogues were evaluated at 50 °C by LCMS analysis.28 Native oxytocin degrades with a half-life of 3.9 days, whereas the synthetic analogues demonstrate significant stability (23a, 18.8 days; 23d, 68.6 days; 23h, 23.1 days) (Figure 6B). The introduction of β-thiolated amino acid residues is simple and straightforward; more importantly, our approach enables the construction of peptides bearing novel disulfide bridges with high stability and structural diversity.
Figure 6.
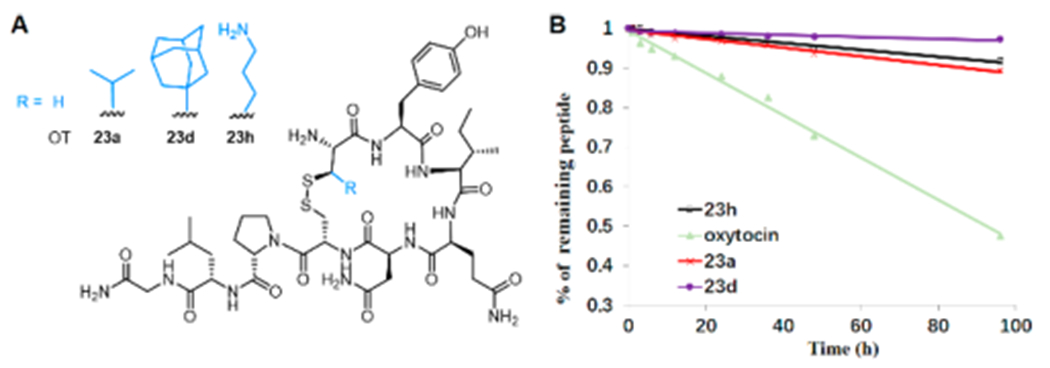
Thermal stability of synthetic oxytocin analogues. (A) Synthetic oxytocin analogues. (B) Thermal stability of oxytocin (OT) and 23a,d,h in water at 50 °C.
This method is not without limitations. For example, the reaction is performed under photoredox conditions, and thus it will be difficult to introduce radical-sensitive groups (cyclopropyl methyl, disulfide). Furthermore, only the trans diastereomer is obtained, which is another limitation of this strategy. Considering the elegant work reported by Knowles and Miller,11b a kinetic- and catalyst-controlled H atom transfer from B to C might serve as an alternative way to the other diastereomer (Figure 2A).
CONCLUSION
In summary, we have developed a practical and general strategy for the diastereoselective preparation of β-thiolated/selenolated amino acids via a photoredox-catalyzed asymmetric Giese reaction. In contrast to existing methods using nucleophilic and electrophilic thiol/selenol reagents to construct C–S/Se bonds, this method unprecedentedly combines the concept of “self-regeneration of chirality centers” with modern photoredox catalysis. Nevertheless, the described convenient approach allowed the scalable and diverse synthesis of proteinogenic amino acid analogues, including β-thiolated amino acids (Leu, Glu, Gln, Lys, Arg, Trp, Met), β-selenolated amino acids (Leu, Lys, Arg, Met), and a broad scope of unnatural β-thiolated/selenolated amino acids via a single-step asymmetric alkylation on a multigram scale. We believe that this methodology will not only significantly expand the utility of the ligation–dechalcogenation strategy but also allow the design and implementation of amino acids at will and the construction of disulfide-engineered peptides and proteins in academic and pharmaceutical settings.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work came from the National Natural Science Foundation of China (91753102, 21672146, and 21907064) and the Shanghai Committee of Science and Technology (17JC1405300). Q.Z. thanks the National Science Foundation (CHE 1710174) and the National Institutes of Health (1R35 GM138336-01) for support.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04994.
Detailed experimental procedures and spectral data(PDF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c04994
The authors declare no competing financial interest.
Contributor Information
Hongli Yin, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, People’s Republic of China.
Mengjie Zheng, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, People’s Republic of China.
Huan Chen, Department of Chemistry, University at Albany, State University of New York, Albany, New York 12222, United States.
Siyao Wang, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, People’s Republic of China.
Qingqing Zhou, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, People’s Republic of China.
Qiang Zhang, Department of Chemistry, University at Albany, State University of New York, Albany, New York 12222, United States.
Ping Wang, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, People’s Republic of China.
REFERENCES
- (1).(a) Mamathambika BS; Bardwell JC Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol 2008, 24, 211–235. [DOI] [PubMed] [Google Scholar]; (b) Metanis N; Hilvert D Strategic use of non-native diselenide bridges to steer oxidative protein folding. Angew. Chem., Int. Ed 2012, 51, 5585–5588. [DOI] [PubMed] [Google Scholar]; (c) Reich HJ; Hondal RJ Why nature chose selenium. ACS Chem. Biol 2016, 11, 821–841. [DOI] [PubMed] [Google Scholar]
- (2).Fass D; Thorpe C Chemistry and enzymology of disulfide cross-linking in proteins. Chem. Rev 2018, 118, 1169–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jacob C; Giles GI; Giles NM; Sies H Sulfur and selenium: the role of oxidation state in protein structure and function. Angew. Chem., Int. Ed 2003, 42, 4742–4758. [DOI] [PubMed] [Google Scholar]
- (4).For classic examples, see:; (a) Davie EAC; Mennen SM; Xu Y; Miller SJ Asymmetric catalysis mediated by synthetic peptides. Chem. Rev 2007, 107, 5759–5812. [DOI] [PubMed] [Google Scholar]; (b) Mousa R; Dardashti RN; Metanis N Selenium and selenocysteine in protein chemistry. Angew. Chem., Int. Ed 2017, 56, 15818–15827. [DOI] [PubMed] [Google Scholar]; An umpolung approach for the chemoselective arylation of selenocysteine in unprotected peptides: [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cohen DT; Zhang CB; Pentelute L; Buchwald SL J. Am. Chem. Soc 2015, 137, 9784–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cohen DT; Zhang C; Fadzen CM; Mijalis AJ; Hie L; Johnson KD; Shriver Z; Plante O; Miller SJ; Buchwald SL; Pentelute BL A chemoselective strategy for late-stage functionalization of complex small molecules with polypeptides and proteins. Nat. Chem 2019, 11, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) de-Gruyter JN; Malins LR; Baran PS Residue-specific peptide modification: a chemist’s guide. Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Li J-B; Tang S; Zheng J-S; Tian C-L; Liu L Removable backbone modification method for the chemical synthesis of membrane proteins. Acc. Chem. Res 2017, 50, 1143–1153. [DOI] [PubMed] [Google Scholar]; (g) Jbara M; Maity SK; Brik A Palladium in the chemical synthesis and modification of proteins. Angew. Chem. Int. Ed 2017, 56, 10644–10655. [DOI] [PubMed] [Google Scholar]; (h) Zhang Y; Zhang Q; Wong C; Li X Chemoselective peptide cyclization and bicyclization directly on unprotected peptides. J. Am. Chem. Soc 2019, 141, 12274–12279. [DOI] [PubMed] [Google Scholar]
- (5).For selected examples, see:; (a) Dawson PE; Muir TW; Clark-Lewis I; Kent SBH Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]; (b) Mcgrath NA; Raines RT Chemoselectivity in chemical biology: acyl transfer reactions with sulfur and selenium. Acc. Chem. Res 2011, 44, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bondalapati S; Jbara M; Brik A Expanding the chemical toolbox for the synthesis of large and uniquely modified proteins. Nat. Chem 2016, 8, 407–418. [DOI] [PubMed] [Google Scholar]; (d) Burke H; McSweeney ML; Scanlan EM Exploring chemoselective S-to-N acyl transfer reactions in synthesis and chemical biology. Nat. Commun 2017, 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Flood DT; Hintzen JCJ; Bird MJ; Cistrone PA; Chen JS; Dawson PE Leveraging the knorr pyrazole synthesis for the facile generation of thioester surrogates for use in native chemical ligation. Angew. Chem., Int. Ed 2018, 57, 11634–11639. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Agouridas V; Mahdi OE; Diemer V; Cargoët M; Monbaliu J-CM; Melnyk O Native chemical ligation and extended methods: mechanisms, catalysis, scope, and limitations. Chem. Rev 2019, 119, 7328–7443. [DOI] [PubMed] [Google Scholar]; (g) Thompson RE; Muir TW Chemoenzymatic semisynthesis of proteins. Chem. Rev 2020, 120, 3051–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Yan LZ; Dawson PE Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization. J. Am. Chem. Soc 2001, 123, 526–533. [DOI] [PubMed] [Google Scholar]; (b) Wan Q; Danishefsky SJ Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides. Angew. Chem. Int. Ed 2007, 46, 9248–9252. [DOI] [PubMed] [Google Scholar]; (c) Metanis N; Keinan E; Dawson PE Traceless ligation of cysteine peptides using selective deselenization. Angew. Chem., Int. Ed 2010, 49, 7049–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jin K; Li T; Chow HY; Liu H; Li X P-B desulfurization: an enabling method for protein chemical synthesis and site-specific deuteration. Angew. Chem., Int. Ed 2017, 56, 14607–14611. [DOI] [PubMed] [Google Scholar]; (e) Malins LR; Mitchell NJ; McGowan S; Payne RJ Oxidative deselenization of selenocysteine: applications for programmed ligation at serine. Angew. Chem., Int. Ed 2015, 54, 12716–12721. [DOI] [PubMed] [Google Scholar]; (f) Dery S; Reddy PS; Dery L; Mousa R; Dardashti RN; Metanis N Insights into the deselenization of selenocysteine into alanine and serine. Chem. Sci 2015, 6, 6207–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wu C; Leroux J-C; Gauthier MA Twin disulfides for orthogonal disulfide pairing and the directed folding of multicyclic peptides. Nat. Chem 2012, 4, 1044–1049. [DOI] [PubMed] [Google Scholar]
- (8).Hruby VJ Design in topographical space of peptide and peptidomimetic ligands that affect behavior. a chemist’s glimpse at the mind-body problem. Acc. Chem. Res 2001, 34, 389–397. [DOI] [PubMed] [Google Scholar]
- (9).(a) Noisier AFM; Albericio F Advance in ligation techniques for peptide and protein synthesis. Amino Acids, Pept. Proteins 2014, 39, 1–20. [Google Scholar]; (b) Moyal T; Hemantha HP; Siman P; Refuaa M; Brik A Highly efficient one-pot ligation and desulfurization. Chem. Sci 2013, 4, 2496–2501. [Google Scholar]; (c) Reimann O; Smet-Nocca C; Hackenberger CPR Traceless purification and desulfurization of tau protein ligation products. Angew. Chem., Int. Ed 2015, 54, 306–310. [DOI] [PubMed] [Google Scholar]; (d) Tang S; Liang L-J; Si Y-Y; Gao S; Wang J-X; Liang J; Mei Z; Zheng J-S; Liu L Practical chemical synthesis of atypical ubiquitin chains by using an isopeptide-linked Ub isomer. Angew. Chem., Int. Ed 2017, 56, 13333–13337. [DOI] [PubMed] [Google Scholar]; (e) Chow HY; Zhang Y; Matheson E; Li X Ligation technologies for the synthesis of cyclic peptides. Chem. Rev 2019, 119, 9971–10001. [DOI] [PubMed] [Google Scholar]; (f) Wang S; Thopate YA; Zhou Q; Wang P Chemical protein synthesis by native chemical ligation and variations thereof. Chin. J. Chem 2019, 37, 1181–1193. [Google Scholar]
- (10).(a) Chen S; Gopalakrishnan R; Schaer T; Marger F; Hovius R; Bertrand D; Pojer F; Heinis C Dithiol amino acids can structurally shape and enhance the ligand-binding properties of polypeptides. Nat. Chem 2014, 6, 1009–1016. [DOI] [PubMed] [Google Scholar]; (b) Murar CE; Ninomiya M; Shimura S; Karakus U; Boyman O; Bode JW Chemical synthesis of interleukin-2 and disulfide stabilizing analogues. Angew. Chem., Int. Ed 2020, 59, 8425–8429. [DOI] [PubMed] [Google Scholar]
- (11).(a) Guerin DJ; Miller SJ Asymmetric azidation-cycloaddition with open-chain peptide-based catalysts. A sequential enantioselective route to triazoles. J. Am. Chem. Soc 2002, 124, 2134–2136. [DOI] [PubMed] [Google Scholar]; (b) Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-driven deracemization enabled by excited-state electron transfer. Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For selected references, see:; (a) Gracia-Vitoria J; Osante I; Cativiela C Stereoselective synthesis of modified cysteines. Tetrahedron: Asymmetry 2017, 28, 215–245. [Google Scholar]; (b) Shang S; Tan Z; Dong S; Danishefsky SJ An advance in proline ligation. J. Am. Chem. Soc 2011, 133, 10784–10786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Townsend SD; Tan Z; Dong S; Shang S; Brailsford JA; Danishefsky SJ Advances in proline ligation. J. Am. Chem. Soc 2012, 134, 3912–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wong CTT; Tung CL; Li X Synthetic cysteine surrogates used in native chemical ligation. Mol. BioSyst 2013, 9, 826–833. [DOI] [PubMed] [Google Scholar]; (e) Kulkarni SS; Sayers J; Premdjee B; Payne RJ Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nature Rev. Chem 2018, 2, 1–17. [Google Scholar]
- (13).An elegant enolate chemistry has been used to build thiol/selenol residues in three to five steps, but this method is limited to Asp, Asn, and Glu scaffolds. For selected reviews and references, see:; (a) Thompson RE; Chan B; Radom L; Jolliffe KA; Payne RJ Chemoselective ligation-desulfurization at aspartate. Angew. Chem., Int. Ed 2013, 52, 9723–9727. [DOI] [PubMed] [Google Scholar]; (b) Mitchell NJ; Sayers J; Kulkarni SS; Clayton D; Goldys AM; Ripoll-Rozada J; Pereira PJB; Chan B; Radom L; Payne RJ Accelerated protein synthesis via one-pot ligation-deselenization chemistry. Chem. 2017, 2, 703–715. [Google Scholar]
- (14).For selected references on the Giese reaction, see:; (a) Giese B; Dupuis J Diastereoselective syntheses of C-glycopyranosides. Angew. Chem., Int. Ed. Engl 1983, 22, 622–623. [Google Scholar]; (b) Giese B Formation of CC bonds by addition of free radicals to alkenes. Angew. Chem., Int. Ed. Engl 1983, 22, 753–764. [Google Scholar]; (c) Jasperse CP; Curran DP; Fevig TL Radical reactions in natural product synthesis. Chem. Rev 1991, 91, 1237–1286. [Google Scholar]; (d) Srikanth GSC; Castle SL Advances in radical conjugate additions. Tetrahedron 2005, 61, 10377–10441. [Google Scholar]
- (15).(a) Polt R; Seebach D Stereoselective alkylation of glycine units in dipeptide derivatives: “chirality transfer” via a pivalaldehyde N,N-acetal center. J. Am. Chem. Soc 1989, 111, 2622–2632. [Google Scholar]; (b) Leonard DJ; Ward JW; Clayden J Asymmetric α-arylation of amino acids. Nature 2018, 562, 105–109. [DOI] [PubMed] [Google Scholar]
- (16).Jeanguenat A; Seebach D Stereoselective chain elongation at C-3 of cysteine through 2,3-dihydrothiazoles, without racemization. preparation of 2-amino-5-hydroxy-3-mercaptoalkanoic acid derivatives. J. Chem. Soc., Perkin Trans 1 1991, 2291–2298. [Google Scholar]
- (17).For selected references, see:; (a) Zuo Z; Cong H; Li W; Choi J; Fu GC; MacMillan DWC Enantioselective decarboxylative arylation of α-amino acids via the merger of photoredox and nickel catalysis. J. Am. Chem. Soc 2016, 138, 1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCarver SJ; Qiao JX; Carpenter J; Borzilleri RM; Poss MA; Eastgate MD; Miller M; MacMillan DWC Decarboxylative peptide macrocyclization through photoredox catalysis. Angew. Chem., Int. Ed 2017, 56, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bloom S; Liu C; Kölmel DK; Qiao JX; Zhang Y; Poss MA; Ewing WR; MacMillan DWC Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials. Nat. Chem 2018, 10, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For selected examples, see:; (a) Ni S; Garrido-Castro AF; Merchant RR; deGruyter JN; Schmitt DC; Mousseau JJ; Gallego GM; Yang S; Collins MR; Qiao JX; Yeung K; Langley DR; Poss MA; Scola PM; Qin T; Baran PS A general amino acid synthesis enabled by innate radical cross-coupling. Angew. Chem., Int. Ed 2018, 57, 14560–14565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pratsch G; Lackner GL; Overman LE Constructing quaternary carbons from N-(acyloxy)phthalimide precursors of tertiary radicals using visible-light photocatalysis. J. Org. Chem 2015, 80, 6025–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Narayanam JMR; Tucker JW; Stephenson CRJ Electron-transfer photoredox catalysis: development of a tin-free reductive dehalogenation reaction. J. Am. Chem. Soc 2009, 131, 8756–8757. [DOI] [PubMed] [Google Scholar]
- (20).Gracia-Vitoria J; Osante I; Cativiela C; Merino P; Tejero T Self-regeneration of chirality with L-cysteine through 1,3-dipolar cycloadditions between diazoalkanes and enantiomerically pure thiazolines: experimental and computational studies. J. Org. Chem 2018, 83, 3960–3972. [DOI] [PubMed] [Google Scholar]
- (21).Schwarz J; König B Metal-free, visible-light-mediated, decarboxylative alkylation of biomass-derived compounds. Green Chem. 2016, 18, 4743–4749. [Google Scholar]
- (22).(a) Wang Z; Xu W; Liu L; Zhu TF A synthetic molecular system capable of mirror-image genetic replication and transcription. Nat. Chem 2016, 8, 698–704. [DOI] [PubMed] [Google Scholar]; (b) Teng P; Ma N; Cerrato DC; She F; Odom T; Wang X; Ming L-J; van der Vaart A; Wojtas L; Xu H; Cai J Right-handed helical foldamers o-AApeptides. J. Am. Chem. Soc 2017, 139, 7363–7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mitchell NJ; Malins LR; Liu X; Thompson RE; Chan B; Radom L; Payne RJ Rapid additive-free selenocystine–selenoester peptide ligation. J. Am. Chem. Soc 2015, 137, 14011–14014. [DOI] [PubMed] [Google Scholar]
- (24).For applications of peptide ligation–desulfurization/deselenization reactions, see the selected reviews:; (a) Ma J; Zeng J; Wan Q Postligation-desulfurization: a general approach for chemical protein synthesis. Top. Curr. Chem 2014, 363, 57–102. [DOI] [PubMed] [Google Scholar]; (b) Kulkarni SS; Sayers J; Premdjee B; Payne RJ Rapid and efficient protein synthesis through expansion of the native chemical ligation concept. Nature Rev. Chem 2018, 2, 1–17. [Google Scholar]
- (25).(a) Berger AA; Völler J; Budisa N; Koksch B Deciphering the fluorine code-the many hats fluorine wears in a protein environment. Acc. Chem. Res 2017, 50, 2093–2103. [DOI] [PubMed] [Google Scholar]; (b) Moschner J; Stulberg V; Fernandes R; Huhmann S; Leppkes J; Koksch B Approaches to obtaining fluorinated α-amino acids. Chem. Rev 2019, 119, 10718–10801. [DOI] [PubMed] [Google Scholar]
- (26).Wikstrom MK Proton pump coupled to cytochrome c oxidase in mitochondria. Nature 1977, 266, 271–273. [DOI] [PubMed] [Google Scholar]
- (27).Wang Y; Chou DH-C A thiol–ene coupling approach to native peptide stapling and macrocyclization. Angew. Chem., Int. Ed 2015, 54, 10931–10934. [DOI] [PubMed] [Google Scholar]
- (28).de Araujo AD; Mobli M; Castro J; Harrington AM; Vetter I; Dekan Z; Muttenthaler M; Wan J; Lewis RJ; King GF; Brierley SM; Alewood PF Selenoether oxytocin analogues have analgesic properties in a mouse model of chronic abdominal pain. Nat. Commun 2014, 5, 1–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.