

Abstract
The basal forebrain cholinergic system (BFCS) has long been implicated in age-related cognitive changes and the pathophysiology of Alzheimer’s disease (AD). Limitations of cholinergic interventions helped to inspire a shift away from BFCS in AD research. A resurgence in interest in the BFCS following methodological and analytical advances has resulted in a call for the BFCS to be examined in novel frameworks. We outline the basic structure and function of the BFCS, its role in supporting cognitive and affective function, and its vulnerability to aging and AD. We consider the BFCS in the context of the amyloid hypothesis and evolving concepts in AD research: resilience and resistance to pathology, selective neuronal vulnerability, trans-synaptic pathology spread and sleep health. We highlight 1) the potential role of the BFCS in cognitive resilience, 2) recent work refining understanding about the selective vulnerability of BFCS to AD, 3) BFCS connectivity that suggests it is related to tau spreading and neurodegeneration and 4) the gap between BFCS involvement in AD and sleep-wake cycles.
Keywords: tau, amyloid, attention, learning, memory, resilience, reserve, basal forebrain, acetylcholine
Introduction
The basal forebrain cholinergic system (BFCS) was implicated in the early pathophysiology of Alzheimer’s disease (AD) by seminal studies completed over 40 years ago (Bartus et al., 1982; Rossor et al., 1982; Whitehouse et al., 1982). While research interest in this system has waxed and waned over the subsequent decades, we are currently in a period in which there is renewed, accelerating interest in the BFCS relevant to aging and AD. Here, we review the basic structure and function of the BFCS and foundational work describing its role in cognition, aging and the pathophysiology of AD. We will discuss the cholinergic hypothesis of AD and how evidence from animal models and humans, including patients living with AD, ultimately shifted the focus of AD research to the amyloid hypothesis. We argue that there is a need for further work to integrate the contribution of the BFCS, especially its role in attention and learning, into the current amyloid-first model of AD that generally focuses on early tau in the medial temporal lobe (MTL) and the resulting episodic memory impairment. Finally, we outline the recent breakthroughs in our understanding of the BFCS through the lens of current concepts in AD research including resistance and resilience to pathology, selective neuronal vulnerability, trans-synaptic spreading of pathology and sleep.
Brief anatomy and development of the basal forebrain cholinergic system
The BFCS projects to prefrontal cortex, hippocampus, entorhinal cortex and amygdala (Mesulam et al., 1983; Woolf, 1991). Detailed accounts of cell types and differential projection patterns of the BF are outside the scope of the present work, but previous reviews cover these topics in depth (Ballinger et al., 2016; Woolf, 1991; Záborszky et al., 2018). In addition, while our focus is on the BFCS, there are excellent reviews which describe cholinergic neurons outside the BF (Beierlein, 2014; Calabresi et al., 2000; Mena-Segovia, 2016; Picciotto et al., 2012; Woolf, 1991).
The BF is comprised of many small nuclei that produce a variety of neurotransmitters. Structures containing choline acetyl transferase (ChAT), the final enzyme in the synthesis cascade for acetylcholine, include the nucleus basalis of Meynert, medial septum, ventral pallidum, vertical and horizontal diagonal band nuclei, substantia innominata/extended amygdala, and peripallidal regions (Woolf, 1991). These cholinergic neurons in the BF make up the BFCS and are the primary source of cholinergic cortical projections. Prefrontal cortex and amygdala receive cholinergic innervation from the nucleus basalis of Meynert and substantia innominata (Mesulam et al., 1983), while the entorhinal cortex and hippocampus receive cholinergic innervation from the medial septum and vertical diagonal band nuclei (Frotscher and Léránth, 1985; Gaykema et al., 1990; Kondo and Zaborszky, 2016). In turn, the BFCS receives reciprocal inputs from prefrontal cortex, amygdala, entorhinal cortex and hippocampus (Krettek and Price, 1978; Mesulam and Mufson, 1984; Russchen et al., 1985), and is also modulated by other subcortical systems including the noradrenergic locus coeruleus, serotonergic dorsal raphe nucleus and the dopaminergic substantia nigra (Giorgi et al., 2017; Russchen et al., 1985; Steinbusch, 1981; Swanson and Hartman, 1975). We refer the reader to reviews mapping BFCS afferents, projections, and topographic patterns of cortical innervation for more detailed information (Ballinger et al., 2016; Záborszky et al., 2018, 1991).
While the cholinergic inputs throughout the cerebral cortex have been described as diffuse, there appears to be remarkable specialization and functional precision among these inputs. The BFCS supports spatially and temporally discreate signaling, which is achieved via spatially-specific mapping of BFCS projections, cholinergic modulation of thalamic inputs to cortex (Obermayer et al., 2017), and multiple signaling modes (Parikh et al., 2007). There is now broad appreciation that acetylcholine transmission is both phasic and tonic (though see Sarter and Lustig, 2020 for discussion of new frameworks for understanding tonic cholinergic action). The development of amperometric recording for measuring transient cholinergic signaling in performing animals (Howe et al., 2013; Parikh et al., 2007), and optogenetic studies driving behavior through phasic acetylcholine release (Gritton et al., 2016) have been critical for redefining the BFCS as a system capable of modulating specific cognitive operations in a spatially and temporally discrete manner. This specificity is further supported by diversity in receptor subtypes, muscarinic (m1–5) and nicotinic acetylcholine receptors (α7; α4β2; α3β4), that are selectively implicated in promoting frequency-specific regional oscillatory activity (Gu et al., 2020; Howe et al., 2017).
Animal studies show that during development BFCS neurons are highly dependent on the neurotrophin nerve growth factor (NGF). NGF, a target-derived neurotrophic factor, is necessary for the proper axon sprouting, guidance, and maturation of acetylcholine-producing neurons (Campenot, 1977; Gallo et al., 1997; Patel et al., 2000; Tuttle and O’Leary, 1998). BFCS neurons are unique in that they continue to rely on NGF postnatally and across the lifespan (Cuello, 1996; Sofroniew et al., 2001), and are the major cell group that expresses NGF receptors (both high affinity tropomyosin-related kinase A (TrkA) and low affinity p75 neurotrophin receptors; Mufson et al., 2019). NGF promotes the gene expression of another trophic factor critical for BFCS development, neuropeptide galanin, deficits in which cause cholinergic loss and cognitive impairment (O’Meara et al., 2000).
Our understanding of the diverse functions of NGF and NGF’s role in the development, protection and repair of cholinergic neurons is an area of active investigation (Aloe et al., 2012; Mufson et al., 2019; Sofroniew et al., 2001). What is clear is that deficits in NGF signaling (particularly via TrkA receptors) leads to vulnerability in the BFCS, impaired structure and function (Parikh et al., 2013; Sarter and Bruno, 2004), reduced acetylcholine signaling (Auld et al., 2001; Knipper et al., 1994; Sala et al., 1998), and disrupted learning and memory (Gutiérrez et al., 1997; Woolf et al., 2001).
Basal forebrain cholinergic system function and cognition
Acetylcholine is consistently implicated in attention, learning and memory in both animal and human studies. Acetylcholine shows patterns of traditional neuromodulatory action critical for basic mechanisms of attention, enhancing gain (Disney et al., 2007), tuning (Zinke et al., 2006), and attentional modulation (Herrero et al., 2008) in sensory cortex. Multiple modes of cholinergic signaling in prefrontal cortex supports the maintenance of attentional control and effort (Berry et al., 2015; Silvestrini et al., 2022; St Peters et al., 2011), as well as rapid attentional shifts and cue detection (Gritton et al., 2016; Howe et al., 2013; Parikh et al., 2007).
Supporting a role of acetylcholine in learning and memory, the entorhinal cortex and hippocampus receive direct BFCS input and acetylcholine release in hippocampus has been linked with long-term potentiation (Blitzer et al., 1990; Bröcher et al., 1992; Patil et al., 1998) as well as long-term depression (Brzosko et al., 2017; Jo et al., 2010; Seol et al., 2007; Williams and Johnston, 1990). Knocking out or blocking cholinergic receptors causes learning and memory impairment (Carli et al., 1997; Douchamps et al., 2013; Wallenstein and Vago, 2001), disruptions in hippocampal theta oscillatory activity (Douchamps et al., 2013; Lu and Henderson, 2010), and reduced theta-gamma locking in the medial entorhinal cortex (Newman et al., 2013).
Bidirectional connections between the BF and amygdala (Carlsen et al., 1985; Russchen et al., 1985) support a role of acetylcholine in fear memories and anxiety-like behavior (Picciotto et al., 2015). Cholinergic receptor blockade modulates depression-like symptoms in rodents (De Pablo et al., 1991; Picciotto et al., 2002). While the role of the BFCS in affective function receives comparatively less research attention relative to other cognitive domains, mood disorders are closely associated with poorer executive function and memory performance (James et al., 2021; Snyder et al., 2015). Relationships between disordered affective function and cognition are likely bidirectional, thus dysregulation cholinergic activity has negative impacts on cognition and mood, which may be magnified by interactions between these affective and cognitive systems.
In humans, evidence for the role of the BFCS in normal cognition has been explored using pharmaceutical interventions and innate biochemical differences arising from common genetic polymorphisms. Administration of scopolamine, an anticholinergic muscarinic antagonist, leads to altered BFCS function in humans measurable as deficits in memory and attention (Broks et al., 1988; Safer and Allen, 1971). Early scopolamine studies were a key factor in linking cholinergic signaling to age-related decreases in memory performance and, eventually, AD (Drachman and Leavitt, 1974). Over decades, research with scopolamine using more specific tasks revealed induced impairment in working memory, selective and sustained attention, and metacognition/subjective awareness (Lenz et al., 2012; Mintzer and Griffiths, 2003). Importantly, while cognitive deficits are observed in both younger and cognitively normal older adults after scopolamine, the deficits produced in older adults are more pronounced, indicating a higher level of vulnerability in the BFCS associated with age (Molchan et al., 1992). There is also a link to β-amyloid (Aβ), one of the hallmark pathologies of AD, such that the response of healthy older adults to scopolamine is moderated by pathology, with larger performance decrements in individuals with higher Aβ (Lim et al., 2015). Human genetics work also supports the critical role of BFCS in attention. A genetic polymorphism that affects choline transporter function has been shown to be associated with measures of distractibility and visual attention in healthy middle aged adults (Berry et al., 2015, 2014; Sarter et al., 2016).
The cholinergic hypothesis and a modern reexamination of the role of BFCS in AD
It has long been suggested that the loss of cholinergic neurons in the BF underlies cognitive decline in memory, learning and attention and that this process is a part of typical aging but is exacerbated by AD (Grothe et al., 2012; Mesulam, 2012; Muir, 1997). The early observations that cholinergic neurons degenerated and that ChAT was reduced in the brains of AD patients combined with studies showing pharmaceutical disruption of the BFCS produced memory and attention deficits, led to the so-called “cholinergic hypothesis” (Bartus et al., 1982; Plotkin and Jarvik, 1986; Whitehouse et al., 1982) (Fig. 1). The cholinergic hypothesis was the focus of much research in the 70s and 80s and placed the BFCS at the center of AD etiology. Subsequently, acetylcholinesterase inhibitors were developed as treatments for AD (Davis et al., 1992; Rogers and Friedhoff, 1996). Over time, it was understood that these treatments had modest effects in some patients (Pa et al., 2013) and no effect in others (see Marucci et al., 2021 for recent review) which, along with evidence supporting the central role of cortical Aβ pathology in AD, cast doubt on the cholinergic hypothesis. Perhaps the most critical evidence that Aβ pathology is a key initiating factor in AD was the identification of Aβ-modulating genetic mutations in the genes APP, PSEN1 and PSEN2 that lead to autosomal dominant AD (Goate et al., 1991; Rogaev et al., 1995; Schellenberg et al., 1992; Van Broeckhoven et al., 1992). Ultimately, the amyloid hypothesis became the predominant model that persists and has guided much of the AD research, especially human biomarker research, in the last 25 years.
Figure 1.
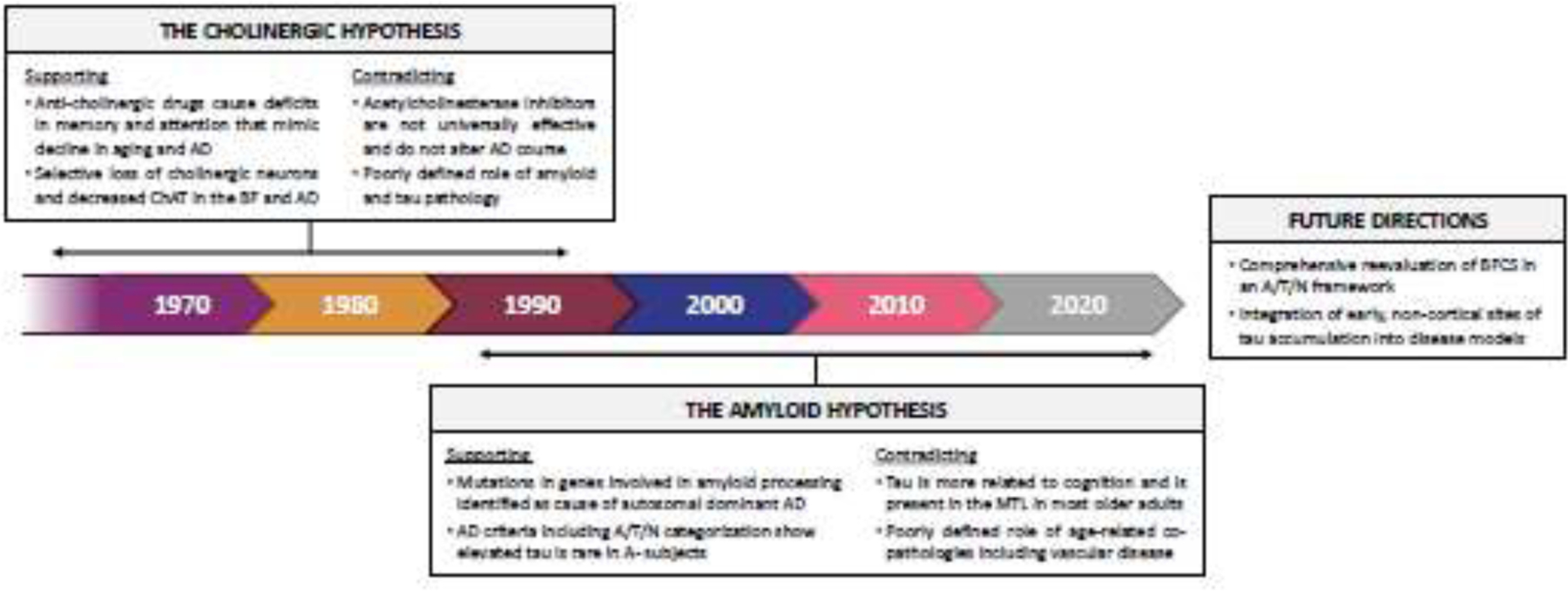
History of cholinergic and amyloid hypotheses of Alzheimer’s disease. We present a timeline illustrating the factors contributing to the evolution of the cholinergic hypothesis and the amyloid hypothesis. We highlight their strengths and limitations, and suggest areas ripe for further theoretical development. AD = Alzheimer’s disease; ChAT = choline acetyltransferase; BF = basal forebrain; A/T/N = amyloid/tau/neurodegeneration; MTL = medial temporal lobe; BFCS = basal forebrain cholinergic system
In this review we focus on the evidence supporting the amyloid hypothesis and the key role amyloid plays in AD pathogenesis, but of course there is also contradictory data that suggests the amyloid hypothesis is flawed or incomplete (Morris et al., 2018). This includes the failure of many anti-Aβ clinical trials (although recent positive data may indicate that there is some hope for this approach (van Dyck et al., 2023) and the fact that Aβ pathology does not universally lead to AD with pathological levels of Aβ observed in cognitively normal older adults.
The amyloid hypothesis of AD pathogenesis posits that Aβ pathology drives cortical spread of tau pathology out of the MTL which in turn results in neurodegeneration and cognitive decline (Jack et al., 2018; Jagust, 2018). While there is abundant evidence to support this model, it has been mostly narrowly applied to a cortex-centric process where tau pathology begins to deposit in MTL regions like the entorhinal cortex (ERC) and hippocampus, affecting episodic memory performance, and then spreads to other cortical regions in a largely stereotyped pattern (Kaufman et al., 2018). There is a need, however, to integrate the role of the BFCS into this model (Hampel et al., 2019). It is critical to bring together the well-known early observations of BFCS involvement in AD and the current literature where the amyloid hypothesis is the foundation and ideas like resilience and resistance to pathology, selective neuronal vulnerability, trans-synaptic spread of tau pathology, and associations with sleep are key research areas. Stitching together the basis of the cholinergic hypothesis and modern understanding of AD pathogenesis will result in a broader, more holistic understanding of AD etiology.
Linking the cholinergic and amyloid hypotheses
There is evidence of interactions between the BFCS and Aβ, which may mechanistically bridge cholinergic and amyloid hypotheses. Here we briefly review evidence that there are bidirectional effects between increasing Aβ and reduced cholinergic integrity which may accelerate pathology spread. There are several reviews focused on this topic where greater detail is available (Auld et al., 2002; Canu et al., 2017; Cattaneo and Calissano, 2012; Do Carmo et al., 2021; Iulita and Cuello, 2014; Mufson et al., 2019; Sarter and Bruno, 2004).
Aβ impacts the BFCS through local plaque accumulation (Arendt et al., 1988) as well as effects of soluble Aβ interacting directly with various components of the cholinergic system. In animal studies, the presence of soluble Aβ causally disrupts cholinergic transmission by blocking nicotinic receptor signaling (Liu et al., 2001; Pettit et al., 2001), and impeding acetylcholine transport with downstream effects on acetylcholine synthesis and release (Auld et al., 2002, 1998; Kar et al., 1998, 1996). Equally, the BFCS impacts Aβ metabolism. Acetylcholine muscarinic receptors are involved in the secretory processing of APP towards the non-amyloidogenic pathway (Nitsch et al., 1992; Seo et al., 2002; Wolf et al., 1995). Experimentally-induced loss of cholinergic neurons or pharmacological reduction in acetylcholine availability impacts APP processing and triggers increases in Aβ deposition (Beach et al., 2001, 2000; Roberson and Harrell, 1997). Thus, the BFCS is actively targeted by Aβ and disruption of cholinergic activity increases Aβ pathology, which suggests a vicious cycle by which reduced BFCS integrity triggers pathological processes, further reducing cholinergic function.
Maintaining trophic support of BFCS may delay Aβ pathological progression by preserving the integrity of cholinergic function despite pathology. NGF is appreciated to be a “survival” factor (Hefti, 1986; Kromer, 1987; Williams et al., 1986) which prevents degeneration following injury. These functions are largely mediated by NGF-TrkA signaling (see Mufson et al., 2019 for review and discussion of the role of p75 receptors). It appears that synthesis of NGF does not change appreciably with age or disease (Fahnestock et al., 1996), but that the BFCS loses trophic support through reduction of TrKA receptors (Ginsberg et al., 2006; Siegel and Chauhan, 2000), breakdown in retrograde transport of target-derived NGF (Mufson et al., 1995), and through disruptions in the conversion of pro-NGF to mature NGF (Allard et al., 2012; Iulita and Cuello, 2014). Emerging research in AD and Down syndrome suggests early increases in Aβ and inflammation drive dysregulation of trophic support and particularly dysregulation of NGF metabolism (Iulita and Cuello, 2014). These findings suggest therapeutically targeting NGF trophic pathways would be useful in delaying disease progression, though direct treatment with NGF has been technically challenging (Mitra et al., 2019) and recent NGF gene transfer trials have shown no evidence of efficacy to date (Rafii et al., 2018, 2014).
The successful maintenance of TrKA receptor density in aging appears to be particularly impactful in maintaining healthy aging trajectories both because of its role in trophic support, but also because TrkA receptors are implicated in steering Aβ metabolism away from pathological pathways (Rossner et al., 1998a). Specifically, in vitro studies reveal TrkA receptor stimulation increases non-amyloidogenic APP secretion and decreases APP mRNA levels (Rossner et al., 1998b, 1998a). Reductions in NGF/TrkA signaling with aging and disease is particularly insidious given its loss directly accelerates pathological processes in addition to the reduction in neuroprotection (Capsoni et al., 2000). Previous reviews have detailed candidate methods for therapeutically leveraging NGF-TrkA interactions to increase neuroprotective functional and stave off pathological processes (Canu et al., 2017; Mufson et al., 2019).
In human studies, BF volume measured with structural MRI is used as a gross measure of BF integrity related to neuronal loss. As such, BF, like many structures in the brain, shows decreased volume with advancing age (Grothe et al., 2012). Longitudinal measures indicate that the BF is particularly vulnerable in aging with rates of atrophy that are similar to other vulnerable regions, like the hippocampus, and higher than global rates of gray matter loss (Grothe et al., 2013). These changes in aging may be linked to preclinical AD pathology: Aβ-positive cognitively normal older adults show greater longitudinal degeneration in the BF compared to Aβ-negative older adults (Schmitz and Nathan Spreng, 2016). In addition, BFCS volumes predict Aβ status and burden in cognitively normal older adults (Grothe et al., 2014; Teipel et al., 2014) and impaired subjects, outperforming hippocampal volume (Teipel et al., 2014). Consistent with early postmortem studies, AD patients show significant BFCS atrophy relative to older adult controls (Teipel et al., 2011). Indeed, diagnostic accuracy for AD using BFCS volumes is on par or better than diagnostic accuracy using hippocampal volume (Grothe et al., 2012; Kilimann et al., 2014). As in preclinical patients, BFCS volume in AD patients is also related to Aβ burden (Kerbler et al., 2015), mirroring animal work linking Aβ deposition to BFCS degeneration.
New perspectives on BFCS in current AD research subfields
Of course, the “amyloid hypothesis of AD” oversimplifies a broad literature with many subfields. As we have already discussed, there are connections between the cholinergic and Aβ-centric models of AD, but the BFCS needs to be better integrated into modern research areas that generally have emerged from work on cortical Aβ, MTL tau and memory deficits (Fig. 2). Here we discuss four such research areas, how their conceptual underpinnings relate to the BFCS, recent studies suggesting a role for BFCS and, finally, future directions that may better elucidate the contributions of the BFCS to each subfield.
Figure 2.
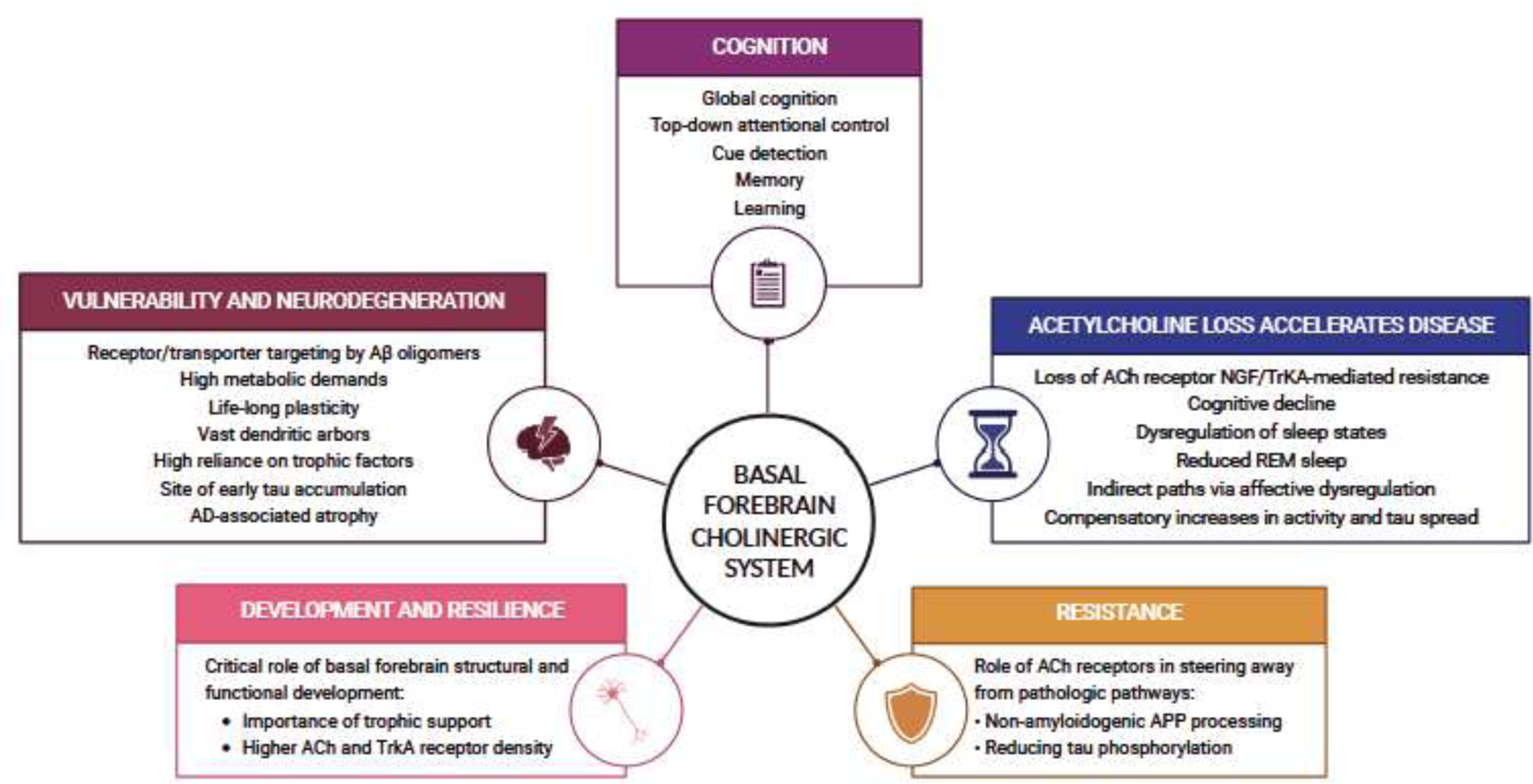
Role of the cholinergic basal forebrain system in Alzheimer’s disease. We highlight the central role of the basal forebrain cholinergic system in supporting optimal cognitive performance and brain health, and highlight its selective vulnerability and contribution to resistance and resilience to Alzheimer’s disease (AD). ACh = acetylcholine; TrkA = tropomyosin receptor kinase A; NGF = nerve growth factor; REM = rapid eye movement; APP = amyloid-beta precursor protein
Resilience and resistance
Neuropathological studies in humans have revealed that individuals who are cognitively normal at death can have significant AD pathology in their brain (Price and Morris, 1999). The concept of cognitive reserve arose from these observations as an explanation for why some people are able to maintain normal cognition in the context of pathology or other brain insults (Stern, 2002). More recently, the terms resilience and resistance have been coined to describe coping with or avoiding pathology, respectively (Arenaza-Urquijo and Vemuri, 2018). These terms have been adopted in neuropathological and in vivo biomarker studies where quantitative measurements of pathology are possible. Resilience, or coping better than predicted with a specific level of pathology, occurs when pathology is present but the usual cognitive decline associated with that pathology is not. In contrast, resistance, or avoiding pathology, is a concept that suggests specific characteristics or factors may make individuals resistant to pathology accumulation.
Animal work suggests that the proper development of the cholinergic system may set the stage for optimal cognitive trajectories (Parikh et al., 2013; Sarter and Bruno, 2004). Equally, it is possible that incompletely matured BFCS or lack of neurotrophic support later in life may limit cognitive capacity and leave individuals vulnerable to cognitive decline following further age-related losses. There is a need for future hypothesis testing to define the extent to which inoptimal BFCS development or reduced NGF trophic support increases one’s vulnerability to dementia and AD. Promising research in this area has leveraged a transgenic mouse model with reduced NGF trophic support (Ruberti et al., 2000). Reduced NGF activity recapitulates major features of AD including cholinergic deficits accompanied by cognitive impairment, elevated age-dependent Aβ pathology, hyperphosphorylated tau and neurodegeneration (Capsoni et al., 2000). Further research is needed to establish whether improper BFCS development is driving these effects (Yegla and Parikh, 2017).
The BFCS innervates prefrontal and temporal lobe regions involved in a broad range of cognitive functions including attention, learning and memory. The concept of cholinergic tone in supporting attention means that cholinergic system dysfunction could impact cognition across domains (Sarter and Bruno, 1997). Indeed, neuromodulator systems like the BFCS represent compelling candidate mechanisms supporting cognitive resilience (Ciampa et al., 2022, 2021) and a robust cholinergic neuromodulatory system could be a mechanism of resilience and protect against cognitive decline. Particularly, cholinergic inputs to prefrontal cortex are implicated in effort-related attentional enhancement via interactions with the dopamine system (Silvestrini et al., 2022; St Peters et al., 2011). Effort-related engagement of prefrontal cortex is diminished in humans with a genetically-determined deficits in cholinergic function (Berry et al., 2015).
Together, the proper development and maintenance of a robust BFCS may imbue cognitive reserve through standard modulatory activity supporting high-order cognitive operations, but also through cholinergically-mediated recruitment of attentional systems to counteract structural and functional losses. In human neuroimaging studies, BFCS atrophy has been shown to relate to worse cognition in older adults but these associations seem to not map onto canonical neuropsychological domains but rather with an overall level of cognition or general intelligence (Grothe et al., 2013; Lammers et al., 2018; Wolf et al., 2014). This supports the idea that an intact BFCS with robust cholinergic signaling could be a mechanism of resilience to age- and pathology-related domain-specific deficits.
Maintenance of an intact BFCS and associated neurotrophic support mechanisms may also underlie resistance to Aβ and tau accumulation. As we have previously discussed, reduced cholinergic signaling and experimental ablation of acetylcholine-producing BF neurons increases Aβ accumulation (Beach et al., 2001, 2000; Roberson and Harrell, 1997; Tropea et al., 2021). Both acetylcholine and NGF are implicated in reducing tau phosphorylation (Hellström-Lindahl, 2000; Nuydens et al., 1997; Sadot et al., 1996). Loss of BFCS and NGF-TrkA signaling appears to dismantle resistance mechanisms and accelerate AD pathologic processes. Future work should probe deeper into the relationship between optimal BFCS function, including supporting attention and learning, and its relationship to 1) preventing pathology deposition and 2) better coping with pathology, especially in the MTL.
Selective vulnerability
The flipside of resilience and resistance is vulnerability. The fact that some neurons are selectively vulnerable to certain pathological processes is a fundamental principle of neurodegenerative disorders that has been incorporated into AD research for many decades. While the idea of selective neuronal vulnerability is not new, modern techniques in both basic science (e.g., single cell profiling, human iPSC models) and neuroimaging (in vivo AD biomarkers, especially for tau) have led to a new era of selective vulnerability research that aims to use preclinical and clinical phenotyping to gain a better understanding why some neurons are susceptible to degeneration in specific disease contexts (Fu et al., 2018).
The vulnerability of the BFCS to AD has been long documented (Davies and Maloney, 1976), but recent work in human tissue has revealed that BFCS is in fact selectively vulnerable to AD-related tauopathy and resilient to tauopathies associated with frontotemporal lobar degeneration (FTLD). Moreover, FTLD with TPD-43-immunoreactive pathology shows virtually no pathology in the BFCS, indicating possible resistance to this pathology (Geula et al., 2021). Taking this a step further, another study recently showed that even within AD subtypes (e.g., hippocampal sparing, limbic-predominant) there are differences in BFCS vulnerability associated with age of onset, APOE4 carrier status and sex (Hanna Al-Shaikh et al., 2020). Emerging research leverages differential gene expression changes in AD animal models and postmortem tissue to identify the molecular mechanisms underlying selective vulnerability of BFCS neurons. Analyses of transcriptomic changes implicate pathways involved in oxidative phosphorylation, oxidative stress, energy metabolism, trophic support, and cell adhesion (Alldred et al., 2021b, 2021a; Beck et al., 2022), which are potential targets for therapeutic intervention.
It has been posited that the vast dendritic arbors and high metabolic demands of lifelong plasticity contribute to BF cholinergic neurons’ vulnerability to AD (Mattson and Magnus, 2006; Wu et al., 2014), and it has long been known that the BFCS is vulnerable to early tau accumulation (Braak and Del Tredici, 2015; Mesulam et al., 2004). Metabolically costly plasticity is a feature the BFCS shares with the MTL, especially the hippocampus (Bartsch and Wulff, 2015). On their face, BF and the MTL seem very different: one is a subcortical structure and the seat of the cholinergic system in the cerebrum and the other is primarily layered cortex serving as the major epicenter of the brain’s episodic memory system. But neurons in BF and MTL structures underlying episodic memory function share broad characteristics including the demands of continual synaptic plasticity due to their involvement in memory and attention (Hasselmo, 2006; Suh et al., 2011; VanderWeele, 2016), and their vulnerability to certain species of pathological tau and AD-related neurodegeneration. Future work should explore whether these shared characteristics mean shared mechanisms of vulnerability to AD pathology. Such cross-system comparisons should also extend to locus coeruleus and dorsal raphe, which are early sites of abnormal tau deposition and have similar characteristics to the BFCS given their role as subcortical neuromodulatory nuclei (Ehrenberg et al., 2017; Grinberg et al., 2009; Matchett et al., 2021).
Trans-synaptic spread of pathology
Current models that posit tau pathology spreads trans-synaptically (Khan et al., 2014; Liu et al., 2012; Wu et al., 2016) have brought the BFCS and the question of its role in AD into a new framework. The extensive projections of BFCS neurons to cortex and the evidence that BFCS is a region of early tau aggregation has led to questions about whether it is a key tau seeding region. Thus, there are two main hypotheses that need to be tested: 1) early tau accumulation in the BF causes neuronal injury and denervation of cholinergic targets resulting in dysfunction of those target regions and 2) early tau in the BF spreads to cholinergic target regions leading to neurotoxicity and degeneration in the target regions. While the field is just beginning to address these hypotheses there are some structural and functional covariance studies in humans that indicate tau in BF may have broader effects across the brain.
Using structural co-variance analyses, it has been shown that BF structure is associated with degeneration in the cortex and amygdala, which indicates that neurogenerative processes in the BFCS and its targets are linked (Schmitz et al., 2018). BFCS volume also predicts longitudinal degeneration of the MTL, while the reverse is not true, and this association is moderated by AD pathology (Fernández-Cabello et al., 2020). Another way to explore the relationship between connectivity and tau spread is using functional connectivity measures from task-free fMRI data, which has been used in part to define components of the human BFCS in vivo (Fritz et al., 2019; Markello et al., 2018). It is possible that compensatory increases in functional connectivity precede disconnection resulting from neuronal loss and that this may speed up the spread of tau pathology. One recent study showed that higher connectivity between BF and amygdala was related to greater AD pathology burden and better memory performance in cognitively healthy older adults (Zeng et al., 2022). More human fMRI research is needed to examine BFCS functional connectivity and tau pathology: functional changes may help explain tau effects on cognition that are not explained by neurodegeneration (Ossenkoppele et al., 2021). Functional imaging and co-variance approaches may also shed light on the degree to which declines in BFCS structure and function is associated with pathological processes in the locus coeruleus and dorsal raphe nucleus, which project directly to the BFCS (Russchen et al., 1985), and are themselves sites of early tau deposition (Ehrenberg et al., 2017; Grinberg et al., 2009; Matchett et al., 2021).
In addition, future complementary research in animal models will be useful to track the degree to which tau seeding in the BFCS affects connected target regions and how BFCS is affected by tau in the entorhinal cortex and locus coeruleus. A recent study in rats examined the impact of tau seeding in the locus coeruleus on connected structures following tonic and phasic locus coeruleus stimulation protocols (Omoluabi et al., 2021). While the methods supporting this research are still under development, application of this approach to the BFCS would be highly novel and promises to shed light on the spatial distribution of tau pathology arising from a BF epicenter, and the firing patterns of BFCS which are most likely to promote pathology spread.
Sleep
In addition to its role in attention, memory and AD the BFCS has also long been implicated in regulating arousal and wakefulness (Gritton et al., 2009; Yamakawa et al., 2016). During sleep, brainstem and BF cholinergic signaling (Baghdoyan et al., 1984; Vazquez and Baghdoyan, 2001) is believed to underlie state transitions and specifically support rapid eye movement (REM) sleep (Irmak and de Lecea, 2014). While NREM sleep is the primary focus in AD sleep research, REM sleep is dysregulated in aging and AD (Zhang et al., 2022), and may contribute to gross disruptions in sleep architecture that impact AD progression. REM sleep decreases with age (Van Cauter et al., 2000), MCI (Sanchez-Espinosa et al., 2014), and AD (Zhang et al., 2022), and has been linked with reduced gray matter volume in AD regions including the BF (Sanchez-Espinosa et al., 2014), and disruptions in blood brain barrier integrity (Gómez-González et al., 2013). Onset to REM sleep is increased in AD patients (Bliwise et al., 1989), which has been recapitulated in mouse models (Huitrón-Reséndiz et al., 2002), and was once promoted as an early AD biomarker. Reduced REM sleep is also a feature of APP/Aβ mouse models (Huitrón-Reséndiz et al., 2002; Sethi et al., 2015; Zhang et al., 2005), combined tau and APP models (Jyoti et al., 2015; Platt et al., 2011), and tau-only models (Holth et al., 2017). Observations that both Aβ-only and tau-only animal models show dysregulation in sleep architecture illustrate that there are multiple interacting pathways connecting AD processes with impoverished sleep.
Recent studies have highlighted the relationship between sleep, aging and AD pathology and sleep has garnered interest as a potentially modifiable risk factor for AD (Lucey et al., 2021; Romanella et al., 2021). Aβ burden in humans is related to changes in sleep duration and non-rapid eye movement (NREM) sleep quality (Mander et al., 2015; Winer et al., 2019). There is also evidence that tau is related to sleep changes in preclinical and early AD (Lucey et al., 2019). Based on these studies, relationships between sleep disruptions and AD pathology appear to be bidirectional. Current models propose accumulation of pathology reduces sleep and, in turn, reduced sleep leads to accelerated pathology deposition by limiting clearance of soluble Aβ and tau (see Wang and Holtzman, 2020 for recent review). There is a need to bridge the gap between the BFCS roles in AD and in sleep and to integrate BFCS into the recent explosion of research showing AD pathology is associated with disordered sleep.
Summary and future directions
In sum, the long, rich history of BFCS in AD research has built a foundation from which to continue to integrate the BFCS into current models of the pathophysiology of AD. Continuing breakthroughs in neuroimaging approaches in humans as well as causal hypothesis testing in animal models promises to further clarify the role of BFCS in AD. In humans, neuroimaging approaches will help to link in vivo pathology, especially tau, to BFCS volume and connectivity. In addition, human PET imaging targeting TrKA receptors has now been established (Bailey et al., 2019) and will enrich our understanding of patterns of cholinergic loss in aging and AD. In animals, tau-seeding studies will help to elucidate the role of BFCS projections in tau pathology spread.
Recent advances in treatments to reduce Aβ plaques using monoclonal antibodies in humans will provide new opportunities to evaluate the impact on BFCS structural and functional integrity. Aβ-reducing interventions may further enliven BFCS-focused research given proposals about Aβ’s negative effect on cholinergic signaling and dysregulation of trophic support. In the future, given the complexity of AD pathophysiology, a combination treatment approach separately targeting Aβ, tau and cholinergic signaling may provide the greatest clinical benefit.
Highlights.
Reexamining basal forebrain in Alzheimer’s is critical to a comprehensive model
Integration of seminal contributions from research in animal models and humans
Discussion of resilience, selective vulnerability, trans-synaptic tau spread, sleep
Acknowledgements:
This research was supported by the National Institutes of Health grants R01-AG074330 (to A.S.B.), K01-AG078443 (to T.M.H.) and R03-AG067033 (to T.M.H). We would like to thank Jordyn L. Cowan for assistance formatting figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests:
The authors have no competing interests to declare.
References
- Allard S, Leon WC, Pakavathkumar P, Bruno MA, Ribeiro-da-Silva A, Cuello AC, 2012. Impact of the NGF Maturation and Degradation Pathway on the Cortical Cholinergic System Phenotype. J. Neurosci 32, 2002–2012. 10.1523/JNEUROSCI.1144-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Lee SH, Stutzmann GE, Ginsberg SD, 2021a. Oxidative Phosphorylation Is Dysregulated Within the Basocortical Circuit in a 6-month old Mouse Model of Down Syndrome and Alzheimer’s Disease. Front Aging Neurosci 13, 707950. 10.3389/fnagi.2021.707950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alldred MJ, Penikalapati SC, Lee SH, Heguy A, Roussos P, Ginsberg SD, 2021b. Profiling Basal Forebrain Cholinergic Neurons Reveals a Molecular Basis for Vulnerability Within the Ts65Dn Model of Down Syndrome and Alzheimer’s Disease. Mol Neurobiol 58, 5141–5162. 10.1007/s12035-021-02453-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloe L, Rocco ML, Bianchi P, Manni L, 2012. Nerve growth factor: from the early discoveries to the potential clinical use. J Transl Med 10, 239. 10.1186/1479-5876-10-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenaza-Urquijo EM, Vemuri P, 2018. Resistance vs resilience to Alzheimer disease: Clarifying terminology for preclinical studies. Neurology 90, 695–703. 10.1212/WNL.0000000000005303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Taubert G, Bigl V, Arendt A, 1988. Amyloid deposition in the nucleus basalis of Meynert complex: a topographic marker for degenerating cell clusters in Alzheimer’s disease. Acta Neuropathol 75, 226–232. 10.1007/BF00690530 [DOI] [PubMed] [Google Scholar]
- Auld DS, Kar S, Quirion R, 1998. β-Amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends in Neurosciences 21, 43–49. 10.1016/S0166-2236(97)01144-2 [DOI] [PubMed] [Google Scholar]
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R, 2002. Alzheimer’s disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 68, 209–245. 10.1016/s0301-0082(02)00079-5 [DOI] [PubMed] [Google Scholar]
- Auld DS, Mennicken F, Quirion R, 2001. Nerve Growth Factor Rapidly Induces Prolonged Acetylcholine Release from Cultured Basal Forebrain Neurons: Differentiation between Neuromodulatory and Neurotrophic Influences. J. Neurosci 21, 3375–3382. 10.1523/JNEUROSCI.21-10-03375.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdoyan HA, Rodrigo-Angulo ML, McCarley RW, Hobson JA, 1984. Site-specific enhancement and suppression of desynchronized sleep signs following cholinergic stimulation of three brainstem regions. Brain Res 306, 39–52. 10.1016/0006-8993(84)90354-8 [DOI] [PubMed] [Google Scholar]
- Bailey JJ, Kaiser L, Lindner S, Wüst M, Thiel A, Soucy J-P, Rosa-Neto P, Scott PJH, Unterrainer M, Kaplan DR, Wängler C, Wängler B, Bartenstein P, Bernard-Gauthier V, Schirrmacher R, 2019. First-in-Human Brain Imaging of [18F]TRACK, a PET tracer for Tropomyosin Receptor Kinases. ACS Chem. Neurosci 10, 2697–2702. 10.1021/acschemneuro.9b00144 [DOI] [PubMed] [Google Scholar]
- Ballinger E, Ananth M, Talmage DA, Role L, 2016. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 91, 1199–1218. 10.1016/j.neuron.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch T, Wulff P, 2015. The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience, Hippocampal vulnerability: from molecules to disease 309, 1–16. 10.1016/j.neuroscience.2015.07.084 [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, Beer B, Lippa AS, 1982. The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. 10.1126/science.7046051 [DOI] [PubMed] [Google Scholar]
- Beach TG, Kuo Y-M, Schwab C, Walker DG, Roher AE, 2001. Reduction of cortical amyloid β levels in guinea pig brain after systemic administration of physostigmine. Neuroscience Letters 310, 21–24. 10.1016/S0304-3940(01)02076-6 [DOI] [PubMed] [Google Scholar]
- Beach TG, Potter PE, Kuo YM, Emmerling MR, Durham RA, Webster SD, Walker DG, Sue LI, Scott S, Layne KJ, Roher AE, 2000. Cholinergic deafferentation of the rabbit cortex: a new animal model of Abeta deposition. Neurosci Lett 283, 9–12. 10.1016/s0304-3940(00)00916-2 [DOI] [PubMed] [Google Scholar]
- Beck JS, Madaj Z, Cheema CT, Kara B, Bennett DA, Schneider JA, Gordon MN, Ginsberg SD, Mufson EJ, Counts SE, 2022. Co-expression network analysis of frontal cortex during the progression of Alzheimer’s disease. Cereb Cortex 32, 5108–5120. 10.1093/cercor/bhac001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, 2014. Synaptic mechanisms underlying cholinergic control of thalamic reticular nucleus neurons. J Physiol 592, 4137–4145. 10.1113/jphysiol.2014.277376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry AS, Blakely RD, Sarter M, Lustig C, 2015. Cholinergic capacity mediates prefrontal engagement during challenges to attention: evidence from imaging genetics. Neuroimage 108, 386–395. 10.1016/j.neuroimage.2014.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry AS, Demeter E, Sabhapathy S, English BA, Blakely RD, Sarter M, Lustig C, 2014. Disposed to distraction: Genetic variation in the cholinergic system influences distractibility but not time-on-task effects. J Cogn Neurosci 26, 1981–1991. 10.1162/jocn_a_00607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM, 1990. Cholinergic stimulation enhances long-term potentiation in the CA1 region of rat hippocampus. Neurosci Lett 119, 207–210. 10.1016/0304-3940(90)90835-w [DOI] [PubMed] [Google Scholar]
- Bliwise DL, Tinklenberg J, Yesavage JA, Davies H, Pursley AM, Petta DE, Widrow L, Guilleminault C, Zarcone VP, Dement WC, 1989. REM latency in Alzheimer’s disease. Biol Psychiatry 25, 320–328. 10.1016/0006-3223(89)90179-0 [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, 2015. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 138, 2814–2833. 10.1093/brain/awv236 [DOI] [PubMed] [Google Scholar]
- Bröcher S, Artola A, Singer W, 1992. Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Research 573, 27–36. 10.1016/0006-8993(92)90110-U [DOI] [PubMed] [Google Scholar]
- Broks P, Preston GC, Traub M, Poppleton P, Ward C, Stahl SM, 1988. Modelling dementia: Effects of scopolamine on memory and attention. Neuropsychologia 26, 685–700. 10.1016/0028-3932(88)90004-8 [DOI] [PubMed] [Google Scholar]
- Brzosko Z, Zannone S, Schultz W, Clopath C, Paulsen O, 2017. Sequential neuromodulation of Hebbian plasticity offers mechanism for effective reward-based navigation. Elife 6, e27756. 10.7554/eLife.27756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G, 2000. Acetylcholine-mediated modulation of striatal function. Trends in Neurosciences 23, 120–126. 10.1016/S0166-2236(99)01501-5 [DOI] [PubMed] [Google Scholar]
- Campenot RB, 1977. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci U S A 74, 4516–4519. 10.1073/pnas.74.10.4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canu N, Amadoro G, Triaca V, Latina V, Sposato V, Corsetti V, Severini C, Ciotti MT, Calissano P, 2017. The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int J Mol Sci 18, 1319. 10.3390/ijms18061319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A, 2000. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proceedings of the National Academy of Sciences 97, 6826–6831. 10.1073/pnas.97.12.6826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carli M, Luschi R, Samanin R, 1997. Dose-related impairment of spatial learning by intrahippocampal scopolamine: antagonism by ondansetron, a 5-HT3 receptor antagonist. Behav Brain Res 82, 185–194. 10.1016/s0166-4328(97)80988-6 [DOI] [PubMed] [Google Scholar]
- Carlsen J, Záborszky L, Heimer L, 1985. Cholinergic projections from the basal forebrain to the basolateral amygdaloid complex: A combined retrograde fluorescent and immunohistochemical study. Journal of Comparative Neurology 234, 155–167. 10.1002/cne.902340203 [DOI] [PubMed] [Google Scholar]
- Cattaneo A, Calissano P, 2012. Nerve Growth Factor and Alzheimer’s Disease: New Facts for an Old Hypothesis. Mol Neurobiol 46, 588–604. 10.1007/s12035-012-8310-9 [DOI] [PubMed] [Google Scholar]
- Ciampa CJ, Parent JH, Harrison TM, Fain RM, Betts MJ, Maass A, Winer JR, Baker SL, Janabi M, Furman DJ, D’Esposito M, Jagust WJ, Berry AS, 2022. Associations among locus coeruleus catecholamines, tau pathology, and memory in aging. Neuropsychopharmacology 47, 1106–1113. 10.1038/s41386-022-01269-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampa CJ, Parent JH, Lapoint MR, Swinnerton KN, Taylor MM, Tennant VR, Whitman AJ, Jagust WJ, Berry AS, 2021. Elevated Dopamine Synthesis as a Mechanism of Cognitive Resilience in Aging. Cereb Cortex bhab 379. 10.1093/cercor/bhab379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello AC, 1996. Effects of trophic factors on the CNS cholinergic phenotype. Prog Brain Res 109, 347–358. 10.1016/s0079-6123(08)62117-2 [DOI] [PubMed] [Google Scholar]
- Davies P, Maloney AJF, 1976. Selective loss of central cholinergic neurons in Alzheimer’s disease. The Lancet 308, 1403. 10.1016/S0140-6736(76)91936-X [DOI] [PubMed] [Google Scholar]
- Davis KL, Thal LJ, Gamzu ER, Davis CS, Woolson RF, Gracon SI, Drachman DA, Schneider LS, Whitehouse PJ, Hoover TM, 1992. A double-blind, placebo-controlled multicenter study of tacrine for Alzheimer’s disease. The Tacrine Collaborative Study Group. N Engl J Med 327, 1253–1259. 10.1056/NEJM199210293271801 [DOI] [PubMed] [Google Scholar]
- De Pablo JM, Ortiz-Caro J, Sanchez-Santed F, Guillamón A, 1991. Effects of diazepam, pentobarbital, scopolamine and the timing of saline injection on learned immobility in rats. Physiol Behav 50, 895–899. 10.1016/0031-9384(91)90411-g [DOI] [PubMed] [Google Scholar]
- Disney AA, Aoki C, Hawken MJ, 2007. Gain Modulation by Nicotine in Macaque V1. Neuron 56, 701–713. 10.1016/j.neuron.2007.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Carmo S, Kannel B, Cuello AC, 2021. The Nerve Growth Factor Metabolic Pathway Dysregulation as Cause of Alzheimer’s Cholinergic Atrophy. Cells 11, 16. 10.3390/cells11010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douchamps V, Jeewajee A, Blundell P, Burgess N, Lever C, 2013. Evidence for Encoding versus Retrieval Scheduling in the Hippocampus by Theta Phase and Acetylcholine. J. Neurosci 33, 8689–8704. 10.1523/JNEUROSCI.4483-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DA, Leavitt J, 1974. Human memory and the cholinergic system. A relationship to aging? Arch Neurol 30, 113–121. 10.1001/archneur.1974.00490320001001 [DOI] [PubMed] [Google Scholar]
- Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, Leite RP, Diehl Rodriguez R, Mejia MB, Rüb U, Farfel JM, de Lucena Ferretti-Rebustini RE, Nascimento CF, Nitrini R, Pasquallucci CA, Jacob-Filho W, Miller B, Seeley WW, Heinsen H, Grinberg LT, 2017. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathol. Appl. Neurobiol 43, 393–408. 10.1111/nan.12387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnestock M, Scott SA, Jetté N, Weingartner JA, Crutcher KA, 1996. Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Brain Res Mol Brain Res 42, 175–178. 10.1016/s0169-328x(96)00193-3 [DOI] [PubMed] [Google Scholar]
- Fernández-Cabello S, Kronbichler M, van Dijk KRA, Goodman JA, Nathan Spreng R, Schmitz TW, 2020. Basal forebrain volume reliably predicts the cortical spread of Alzheimer’s degeneration. Brain 143, 993–1009. 10.1093/brain/awaa012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz HJ, Ray N, Dyrba M, Sorg C, Teipel S, Grothe MJ, 2019. The corticotopic organization of the human basal forebrain as revealed by regionally selective functional connectivity profiles. Human Brain Mapping 40, 868. 10.1002/HBM.24417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Léránth C, 1985. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: A combined light and electron microscopic study. Journal of Comparative Neurology 239, 237–246. 10.1002/cne.902390210 [DOI] [PubMed] [Google Scholar]
- Fu H, Hardy J, Duff KE, 2018. Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21, 1350–1358. 10.1038/s41593-018-0221-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G, Lefcort FB, Letourneau PC, 1997. The trkA Receptor Mediates Growth Cone Turning toward a Localized Source of Nerve Growth Factor. J. Neurosci 17, 5445–5454. 10.1523/JNEUROSCI.17-14-05445.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaykema RPA, Luiten PGM, Nyakas C, Traber J, 1990. Cortical projection patterns of the medial septum-diagonal band complex. Journal of Comparative Neurology 293, 103–124. 10.1002/cne.902930109 [DOI] [PubMed] [Google Scholar]
- Geula C, Dunlop SR, Ayala I, Kawles AS, Flanagan ME, Gefen T, Mesulam M-M, 2021. Basal forebrain cholinergic system in the dementias: Vulnerability, resilience, and resistance. Journal of Neurochemistry 158, 1394–1411. 10.1111/JNC.15471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ, 2006. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. Journal of Neurochemistry 97, 475–487. 10.1111/j.1471-4159.2006.03764.x [DOI] [PubMed] [Google Scholar]
- Giorgi FS, Ryskalin L, Ruffoli R, Biagioni F, Limanaqi F, Ferrucci M, Busceti CL, Bonuccelli U, Fornai F, 2017. The Neuroanatomy of the Reticular Nucleus Locus Coeruleus in Alzheimer’s Disease. Frontiers in Neuroanatomy 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–6. 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- Gómez-González B, Hurtado-Alvarado G, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez-Moctezuma J, 2013. REM sleep loss and recovery regulates blood-brain barrier function. Curr Neurovasc Res 10, 197–207. 10.2174/15672026113109990002 [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Rüb U, Ferretti REL, Nitrini R, Farfel JM, Polichiso L, Gierga K, Jacob-Filho W, Heinsen H, Brazilian Brain Bank Study Group, 2009. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol 35, 406–416. 10.1111/j.1365-2990.2009.00997.x [DOI] [PubMed] [Google Scholar]
- Gritton HJ, Howe WM, Mallory CS, Hetrick VL, Berke JD, Sarter M, 2016. Cortical cholinergic signaling controls the detection of cues. Proc Natl Acad Sci U S A 113, E1089–1097. 10.1073/pnas.1516134113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritton HJ, Sutton BC, Martinez V, Sarter M, Lee TM, 2009. Interactions between cognition and circadian rhythms: Attentional demands modify circadian entrainment. Behavioral Neuroscience 123, 937–948. 10.1037/a0017128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel S, 2013. Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer’s disease. Neurobiology of Aging 34, 1210–1220. 10.1016/j.neurobiolaging.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M, Heinsen H, Teipel SJ, 2012. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol Psychiatry 71, 805–813. 10.1016/j.biopsych.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Ewers M, Krause B, Heinsen H, Teipel SJ, 2014. Basal forebrain atrophy and cortical amyloid deposition in nondemented elderly subjects. Alzheimers Dement 10, S344–S353. 10.1016/j.jalz.2013.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Smith KG, Alexander GM, Guerreiro I, Dudek SM, Gutkin B, Jensen P, Yakel JL, 2020. Hippocampal Interneuronal α7 nAChRs Modulate Theta Oscillations in Freely Moving Mice. Cell Rep 31, 107740. 10.1016/j.celrep.2020.107740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez H, Miranda MI, Bermúdez-Rattoni F, 1997. Learning Impairment and Cholinergic Deafferentation after Cortical Nerve Growth Factor Deprivation. J. Neurosci 17, 3796–3803. 10.1523/JNEUROSCI.17-10-03796.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Mesulam M-M, Cuello AC, Khachaturian AS, Vergallo A, Farlow MR, Snyder PJ, Giacobini E, Khachaturian ZS, Cholinergic System Working Group, and for the A.P.M.I. (APMI), 2019. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J Prev Alzheimers Dis 6, 2–15. 10.14283/jpad.2018.43 [DOI] [PubMed] [Google Scholar]
- Hanna Al-Shaikh FS, Duara R, Crook JE, Lesser ER, Schaeverbeke J, Hinkle KM, Ross OA, Ertekin-Taner N, Pedraza O, Dickson DW, Graff-Radford NR, Murray ME, 2020. Selective Vulnerability of the Nucleus Basalis of Meynert Among Neuropathologic Subtypes of Alzheimer Disease. JAMA Neurol 77, 225–233. 10.1001/jamaneurol.2019.3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, 2006. The Role of Acetylcholine in Learning and Memory. Current opinion in neurobiology 16, 710. 10.1016/J.CONB.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti F, 1986. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci 6, 2155–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström-Lindahl E, 2000. Modulation of β-amyloid precursor protein processing and tau phosphorylation by acetylcholine receptors. European Journal of Pharmacology 393, 255–263. 10.1016/S0014-2999(00)00028-5 [DOI] [PubMed] [Google Scholar]
- Herrero JL, Roberts MJ, Delicato LS, Gieselmann MA, Dayan P, Thiele A, 2008. Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature 454, 1110–1114. 10.1038/nature07141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, Mahan TE, Robinson GO, Rocha A, Holtzman DM, 2017. Altered sleep and EEG power in the P301S Tau transgenic mouse model. Ann Clin Transl Neurol 4, 180–190. 10.1002/acn3.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe WM, Berry AS, Francois J, Gilmour G, Carp JM, Tricklebank M, Lustig C, Sarter M, 2013. Prefrontal cholinergic mechanisms instigating shifts from monitoring for cues to cue-guided performance: converging electrochemical and fMRI evidence from rats and humans. J. Neurosci 33, 8742–8752. 10.1523/JNEUROSCI.5809-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe WM, Gritton HJ, Lusk NA, Roberts EA, Hetrick VL, Berke JD, Sarter M, 2017. Acetylcholine Release in Prefrontal Cortex Promotes Gamma Oscillations and Theta-Gamma Coupling during Cue Detection. J Neurosci 37, 3215–3230. 10.1523/JNEUROSCI.2737-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitrón-Reséndiz S, Sánchez-Alavez M, Gallegos R, Berg G, Crawford E, Giacchino JL, Games D, Henriksen SJ, Criado JR, 2002. Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Res 928, 126–137. 10.1016/s0006-8993(01)03373-x [DOI] [PubMed] [Google Scholar]
- Irmak SO, de Lecea L, 2014. Basal forebrain cholinergic modulation of sleep transitions. Sleep 37, 1941–1951. 10.5665/sleep.4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iulita MF, Cuello AC, 2014. Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol Sci 35, 338–348. 10.1016/j.tips.2014.04.010 [DOI] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Elliott C, Masliah E, Ryan L, Silverberg N, 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & Dementia 14, 535–562. 10.1016/J.JALZ.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W, 2018. Imaging the evolution and pathophysiology of Alzheimer disease. Nature Reviews Neuroscience 19, 687–700. 10.1038/s41583-018-0067-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TA, Weiss-Cowie S, Hopton Z, Verhaeghen P, Dotson VM, Duarte A, 2021. Depression and episodic memory across the adult lifespan: A meta-analytic review. Psychol Bull 147, 1184–1214. 10.1037/bul0000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Son GH, Winters BL, Kim MJ, Whitcomb DJ, Dickinson BA, Lee Y-B, Futai K, Amici M, Sheng M, Collingridge GL, Cho K, 2010. Muscarinic receptors induce LTD of NMDAR EPSCs via a mechanism involving hippocalcin, AP2 and PSD-95. Nat Neurosci 13, 1216–1224. 10.1038/nn.2636 [DOI] [PubMed] [Google Scholar]
- Jyoti A, Plano A, Riedel G, Platt B, 2015. Progressive age-related changes in sleep and EEG profiles in the PLB1Triple mouse model of Alzheimer’s disease. Neurobiol Aging 36, 2768–2784. 10.1016/j.neurobiolaging.2015.07.001 [DOI] [PubMed] [Google Scholar]
- Kar S, Issa AM, Seto D, Auld DS, Collier B, Quirion R, 1998. Amyloid β-Peptide Inhibits High-Affinity Choline Uptake and Acetylcholine Release in Rat Hippocampal Slices. Journal of Neurochemistry 70, 2179–2187. 10.1046/j.1471-4159.1998.70052179.x [DOI] [PubMed] [Google Scholar]
- Kar S, Seto D, Gaudreau P, Quirion R, 1996. Beta-amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal slices. J. Neurosci 16, 1034–1040. 10.1523/JNEUROSCI.16-03-01034.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI, 2018. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathologica 136, 57–67. 10.1007/s00401-018-1855-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbler GM, Fripp J, Rowe CC, Villemagne VL, Salvado O, Rose S, Coulson EJ, Alzheimer’s Disease Neuroimaging Initiative, 2015. Basal forebrain atrophy correlates with amyloid β burden in Alzheimer’s disease. Neuroimage Clin 7, 105–113. 10.1016/j.nicl.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, Mayeux R, Duff KE, Small SA, 2014. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nature Neuroscience 17, 304–311. 10.1038/nn.3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilimann I, Grothe M, Heinsen H, Alho EJL, Grinberg L, Amaro E, Dos Santos GAB, da Silva RE, Mitchell AJ, Frisoni GB, Bokde ALW, Fellgiebel A, Filippi M, Hampel H, Klöppel S, Teipel SJ, 2014. Subregional basal forebrain atrophy in Alzheimer’s disease: a multicenter study. J Alzheimers Dis 40, 687–700. 10.3233/JAD-132345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipper M, da Penha Berzaghi M, Blöchl A, Breer H, Thoenen H, Lindholm D, 1994. Positive Feedback between Acetylcholine and the Neurotrophins Nerve Growth Factor and Brain-derived Neurotrophic Factor in the Rat Hippocampus. European Journal of Neuroscience 6, 668–671. 10.1111/j.1460-9568.1994.tb00312.x [DOI] [PubMed] [Google Scholar]
- Kondo H, Zaborszky L, 2016. Topographic organization of the basal forebrain projections to the perirhinal, postrhinal, and entorhinal cortex in rats. J Comp Neurol 524, 2503–2515. 10.1002/cne.23967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krettek JE, Price JL, 1978. Amygdaloid projections to subcortical structures within the basal forebrain and brainstem in the rat and cat. J Comp Neurol 178, 225–254. 10.1002/cne.901780204 [DOI] [PubMed] [Google Scholar]
- Kromer LF, 1987. Nerve growth factor treatment after brain injury prevents neuronal death. Science 235, 214–216. 10.1126/science.3798108 [DOI] [PubMed] [Google Scholar]
- Lammers F, Borchers F, Feinkohl I, Hendrikse J, Kant IMJ, Kozma P, Pischon T, Slooter AJC, Spies C, van Montfort SJT, Zacharias N, Zaborszky L, Winterer G, 2018. Basal forebrain cholinergic system volume is associated with general cognitive ability in the elderly. Neuropsychologia 119, 145–156. 10.1016/J.NEUROPSYCHOLOGIA.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz RA, Baker JD, Locke C, Rueter LE, Mohler EG, Wesnes K, Abi-Saab W, Saltarelli MD, 2012. The scopolamine model as a pharmacodynamic marker in early drug development. Psychopharmacology 220, 97–107. 10.1007/s00213-011-2456-4 [DOI] [PubMed] [Google Scholar]
- Lim YY, Maruff P, Schindler R, Ott BR, Salloway S, Yoo DC, Noto RB, Santos CY, Snyder PJ, 2015. Disruption of cholinergic neurotransmission exacerbates Aβ-related cognitive impairment in preclinical Alzheimer’s disease. Neurobiol Aging 36, 2709–2715. 10.1016/j.neurobiolaging.2015.07.009 [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K, 2012. Trans-synaptic spread of tau pathology in vivo. PloS one 7, e31302. 10.1371/journal.pone.0031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK, 2001. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A 98, 4734–4739. 10.1073/pnas.081553598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CB, Henderson Z, 2010. Nicotine induction of theta frequency oscillations in rodent hippocampus in vitro. Neuroscience 166, 84–93. 10.1016/j.neuroscience.2009.11.072 [DOI] [PubMed] [Google Scholar]
- Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TLS, Holtzman DM, 2019. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci Transl Med 11, eaau6550. 10.1126/scitranslmed.aau6550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey BP, Wisch J, Boerwinkle AH, Landsness EC, Toedebusch CD, McLeland JS, Butt OH, Hassenstab J, Morris JC, Ances BM, Holtzman DM, 2021. Sleep and longitudinal cognitive performance in preclinical and early symptomatic Alzheimer’s disease. Brain 144, 2852–2862. 10.1093/brain/awab272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP, 2015. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci 18, 1051–1057. 10.1038/nn.4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markello RD, Spreng RN, Luh WM, Anderson AK, De Rosa E, 2018. Segregation of the human basal forebrain using resting state functional MRI. NeuroImage 173, 287–297. 10.1016/J.NEUROIMAGE.2018.02.042 [DOI] [PubMed] [Google Scholar]
- Marucci G, Buccioni M, Ben DD, Lambertucci C, Volpini R, Amenta F, 2021. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 190, 108352. 10.1016/j.neuropharm.2020.108352 [DOI] [PubMed] [Google Scholar]
- Matchett BJ, Grinberg LT, Theofilas P, Murray ME, 2021. The mechanistic link between selective vulnerability of the locus coeruleus and neurodegeneration in Alzheimer’s disease. Acta Neuropathol 10.1007/s00401-020-02248-1 [DOI] [PMC free article] [PubMed]
- Mattson MP, Magnus T, 2006. Ageing and neuronal vulnerability. Nature Reviews Neuroscience 2006 7:4 7, 278–294. 10.1038/nrn1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena-Segovia J, 2016. Structural and functional considerations of the cholinergic brainstem. J Neural Transm (Vienna) 123, 731–736. 10.1007/s00702-016-1530-9 [DOI] [PubMed] [Google Scholar]
- Mesulam M, 2012. Cholinergic Aspects of Aging and Alzheimer’s Disease. Biol Psychiatry 71, 760–761. 10.1016/j.biopsych.2012.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M, Shaw P, Mash D, Weintraub S, 2004. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Annals of Neurology 55, 815–828. 10.1002/ana.20100 [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, 1984. Neural inputs into the nucleus basalis of the substantia innominata (Ch4) in the rhesus monkey. Brain 107 ( Pt 1), 253–274. 10.1093/brain/107.1.253 [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH, 1983. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol 214, 170–197. 10.1002/cne.902140206 [DOI] [PubMed] [Google Scholar]
- Mintzer MZ, Griffiths RR, 2003. Lorazepam and scopolamine: A single-dose comparison of effects on human memory and attentional processes. Experimental and Clinical Psychopharmacology 11, 56–72. 10.1037/1064-1297.11.1.56 [DOI] [PubMed] [Google Scholar]
- Mitra S, Behbahani H, Eriksdotter M, 2019. Innovative Therapy for Alzheimer’s Disease-With Focus on Biodelivery of NGF. Frontiers in Neuroscience 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molchan SE, Martinez RA, Hill JL, Weingartner HJ, Thompson K, Vitiello B, Sunderland T, 1992. Increased cognitive sensitivity to scopolamine with age and a perspective on the scopolamine model. Brain Research Reviews 17, 215–226. 10.1016/0165-0173(92)90017-G [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel B, 2018. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol 136, 663–689. 10.1007/s00401-018-1918-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Conner JM, Kordower JH, 1995. Nerve growth factor in Alzheimer’s disease: defective retrograde transport to nucleus basalis. Neuroreport 6, 1063–1066. 10.1097/00001756-199505090-00028 [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Ginsberg SD, Mahady L, Perez SE, Massa SM, Longo FM, Ikonomovic MD, 2019. Nerve Growth Factor Pathobiology During the Progression of Alzheimer’s Disease. Front Neurosci 13, 533. 10.3389/fnins.2019.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir JL, 1997. Acetylcholine, aging, and Alzheimer’s disease. Pharmacol Biochem Behav 56, 687–696. 10.1016/s0091-3057(96)00431-5 [DOI] [PubMed] [Google Scholar]
- Newman EL, Gillet SN, Climer JR, Hasselmo ME, 2013. Cholinergic Blockade Reduces Theta-Gamma Phase Amplitude Coupling and Speed Modulation of Theta Frequency Consistent with Behavioral Effects on Encoding. J Neurosci 33, 19635–19646. 10.1523/JNEUROSCI.2586-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH, 1992. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307. 10.1126/science.1411529 [DOI] [PubMed] [Google Scholar]
- Nuydens R, Dispersyn G, de Jong M, van den Kieboom G, Borgers M, Geerts H, 1997. Aberrant tau phosphorylation and neurite retraction during NGF deprivation in PC12 cells. Biochem Biophys Res Commun 240, 687–691. 10.1006/bbrc.1997.7721 [DOI] [PubMed] [Google Scholar]
- Obermayer J, Verhoog MB, Luchicchi A, Mansvelder HD, 2017. Cholinergic Modulation of Cortical Microcircuits Is Layer-Specific: Evidence from Rodent, Monkey and Human Brain. Frontiers in Neural Circuits 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara G, Coumis U, Ma SY, Kehr J, Mahoney S, Bacon A, Allen SJ, Holmes F, Kahl U, Wang FH, Kearns IR, Ove-Ogren S, Dawbarn D, Mufson EJ, Davies C, Dawson G, Wynick D, 2000. Galanin regulates the postnatal survival of a subset of basal forebrain cholinergic neurons. Proc Natl Acad Sci U S A 97, 11569–11574. 10.1073/pnas.210254597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoluabi T, Torraville SE, Maziar A, Ghosh A, Power KD, Reinhardt C, Harley CW, Yuan Q, 2021. Novelty-like activation of locus coeruleus protects against deleterious human pretangle tau effects while stress-inducing activation worsens its effects. Alzheimers Dement (N Y) 7, e12231. 10.1002/trc2.12231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Smith R, Mattsson-Carlgren N, Groot C, Leuzy A, Strandberg O, Palmqvist S, Olsson T, Jögi J, Stormrud E, Cho H, Ryu YH, Choi JY, Boxer AL, Gorno-Tempini ML, Miller BL, Soleimani-Meigooni D, Iaccarino L, La Joie R, Baker S, Borroni E, Klein G, Pontecorvo MJ, Devous MD, Jagust WJ, Lyoo CH, Rabinovici GD, Hansson O, 2021. Accuracy of Tau Positron Emission Tomography as a Prognostic Marker in Preclinical and Prodromal Alzheimer Disease: A Head-to-Head Comparison against Amyloid Positron Emission Tomography and Magnetic Resonance Imaging. JAMA Neurology 78, 961–971. 10.1001/jamaneurol.2021.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pa J, Berry AS, Compagnone M, Boccanfuso J, Greenhouse I, Rubens MT, Johnson JK, Gazzaley A, 2013. Cholinergic enhancement of functional networks in older adults with mild cognitive impairment. Ann Neurol 73, 762–773. 10.1002/ana.23874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Howe WM, Welchko RM, Naughton SX, D’Amore DE, Han DH, Deo M, Turner DL, Sarter M, 2013. Diminished trkA receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur J Neurosci 37, 278–293. 10.1111/ejn.12090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M, 2007. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron 56, 141–154. 10.1016/j.neuron.2007.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD, 2000. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron 25, 345–357. 10.1016/s0896-6273(00)80899-5 [DOI] [PubMed] [Google Scholar]
- Patil MM, Linster C, Lubenov E, Hasselmo ME, 1998. Cholinergic agonist carbachol enables associative long-term potentiation in piriform cortex slices. J Neurophysiol 80, 2467–2474. 10.1152/jn.1998.80.5.2467 [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL, 2001. β-Amyloid 1–42 Peptide Directly Modulates Nicotinic Receptors in the Rat Hippocampal Slice. J. Neurosci 21, RC120–RC120. 10.1523/JNEUROSCI.21-01-j0003.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Brunzell DH, Caldarone BJ, 2002. Effect of nicotine and nicotinic receptors on anxiety and depression. NeuroReport 13, 1097–1106. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Higley MJ, Mineur YS, 2012. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. 10.1016/j.neuron.2012.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Lewis AS, van Schalkwyk GI, Mineur YS, 2015. Mood and anxiety regulation by nicotinic acetylcholine receptors: a potential pathway to modulate aggression and related behavioral states. Neuropharmacology 96, 235–243. 10.1016/j.neuropharm.2014.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt B, Drever B, Koss D, Stoppelkamp S, Jyoti A, Plano A, Utan A, Merrick G, Ryan D, Melis V, Wan H, Mingarelli M, Porcu E, Scrocchi L, Welch A, Riedel G, 2011. Abnormal Cognition, Sleep, EEG and Brain Metabolism in a Novel Knock-In Alzheimer Mouse, PLB1. PLoS One 6, e27068. 10.1371/journal.pone.0027068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin DA, Jarvik LF, 1986. Cholinergic Dysfunction in Alzheimer Disease: Cause or Effect?**This research was supported in part by National Institute of Mental Health grants MH17251 and MH36205, the Henry J. Kaiser Foundation, and the Veterans Administration., in: Van Ree JM, Matthysse S (Eds.), Progress in Brain Research Elsevier, pp. 91–103. 10.1016/S0079-6123(08)60644-5 [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC, 1999. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Annals of Neurology 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Rafii MS, Baumann TL, Bakay RAE, Ostrove JM, Siffert J, Fleisher AS, Herzog CD, Barba D, Pay M, Salmon DP, Chu Y, Kordower JH, Bishop K, Keator D, Potkin S, Bartus RT, 2014. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimer’s & Dementia 10, 571–581. 10.1016/j.jalz.2013.09.004 [DOI] [PubMed] [Google Scholar]
- Rafii MS, Tuszynski MH, Thomas RG, Barba D, Brewer JB, Rissman RA, Siffert J, Aisen PS, for the AAV2-NGF Study Team, 2018. Adeno-Associated Viral Vector (Serotype 2)–Nerve Growth Factor for Patients With Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurology 75, 834–841. 10.1001/jamaneurol.2018.0233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson MR, Harrell LE, 1997. Cholinergic activity and amyloid precursor protein metabolism. Brain Res Brain Res Rev 25, 50–69. 10.1016/s0165-0173(97)00016-7 [DOI] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, 1995. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–8. 10.1038/376775a0 [DOI] [PubMed] [Google Scholar]
- Rogers SL, Friedhoff LT, 1996. The efficacy and safety of donepezil in patients with Alzheimer’s disease: results of a US Multicentre, Randomized, Double-Blind, Placebo-Controlled Trial. The Donepezil Study Group. Dementia 7, 293–303. 10.1159/000106895 [DOI] [PubMed] [Google Scholar]
- Romanella SM, Roe D, Tatti E, Cappon D, Paciorek R, Testani E, Rossi A, Rossi S, Santarnecchi E, 2021. The Sleep Side of Aging and Alzheimer’s Disease. Sleep Med 77, 209–225. 10.1016/j.sleep.2020.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V, 1998a. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog Neurobiol 56, 541–569. 10.1016/s0301-0082(98)00044-6 [DOI] [PubMed] [Google Scholar]
- Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V, 1998b. p75 and TrkA receptor signaling independently regulate amyloid precursor protein mRNA expression, isoform composition, and protein secretion in PC12 cells. J Neurochem 71, 757–766. 10.1046/j.1471-4159.1998.71020757.x [DOI] [PubMed] [Google Scholar]
- ROSSOR MN, GARRETT NJ, JOHNSON AL, MOUNTJOY CQ, ROTH M, IVERSEN LL, 1982. A POST-MORTEM STUDY OF THE CHOLINERGIC AND GABA SYSTEMS IN SENILE DEMENTIA. Brain 105, 313–330. 10.1093/brain/105.2.313 [DOI] [PubMed] [Google Scholar]
- Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J, Gonfloni S, Rossi G, Berardi N, Cattaneo A, 2000. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J Neurosci 20, 2589–2601. 10.1523/JNEUROSCI.20-07-02589.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russchen FT, Amaral DG, Price JL, 1985. The afferent connections of the substantia innominata in the monkey, Macaca fascicularis. J Comp Neurol 242, 1–27. 10.1002/cne.902420102 [DOI] [PubMed] [Google Scholar]
- Sadot E, Gurwitz D, Barg J, Behar L, Ginzburg I, Fisher A, 1996. Activation of m1 Muscarinic Acetylcholine Receptor Regulates τ Phosphorylation in Transfected PC12 Cells. Journal of Neurochemistry 66, 877–880. 10.1046/j.1471-4159.1996.66020877.x [DOI] [PubMed] [Google Scholar]
- Safer DJ, Allen RP, 1971. The central effects of scopolamine in man. Biol Psychiatry 3, 347–355. [PubMed] [Google Scholar]
- Sala R, Viegi A, Rossi FM, Pizzorusso T, Bonanno G, Raiteri M, Maffei L, 1998. Nerve growth factor and brain-derived neurotrophic factor increase neurotransmitter release in the rat visual cortex. European Journal of Neuroscience 10, 2185–2191. 10.1046/j.1460-9568.1998.00227.x [DOI] [PubMed] [Google Scholar]
- Sanchez-Espinosa MP, Atienza M, Cantero JL, 2014. Sleep deficits in mild cognitive impairment are related to increased levels of plasma amyloid-β and cortical thinning. Neuroimage 98, 395–404. 10.1016/j.neuroimage.2014.05.027 [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno JP, 2004. Developmental origins of the age-related decline in cortical cholinergic function and associated cognitive abilities. Neurobiol Aging 25, 1127–1139. 10.1016/j.neurobiolaging.2003.11.011 [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno JP, 1997. Cognitive functions of cortical acetylcholine: toward a unifying hypothesis. Brain Research Reviews 23, 28–46. 10.1016/S0165-0173(96)00009-4 [DOI] [PubMed] [Google Scholar]
- Sarter M, Lustig C, 2020. Forebrain Cholinergic Signaling: Wired and Phasic, Not Tonic, and Causing Behavior. J. Neurosci 40, 712–719. 10.1523/JNEUROSCI.1305-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C, Blakely RD, Koshy Cherian A, 2016. Cholinergic genetics of visual attention: Human and mouse choline transporter capacity variants influence distractibility. J Physiol Paris 110, 10–18. 10.1016/j.jphysparis.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberg G, Bird T, Wijsman E, Orr H, Anderson L, Nemens E, White J, Bonnycastle L, Weber J, Alonso M, et al. , 1992. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 258, 668–671. 10.1126/science.1411576 [DOI] [PubMed] [Google Scholar]
- Schmitz TW, Mur M, Aghourian M, Bedard MA, Spreng RN, 2018. Longitudinal Alzheimer’s Degeneration Reflects the Spatial Topography of Cholinergic Basal Forebrain Projections. Cell Reports 24, 38–46. 10.1016/J.CELREP.2018.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz TW, Nathan Spreng R, 2016. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nature Communications 7. 10.1038/ncomms13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H, Ferree AW, Isacson O, 2002. Cortico-hippocampal APP and NGF levels are dynamically altered by cholinergic muscarinic antagonist or M1 agonist treatment in normal mice: Change of APP and NGF levels by muscarinic stimulation. European Journal of Neuroscience 15, 498–506. 10.1046/j.0953-816x.2001.01884.x [DOI] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee H-K, Kirkwood A, 2007. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron 55, 919–929. 10.1016/j.neuron.2007.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi M, Joshi SS, Webb RL, Beckett TL, Donohue KD, Murphy MP, O’Hara BF, Duncan MJ, 2015. Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer’s disease. Neuroscience 290, 80–89. 10.1016/j.neuroscience.2015.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel GJ, Chauhan NB, 2000. Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Research Reviews 33, 199–227. 10.1016/S0165-0173(00)00030-8 [DOI] [PubMed] [Google Scholar]
- Silvestrini N, Musslick S, Berry AS, Vassena E, 2022. An integrative effort: Bridging motivational intensity theory and recent neurocomputational and neuronal models of effort and control allocation. Psychol Rev 10.1037/rev0000372 [DOI] [PubMed]
- Snyder HR, Miyake A, Hankin BL, 2015. Advancing understanding of executive function impairments and psychopathology: Bridging the gap between clinical and cognitive approaches. Frontiers in Psychology 6. 10.3389/fpsyg.2015.00328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC, 2001. Nerve Growth Factor Signaling, Neuroprotection, and Neural Repair. Annual Review of Neuroscience 24, 1217–1281. 10.1146/annurev.neuro.24.1.1217 [DOI] [PubMed] [Google Scholar]
- St Peters M, Demeter E, Lustig C, Bruno JP, Sarter M, 2011. Enhanced control of attention by stimulating mesolimbic-corticopetal cholinergic circuitry. J Neurosci 31, 9760–9771. 10.1523/JNEUROSCI.1902-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbusch HW, 1981. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience 6, 557–618. 10.1016/0306-4522(81)90146-9 [DOI] [PubMed] [Google Scholar]
- Stern Y, 2002. What is cognitive reserve? Theory and research application of the reserve concept. Journal of the International Neuropsychological Society : JINS 8, 448–60. [PubMed] [Google Scholar]
- Suh J, Rivest AJ, Nakashiba T, Tominaga T, Tonegawa S, 2011. Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science 334, 1415–1420. 10.1126/science.1210125 [DOI] [PubMed] [Google Scholar]
- Swanson LW, Hartman BK, 1975. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol 163, 467–505. 10.1002/cne.901630406 [DOI] [PubMed] [Google Scholar]
- Teipel S, Heinsen H, Amaro E, Grinberg LT, Krause B, Grothe M, Alzheimer’s Disease Neuroimaging Initiative, 2014. Cholinergic basal forebrain atrophy predicts amyloid burden in Alzheimer’s disease. Neurobiol Aging 35, 482–491. 10.1016/j.neurobiolaging.2013.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teipel SJ, Meindl T, Grinberg L, Grothe M, Cantero JL, Reiser MF, Möller HJ, Heinsen H, Hampel H, 2011. The cholinergic system in mild cognitive impairment and Alzheimer’s disease: an in vivo MRI and DTI study. Hum Brain Mapp 32, 1349–1362. 10.1002/hbm.21111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea MR, Li Puma DD, Melone M, Gulisano W, Arancio O, Grassi C, Conti F, Puzzo D, 2021. Genetic deletion of α7 nicotinic acetylcholine receptors induces an age-dependent Alzheimer’s disease-like pathology. Progress in Neurobiology 206, 102154. 10.1016/j.pneurobio.2021.102154 [DOI] [PubMed] [Google Scholar]
- Tuttle R, O’Leary DD, 1998. Neurotrophins rapidly modulate growth cone response to the axon guidance molecule, collapsin-1. Mol Cell Neurosci 11, 1–8. 10.1006/mcne.1998.0671 [DOI] [PubMed] [Google Scholar]
- Van Broeckhoven C, Backhovens H, Cruts M, De Winter G, Bruyland M, Cras P, Martin JJ, 1992. Mapping of a gene predisposing to early-onset Alzheimer’s disease to chromosome 14q24.3. Nat Genet 2, 335–339. 10.1038/ng1292-335 [DOI] [PubMed] [Google Scholar]
- Van Cauter E, Leproult R, Plat L, 2000. Age-related changes in slow wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA 284, 861–868. 10.1001/jama.284.7.861 [DOI] [PubMed] [Google Scholar]
- van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, Froelich L, Katayama S, Sabbagh M, Vellas B, Watson D, Dhadda S, Irizarry M, Kramer LD, Iwatsubo T, 2023. Lecanemab in Early Alzheimer’s Disease. New England Journal of Medicine 388, 9–21. 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- VanderWeele TJ, 2016. Mediation Analysis: A Practitioner’s Guide 10.1146/annurev-publhealth-032315-021402 37, 17–32. [DOI] [PubMed] [Google Scholar]
- Vazquez J, Baghdoyan HA, 2001. Basal forebrain acetylcholine release during REM sleep is significantly greater than during waking. Am J Physiol Regul Integr Comp Physiol 280, R598–601. 10.1152/ajpregu.2001.280.2.R598 [DOI] [PubMed] [Google Scholar]
- Wallenstein GV, Vago DR, 2001. Intrahippocampal scopolamine impairs both acquisition and consolidation of contextual fear conditioning. Neurobiol Learn Mem 75, 245–252. 10.1006/nlme.2001.4005 [DOI] [PubMed] [Google Scholar]
- Wang C, Holtzman DM, 2020. Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology 45, 104–120. 10.1038/s41386-019-0478-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse P, Price D, Struble R, Clark A, Coyle J, DeLong MR, 1982. Alzheimer’s Disease and Senile Dementia: Loss of Neurons in the Basal Forebrain. Science 215, 1237–1239. 10.1126/science.7058341 [DOI] [PubMed] [Google Scholar]
- Williams LR, Varon S, Peterson GM, Wictorin K, Fischer W, Bjorklund A, Gage FH, 1986. Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc Natl Acad Sci U S A 83, 9231–9235. 10.1073/pnas.83.23.9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S, Johnston D, 1990. Muscarinic depression of synaptic transmission at the hippocampal mossy fiber synapse. J Neurophysiol 64, 1089–1097. 10.1152/jn.1990.64.4.1089 [DOI] [PubMed] [Google Scholar]
- Winer JR, Mander BA, Helfrich RF, Maass A, Harrison TM, Baker SL, Knight RT, Jagust WJ, Walker MP, 2019. Sleep as a Potential Biomarker of Tau and β-Amyloid Burden in the Human Brain. The Journal of neuroscience : the official journal of the Society for Neuroscience 39, 6315–6324. 10.1523/JNEUROSCI.0503-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf BA, Wertkin AM, Jolly YC, Yasuda RP, Wolfe BB, Konrad RJ, Manning D, Ravi S, Williamson JR, Lee VM-Y, 1995. Muscarinic Regulation of Alzheimer’s Disease Amyloid Precursor Protein Secretion and Amyloid β-Protein Production in Human Neuronal NT2N Cells (∗). Journal of Biological Chemistry 270, 4916–4922. 10.1074/jbc.270.9.4916 [DOI] [PubMed] [Google Scholar]
- Wolf D, Grothe M, Fischer FU, Heinsen H, Kilimann I, Teipel S, Fellgiebel A, 2014. Association of basal forebrain volumes and cognition in normal aging. Neuropsychologia 53, 54–63. 10.1016/j.neuropsychologia.2013.11.002 [DOI] [PubMed] [Google Scholar]
- Woolf NJ, 1991. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol 37, 475–524. 10.1016/0301-0082(91)90006-m [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Milov AM, Schweitzer ES, Roghani A, 2001. Elevation of Nerve Growth Factor and Antisense Knockdown of TrkA Receptor during Contextual Memory Consolidation. J. Neurosci 21, 1047–1055. 10.1523/JNEUROSCI.21-03-01047.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J, 2014. Complete morphologies of basal forebrain cholinergic neurons in the mouse. eLife 2014. 10.7554/eLife.02444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RACM, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE, 2016. Neuronal activity enhances tau propagation and tau pathology in vivo. Nature Neuroscience 19, 1085–1092. 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa GR, Basu P, Cortese F, MacDonnell J, Whalley D, Smith VM, Antle MC, 2016. The cholinergic forebrain arousal system acts directly on the circadian pacemaker. Proc Natl Acad Sci U S A 113, 13498–13503. 10.1073/pnas.1610342113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegla B, Parikh V, 2017. Developmental suppression of forebrain trkA receptors and attentional capacities in aging rats: A longitudinal study. Behav Brain Res 335, 111–121. 10.1016/j.bbr.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Záborszky L, Cullinan WE, Braun A, 1991. Afferents to basal forebrain cholinergic projection neurons: an update. Adv Exp Med Biol 295, 43–100. 10.1007/978-1-4757-0145-6_2 [DOI] [PubMed] [Google Scholar]
- Záborszky L, Gombkoto P, Varsanyi P, Gielow MR, Poe G, Role LW, Ananth M, Rajebhosale P, Talmage DA, Hasselmo ME, Dannenberg H, Minces VH, Chiba AA, 2018. Specific Basal Forebrain-Cortical Cholinergic Circuits Coordinate Cognitive Operations. J Neurosci 38, 9446–9458. 10.1523/JNEUROSCI.1676-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Qiu T, Li K, Luo X, Wang S, Xu X, Liu X, Hong L, Li J, Huang P, Zhang M, 2022. Increased functional connectivity between nucleus basalis of Meynert and amygdala in cognitively intact elderly along the Alzheimer’s continuum. NeuroImage: Clinical 36, 103256. 10.1016/j.nicl.2022.103256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Veasey SC, Wood MA, Leng LZ, Kaminski C, Leight S, Abel T, Lee VM-Y, Trojanowski JQ, 2005. Impaired rapid eye movement sleep in the Tg2576 APP murine model of Alzheimer’s disease with injury to pedunculopontine cholinergic neurons. Am J Pathol 167, 1361–1369. 10.1016/S0002-9440(10)61223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ren R, Yang L, Zhang H, Shi Y, Okhravi HR, Vitiello MV, Sanford LD, Tang X, 2022. Sleep in Alzheimer’s disease: a systematic review and meta-analysis of polysomnographic findings. Transl Psychiatry 12, 1–12. 10.1038/s41398-022-01897-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke W, Roberts MJ, Guo K, McDonald JS, Robertson R, Thiele A, 2006. Cholinergic modulation of response properties and orientation tuning of neurons in primary visual cortex of anaesthetized Marmoset monkeys. Eur J Neurosci 24, 314–328. 10.1111/j.1460-9568.2006.04882.x [DOI] [PMC free article] [PubMed] [Google Scholar]