

Abstract

In this study, two new series of 3-cyanopyridinones (3a–e) and 3-cyanopyridines (4a–e) were synthesized and evaluated for their cytotoxicity and Pim-1 kinase inhibitory activity adopting 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay and in vitro Pim-1 kinase inhibition assay, respectively. Most of the tested compounds revealed promising cytotoxicity against HepG-2, HCT-116, MCF-7, and PC-3 cell lines. Among them, compounds 4c and 4d showed more potent cytotoxicity against the HePG2 cell line with IC50 = 8.02 ± 0.38 and 6.95 ± 0.34 μM, respectively, than that of the reference 5-FU (IC50 = 9.42 ± 0.46 μM). Moreover, compound 4c was more potent against HCT-116 (IC50 = 7.15 ± 0.35 μM) than 5-FU (IC50 = 8.01 ± 0.39 μM), while compound 4d with IC50 = 8.35 ± 0.42 μM displayed comparable activity to that of the reference drug. Furthermore, high cytotoxic activity was manifested by compounds 4c and 4d against MCF-7 and PC3 cell lines. Our results have also indicated that compounds 4b, 4c, and 4d elicited remarkable inhibition of Pim-1 kinase; 4b and 4c showed equipotent inhibitory activity to that of the reference quercetagetin. Meanwhile, 4d displayed IC50 = 0.46 ± 0.02 μM, showed the best inhibitory activity among the tested compounds, and was more potent than quercetagetin (IC50 = 0.56 ± 0.03 μM). For optimization of the results, docking study of the most potent compounds 4c and 4d in the Pim-1 kinase active site was carried out and compared with both quercetagetin and the reported Pim-1 inhibitor A (VRV), and the results were consistent with those of the biological study. Consequently, compounds 4c and 4d are worthy of further investigations toward the discovery of Pim-1 kinase inhibitors as drug candidates for cancer therapy. Compound 4b was successfully radiolabeled with radioiodine-131, and its biodistribution in Ehrlich ascites carcinoma (EAC)-bearing mice showed more observable uptake in tumor sites, and hence, it can be introduced as a new radiolabeled agent for tumor imaging and therapy.
1. Introduction
Cancer is a complex disease characterized by uncontrolled proliferation and circulation of cells described as metastasis, and it remains the world’s leading cause of death.1 Different classes of chemotherapeutic agents, either alone or in combination, are being used for treating this deadly disease. However, the side effects associated with the conventional chemotherapy along with the poor bioavailability profile have led to an urgent need for target-based chemotherapeutic drugs.2,3 The oncogenic serine/threonine kinase (Pim-1) plays a pivotal role in phosphorylating and regulating the activity of many proteins involved in cell survival, proliferation, and apoptosis, so Pim-1 kinase is linked to many cancer types.4 It has been reported to be overexpressed in several types of solid cancers such as prostate as well as hematological cancers such as leukemia, multiple myeloma, and diffuse large B cell lymphomas (DLBCL).5,6 Downregulation of Pim-1 expression by Pim-1 inhibitors has been reported to cause cell cycle arrest and increase apoptosis in some types of cancer including prostate,5 breast,7 colon,8 hepatic,9 and pancreatic cancers10 through different pathways, making it an attractive therapeutic target in cancer therapy. Many reported studies are concerned with discovering various classes of heterocyclic and fused heterocyclic compounds to target and inhibit Pim-1 kinase as a promising tool for fighting cancer.11−14 Among different recognized classes, 3-cyanopyridine-based compounds are known to have significant cytotoxic effects owing to their ability to interact with different types of biological targets including the Pim-1 kinase enzyme.15−18 For example, 6-(5-bromo-2-hydroxy) phenyl-2-oxo-4-phenyl-3- pyridine carbonitrile A (VRV),16 compound B,16 compound C,19 and compound D(19) (Figure 1) have been found to be inhibitors of Pim-1 kinase. Furthermore, cyanopyridine derivatives with higher lipophilic properties as in compound E (CLogP: 4.49652)20,21 (Figure 1) inhibit survivin, which is highly expressed in most human tumors.22 However, the reported cyanopyridine derivative F has the ability to inhibit β-tubulin polymerization23 (Figure 1).
Figure 1.

Structures of the reported anticancer lead compounds (A–F).
Inspired by the previous findings, the goal of this study was to design new Pim-1 inhibitors with enhanced cytotoxic activity via modifications of the structure of the reported Pim-1 inhibitor A (VRV). A new series was designed and synthesized by keeping the 6-phenyl cyanopyridinone scaffold of the lead compound A with structural modifications at positions 4 and 6 (Figure 2). Modification of the 4-aryl moiety was achieved via the incorporation of a lipophilic 2-chloroquinolin-3-yl pharmacophoric moiety with documented anticancer24−27 and Pim-1 inhibitory activities.28,29 In addition, introduction of different substituents at the phenyl ring at the 6-position was achieved, and such substituents were selected to offer variable an electronic, lipophilic, and steric environment that could affect the targeted biological activity. Aromatization of the cyanopyridinone ring was considered via converting the electron-releasing group OH (C=O) at position 2 of the pyridine ring into an electron-withdrawing group Cl functionality to study the effect of this structural variation on the anticancer activity. Accordingly, two new series including cyanopyridinones and the corresponding chloro derivatives (cyanopyridines) (Figure 2) were designed to be synthesized to compare their cytotoxic activities. MTT assay was used to study the cytotoxic activities of the designed compounds against four different human tumor cell lines HepG-2, HCT-116, MCF-7, and PC-3 using 5-FU as standard antitumor agents. Moreover, their potential Pim-1 inhibitory activity was revealed via enzyme inhibition assay and molecular docking analysis.
Figure 2.

Rational design of the newly synthesized compounds.
2. Results and Discussion
2.1. Chemistry
The reaction sequences employed for the synthesis of the targeted compounds are illustrated in Scheme 1.
Scheme 1. Synthesis of Cyanopyridinones 3a–e and Cyanopyridines 4a–e.

2.1.1. Synthesis of Compounds 3
The reported synthetic intermediate 2-chloroquinoline-3-carbaldehyde (1) has received considerable attention in the synthesis of large numbers of heterocyclic systems.30 It was synthesized by cyclization of N-phenylacetamide with Vilsmeier′s reagent DMF/POCl3 via a multicomponent reaction that involves chlorination, formylation, and cyclization. A one-pot four-component reaction was adopted in the synthesis of 6-((un)substituted phenyl)-4-(2-chloroquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile derivatives (3a–e). Hence, the key intermediate (1), different acetophenone derivatives (2a–e), ethyl cyanoacetate, and ammonium acetate were refluxed in ethanol to afford the new targeted compounds in a yield ranging from 68 to 88%.
A plausible mechanism for this reaction is indicated in Figure 3.31 The mechanism was presupposed to proceed through condensation of 2-chloroquinoline-3-carbaldehyde (1) with the more reactive methylene group in ethyl cyanoacetate rather than with the less reactive methyl group in acetophenone derivatives 2 to produce cyano intermediates. The Michael addition of compound 2 on the produced cyano intermediates takes place, followed by the replacement of enolic OH by NH2, cyclization, elimination of ethanol, and finally dehydrogenation to produce the tautomer structures of compounds 3.
Figure 3.

Mechanism of the one-pot four-component reaction involved in the synthesis of cyanopyridinone derivative 3.
Spectroscopic techniques and elemental analyses were employed to secure the structures of the carbonitrile derivative 3. The IR spectra of compounds 3 showed absorption bands at 3370–3449 cm–1 representing the NH group and bands assignable for the nitrile (CN) stretching vibration absorption at 2215–2224 cm–1. Additionally, sharp and strong absorption bands in the range of 1648–1652 cm–1 were observed in the IR spectra, which were attributable to the carbonyl group. The relatively lower values of the carbonyl stretching absorptions than those of the typical carbonyl stretching may be due to the single-bond character of the tautomeric enol form, leading to lower absorption frequency. The 1H NMR spectra of compounds 3 confirmed the presence of their keto–enol (lactam–lactim) tautomer mixtures, which comprise predominantly the keto form in different ratios over the enol form. For example, two singlet peaks resonated at δ 6.97 and δ 8.27 ppm, in the1H NMR spectrum of compound 3c, representing pyridinone-H and pyridine-H in 92:8 ratio, respectively. This feature was also supported by the presence of two D2O exchangeable singlets at δ 3.97 and δ 12.21 ppm corresponding to NH and OH protons, respectively, in the 1H NMR spectrum of compound 3c.
2.1.2. Synthesis of Compounds 4
Deoxychlorination of the cyanopyridinone derivatives 3a–e was achieved by reaction with phosphorous oxychloride (POCl3) under solvent-free and reflux conditions to afford the corresponding 2-chloro-cyanopyridine derivatives 4a–e according to the suggested mechanism illustrated in Figure 4.32
Figure 4.

Mechanism of the formation of 2-chloro-cyanopyridine derivatives 4.
The structures of compounds 4 were substantiated from their elemental and spectral analyses. The characteristic feature is the absence of absorption bands and peaks characteristic for NH, CO, and OH groups of compounds 3 in their IR and 1H NMR spectra, respectively. The 1H NMR spectrum of compound 4e was characterized by the presence of the pyridine proton 5′-H that resonated completely (100%) at δ 8.83 ppm as a singlet peak, indicating aromatization of the pyridone ring and the absence of the keto–enol isomers. The structure was additionally confirmed by 13C NMR spectroscopy that showed signals, which are determined to be in accordance with the recommended molecular structure. In mass spectrometry, for example, 4b chart, there are three consecutive peaks (Figure S13), (390.8≈391), (392.9≈393), and 395, and their relative intensity is 10:6:1; this is sharp evidence that we have a dichlorinated compound.
2.2. Biological Screening
2.2.1. In Vitro Cytotoxicity Assay
The in vitro cytotoxicity study of the newly synthesized compounds was performed on four different cell lines, namely, HepG2, MCF-7, PC3, and HCT-116 cell lines by employing MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay using 5-FU as a standard antitumor agent. The tested compounds exhibited different degrees of cytotoxic activity against the tested cell lines, and the results were displayed as IC50 (Table 1).
Table 1. In Vitro Cytotoxic Activity of the Designed Compounds.
| IC50 (μM)a |
||||
|---|---|---|---|---|
| comp. no | HePG2b | MCF-7c | PC3d | HCT-116e |
| 5-FUf | 9.42 ± 0.46 | 7.75 ± 0.37 | 10.34 ± 0.50 | 8.01 ± 0.39 |
| 3a | 11.64 ± 0.57 | 60.03 ± 2.93 | 26.91 ± 1.28 | 49.92 ± 2.36 |
| 3b | 12.36 ± 0.59 | 42.36 ± 2.13 | 18.36 ± 0.89 | 30.86 ± 1.52 |
| 3c | 14.12 ± 0.68 | 25.38 ± 1.20 | 25.80 ± 1.30 | 18.30 ± 0.89 |
| 3d | 16.21 ± 0.77 | 19.88 ± 0.96 | 20.49 ± 0.97 | 20.02 ± 0.99 |
| 3e | 11.60 ± 0.57 | 45.94 ± 2.18 | 25.87 ± 1.25 | 50.74 ± 2.50 |
| 4a | 10.01 ± 0.49 | 75.68 ± 3.74 | 35.27 ± 1.67 | 50.02 ± 2.44 |
| 4b | 11.95 ± 0.59 | 19.18 ± 0.94 | 15.00 ± 0.74 | 20.19 ± 0.96 |
| 4c | 8.02 ± 0.38 | 15.74 ± 0.78 | 13.64 ± 0.67 | 7.15 ± 0.35 |
| 4d | 6.95 ± 0.34 | 8.50 ± 0.42 | 14.08 ± 0.70 | 8.35 ± 0.42 |
| 4e | 11.50 ± 0.56 | 55.48 ± 2.71 | 35.08 ± 1.73 | 65.09 ± 3.08 |
IC50 (μM): Expressed as mean ± S.D. 1–10 (very strong), 11–20 (strong), 21–50 (moderate), 51–100 (weak), and above 100 (noncytotoxic).
Human hepatocellular carcinoma cell line (HepG2).
Human breast adenocarcinoma cell line (MCF-7).
Human prostate cancer cell line (PC3).
Human colorectal carcinoma cell line (HCT-116).
5-FU: 5-fluorouracil.
Generally, most of the tested compounds exhibited promising cytotoxic activity against the examined cancer cell lines, especially against the HepG-2 cell line and with the cyanopyridine series (4). With respect to the HepG-2 cell line, a significant increase in the cytotoxicity of the cyanopyridine derivatives (4) was noticed in comparison with the activity of the corresponding cyanopyridones (3) (Table 1). Also, the cyanopyridine derivatives bearing 3-aminophenyl (4b), 4-methoxyphenyl (4c), and 4-bromophenyl (4d) at position 6 of the pyridine ring displayed more cytotoxicity against MCF-7, PC3, and HCT-116 cell lines than that of their corresponding cyanopyridone derivatives 3b, 3c, and 3d. However, the cyanopyridone derivatives bearing unsubstituted phenyl (3a) and 2,4-dichlorophenyl (3e) showed more cytotoxicity than that of their corresponding cyanopyridines 4a and 4e, respectively, against MCF-7, PC3, and HCT-116 cell lines. Compounds 4b, 4c, and 4d displayed significant and broad spectrum cytotoxic activity against the four examined cancer cell lines HepG2, MCF-7, PC3, and HCT-116, with IC50 values ranging from 6.95 ± 0.34 to 20.19 ± 0.96 μM. Compound 4c exhibited cytotoxicity against the HepG2 cell line with an IC50 value of 8.02 ± 0.38 μM, which is better than that of the reference drug 5-FU (IC50 = 9.42 ± 0.46 μM). Also, compound 4c showed selective activity against the HCT-116 cell line with an IC50 value of 7.15 ± 0.35 μM, which is better than that of the reference drug and the tested designed compounds. In addition, compound 4c showed the highest activity against the PC3 cell line and very strong activity against the MCF-7 cell line with IC50 values of 13.64 ± 0.67 and 15.74 ± 0.78 μM, respectively. Concerning the activity of the tested compounds against the HepG-2 cell line, compound 4d displayed the highest and selective activity, which exceeded that of 5-FU, as it showed an IC50 value of 6.95 ± 0.34 μM. However, it exerted approximate equipotent activity to that of 5-FU (IC50 = 8.01 ± 0.39 μM) against HCT-116 with an IC50 value of 8.35 ± 0.42 μM. In addition, compound 4d showed a remarkable effect on the viability of MCF-7 and PC3 cell lines with IC50 values of 8.50 ± 0.42 and 14.08 ± 0.70 μM, respectively, and was identified as the most active among the tested compounds against the MCF-7 cell line.
2.2.2. Pim-1 Kinase Inhibition Assay
To investigate whether the antitumor activity of the designed compounds was related to inhibition of Pim-1 kinase, a luminescent kinase assay was performed. The 10 new synthesized compounds were evaluated using the flavonol quercetagetin (3,3′,4′,5,6,7-hexahydroxyflavone) as a positive control and compound A as a template of 4,6-diaryl-3-cyanopyridine reported as a Pim-1 kinase inhibitor. IC50 values of the tested compounds were calculated and are listed in Table 2 and graphically represented in Figure 5. All the tested cyanopyridines 4, except compound 4a, showed higher inhibition of Pim-1 kinase than that of their corresponding cyanopyridone derivatives 3. Compounds 3a–e showed a certain level of inhibition of Pim-1 kinase with IC50 values ranging from 2.31 ± 0.11 to 0.72 ± 0.03 μM and are less potent than the reference drug quercetagetin (IC50 = 0.56 ± 0.03 μM). However, compounds 3a–d displayed more inhibitory activity than that of the reported compound A (IC50 = 0.93 ± 0.05 μM). Regarding the inhibitory activity of cyanopyridine series, compounds 4a and 4e are about 2- and 2.5-fold less potent than quercetagetin, respectively, and showed comparable activity with A. Compounds 4b and 4c displayed approximate equipotent inhibitory activity to that of quercetagetin in a sub-micromolar range, IC50 = 0.63 ± 0.03 and 0.61 ± 0.03 μM, respectively. In addition, the later compounds showed inhibitory activity, which exceeded that of A. Compound 4d with IC50 value = 0.46 ± 0.02 μM showed the highest and remarkable Pim-1 kinase inhibitory activity among all the tested compounds, which is better than that of quercetagetin (IC50 = 0.56 ± 0.03 μM) and 2 times more potent than A (IC50 = 0.93 ± 0.05 μM).
Table 2. Results of the In Vitro Pim-1 Inhibition Assay and Docking Interaction Energy.
| comp. no | IC50 (μM),a Pim-1 inhibition | docking interaction energy (kcal/mol), PDB ID: 2OBJ | docking interaction energy (kcal/mol), PDB ID: 2O64 |
|---|---|---|---|
| quercetagetin | 0.56 ± 0.03 | NTb | –14.53 |
| compound A | 0.93 ± 0.05 | –10.67 | NTb |
| 3a | 0.90 ± 0.04 | –11.68 | –11.60 |
| 3b | 0.85 ± 0.04 | –11.57 | –11.97 |
| 3c | 0.79 ± 0.04 | –11.16 | –11.80 |
| 3d | 0.72 ± 0.03 | –11.37 | –11.03 |
| 3e | 2.31 ± 0.11 | –11.48 | –11.08 |
| 4a | 1.24 ± 0.06 | –11.71 | –11.20 |
| 4b | 0.63 ± 0.03 | –11.64 | –11.81 |
| 4c | 0.61 ± 0.03 | –12.01 | –12.23 |
| 4d | 0.46 ± 0.02 | –12.04 | –12.43 |
| 4e | 1.42 ± 0.07 | –11.58 | –11.25 |
IC50 (μM): expressed as mean ± S.D.
NT: not tested.
Figure 5.

Pim-1 kinase inhibition chart of the tested compounds versus A and quercetagetin (reference drug) expressed as IC50 (uM).
2.3. Molecular Modeling Simulation
Molecular docking as a computational procedure was conducted to understand the binding efficiency of a ligand to its macromolecular target (receptor). In consequence of that all our newly synthesized compounds belong to the organic class of phenyl pyridines, which are polycyclic aromatic compounds that contain a benzene ring linked to a pyridine ring via a C–C bond at position 6, we have chosen to compare them with compound A as a standard Pim-1 inhibitor from the same class. Herein, molecular docking was carried out for all the newly synthesized compounds onto the binding site of pyridin-2(1H)-one-based A (VRV), internal poison, of the crystal structure of human Pim-1 kinase (PDB: 2OBJ) as illustrated in Figure 6.
Figure 6.

2D and 3D interactions
of 4c (upper panel), compounds 4b (middle
panel), and compound A (lower panel)
with the VRV binding site of the crystal structure of human Pim-1
kinase (PDB: 2OBJ).16 polar,
polar,  greasy,
greasy,  backbone acceptor,
backbone acceptor,  ligand exposure,
ligand exposure,  acidic,
acidic,  side chain acceptor,
side chain acceptor,  backbone donor,
backbone donor,  receptor exposure,
receptor exposure,  basic,
basic,  side chain donor, and
side chain donor, and  arene–H bond.
arene–H bond.
Docking results showed that compounds 4c and 4d scored the highest free binding energies to the active site of the receptor with −12.0116577 and −12.0388889 kcal/mol, respectively. In the case of the compound 4c binding mode, both the p-methoxylated phenyl and pyridine ring as aromatic systems acted as nonclassical Lewis bases and formed arene–H bonds with the conserved amino acids Ile185 and Val52, respectively. Moreover, the Cl atom at position 2 of the pyridine ring constructed a halogen bond with Glu121, while the sp-hybridized nitrogen in the cyano group at position 3 of pyridine acted as a H-bond acceptor and built up a H-bond with the H-bond donor Pro123. Furthermore, the cyan shadow of the conserved amino acids Leu44, Val52, Val126, Leu174, and Ile185 from the receptor side indicated strong hydrophobic/hydrophilic interactions with the blue-shadowed methyl of the p-methoxy group and fused benzene ring of the quinolin-3-yl moiety from the ligand side.
While docking results of compound 4d revealed two different types of bifurcated bonds, the Cl atom at position 2 of the quinolin-3-yl moiety formed a bifurcated halogen bond with Asn172 and Asp186, whereas the nitril nitrogen constructed a bifurcated H-bond with Gly47 and Phe49. Additionally, the H-acceptor nitrogen of the pyridine ring in the quinolin-3-yl moiety participated in H-bond formation with the H-donor Lys169. Eventually, the hydrophobic/hydrophilic interactions appeared from the cyan shadow of the conserved amino acids from the receptor side and deep blue shadow of all ligand moieties, improved the overall recognition, and enhanced the receptor/ligand complex stability.
On the other hand, our newly synthesized compounds comprise a quinoline moiety (benzopyridine) and quercetagetin, the standard Pim-1 inhibitor, is a member of the benzopyran class, so these both fused heterocycles are isosters. Due to structural similarity, quinolines versus quercetagetin as Pim-1 inhibitors had been investigated before.33 Hence, we have docked all the newly synthesized compounds onto the quercetagetin-binding site of the crystal structure of Pim-1 (PDB: 2O64),34 and surprisingly, the same aforementioned two compounds 4c and 4d scored the highest free binding energies.
As shown in Figure 7, compound 4c interacted with the active site of the receptor via its Cl atom at position 2 of the pyridine ring, which formed a halogen bond with Glu121, and its sp-hybridized nitrogen atom of the cyano group at position 3 of the pyridine ring that established a bifurcated H-bond with the conserved amino acids Arg122 and Pro123. The stability of the receptor/ligand complex has scored −12.234746 kcal/mol. However, compound 4d featured miscellaneous and versatile bonding patterns. The two pyridine rings of the ligand shared interactions with the pocket of the receptor, as they acted as nonclassical Lewis bases; the pyridine of cyanopyridine constructed an arene–H bond with the H-donor Val52, while the pyridine of the quinolin-3-yl moiety built up an arene–arene bond with the aromatic amino acid Phe49. Also, the two halogens of the ligand displayed a characteristic role in the interaction with the receptor pocket; Br at the p-position formed a halogen bond with Pro123, while the Cl atom at position 2 of the quinolin-3-yl moiety constructed a bifurcated halogen bond with Asn172 and Asp186. Besides, the nitril nitrogen formed a H-bond with the H-donor Lys67 that increased the stability of the receptor/ligand complex to a score of −12.4264545 kcal/mol.
Figure 7.

2D and 3D interactions
of 4c (upper panel), compounds 4d (middle
panel), and quercetagetin (lower panel) with the
quercetagetin-binding site of the crystal structure of Pim-1 (PDB: 2O64).34 polar,
polar,  greasy,
greasy,  backbone acceptor,
backbone acceptor,  ligand exposure,
ligand exposure,  acidic,
acidic,  side chain acceptor,
side chain acceptor,  backbone donor,
backbone donor,  receptor exposure,
receptor exposure,  basic,
basic,  side chain donor, and
side chain donor, and  arene–H bond.
arene–H bond.
Of note, upon docking of our new compounds on human Pim-1 kinase (PDB: 2OBJ), the CN group at position 3 of the pyridin-2(1H)-one and/or pyridine rings played an indispensable role in fixing the ligand inside the active site of the receptor, as it was found to be involved in H-bond formation in the entire set of the docked compounds. Besides, the pyridine ring of cyanopyridin-2(1H)-one and/or cyanopyridine contributed significantly in the pocket/ligand interactions whether via its steric effect or by formation of arene–H or arene–arene bonds, through acting as a nonclassical Lewis base. However, replacement of 2-oxo with 2-chloro at the pyridine ring was found to be very advantageous, as all members of the series 4a–e had scored higher free binding energies rather than their counterparts in the series 3a–e. This can be attributed to the big leap toward lipophilicity in the series 4a–e based on aromatization of the pyridine ring and substitution with a bigger steric Cl atom than the oxygen atom and higher affinity of Cl to be involved in hydrophobic/hydrophilic interactions as shown in 4a, 4b, and 4d poses. Likewise, substitution of the phenyl ring as in the case of compound A at position 4 of pyridine in cyanopyridin-2(1H)-one and/or cyanopyridine rings, with the quinoline moiety, was found to be very gainful; this finding confirms and agrees with the concept that the quinoline moiety has a noteworthy Pim-1 inhibition activity.28,29 Eventually, the substitution on the benzene ring at position 6 of the pyridine ring of cyanopyridin-2(1H)-one and/or cyanopyridine noticeably affects the free binding energies of the ligands (4a–e) into the receptor pocket; unsubstitution, as in 4a, was found to be better than 2,4-disubtitution with Cl (4e). Undeniably, 4d, 4c, and 4b are in the decreasing order of lipophilicity, and p-substitution with the steric atom Br with its three lone pairs of electrons (4d), acting as a nonclassical Lewis base, displayed the best binding energy rather than 4c and 4b with p-methoxy and m-amino substitution, respectively.
2.4. Radiolabeling Study
2.4.1. Radiolabeling of Compound 4b
The potent cytotoxic and Pim-1 kinase inhibitors 4b–d were chosen to be radioiodinated via electrophilic substitution using radioiodine-131 and chloramine-T (CAT) as an oxidizing agent. Out of the tested compounds, compound 4b showed the highest radiochemical yield (131I-4b, 86%), and hence, 131I-4b has been chosen for further analysis.
2.4.2. Radiochemical Analysis of 131I-4b
The radiochemical yield was determined using both paper chromatography and paper electrophoresis techniques. The effect of various parameters and conditions on radiolabeling efficiency, such as the amount of the oxidizing agent (CAT), amount of the substrate, pH of the reaction, and reaction time, were studied to maximize the radiochemical yield.
2.4.2.1. Paper Chromatography
131I-4b moved with the mobile phase to Rf 0.8–1, while free iodide stayed near the point of spotting with Rf 0.0–0.1.
2.4.2.2. Paper Electrophoresis
131I-4b was detected near the point of spotting, while free iodide moved toward the anode, Figure 8. The percentage of radiochemical yield was calculated according to the following equation:35,36 % radiochemical yield = fraction of activity/total activity × 100.
Figure 8.

Electrophoresis analysis of 131I-4b.
2.4.3. Factors Affecting the Labeling Yield
2.4.3.1. Effect of the CAT Content on the Radiochemical Yield of 131I-4b
It was observed that the radiochemical yield of 131I-4b increased with increasing CAT content from 50 μg till it reached the maximum yield at 500 μg. Above 500 μg of CAT, the radiochemical yield of 131I-4b was slightly decreased, Figure 9A.
Figure 9.
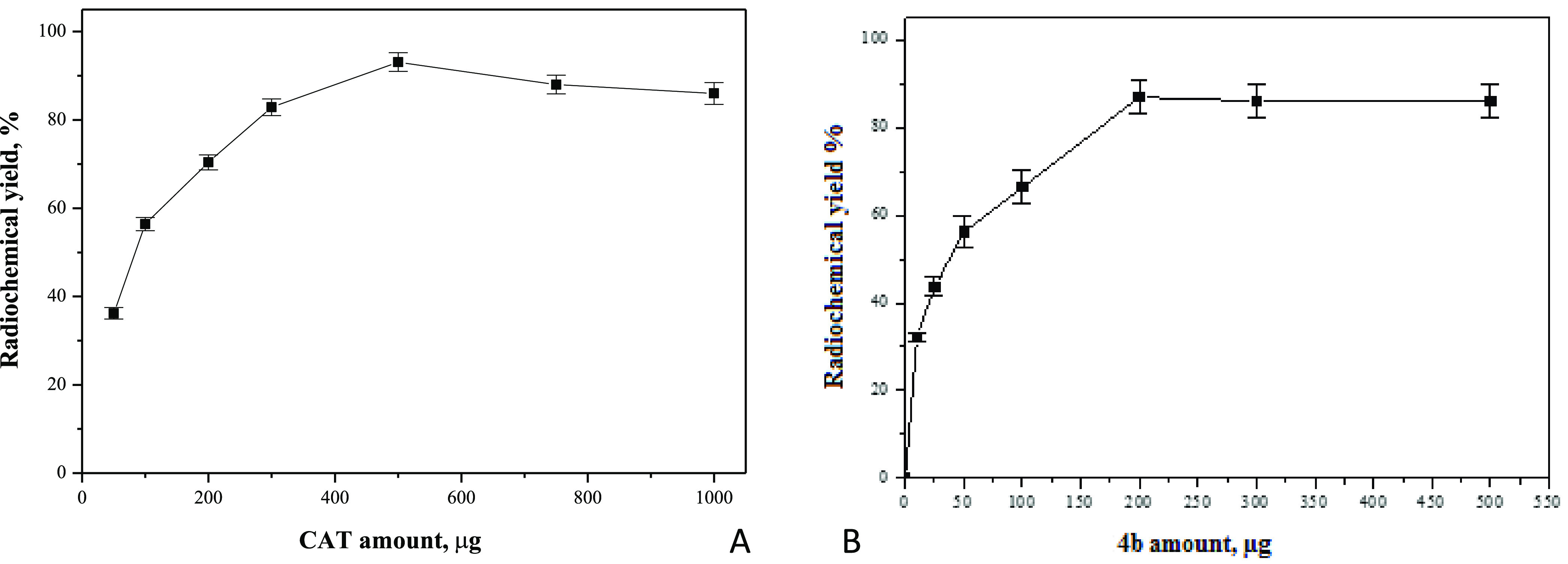
(A) Effect of the CAT content on the radiochemical yield of 131I-4b. (B) Effect of the substrate amount on the radiochemical yield of 131I-4b.
2.4.3.2. Effect of the Substrate Amount
200 μg of compound 4b was the optimum amount to get the maximum labeling yield; below 200 μg, the yield was decreased, while above 200 μg, the yield was not affected (Figure 9B).
2.4.3.3. Effect of Reaction Time on the Radiochemical Yield of 131I-4b
Reaction time was an essential factor in the labeling procedures, as below the optimum time, there was an incomplete labeling and with time, the yield was improved and reached 93.1% at 1 h. By extending the time to more than 60 min, the yield was decreased, Figure 10A.
Figure 10.

(A) Effect of reaction time on the radiochemical yield of 131I-4b. (B) Effect of pH on the radiochemical yield of 131I-4b.
2.4.3.4. Effect of pH on the Radiochemical Yield of 131I-4b
The effect of pH of reaction media on the labeling yield is presented in Figure 10B. pH 4 was the optimum medium for labeling, and at pH 3, the yield was 61.8%. However, at pH above 5, the yield was decreased to 74.6, 60.4, and 50.6% at pH 6.5, 8, and 10, respectively.
2.4.3.5. Impact of Time on the In Vitro Stability of 131I-4b
The labeled compound was stable up to 12 h post-labeling; then, the yield decreased to 80% at the subsequent 12 h, Table 3.
Table 3. In Vitro Stability of 131I-4ba.
| time, h | labeled compound, % | free iodide, % |
|---|---|---|
| 1 | 97.4 ± 2.3 | 2.6 ± 0.5 |
| 2 | 95.4 ± 2.5 | 4.6 ± 0.3 |
| 4 | 93.2 ± 2.3 | 6.8 ± 0.4 |
| 12 | 93.1± 2.1 | 6.9 ± 0.5 |
| 24 | 80.4 ± 2.4 | 13.6 ± 0.8 |
Values expressed as mean ± SEM, n = 3.
2.4.4. Biodistribution Study of 131I-4b
2.4.4.1. In Normal Mice
131I-4b was distributed in the blood, liver, stomach, and intestine at 30 min post-injection at 7.1, 5.4, 12.3, and 5.1, respectively. At 1 h post-injection, 131I-4b uptake was increased in the stomach, lungs, and kidneys; at 2 and 4 h post-injection, most tissues and organs showed a decrease in the 131I-4b uptake except the urine and thyroid, Table 4. A rapid uptake was observed in the stomach, which may be due to the high proliferation rate in this organ.
Table 4. Biodistribution of 131I-4b in Normal Micea.
| injected dose/gram tissue at different time intervals, % |
||||
|---|---|---|---|---|
| organs and body fluids | 1/2 h | 1 h | 2 h | 4 h |
| blood | 7.1 ± 0.5b | 6.9 ± 0.3b | 4.4 ± 0.3b | 1.8 ± 0.3b |
| bone | 1.1 ± 0.03b | 1.2 ± 0.02b | 0.8 ± 0.7b | 0.8 ± 0.02b |
| liver | 5.4 ± 0.5b | 6.9 ± 0.4b | 8.4 ± 0.7 | 2.7 ± 0.02 |
| stomach | 12.3 ± 1.2 | 16.0 ± 1.6b | 14.1 ± 1.2b | 10.2 ± 0.9b |
| intestine | 5.1 ± 0.3b | 4.4 ± 0.3b | 3.1 ± 0.3b | 2.9 ± 0.2b |
| lung | 3.9 ± 0.3b | 6.4 ± 0.6b | 4.6 ± 0.5b | 3.2 ± 0.2b |
| heart | 5.2 ± 0.3b | 5.0 ± 0.4b | 3.6 ± 0.3b | 1.7 ± 0.01b |
| spleen | 3.1 ± 0.2b | 2.7 ± 0.2 | 2.3 ± 0.2b | 1.1 ± 0.02 |
| kidney | 3.9 ± 0.4b | 6.2 ± 0.5b | 5.8 ± 0.6b | 4.5 ± 0.3b |
| urine | 5.1 ± 0.6b | 7.5 ± 0.5b | 14.3 ± 1.2b | 18.1 ± 1.3b |
| brain | 0.4 ± 0.06b | 0.5 ± 0.03b | 0.6 ± 0.06b | 0.7 ± 0.03b |
| thyroid | 2.7 ± 0.2b | 4.8 ± 0.4 | 7.6 ± 0.6b | 9.4 ± 1.2 |
| normal muscle | 1.4 ±0.07b | 1.6 ± 0.08 | 1.5 ± 0.05b | 1.9 ± 0.08 |
Values expressed as mean ± SEM, n = 3.
Significantly different from each previous value of each organ using unpaired Student’s t-test (P ≤ 0.05).
2.4.4.2. In Solid Tumor-Bearing Mice
Data of biodistribution of 131I-4b in solid tumor-bearing mice are represented in Table 5. Tumor uptake of 131I-4b in the tumor muscle was observed and was about 6.2 and 7.1 times that in normal muscle at 1 and 2 h post-injection, respectively. This refers to the orientation of the 131I-4b to the tumor site with a marked extent that was enough to distinguish between the tumor and normal muscle. Thyroid uptake increased with time, which may be due to the dehalogenation metabolism in the liver. The T/NT ratio showed a marked increase of 131I-4b in the tumor muscle than in the normal one. The T/NT ratio was 3.2 ± 0.2 at 30 min, which increased with time till it reached 6.2 ± 0.5 at 60 min post-injection. The uptake of 131I-4b was higher than that of recently developed radiopharmaceuticals.36−38
Table 5. Biodistribution of 131I-4b in Solid Tumor-Bearing Micea.
| injected dose/gram tissue at different time intervals, % |
||||
|---|---|---|---|---|
| organs and body fluids | 1/2 h | 1 h | 2 h | 4 h |
| blood | 7.1 ± 0.5c | 6.1 ± 0.4c | 4.0 ± 0.3c | 1.5 ± 0.03c |
| bone | 1.1 ± 0.03c | 0.9 ± 0.02c | 0.6 ± 0.01c | 0.8 ± 0.02c |
| liver | 5.5 ± 0.5c | 6.2 ± 0.4c | 8.0 ± 0.7 | 2.1 ± 0.2 |
| stomach | 13.3 ± 1.4 | 15.0 ± 1.3c | 14.1 ± 1.2c | 10.2 ± 1.1c |
| intestine | 5.5 ± 0.3c | 4.5 ± 0.2c | 3.3 ± 0.3c | 2.8 ± 0.2c |
| lung | 3.8 ± 0.3c | 11.6 ± 1.1c | 6.3 ± 0.5c | 3.1 ± 0.2c |
| heart | 5.6 ± 0.3c | 4.0 ± 0.3c | 3.6 ± 0.3c | 1.5 ± 0.01c |
| spleen | 3.3 ± 0.1c | 2.5 ± 0.02 | 2.3 ± 0.05c | 1.1 ± 0.02 |
| kidney | 3.6 ± 0.3c | 4.6 ± 0.3c | 6.6 ± 0.6c | 2.5 ± 0.3c |
| urine | 3.4 ± 0.4c | 7.5 ± 0.5c | 11.3 ± 1.1c | 14.1 ± 1.2c |
| brain | 0.3 ± 0.02c | 0.4 ± 0.03c | 0.6 ± 0.06c | 0.7 ± 0.03c |
| thyroid | 3.7 ± 0.5c | 5.8 ± 0.5 | 7.6 ± 0.6c | 9.4 ± 0.8 |
| normal muscle | 1.1 ± 0.04c | 1.2 ± 0.05 | 1.4 ± 0.09c | 1.3 ± 0.07 |
| tumor muscle | 3.6 ± 0.3c | 7.5 ± 0.6 | 10.0 ± 1.1c | 3.1 ± 0.2 |
| T/NT ratiob | 3.2 ± 0.2c | 6.2 ± 0.5 | 7.1 ± 0.6c | 2.4 ± 0.2 |
| T/blood ratio | 0.5 ± 0.03c | 1.2 ± 0.02 | 2.5 ± 0.3c | 2.1 ± 0.2 |
Values expressed as mean ± SEM, n = 3.
T/NT: tumored (muscle)/nontumored (muscle).
Significantly different from each previous value of each organ using unpaired Student’s t-test (P ≤ 0.05).
3. Conclusions
Two series of cyanopyridine-based compounds have been designed, synthesized, characterized, and screened for their cytotoxicity and Pim-1 kinase inhibition activity. The cytotoxic activity of most of the tested compounds belonging to the cyanopyridine series 4 is higher than that of their corresponding cyanopyridones 3. Compounds 4c and 4d with OCH3 or Br substituents at the p-position of the phenyl ring, respectively, showed remarkable cytotoxicity against HepG2 and HCT-116 cancer cell lines with IC50 values ranging from 6.95 ± 0.34 to 8.35 ± 0.42 μM, which is superior to that of the reference drug 5-FU and comparable in the case of compound 4d against the HCT-116 cell line. Also, compound 4b with an amino substituent at the m-position of the phenyl ring showed strong activity against the four tested cancer cell lines but lesser than that of 4c and 4d. Meanwhile, the overall cytotoxic activity of compound 4b was greater than that of compounds with unsubstituted and disubstituted phenyl rings as proved from the IC50 values. The in vitro Pim-1 kinase assay was carried out to explore the Pim-1 inhibitory activity of the targeted compounds and validate the design of this study. Compounds 4b, 4c, and 4d elicited remarkable inhibition of Pim-1 kinase with IC50 values ranging from 0.46 ± 0.02 to 0.63 ± 0.03 μM in comparison with both quercetagetin (IC50 = 0.56 ± 0.03 μM) and the reported Pim-1 kinase inhibitor A (IC50 = 0.93 ± 0.05 μM) with good agreement with the biological results. From docking results, it was observed that the series 4a–e with enhanced lipophilicity by aromatization of the pyridine ring and substitution of 2-oxo with 2-chloro showed better binding profiles and energies than those of 3a–e. Moreover, p-substitution at the phenyl ring at position 6 of pyridine, whether with the Br or methoxy group, have remarkably enhanced the free binding energies of 4d (−12.04 kcal/mol) and 4c (−12.01 kcal/mol), respectively. Consequently, compounds 4c and 4d can serve as lead compounds for optimization to speed up the development of drugs targeting Pim-1 kinase. Compound 4b was successfully radiolabeled with radioiodine-131, and 123I-4b was found to biodistributed and highly localized in tumor sites, which was considered as an ideal vector to carry radioiodine to the nucleus of tumor cells and facilitates tumor imaging and diagnosis, especially in the stomach. These results encourage further application to evaluate this radiolabeled compound 123I-4bin vivo and in vitro on cancer cell lines.
4. Experimental Section
4.1. Chemistry
All chemicals and solvents were reagent grade and were used without further purification. Melting points (°C) were recorded using Stuart melting point apparatus and are uncorrected. IR spectra were recorded on a Shimadzu IR-470 spectrometer (υ′ in cm–1) using a KBr disk at the Faculty of Pharmacy, Mansoura University, Egypt. 1H NMR and 13C NMR spectra were recorded on a Jeol spectrometer at 500 and 125 MHz, respectively, in DMSO-d6 with TMS as an internal standard, at the Faculty of Science, Mansoura University, Egypt, and a Bruker spectrometer at 400 and 100 MHz, respectively, in DMSO-d6 with TMS as an internal standard, at the Faculty of Pharmacy, Mansoura University, Egypt. MS analyses were performed on a 1260 Infinity II Prime LC system coupled to the LC/MSD iQ. (a single quadrupole mass spectrometer), at the microanalytical center, Faculty of Science, Mansoura University, Egypt. Elemental analysis was carried out for C, H, and N at the Microanalytical Centre of Cairo University, and it agreed with the proposed structures within ±0.4% of the calculated values. Column chromatography was carried out on silica gel G60 (70–230 mesh, ASTM; Merck and 230–400 mesh, Silicycle Inc.). The completion of reactions was monitored using thin-layer chromatography (TLC) on silica gel G60 F-245 (Merck), and the spots were visualized using UV (366, 245 nm). 2-Chloroquinoline-3-carbaldehyde (1) was prepared according to the previous report,39 and 4-(2-chloroquinolin-3-yl)-6-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (3c) was synthesized as reported, (reported m.p. >300 °C).40
4.1.1. Procedure for the Synthesis of 6-((Un)substituted phenyl)-4-(2-chloroquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitriles (3a–e)
A mixture of the appropriate acetophenone derivative (2a–e) (1 mmol) and ammonium acetate (8 mmol, 0.617g) in ethanol (30 mL) was stirred at room temperature for 10 min. Then, 2-chloroquinoline-3-carbaldehyde (1) (1 mmol, 0.191g) and ethyl cyanoacetate (1 mmol, 0.113 g) were added. The reaction mixture was heated under reflux for 12 h and then allowed to cool. The solvent was evaporated in vacuo to give a yellow residue, which was then purified by column chromatography using an n-hexane/ethylacetate (6.5:3.5) solvent system to afford the titled compounds 3a–e.
4.1.1.1. 4-(2-Chloroquinolin-3-yl)-2-oxo-6-phenyl-1,2-dihydropyridine-3-carbonitrile (3a)
Pale-yellow crystals (EtOH), m.p. 292–294 °C, yield (68%), IR (KBr, cm–1): 1580 (ArC=C), 1652 (C=O), 2221 (C≡N), 3425 (OH), 3448 (NH). 1H NMR (500 MHz, DMSO-d6); δ 7.08 (s, 1H, pyridinone-H, 75%), 7.31 (t, 1H, J = 7.4 Hz, 3″-H), 7.42 (d, 1H, J = 7.4 Hz, 4″-H), 7.62 (dd, 3H, J = 21.2, J =11.0 Hz, 5″-H), 7.82 (t, 1H, J = 7.2 Hz, 6-H), 7.99 (m, 3H, 2″-H, 6″-H, 7-H), 8.13 (d, 1H, J = 8.4 Hz, 8-H), 8.19 (d, 1H, J = 8.0 Hz, 5-H), 8.35 (s, 1H, pyridine-H, 25%), 8.79 (s, 1H, 4-H), 11.42 (s, 1H, NH exchangeable by D2O), 12.23 (s, 1H, OH exchangeable by D2O). 13C NMR (125 MHz, DMSO-d6); δ 105.2(C-5′), 116.3(C-CN), 121.5(C-3), 126.3(C-4a), 127.2(C-6), 127.6(C-8), 127.7(C-4″), 128.3(C-5), 128.4(C-2″, C-6″), 128.9(C-3″, C-5″), 130.8(C-3), 131.1(C-7), 131.5(C-1″), 136.1(C-4), 146.9(C-8a), 149.5(C-2), 159.7(C-2′), 160.5(C-6′), 166.8(C-4′). Anal. Calc. for C21H12ClN3O calculated %; C: 70.50, H: 3.38, N: 11.74. Found %; C: 70.49, H: 3.40, N: 11.72.
4.1.1.2. 6-(3-Aminophenyl)-4-(2-chloroquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (3b)
Yellow crystals (EtOH), m.p. 294–296 °C, yield (83%), IR (KBr, cm–1): (1588 (ArC=C), 1649 (C=O), 2220 (C≡N), 3370 (NH), 3460 (NH2,). 1H NMR (500 MHz, DMSO-d6); δ 1.04 (s, 1H, NH exchangeable by D2O), 5.37 (s, 2H, NH2 exchangeable by D2O), 6.74 (d, 1H, J = 7.6 Hz, 6″-H), 6.84 (s, 1H, pyridinone-H, 90%), 6.90–7.03 (m, 2H, 2″-H, 4″-H), 7.16 (t, 1H, J = 7.7 Hz, 5″-H), 8.28 (s, 1H, pyridine-H, 10%), 7.76 (t, 1H, J = 7.3 Hz, 6-H), 7.94 (t, 1H, J = 7.5 Hz, 7-H), 8.07 (d, 1H, J = 8.4 Hz, 8-H), 8.12 (d, 1H, J = 8.0 Hz, 5-H), 8.72 (s, 1H, 4-H), 12.22 (s, 1H, OH exchangeable by D2O).). 13C NMR (125 MHz, DMSO-d6); δ 105.1(C-5′), 111.1(C-2″), 114.5(C-5″), 116.1(C-CN), 118.5(C-6″), 121.4(C-3′), 126.6(C-4a), 127.4(C-6, C-8), 128.3(C-5), 130.7(C-3), 131.0(C-7), 134.1(C-5″), 135.3(C-1″), 135.9(C-4), 147.1(C-8a), 148.5(C-3″), 149.5(C-2), 159.5(C-2′), 160.3(C-6′), 166.7(C-4′). Anal. Calc. for C21H13ClN4O calculated %; C: 67.66, H: 3.51, N: 15.03. Found %; C: 67.65, H: 3.53, N: 15.02.
4.1.1.3. 4-(2-Chloroquinolin-3-yl)-6-(4-methoxyphenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (3c)
Canary-yellow crystals (EtOH), m.p. 297–299 °C, yield (73%),IR (KBr, cm–1): 1544 (ArC=C), 1648 (C=O), 2215 (C≡N), 2937 (C–H aliphatic), 3404 (NH). 1H NMR (500 MHz, DMSO-d6); δ 3.83 (s, 3H, 4″-CH3), 3.97 (s, 1H, NH exchangeable by D2O), 6.97 (s, 1H, pyridinone-H, 92%), 7.08 (d, 2H, J = 8.9 Hz, 3″-H, 5″-H), 7.76 (dd, 1H, J = 11.4 Hz, J = 4.3 Hz, 6-H), 7.86–7.93 (m, 2H, 2″-H, 6″-H), 7.95 (dd, 1H, J = 8.5 Hz, J = 1.0 Hz, 7-H), 8.07 (d, 1H, J = 8.5 Hz, 8-H), 8.13 (d, 1H, J = 7.8 Hz, 5-H), 8.27 (s, 1H, pyridine-H, 8%), 8.72 (s, 1H, 4-H), 12.21 (s, 1H, OH exchangeable by D2O). 13C NMR (125 MHz, DMSO-d6); δ 55.5(C-CH3), 105.2(C-5′), 116.0(C-CN), 121.3(C-3″, C-5″), 121.6(C-3′), 123.3(C-1″), 126.7(C-4a), 127.6(C-6), 127.9(C-8), 128.2(C-5), 130.0(C-2″, C-6″), 130.7(C-3), 131.2(C-7), 135.9(C-4), 146.9(C-8a), 149.5(C-2), 159.7(C-2′, C-4″), 160.4(C-6″), 166.7(C-4′). Anal. Calc. for C22H14ClN3O2 calculated %; C: 68.13, H: 3.64, N: 10.84. Found %; C: 68.15, H: 3.64, N: 10.82.
4.1.1.4. 6-(4-Bromophenyl)-4-(2-chloroquinolin-3-yl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (3d)
Pale-canary yellow crystals (EtOH), m.p. > 300 °C, yield (84%), IR (KBr, cm–1): 1600 (ArC=C), 1650 (C=O), 2218 (C≡N), 3449 (NH). 1H NMR (500 MHz, DMSO-d6); δ 4.93(s, 1H, NH exchangeable by D2O), 7.06 (s, 1H, pyridinone-H, 90%), 7.75–7.79 (m, 3H, 2″-H, 6″-H, 5″-H,), 7.81–7.89 (m, 2H, 3″-H, 6-H), 7.96 (dt, 1H, J = 1.1Hz, J = 7.5 Hz, 7-H), 8.08 (d, 1H, J = 8.4 Hz, 8-H), 8.14 (d, 1H, J = 8.0 Hz, 5-H), 8.29 (s, 1H, pyridine-H, 10%), 8.73 (s, 1H, 4-H), 12.24 (s, 1H, OH exchangeable by D2O). 13C NMR (125 MHz, DMSO-d6); δ 105.2(C-5′), 116.1(C-CN), 121.4(C-3′), 122.5(C-4″), 126.7(C-4a), 127.3(C-6, C-8), 128.3(C-5), 128.9(C-2″, C-6″), 130.0(C-1″), 130.7(C-3), 131.2(C-7), 132.0(C-3″, C-5″), 135.9(C-4), 147.2(C-8a), 149.7(C-2), 159.6(C-2′), 160.5(C-6′), 166.7(C-4′). Anal. Calc. for C22H14ClN3O2 calculated %; C: 57.76, H: 2.54, N: 9.62. Found %; C: 57.78, H: 2.55, N: 9.61.
4.1.1.5. 4-(2-Chloroquinolin-3-yl)-6-(2,4-dichlorophenyl)-2-oxo-1,2-dihydropyridine-3-carbonitrile (3e)
Canary-yellow crystals (EtOH), m.p. 295–297 °C, yield (88%) IR (KBr, cm–1): 1604 (ArC=C), 1650 (C=O), 2224 (C≡N), 2920 (C–H aliphatic), 3425 (NH). 1H NMR (500 MHz, DMSO-d6); 1.91 (s, 1H, NH exchangeable by D2O), 6.74 (s, 1H, pyridinone-H, 92%), 7.62 (dd,1H, J = 8.3 Hz, J =1.9 Hz, 6″-H), 7.67 (d, 1H, J = 8.3 Hz, 5″-H), 7.77 (dt, 1H, J = 7.0 Hz, J = 1.5 Hz, 6-H), 7.87 (d, 1H J = 1.8 Hz, 3″-H), 7.95 (dt, 1H, J = 8.5 Hz, J = 1.5 Hz, 7-H), 8.07 (d, 1H, J = 8.4 Hz, 8-H), 8.15 (d, 1H, J = 7.9 Hz, 5-H), 8.30 (s, 1H, pyridine-H, 8%), 8.74(s, 1H, 4-H), 12.24 (s, 1H, OH exchangeable by D2O). 13C NMR (125 MHz, DMSO-d6); δ 104.9(C-5′), 116.2(C-CN), 121.5(C-3′), 125.5(C-4″), 126.8(C-4a), 127.1(C-5″), 127.6(C-6, C-8), 128.3(C-5), 128.7(C-3″), 129.1(C-6″), 130.2(C-3), 131.0(C-7), 132.3(C-2″), 132.9(C-1″), 135.9(C-4), 147.1(C-8a), 149.5(C-2), 156.5(C-6′), 159.6(C-2′), 166.8(C-4′). Anal. Calc. for C22H14ClN3O2 calculated %; C: 59.11, H: 2.36, N: 9.85. Found %; C: 59.09, H: 2.35, N: 9.87.
4.1.2. Procedure for the Synthesis of 2-Chloro-4-(2-chloroquinolin-3-yl)-6-phenylnicotinonitriles (4a–e)
A mixture of the carbonitrile derivatives (3a–e) (29 mmol) and phosphorus oxychloride (14 mL, 150 mmol) was stirred at 105 °C overnight. The mixture was poured onto crushed ice (200 g) and neutralized with dilute ammonium hydroxide. The formed precipitate was filtered, washed with water, 5% aqueous sodium bicarbonate, and again water and recrystallized from ethanol to afford the targeted compounds 4a–e.
4.1.2.1. 2-Chloro-4-(2-chloroquinolin-3-yl)-6-phenylnicotinonitrile (4a)
Pale-beige crystals, m.p. 185–187 °C, yield (78%), IR (KBr, cm–1): 756 (C–Cl), 1586 (ArC=C), 2229 (C≡N). 1H NMR (500 MHz, DMSO-d6); δ 7.60 (t, 3H, J = 16.50 Hz, 3″-H, 4″-H, 5″-H), 7.80 (t, 1H, J = 9.50 Hz, 6-H), 7.99 (t, 1H, J = 10.00 Hz, 7-H), 8.10 (d, 1H, J = 11.00 Hz, 8-H), 8.15 (d, 1H,, J = 10.00 Hz, 5-H), 8.20 (d, 2H, J = 6.00 Hz, 2″-H, 6″-H), 8.44 (s,1H, 4-H), 8.75 (s, 1H, 5′-H). 13C NMR (125 MHz, DMSO-d6); δ 108.7(C-3′), 114.9(CN), 121.5(C-5′), 126.6(C-6), 128.2(C-4″, C-2″), 128.3(C-6″), 128.9(C-8), 129.1(C-3″), 129.5(C-5″), 129.8(C-7, C-4a), 132.2(C-5), 133.0(C-3), 135.6(C-4), 141.3(C-1″), 147.0(C-8a), 147.7(C-2), 152.1(C-2′), 153.3(C-4′), 160.0(C-6′). Anal. Calc. for C21H11Cl2N3 calculated %; C: 67.04, H: 2.95, N: 11.17. Found %; C: 67.03, H: 2.97, N: 11.15.
4.1.2.2. 6-(3-Aminophenyl)-2-chloro-4-(2-chloroquinolin-3-yl) nicotinonitrile (4b)
Beige crystals, m.p. 277–280 °C, yield (76%), IR (KBr, cm–1): 795(C–Cl), 1585 (ArC=C), 2229 (C≡N), 3420 (NH2,). 1H NMR (400 MHz, DMSO-d6); δ 4.29(s, 2H, NH2 exchangeable by D2O), 7.20 (d, 1H, J = 10.00 Hz, 4″-H), 7.37 (d, 1H, J = 7.50 Hz, 2″-H), 7.46 (t, 1H, J = 10.00 Hz, 5″-H), 7.82 (t, 1H, J = 9.50 Hz, 6-H), 7.89 (d, 1H, J = 10.50 Hz, 6″-H), 8.00(t, 1H, J = 9.50 Hz, 7-H), 8.15 (d, 2H, J = 8.00, C-5, C-8), 8.50 (s, 1H, 4-H), 8.82 (s, 1H, 5′-H). 13C NMR (100 MHz, DMSO-d6); δ 109.0(C-3′), 114.9(C-2″), 121.5(CN, C-4″), 126.6(C-6″), 128.3(C-5′, C-6), 128.8(C-8), 128.9(C-5″, C-7), 129.1(C-5, C-4a), 130.8(C-3), 133.0(C-4), 136.6(C-1″), 141.3(C-8a), 147.0(C-3″), 147.7(C-2), 152.0(C-2′), 153.2(C-4′), 159.6(C-6). MS (m/z): 391 (100%, [M + 1]+), 393 (60.7%, [M + 1]+ +2), 395(11.3%, [M + 1]+ +4). Anal. Calc. for C21H12Cl2N4 calculated %; C: 64.47, H: 3.09, N: 14.32. Found %; C: 64.44, H: 3.12, N: 14.31.
4.1.2.3. 2-Chloro-4-(2-chloroquinolin-3-yl)-6-(4-methoxyphenyl) nicotinonitrile (4c)
Beige crystals, m.p. 194–196 °C, yield (81%), IR (KBr, cm–1):761(C–Cl), 1228 (C–O), 1585 (ArC=C), 2228 (C≡N), 2937 (C–H aliphatic). 1H NMR (400 MHz, DMSO-d6); δ 3.88 (s, 3H, OCH3), 7.15(d, 2H, J = 11.00 Hz, 3″-H, 5″-H), 7.82 (t, 1H, J = 9.50 Hz, 6-H), 8.01(t, 1H, J = 9.50 Hz, 7-H), 8.14 (d, 1H, J = 10.00 Hz, 8-H), 8.17 (d, 1H, J = 10.00 Hz, 5-H), 8.25 (d, 2H, J = 10.50 Hz. 2″-H, 5″-H), 8.48 (s, 1H, 4-H), 8.81(s, 1H, 5′-H). 13C NMR (100 MHz, DMSO-d6); δ 55.6 (C-CH3), 106.0(C-3′), 115.2(C-3″, C-5″), 117.8(C-CN), 122.2(C-5′), 127.6(C-6), 128.3(C-8), 129.1(C-2″, C-6″), 131.4(C-7), 131.8(C-1″), 132.1(C-4a), 132.4(C-5), 135.1(C-3), 137.2(C-4), 146.0(C-8a), 150.3(C-2), 153.1(C-2′), 153.9(C-4′), 159.6(C-4″), 160.2(C-6′). Anal. Calc. for C22H13Cl2N3O calculated %; C: 65.04, H: 3.23, N: 10.34. Found %; C: 65.01, H: 3.27, N: 10.32.
4.1.2.4. 6-(4-Bromophenyl)-2-chloro-4-(2-chloroquinolin-3-yl) nicotinonitrile (4d)
Pale-beige crystals, m.p. 245–247 °C, yield (85%), IR (KBr, cm–1): 762 (C–Cl), 1587 (ArC=C), 2229 (C≡N). 1H NMR (400 MHz, DMSO-d6); δ 7.82(d, 2H, J = 10.50 Hz, 3″-H, 5″-H), 8.01(t, 1H, J = 9.50 Hz, 6-H), 8.15(t, 1H, J = 9.50 Hz, 7-H), 8.19(d, 2H, J = 6.00 Hz, 5-H, 8-H), 8.21(d, 2H, J = 10.50 Hz, 2″-H, 6″-H), 8.61(s, 1H, 4-H), 8.12(s, 1H, 5′-H). 13C NMR (100 MHz, DMSO-d6); δ 106.4(C-3′), 117.2(C-CN), 121.4(C-5′) 121.9(C-4″), 127.5(C-6), 128.2(C-8), 128.9(C-2″, C-6″), 131.3(C-7), 131.7(C-4a), 132.2(C-5), 132.6(C-3″, C-5″), 135.0(C-3), 137.1(C-4), 138.3(C-1″), 146.1(C-8a), 150.2(C-2), 153.0(C-2′), 153.9(C-4′), 160.2(C-6′). Anal. Calc. for C21H10BrCl2N3 calculated %; C: 55.42, H: 2.21, N: 9.23. Found %; C: 55.39, H: 2.24, N: 9.22.
4.1.2.5. 2-Chloro-4-(2-chloroquinolin-3-yl)-6-(2,4-dichlorophenyl) nicotinonitrile (4e)
Pale-beige crystals, m.p. 204–206 °C, yield (78%) IR (KBr, cm–1): 756(C–Cl), 1583 (ArC=C), 2228 (C≡N). 1H NMR (400 MHz, DMSO-d6); δ 7.68 (dd,1H, J = 10.25 Hz, J = 3.00 Hz, 5″-H), 7.78 (d, 1H, J = 10.50 Hz, 6″-H), 7.82 (dt, 1H, J = 10.75 Hz, J = 1.50 Hz, 6-H), 7.90 (d,1H, J = 2.50 Hz, 3″-H), 8.00 (dt, 1H, J = 10.75 Hz, J = 2.00 Hz, 7-H), 8.13(d,1H, J = 10.50 Hz, 8-H), 8.17(d, 1H, J = 10.00 Hz, 5-H), 8.28 (s, 1H, 4-H), 8.83(s, 1H, 5′-H). 13C NMR (100 MHz, DMSO-d6); δ 109.9 (C-3′), 114.6 (C-5′), 126.1(CN), 126.6 (C-6),128.3 (C-8, C-5″), 128.6 (C-6″), 128.9 (C-3″), 129.1(C-7), 130.4 (C-1″), 133.0(C-4a, C-5), 133.7 (C-2″), 135.0 (C-4″), 136.3(C-3), 141.4 (C-4), 146.9 (C-8a), 147.7 (C-2), 151.8 (C-2′), 152.6 (C-4′), 158.7 (C-6′). Anal. Calc. for C21H9Cl4N3 calculated %; C: 56.67, H: 2.04, N: 9.44. Found %; C: 56.68, H: 2.06, N: 9.42.
4.2. Biological Screening
4.2.1. Materials
Four human tumor cell lines, namely, hepatocellular carcinoma (HePG-2), mammary gland breast cancer (MCF-7), colorectal carcinoma (HCT-116), and human prostate carcinoma (PC3) were used. The cell lines were obtained from ATCC via Holding Company for biological products and vaccines (VACSERA), Cairo, Egypt. The reagents RPMI-1640 medium, MTT, dimethyl sulfoxide (DMSO), 5-FU (Sigma Co., St. Louis), fetal bovine serum (GIBCO, U.K.) were used. 5-FU was used as a standard anticancer drug for comparison.
4.2.2. In Vitro Antitumor Evaluation (MTT Assay)
The different cell lines mentioned above were used to determine the inhibitory effects of compounds on cell growth using the MTT assay41,42 conducted in triplicates. This colorimetric assay is based on the conversion of the yellow tetrazolium bromide (MTT) to a purple formazan derivative using mitochondrial succinate dehydrogenase in viable cells. The cells were cultured in RPMI-1640 medium with 10% fetal bovine serum. Antibiotics added were 100 units/mL penicillin and 100 μg/mL streptomycin at 37 °C in a 5% CO2 incubator. The cells were seeded in a 96-well plate at a density of 1.0 × 104 cells/well, at 37 °C for 48 h under 5% CO2. After incubation, the cells were treated with different concentrations of compounds and incubated for 24 h. After 24 h of drug treatment, 20 μL of MTT solution at 5 mg/mL was added and incubated for 4 h. DMSO in a volume of 100 μL was added into each well to dissolve the purple formazan formed. The colorimetric assay is carried out and recorded at an absorbance of 570 nm using a plate reader (EXL 800), and the IC50 values were calculated.
4.2.3. Pim-1 Enzyme Assay
The luminescent kinase assay was performed to measure the Pim-1 kinase inhibitory activity of all the synthesized compounds 3 and 4 as well as compound A tested for their ability to inhibit Pim-1 kinase in comparison with quercetagetin. Activity was measured following the manufacturer’s instructions; ADP-Glo kinase assay43 measures ADP formed from a kinase reaction; ADP is converted into ATP, which is converted into light using Ultra-Glo luciferase. The luminescence signal positively correlates with the ADP amount and kinase activity using up to 1 mM ATP. The assay was performed in 384-well plates using 1 mL of the tested compound or 5% DMSO, 2 mL of enzyme, and 2 mL of substrate/ATP mix in kinase buffer. It was incubated at room temperature for 60 min; then, 5 mL of the ADP-Glo reagent was added and incubated at room temperature for 40 min. Finally, 10 mL of kinase detection reagent was added and incubated at room temperature for 30 min. Luminescence was recorded using a luminometer.
4.3. Molecular Modeling Simulation
The crystal structure of human Pim-1 kinase in complex with inhibitor A (PDB ID: 2OBJ)16 with a resolution of 2.50 Å and the crystal structure of Pim-1 with the quercetagetin inhibitor (PDB: 2O64)34 with a resolution of 2.44 Å were retrieved from the Protein Data Bank. All hydrogen atoms were added to the crystal 3D structure of the protein with their standard geometry, followed by their energy minimization. The investigated compounds, Pim-1 inhibitor 6-(5-bromo-2-hydroxy) phenyl-2-oxo-4phenyl-3-pyridine carbonitrile A and quercetagetin, were drawn into Marvin Sketch of the Marvin suite (http://www.chemaxon.com) to generate the lowest energy conformer. The dock module of MOE (Molecular Operating Environment) version MOE 2019.0102,244 on a computer having Pentium 1.6 GHz workstation, 512 MB memory, using the Windows operating system, was utilized in docking studies. Our tested compounds were docked into the rigid binding pocket of both the active site of VRV and quercetagetin active site of the proteins (PDB ID: 2OBJ, and PDB: 2O64, respectively), using flexible ligand mode. From ligand conformations, the placement phase generates poses. The free energy of binding of the ligand from a given pose is assessed using the GBVI/WSA ΔG as a force field-based scoring function.45
4.4. Radiolabeling Study
4.4.1. Radiolabeling Procedure
Different amounts of compound 4b (10–500 μg) in dimethyl sulfoxide (DMSO) were added to an amber color vial. Then, 20 μL of the freshly prepared chloramine-T (CAT) solution in ethanol containing (50–1000 μg) CAT was added. Then, 10 μL of Na131I (7.2 MBq) was added to the reaction mixture and pH was adjusted by using 150 μL of buffer solutions (3–10). The reaction mixture was vortexed and left at ambient temperature for 5–120 min. A drop of saturated sodium thiosulfate solution (10 mg/mL H2O) was added to quench the reaction by reducing iodine and iodonium to iodide (I–).46
4.4.2. Radiochemical Analysis of 131I-4b
4.4.2.1. Paper Chromatography
Ascending techniques were applied using a solvent system, CHCl3 and ethanol (9:1), on a strand of Whatman paper (1 cm width and 12 cm long).47
4.4.2.2. Paper Electrophoresis
The procedure was done using Whatman paper (2 cm width and 47 cm length), where 20 μL of the reaction mixture was spotted 12 cm away from the cathode. The process was carried out for 1.5 h at 300 V using physiological saline (0.9% w/v NaCl solution) as the electrolyte source solution.48 After complete development, the paper was dried and cut into 1 cm strips; then, each strip was counted using a NaI(Tl) γ-ray scintillation counter, and the percentage of the radiochemical yield was calculated.
4.4.3. Factors Affecting the Labeling Yield
Many factors affecting the labeling yield such as CAT content, substrate amount, reaction time, and pH of reaction medium were investigated. Initially, trials and errors were applied in radiolabeling to get the maximum labeling yield; then, upon studying one factor, other factors are kept at optimum conditions at which the maximum yield was obtained.
4.4.3.1. Effect of the CAT Content on the Radiochemical Yield of 131I-4b
The effect of the CAT content on the radiochemical yield of 131I-4b was studied using 200 μg of compound 4b (100 μL), different amounts of CAT (X), and 10 μL of NaI for 1 h at pH 4, at 25 °C; n = 3.
4.4.3.2. Effect of the Substrate Amount
The effect of the used amount of compound 4b was examined using different amounts of 4b (X μg), 500 μg of CAT (50 μL), and 10 μL of NaI for 1 h at pH 4 and at 25 °C; n = 3.
4.4.3.3. Effect of Reaction Time on the Radiochemical Yield of 131I-4b
The effect of reaction time on the radiochemical yield of 131I-4b was studied using 200 μg of compound 4b (100 μL), 500 μg of CAT (50 μL), and 10 μL of NaI for 1 h at pH 4 and at 25 °C; n = 3.
4.4.3.4. Effect of pH on the Radiochemical Yield of 131I-4b
The radiochemical yield of 200 μg of compound 4b (100 μL), 500 μg of CAT (50 μL), and 10 μL of NaI for 1 h at different pH values, at 25 °C; n = 3 were studied.
4.4.3.5. Impact of Time on the In Vitro Stability of 131I-4b
This experiment was conducted to know the stability of the labeled compound and to know the shelf life of it. After the optimum time for the reaction, a drop of saturated sodium thiosulfate solution (10 mg/mL H2O) was added to stop the reaction and convert excess iodonium to iodide ions. Then, 1–2 μL of samples was taken from the reaction mixtures of 131I-4b at different time intervals 1, 2, 4, 12, and 24 h. The radiochemical yields of the samples were measured using paper chromatography.
4.4.4. Biodistribution of 131I-4b
4.4.4.1. Tumor Transplantation
Ehrlich ascites carcinoma (EAC) was presented as a model in cancer research. By weekly intraperitoneal (IP) transplantation, EAC was maintained in female Swiss albino mice.49 EAC cells were obtained by needle aspiration under aseptic conditions. The ascetic fluid was diluted with sterile saline (0.1 mL contains 2.5 × 106 cells); then, 0.1 mL of this solution was injected intramuscularly in the muscle of the right leg of the mice to produce solid tumor, keeping the left leg as a control. Mice were kept for 7–10 days on a normal diet in a metabolic cage till the tumor was observed.
4.4.4.2. Biodistribution of 131I-4b in Normal and Solid Tumor-Bearing Mice
Biodistribution assay was carried out in normal and solid tumor-bearing mice. Each group consisted of 16 mice divided to 4 subgroups (4 mice each). Injection of 0.2 mL (74 kBq) of 131I-4b was done via the tail vein in mice. Mice were sacrificed at 15, 30, 60, or 120 min by cervical dislocation after anesthesia with chloroform. The organs and fluids of interest were removed, weighed, and counted in a γ-scintillation counter to determine their uptake of 131I-4b. Samples of blood and organs of interest were removed, weighed, and assayed for radioactivity, and the percentages of the injected dose per gram (% ID/g) were calculated. Blood, bone, and muscle weights were taken as 7, 10, and 40% of the total body weight, respectively.49 Solid tumor to normal muscle (T/NT) was calculated as % ID/g for solid tumor and normal muscles.
This study was approved by the animal ethics committee and was conducted according to the regulations established by the Faculty of Pharmacy, Cairo University (PT 4.2.2), and the guidelines of the animal ethics committee of the Labeled Compounds Department of the Egyptian Atomic Energy Authority, Egypt. All the solid waste and liquid waste generated had low radioactivity and were sent to the radioactive waste management unit.
Acknowledgments
The authors thank the Holding Company for Biological Products and Vaccines (VACSERA), Cairo, Egypt, for performing the biological screening.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c08304.
Spectral charts (IR, 1H NMR, 13C NMR, and MS) of some new compounds and resultant 2D and 3D poses and free binding energies from docking both entire sets (3a–e and 4a–e) against crystal structure of human Pim-1 kinase in complex with the inhibitor (VRV) (PDB: 2OBJ) target and on the crystal structure of human Pim-1 kinase with the inhibitor quercetagetin (PDB: 2O64) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Su Z.; Yang Z.; Xu Y.; Chen Y.; Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. 10.1186/s12943-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowski K.; Kciuk M.; Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. 10.3390/ijms21093233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanpour S. H.; Dehghani M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129. 10.1016/j.jcrpr.2017.07.001. [DOI] [Google Scholar]
- Narlik-Grassow M.; Blanco-Aparicio C.; Carnero A. The PIM family of serine/threonine kinases in cancer. Med. Res. Rev. 2014, 34, 136–159. 10.1002/med.21284. [DOI] [PubMed] [Google Scholar]
- Brault L.; Gasser C.; Bracher F.; Huber K.; Knapp S.; Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010, 95, 1004. 10.3324/haematol.2009.017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane N. A.; Reidy M.; Natoni A.; Raab M.; O’dwyer M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015, 5, e325 10.1038/bcj.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nafie M. S.; Amer A. M.; Mohamed A. K.; Tantawy E. S. Discovery of novel pyrazolo [3, 4-b] pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells. Bioorg. Med. Chem. 2020, 28, 115828 10.1016/j.bmc.2020.115828. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Liu T.; Sun H.; Weng W.; Zhang Q.; Liu C.; Han Y.; Sheng W. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Cancer Sci. 2018, 109, 1468–1479. 10.1111/cas.13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung M. S.; Chan K. K. S.; Dai W. J.; Wong C. Y.; Au K. Y.; Wong P. Y.; Wong C. C. L.; Lee T. K. W.; Ng I. O. L.; Kao W. J.; Lo R. C. Anti-tumour effects of PIM kinase inhibition on progression and chemoresistance of hepatocellular carcinoma. J. Pathol. 2020, 252, 65–76. 10.1002/path.5492. [DOI] [PubMed] [Google Scholar]
- Xu J.; Xiong G.; Cao Z.; Huang H.; Wang T.; You L.; Zhou L.; Zheng L.; Hu Y.; Zhang T. PIM-1 contributes to the malignancy of pancreatic cancer and displays diagnostic and prognostic value. J. Exp. Clin. Cancer Res. 2016, 35, 133. 10.1186/s13046-016-0406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekky A. E. M.; Sanad S. M.; Said A. Y.; Elneairy M. A. Synthesis, cytotoxicity, in-vitro antibacterial screening and in-silico study of novel thieno [2, 3-b] pyridines as potential pim-1 inhibitors. Synth. Commun. 2020, 50, 2376–2389. 10.1080/00397911.2020.1778033. [DOI] [Google Scholar]
- Mohareb R. M.; Ibrahim R. A.; Elmetwally A. M.; Gamaan M. S. Synthesis of Fused Quinoline Derivatives With Antiproliferative Activities and Tyrosine Kinases, Pim-1 Kinase Inhibitions. Acta Chim. Slov. 2022, 69, 13–29. 10.17344/acsi.2021.6733. [DOI] [PubMed] [Google Scholar]
- Khamees H. A.; Srinivas M. S.; Nagaraja O.; Madegowda M.; Vahini V.; Chaluvaiah K.; Dasappa J. P.; Warad I. Studies on New Imidazo [2, 1-b][1, 3, 4] thiadiazole Derivatives: Molecular Structure, Quantum Chemical Computational, and In silico Study of Inhibitory Activity Against Pim-1 Protein by using Molecular Modelling Methods and ADMET Profiling. J. Mol. Struct. 2023, 1272, 134161 10.1016/j.molstruc.2022.134161. [DOI] [Google Scholar]
- Castanet A.-S.; Nafie M. S.; Said S. A.; Arafa R. K. Discovery of PIM-1 kinase inhibitors based on the 2, 5-disubstituted 1, 3, 4-oxadiazole scaffold against prostate cancer: Design, synthesis, in vitro and in vivo cytotoxicity investigation. Eur. J. Med. Chem. 2023, 250, 115220 10.1016/j.ejmech.2023.115220. [DOI] [PubMed] [Google Scholar]
- Bass A. K.; Abdelhafez E.; El-Zoghbi M.; Mohamed M. F.; Badr M.; Abuo-Rahma G. E.-D. A. 3-Cyano-2-oxa-pyridines: a promising template for diverse pharmacological activities. J. Adv. Biomed. Pharm. Sci. 2021, 4, 81–86. 10.21608/jabps.2020.52641.1113. [DOI] [Google Scholar]
- Cheney I. W.; Yan S.; Appleby T.; Walker H.; Vo T.; Yao N.; Hamatake R.; Hong Z.; Wu J. Z. Identification and structure–activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg. Med. Chem. Lett. 2007, 17, 1679–1683. 10.1016/j.bmcl.2006.12.086. [DOI] [PubMed] [Google Scholar]
- Farrag A. M.; Ibrahim M. H.; Mehany A. B. M.; Ismail M. M. F. New cyanopyridine-based scaffold as PIM-1 inhibitors and apoptotic inducers: Synthesis and SARs study. Bioorg. Chem. 2020, 105, 104378 10.1016/j.bioorg.2020.104378. [DOI] [PubMed] [Google Scholar]
- Bass A. K.; Nageeb E.-S. M.; El-Zoghbi M. S.; Mohamed M. F.; Badr M.; Abuo-Rahma G. E.-D. A. Utilization of cyanopyridine in design and synthesis of first-in-class anticancer dual acting PIM-1 kinase/HDAC inhibitors. Bioorg. Chem. 2022, 119, 105564 10.1016/j.bioorg.2021.105564. [DOI] [PubMed] [Google Scholar]
- Abouzid K. A.; Al-Ansary G. H.; El-Naggar A. M. Eco-friendly synthesis of novel cyanopyridine derivatives and their anticancer and PIM-1 kinase inhibitory activities. Eur. J. Med. Chem. 2017, 134, 357–365. 10.1016/j.ejmech.2017.04.024. [DOI] [PubMed] [Google Scholar]
- Sabour R.; Harras M. F.; Mehany A. B. Design, synthesis, cytotoxicity screening and molecular docking of new 3-cyanopyridines as survivin inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 94, 103358 10.1016/j.bioorg.2019.103358. [DOI] [PubMed] [Google Scholar]
- Wendt M. D.; Sun C.; Kunzer A.; Sauer D.; Sarris K.; Hoff E.; Yu L.; Nettesheim D. G.; Chen J.; Jin S.; et al. Discovery of a novel small molecule binding site of human survivin. Bioorg. Med. Chem. Lett. 2007, 17, 3122–3129. 10.1016/j.bmcl.2007.03.042. [DOI] [PubMed] [Google Scholar]
- Aqui N. A.; Vonderheide R. H. Survivin as a universal tumor antigen for novel cancer immunotherapy: functions of a killer clone. Cancer Biol. Ther. 2008, 7, 1888–1889. 10.4161/cbt.7.12.7219. [DOI] [PubMed] [Google Scholar]
- Abd Elhameid M. K.; Ryad N.; Al-Shorbagy M. Y.; Mohammed M. R.; Ismail M. M.; El Meligie S. Design, synthesis and screening of 4, 6-diaryl pyridine and pyrimidine derivatives as potential cytotoxic molecules. Chem. Pharm. Bull. 2018, 66, 939. 10.1248/cpb.c18-00269. [DOI] [PubMed] [Google Scholar]
- Mansour B.; Henen M. A.; Bayoumi W. A.; El-Sayed M. A.; Massoud M. A. In colorectal cancer; NMR-monitored β-Catenin inhibition by a Quinoline derivative using Water-LOGSY technique. J. Mol. Struct. 2021, 1246, 131151 10.1016/j.molstruc.2021.131151. [DOI] [Google Scholar]
- Afzal O.; Kumar S.; Haider M. R.; Ali M. R.; Kumar R.; Jaggi M.; Bawa S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. 10.1016/j.ejmech.2014.07.044. [DOI] [PubMed] [Google Scholar]
- Mansour B.; Bayoumi W. A.; El-Sayed M. A.; Abouzeid L. A.; Massoud M. A. M. In vitro cytotoxicity and docking study of novel symmetric and asymmetric dihydropyridines and pyridines as EGFR tyrosine kinase inhibitors. Chem. Biol. Drug Des. 2022, 100, 121. 10.1111/cbdd.14058. [DOI] [PubMed] [Google Scholar]
- Massoud M. A.; El-Sayed M. A.; Bayoumi W. A.; Mansour B. Cytotoxicity and molecular targeting study of novel 2-chloro-3-substituted quinoline derivatives as antitumor agents. Lett. Drug Des. Discovery 2019, 16, 273–283. 10.2174/1570180815666180604090924. [DOI] [Google Scholar]
- Ammar Y. A.; Elhagali G. A.; Abusaif M. S.; Selim M. R.; Zahran M. A.; Naser T.; Mehany A.; Fayed E. A. Carboxamide appended quinoline moieties as potential anti-proliferative agents, apoptotic inducers and Pim-1 kinase inhibitors. Med. Chem. Res. 2021, 30, 1649–1668. 10.1007/s00044-021-02765-y. [DOI] [Google Scholar]
- Li K.; Li Y.; Zhou D.; Fan Y.; Guo H.; Ma T.; Wen J.; Liu D.; Zhao L. Synthesis and biological evaluation of quinoline derivatives as potential anti-prostate cancer agents and Pim-1 kinase inhibitors. Bioorg. Med. Chem. 2016, 24, 1889–1897. 10.1016/j.bmc.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Massoud M.; Bayoumi W.; Farahat A.; Elsayed M.; Mansour B. Chloroquinoline-3-carbaldehydes: synthesis and reactions (2012-2017). Arkivoc 2018, 2018, 244. 10.24820/ark.5550190.p010.399. [DOI] [Google Scholar]
- Kotb E. R.; El-Hashash M.; Salama M. A.; Kalf H. S.; Abdel Wahed N. A. Synthesis and reactions of some novel nicotinonitrile derivatives for anticancer and antimicrobial evaluation. Acta Chim. Slov. 2009, 56, 908–919. [Google Scholar]
- Arnott E. A.; Chan L. C.; Cox B. G.; Meyrick B.; Phillips A. POCl3 chlorination of 4-quinazolones. J. Org. Chem. 2011, 76, 1653–1661. 10.1021/jo102262k. [DOI] [PubMed] [Google Scholar]
- Sliman F.; Blairvacq M.; Durieu E.; Meijer L.; Rodrigo J.; Desmaële D. Identification and structure–activity relationship of 8-hydroxy-quinoline-7-carboxylic acid derivatives as inhibitors of Pim-1 kinase. Bioorg. Med. Chem. Lett. 2010, 20, 2801–2805. 10.1016/j.bmcl.2010.03.061. [DOI] [PubMed] [Google Scholar]
- Holder S.; Zemskova M.; Zhang C.; Tabrizizad M.; Bremer R.; Neidigh J. W.; Lilly M. B. Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol. Cancer Ther. 2007, 6, 163–172. 10.1158/1535-7163.MCT-06-0397. [DOI] [PubMed] [Google Scholar]
- Moustapha M. E.; Motaleb M.; Ibrahim I.; Moustafa M. Oxidative radioiodination of aripiprazole by chloramine-T as a route to a potential brain imaging agent: a mechanistic approach. Radiochemistry 2013, 55, 116–122. 10.1134/S1066362213010232. [DOI] [Google Scholar]
- Motaleb M. A.; El-Kolaly M.; Rashed H.; El-Bary A. Radioiodinated paroxetine, a novel potential radiopharmaceutical for lung perfusion scan. J. Radioanal. Nucl. Chem. 2012, 292, 629–635. 10.1007/s10967-011-1499-7. [DOI] [Google Scholar]
- Sakr T. M.; Ibrahim I.; Abd-Alla W. H. Molecular modeling and preclinical evaluation of radioiodinated tenoxicam for inflammatory disease diagnosis. J. Radioanal. Nucl. Chem. 2018, 316, 233–246. 10.1007/s10967-018-5770-z. [DOI] [Google Scholar]
- Aboumanei M. H.; Abdelbary A. A.; Ibrahim I. T.; Tadros M. I.; El-Kolaly M. T. Design and development of microemulsion systems of a new antineoplaston A10 analog for enhanced intravenous antitumor activity: In vitro characterization, molecular docking, 125I-radiolabeling and in vivo biodistribution studies. Int. J. Pharm. 2018, 545, 240–253. 10.1016/j.ijpharm.2018.05.010. [DOI] [PubMed] [Google Scholar]
- Meth-Cohn O.; Narine B.; Tarnowski B. A versatile new synthesis of quinolines and related fused pyridines, Part 5. The synthesis of 2-chloroquinoline-3-carbaldehydes. J. Chem. Soc., Perkin Trans. 1 1981, 1520–1530. 10.1039/p19810001520. [DOI] [Google Scholar]
- Ladraa S.; Chioua M.; Belfaitah A. A Simple and Ecofriendly One-Pot Synthesis of Highly Substituted 3-Cyanopyridine-Quinoline Hybrids via a Triphenyphosphine-Catalyzed Multicomponent Reaction Under Mild Conditions. J. Heterocycl. Chem. 2017, 54, 603–609. 10.1002/jhet.2631. [DOI] [Google Scholar]
- Denizot F.; Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- AboulMagd A. M.; Hassan H. M.; Sayed A. M.; Abdelmohsen U. R.; Abdel-Rahman H. M. Saccharomonosporine A inspiration; synthesis of potent analogues as potential PIM kinase inhibitors. RSC Adv. 2020, 10, 6752–6762. 10.1039/c9ra10216g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemical Computing Group Inc., 1010 Sherbooke Street, West, Suite# 910, Montreal, QC, Canada, 2019.
- Labute P. The generalized Born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
- Durante A. C. R.; Sobral D.; Miranda A. C.; de Almeida É.; Fuscaldi L.; de Barboza M.; Malavolta L. Comparative study of two oxidizing agents, chloramine T and Iodo-Gen, for the radiolabeling of β-CIT with iodine-131: Relevance for Parkinson’s disease. Pharmaceuticals 2019, 12, 25. 10.3390/ph12010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K.; Ghosh A. Radiochemistry, production processes, labeling methods, and immunoPET imaging pharmaceuticals of iodine-124. Molecules 2021, 26, 414. 10.3390/molecules26020414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motaleb M. A.; Ibrahem I.; Ayoub V.; Geneidi A. Preparation and biological evaluation of 99mTc–ropinirole as a novel radiopharmaceutical for brain imaging. J. Label. Compd. Radiopharm. 2016, 59, 147–152. 10.1002/jlcr.3380. [DOI] [PubMed] [Google Scholar]
- Waly M. A.; El-Gogary S. R.; El-Sepelgy O. Z. One-Pot New Synthetic Method for 3-Amino-2-quinoxalinecarbonitrile. Synth. Commun. 2010, 40, 739–743. 10.1080/00397910903013721. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.