

Abstract
(+)-Matrine and (+)-isomatrine are tetracyclic alkaloids isolated from the plant Sophora flavescens, the roots of which are used in traditional Chinese medicine. Biosynthetically, these alkaloids are proposed to derive from three molecules of (–)-lysine via the intermediacy of the unstable cyclic imine Δ1-piperidine. Inspired by the biosynthesis, a new dearomative annulation reaction has been developed that leverages pyridine as a stable surrogate for Δ1-piperidine. In this key transformation, two molecules of pyridine are joined with a molecule of glutaryl chloride to give the complete tetracyclic framework of the matrine alkaloids in a single step. Using this dearomative annulation, isomatrine is synthesized in four steps from inexpensive commercially available chemicals. Isomatrine then serves as the precursor to additional lupin alkaloids, including matrine, allomatrine, isosophoridine, and sophoridine.
Graphical Abstract

The lupin alkaloids are a structurally diverse class of quinolizidine-containing natural products isolated from plants in the Lupinus genus (Fig. 1A).1 (+)-Matrine (2), the primary component of Chinese Kushen injection, inhibits proliferation in metastatic cancer cell lines and has also been investigated as a therapeutic agent against encephalomyelitis, asthma, arthritis, and osteoporosis.2,3 (–)-Sophoridine (4) is an approved chemotherapeutic in China, which has also demonstrated antibiotic activity.4 Little is known about the pharmacological properties of (+)-isomatrine (1) and (+)-isosophoridine (5), which likely reflects their limited accessibility from commercial vendors.5
Figure 1.

(A) Chemical structures of matrine-type lupin alkaloids. (B) Proposed biosynthesis of matrine. (C) Retrosynthetic analysis of isomatrine.
Although the detailed enzymatic pathway has not been fully annotated, the biosynthesis of matrine is proposed to initiate with the enzymatic conversion of (–)-lysine (6) to Δ1-piperidine (7) (Fig. 1B).6,7 Subsequent dimerization of 7 followed by oxidation and isomerization is proposed to yield quinolizidine 8, a shared biosynthetic precursor to several lupin alkaloids.8,9 Mannich addition of 8 to a third equivalent of 7 and cyclization with the pendant aldehyde is proposed to generate the oxidized tetracycle 9, which upon reduction gives (+)-matridine (10). Seminal studies by Abdusalamov demonstrated that feeding 14C-labeled (+)-10 to Goebelia Pachycarpa resulted in the isolation of radio-labelled (+)-2, suggesting that the final step in the biosynthesis of 2 is a site-selective C–H oxidation.10,11
Synthetically, most of the work prior to 2022 had focused on matrine (2), with four reported total syntheses.12,13,14,15 Synthetic access to the minor congeners is far more limited: until a recent report by Sherburn and coworkers,16 there was a single total synthesis each of allomatrine (3) and isosophoridine (5), and no reported total syntheses of isomatrine or sophoridine (4).17,18 We sought to devise a unified synthesis that could provide access to the series of matrine-type alkaloids shown in Fig. 1A. Inspired by the proposed biosynthesis, it was envisioned that pyridine (14) could serve as a stable, inexpensive synthon for Δ1-piperidine (7), and the remaining five carbons of the tetracyclic matrine framework could derive from glutaryl chloride (15, Fig. 1C). In the key step, we proposed a dearomative annulation via bis-acyl pyridinium salt 17 to form tetracycle 12, a molecule that contains all the carbon and nitrogen atoms of 1.19 Tetracycle 12 could be elaborated to 1 by global reduction followed by a site-selective oxidation of isomatridine (11) reminiscent of the proposed biosynthesis of matrine (2). Isomatrine (1) is the least thermodynamically stable lupin alkaloid and its isomerization to both 2 and 3 has been previously reported.20 We therefore anticipated that access to 1 could enable the synthesis of additional lupin alkaloids.21 This type of late-stage isomerization strategy was also deployed in the 2022 Sherburn synthesis of several matrine alkaloids.16
Our studies commenced with the investigation of the dearomative annulation (Fig. 2A). Addition of glutaryl chloride (15) to pyridine (14) in dichloromethane at −50 °C followed by warming to 20 °C resulted in clean formation of (±)-tetracycle 12 in 62% yield (10 g scale). The reaction was highly robust and could be carried out on one mole scale to produce over 160 grams (67% yield) of (±)-tetracycle 12 in a single batch. The product was isolated by precipitation from the crude reaction mixture, alleviating the need for a workup or column chromatography. Given the cost of pyridine ($7/mol), glutaryl chloride ($211/mol), and all solvents ($31/mol), the raw materials cost $398/mol of product formed.22 Recrystallization of (±)-12 enabled single crystal X-ray diffraction, which confirmed the syn-syn relative stereochemistry.23
Figure 2.
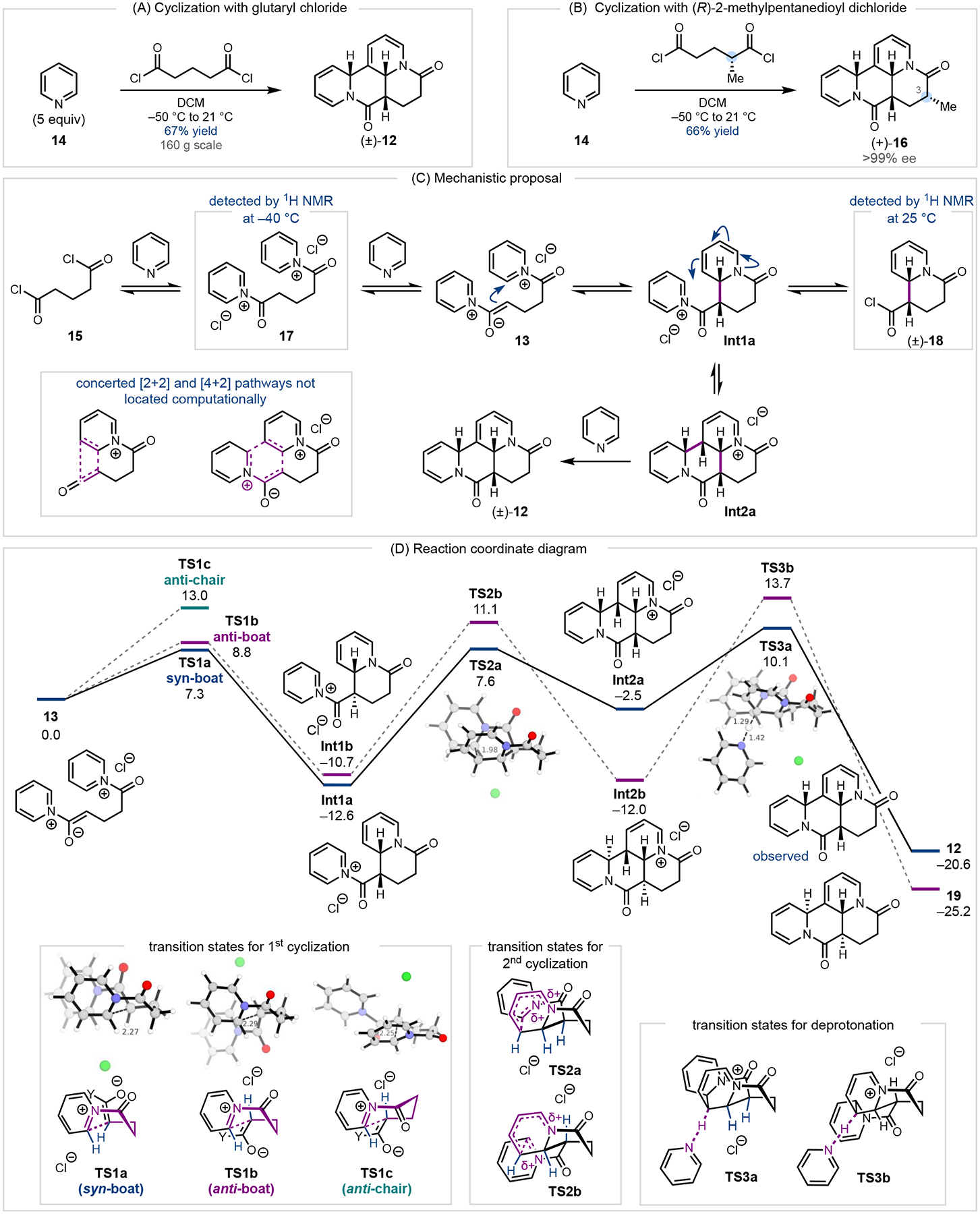
(A) Diastereoselective cyclization of pyridine and glutaryl chloride. (B) Diastereoselective cyclization of (R)-2-methylpentandioyl chloride. (C) Proposed mechanism for the diastereoselective cyclization. (D) Reaction coordinate diagram – performed with Gaussian using ωB97XD/def2-TZVP/SMD(DCM).
To elucidate the reaction pathway, mechanistic and computational studies were undertaken. Monitoring the reaction between 14 and 15 by 1H NMR determined that the major species at −40 °C was bis-acyl pyridinium salt 17 (Fig. 2C). After warming to 25 °C, acid chloride 18 resulting from mono-cyclization was observed. Presumably the acyl pyridinium salt Int1a and acyl chloride 18 are in equilibrium, but the acyl chloride is the major species at 25 °C. A second, minor species assigned as the acid chloride resulting from Int1b (vide infra) was also observed; this species was consumed as the reaction progressed to full conversion.23 Although deprotonation of acyl pyridinium salts or acyl chlorides can give rise to ketene intermediates,24 no such species was detected by 1H NMR or by reactIR. Attempts to calculate a pathway involving ketene intermediates failed to locate a transition state (TS) for a concerted [2+2] cycloaddition. Similarly, no TS for the concerted [4+2] cycloaddition of bis-acyl pyridinium salt 13 could be located. Investigation of a stepwise pathway determined that the lowest-energy TS for the first cyclization involves a boat-like conformation to form the syn product (TS1a, ΔGTS = 7.3 kcal/mol) (Fig. 2D). Attempts to find the analogous chair-like TS were unsuccessful and led instead to conversion to the boat-like TS. The pathways leading to the anti mono-cyclization product (Int1b) are higher in energy (see TS1b and TS1c). The preference for the syn boat compared to the anti boat TS is likely due to favorable dispersive interactions between the heteroaryl ring and the oxygen-bearing carbon of the enolate, as well as minimization of the dipole moment in the syn TS. In order to test the importance of dispersive interactions in these TSs, the TSs were recomputed with B3LYP, a functional known to lack dispersion. Indeed, with this functional, the difference between the two transition states was only 0.1 kcal/mol. Inclusion of dispersion with Grimme’s D3 correction restored the energy difference to 1.4 kcal/mol in favor of the syn boat transition state.
These TSs lead to two intermediates: syn intermediate Int1a (–12.6 kcal/mol) and anti intermediate Int1b (–10.7 kcal/mol). The TS for the second C–C bond formation (TS2a) is most favorable for the syn-syn intermediate (Int2a), with a barrier of 20.2 kcal/mol. The second lowest-energy pathway proceeds via TS2b leading to Int2b, which gives rise to the anti-syn-anti configuration at the ring fusions. The transition states leading to the other four potential diastereomers are higher in energy. Formation of Int2a and Int2b is followed by deprotonation by pyridine. While Int2b is lower in energy than Int2a, the deprotonation of Int2a to give syn-syn (±)-12 follows the lowest-energy pathway. Thus, the selectivity-determining step is the final deprotonation (TS3a) and syn-syn (±)-12 is favored, even though it is thermodynamically less stable than anti-anti 19. These results are consistent with the experimentally observed formation of product (±)-12 as a single diastereomer, despite the initial mixture of monocyclization products. When enantiopure (R)-2-methylpentandioyl chloride was employed, C3-methyl tetracycle (+)-16 was obtained in 66% yield as a single diastereomer and in >99% ee (Fig. 2B). This stereochemical outcome is consistent with the calculations, where the pathway initiating with a syn boat transition state bearing the methyl group in a pseudo-equatorial position is favored.
At this stage, attention turned to elaborating (±)-12 to isomatrine (1) (Fig. 3A). Hydrogenation of tetraene 12 proceeded smoothly followed by reduction with alane to give (±)-isomatridine (11) in 60% yield over two steps. Purification was readily accomplished by generating the hydrogen oxalate salt followed by trituration in acetone, obviating the need for column chromatography. At this stage, resolution of diamine (±)-11 can be achieved by recrystallization of the di-p-toluoyl tartaric acid salt to give 24% recovery (46% theoretical yield) of the desired (+)-diamine 11 in 90% ee.
Figure 3.

(A) Complete total synthesis of isomatrine. (B) Attempted oxidation reactions.
Inspired by the proposed biosynthesis,25 we initially investigated the enzymatic oxidation of (+)-11 to give (+)-1. Unfortunately, a screen of >180 bacterially derived P450 enzymes (both wild type and mutants) failed to produce any promising leads. As a result, our focus turned towards non-enzymatic methods for the selective oxidation of C15. Analysis of the X-ray structure of diamine 11 revealed that N1 points into the cavity of the molecule while the N2 lone pair points outwards. Selective oxidation of N2 was achieved by treatment of diamine 11 with peracetic acid to yield the expected N-oxide 20 in a 54% yield. However, attempts to advance 20 via Polonovski reaction (acetic anhydride/di-tert-butyl-4-methyl pyridine (DTBMP)) led to formation of the undesired enamine 21.26 Analysis of the X-ray structure of 20 confirmed that antiperiplanar alignment of H17 with the N–O bond is ideally suited to regioselectively form the undesired elimination product 21.
We became interested in a report by Kessar and coworkers demonstrating that amine–BF3 adducts could undergo deprotonation using mixtures of tert-butyl lithium (t-BuLi) and potassium tert-butoxide (t-BuOK).27 Consistent with the selectivity in the N-oxide formation, treatment of diamine (+)-11 with BF3·OEt2 quantitatively formed the Lewis acid-base complex. Deprotonation of the BF3 complex of (+)-11 with a mixture of t-BuLi and t-BuOK in N,N,N,N-tetramethylethylenediamine (TMEDA) occurred with good selectivity for the less sterically encumbered C15 over C17 (10:1), as determined by trapping with deuterated methanol (22, Fig. 3B).28 Unfortunately, trapping of this anion with other electrophiles proved more challenging. For example, deprotonation followed by quenching with TMSCl provided silylated diamine 23 in only 35% yield, while trapping with methyl benzoate gave unstable phenyl ketone 25 in 27% yield. The best yield of C15-functionalized product was obtained when 11 was deprotonated and then trapped with trimethyl borate; oxidation with hydrogen peroxide and trapping of the resultant enamine with HCN gave aminonitrile 24 in 55% yield. Aerobic oxidation of 24 provided isomatrine in 46% yield (25% yield over three steps from 11).23, 29 Alternatively, deprotonation of (+)-11, trapping with methyl benzoate, and aerobic oxidation could be carried out in a single reaction flask to give (+)-1 directly in 18–26% yield, depending on the scale.30 This route provides access to (±)-isomatrine in four steps, and (+)-isomatrine can be easily accessed by incorporating the resolution of diamine 11. To date, >1 gram of (+)-isomatrine has been prepared.
Initial attempts to reproduce Okuda’s Pt-catalyzed isomerization of (+)-isomatrine failed to provide the reported yields of (+)-matrine (2) and (+)-allomatrine (3), and instead produced a mixture of five compounds.20 To improve the yield of 2 and 3, while also broadening the synthetic access to other congeners, an investigation of several isomerization catalysts was carried out. Use of Rh/C provided the best yields of (+)-2 (32% yield, Fig. 4), while (+)-3 could be obtained in 83% yield when Pd/C was used. Isomerization with Pt/C provided (+)-isosophoridine (5) in 55% yield. Finally, use of PtO2 at 98 °C for 15 minutes furnished (–)-sophoridine (4) in 10% yield, together with the other isomers.31,32 When the reaction with PtO2 was conducted at 80 °C for 24 hours, (–)-isomer 26 was isolated in 40% yield. To our knowledge, 26 has not yet been isolated from natural sources.
Figure 4.

Isomerization of (+)-isomatrine to additional matrine-type lupin alkaloids. Highlighted values are the isolated yields of the indicated products.
The bioinspired dearomative annulation between pyridine and glutaryl chloride developed here has enabled the first total synthesis of the lupin alkaloid (–)-sophoridine, and the shortest syntheses of (+)-isomatrine, (+)-matrine, (+)-allomatrine, and (+)-isosophoridine reported to date. The brevity of this route results from the ability to construct the entire carbon framework of the matrine-type alkaloids in a single step. The diversity of matrine-type alkaloids prepared from commodity chemicals is anticipated to support future pharmacological investigations.
Supplementary Material
ACKNOWLEDGMENTS
The California Institute of Technology Center for Catalysis and Chemical Synthesis is gratefully acknowledged for access to analytical equipment. We thank the Dow Next Generation Educator Funds and Instrumentation Grants for their support of the Beckman Institute X-ray Crystallography Facility at Caltech, as well as the Caltech CCE NMR facility and Multiuser Mass Spectrometry Laboratory, which is also supported by the NSF CRIF program (CHE-0541745). Dr. M. Shahgholi is acknowledged for acquisition of HRMS data. Dr. Michael Takase and Larry Henling are acknowledged for acquiring the X-ray diffraction data, and Yujia Tao is thanked for assistance in solving X-ray structures. Dr. David Miller of the Arnold lab at Caltech is acknowledged for his help in running the P450 enzyme oxidation screen. Fellowship support was provided by the Natural Sciences and Engineering Research Council (NSERC) of Canada (PGS-D fellowship to J. K. K., grant PGSD3-532535-2019), and the National Institutes of Health (NIH) (F32GM134709 to A. T.). S.E.R. acknowledges financial support from the NIH (R35GM118191), and K. N. H. acknowledges the National Science Foundation (CHE-1764328).
Footnotes
Supporting Information
The Supporting Information is available free of charge at DOI: Experimental procedures, characterization data (1H and 13C NMR, HRMS, FTIR) for all new compounds (PDF), coordination geometries for DFT optimized compounds. X-ray structural data are available free of charge from the Cambridge Structural Database under CCDC 2159764-2159768, 2159770-2159774, and 2163777.
REFERENCES
- (1).Ohmiya S; Saito K; Murakoshi I Chapter 1 Lupine alkaloids. In The Alkaloids: Chemistry and Pharmacology. Cordell GA, Ed.; Academic Press, 1995; pp 1–114. [Google Scholar]
- (2).Zhang H; Chen L; Sun X; Yang Q; Wan L; Guo C Matrine: A promising natural product with various pharmacological activities. Front. Pharmacol 2020, 11, 588. DOI: 10.3389/fphar.2020.00588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).You L; Yang C; Du Y; Wang W; Sun M; Liu J; Ma B; Pang L; Zeng Y; Zhang Z; Dong X; Yin X; Ni J A systematic review of the pharmacology, toxicology and pharmacokinetics of matrine. Front. Pharmacol 2020, 11, 01067. DOI: 10.3389/fphar.2020.01067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wang Q; Li Y; Li K-W; Zhou C-Z Sophoridine: A review of its pharmacology, pharmacokinetics and toxicity. Phytomedicine 2022, 95, 153756. DOI: 10.1016/j.phymed.2021.153756 [DOI] [PubMed] [Google Scholar]
- (5).No vendors were found to stock either (+)-isomatrine or (+)-isosophoridine as found by a CAS SciFinder supplier search (06/14/2022) and an eMolecule supplier search (06/14/2022).
- (6).Bunsupa S; Katayama K; Ikeura E; Oikawa A; Toyooka T; Saito K; Yamazaki M Lysine decarboxylase catalyzes the first step of quinolizidine alkaloid biosynthesis and coevolved with alkaloid production in leguminosae. The Plant Cell 2012, 24, 1202–1216. DOI: 10.1105/tpc.112.095885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yang T; Nagy I; Mancinotti D; Otterbach SL; Andersen TB; Motawia MS; Asp T; Geu-Flores F Transcript profiling of a bitter variety of narrow-leafed lupin to discover alkaloid biosynthetic genes. J. Exp. Bot 2017, 68, 5527–5537. DOI: 10.1093/jxb/erx362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Golebiewski WM; Spenser ID Biosynthesis of the lupine alkaloids. II. Sparteine and lupanine. Can. J. Chem 1988, 66, 1734–1748. DOI: 10.1139/v88-280 [DOI] [Google Scholar]
- (9).Mancinotti D; Frick KM; Geu-Flores F Nat. Prod. Rep 2022, 39, 1423–1437. [DOI] [PubMed] [Google Scholar]
- (10).Abdusalamov BA Biosynthesis and metabolism of some matrine alkaloids in Goebelia pachycarpa. Chem. Nat. Compd 1984, 20, 1–9. DOI: 10.1007/BF00574779 [DOI] [Google Scholar]
- (11).Leeper FJ; Grue-Sørensen G; Spenser ID Biosynthesis of the quinolizidine alkaloids. Incorporation of Δ1-piperidine into matrine. Can. J. Chem 1981, 59, 106–115. DOI: 10.1139/v81-017 [DOI] [Google Scholar]
- (12).Mandell L; Singh KP; Gresham JT; Freeman W Total synthesis of d,l-matrine. J. Am. Chem. Soc 1963, 85, 2682–2683. DOI: 10.1021/ja00900a048 [DOI] [PubMed] [Google Scholar]
- (13).Okuda S; Yoshimoto M; Tsuda K Studies on lupin alkaloids. IV. Total syntheses of optically active matrine and allomatrine. Chem. Pharm. Bull 1966, 14, 275–279. DOI: 10.1248/cpb.14.275 [DOI] [PubMed] [Google Scholar]
- (14).Chen J; Browne LJ; Gonnela NC Total synthesis of (±)-matrine. J. Chem. Soc., Chem. Commun 1986, 905–907. DOI: 10.1039/C39860000905 [DOI] [Google Scholar]
- (15).Boiteau L; Boivin J; Liard A; Quiclet-Sire B; Zard SZ A short synthesis of (±)-matrine. Angew. Chem. Int. Ed 1998, 37, 1128–1131. DOI: 10.1002/(SICI)1521-3773(19980504)37:8<1128::AID-ANIE1128>3.0.CO;2-P [DOI] [PubMed] [Google Scholar]
- (16).Shortly after the deposition of this work on ChemRxiv (DOI: 10.26434/chemrxiv-2022-0v94t), an elegant approach to the matrine alkaloids was disclosed: Magann NL; Westley E; Sowden MJ; Gardiner MG; Sherburn MS Total synthesis of matrine alkaloids ChemRxiv, 2022, DOI: (accessed 2022/06/07). [DOI] [PubMed] [Google Scholar]
- (17).Watkin SV; Camp NP; Brown RCD Total synthesis of the tetracyclic lupin alkaloid (+)-allomatrine. Org. Lett 2013, 15, 4596–4599. DOI: 10.1021/ol402198n [DOI] [PubMed] [Google Scholar]
- (18).Lyu X Stereoselective total synthesis of lupin alkaloids. Ph.D. Dissertation, University of Southampton, Southampton, UK, 2018. Eprints.soton.ac.uk/429608/ (accessed 2022-02-15). [Google Scholar]
- (19).There is precedent for the first step of this synthesis, see: Warneke J; Plaumann M; Wang Z; Böhler E; Kemken D; Kelm S; Dieter L; Azov VA Tetrahedron Lett. 2015, 56, 1124–1127. [Google Scholar]
- (20).Ueno A; Morinaga K; Fukushima S; Iitaka Y; Koiso Y; Okuda S Studies on lupin alkaloids. VI. Isolation and structure of (+)-isomatrine. Chem. Pharm. Bull 1975, 23, 2560–2566. DOI: 10.1248/cpb.23.2560 [DOI] [Google Scholar]
- (21).Galasso V; Asaro F; Berti F; Pergolese B; Kovač B; Pichierri F On the molecular and electronic structure of matrine-type alkaloids. Chem. Phys 2006, 330, 457–468. DOI: 10.1016/j.chemphys.2006.09.017 [DOI] [Google Scholar]
- (22). MilliporeSigma; Pyridine. Product No. P57506-4L (Accessed 2022/06/16). Oakwood Chemical; Glutaryl chloride. Product No. 095828-100g (Accessed 2022/06/16). Fischer Scientific; Dichloromethane. Product No. D37-20 (Accessed 2022/06/16) Fischer Scientific; Methanol. Product No. A412SK-20 (Accessed 2022/06/16)
- (23).See Supporting Information for more details.
- (24).Paull DH; Weatherwax A; Lectka T Catalytic, asymmetric reactions of ketenes and ketene enolates. Tetrahedron 2009, 65, 6771–6803. DOI: 10.1016/j.tet.2009.05.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wink M; Hartmann T Enzymatic synthesis of quinolizidine alkaloids in lupin chloroplasts. Z. Naturforsch 1980, 35, 93–97. DOI: 10.1515/znc-1980-1-218 [DOI] [Google Scholar]
- (26).Grierson D The Polonovski reaction. Org. React 1990, 39, 85–295. DOI: 10.1002/0471264180.or039.02 [DOI] [Google Scholar]
- (27).Kessar SV; Singh P; Singh PKN; Singh SK Facile α-deprotonation-electrophilic substitution of quinuclidine and DABCO. Chem. Commun 1999, 1927–1928. DOI: 10.1039/A905359J [DOI] [Google Scholar]
- (28). Supporting Information tables S1 – S4..
- (29).Chuang T-H; Yang C-C; Chang C-J; Fang J-M Base-catalyzed autoxidation of α-aminonitriles. An efficient method for conversion of aldehydes to amides and 2-amino-2-sulfenylacetonitrile to carbamates. Synlett 1990, 12, 733–734. DOI: 10.1055/s-1990-21229 [DOI] [Google Scholar]
- (30).García-Valverde M; Pedrosa R; Vicente M A novel and efficient oxidation of 1,2-amino alcohols to dialkylamides. Synlett 2002, 12, 2092–2094. DOI: 10.1055/s-2002-35602 [DOI] [Google Scholar]
- (31).Ibragimov BT; Tischenko GN; Kushmuradov YK; Aripov TF Molecular and crystal structure of sophoridine. Chem. Nat. Compd 1979, 15, 308–314. DOI: 10.1007/BF00566082 [DOI] [Google Scholar]
- (32).See Supporting Information table S20 for more details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.