

Abstract
Nicotine is the principal psychoactive component in tobacco that drives addiction through its action on neuronal nicotinic acetylcholine receptors (nAChR). The nicotinic receptor gene CHRNA5, which encodes the α5 subunit, is associated with nicotine use and dependence. In humans, the CHRNA5 missense variant rs16969968 (G>A) is associated with increased risk for nicotine dependence and other smoking-related phenotypes. In rodents, α5-containing nAChRs in dopamine (DA) neurons within the ventral tegmental area (VTA) powerfully modulate nicotine reward and reinforcement. Although the neuroadaptations caused by long-term nicotine exposure are being actively delineated at both the synaptic and behavioral levels, the contribution of α5-containing nAChRs to the cellular adaptations associated with long-term nicotine exposure remain largely unknown. To gain insight into the mechanisms behind the influence of α5-containing nAChRs and the rs16969968 polymorphism on nicotine use and dependence, we used electrophysiological approaches to examine changes in nAChR function arising in VTA neurons during chronic nicotine exposure and multiple stages of nicotine withdrawal. Our results demonstrate that CHRNA5 mutation leads to profound changes in VTA nAChR function at baseline, during chronic nicotine exposure, and during short-term and prolonged withdrawal. Whereas nAChR function was suppressed in DA neurons from WT mice undergoing withdrawal relative to drug-naive or nicotine-drinking mice, α5-null mice exhibited an increase in nAChR function during nicotine exposure that persisted throughout 5-10 weeks of withdrawal. Re-expressing the hypofunctional rs16969968 CHRNA5 variant in α5-null VTA DA neurons did not rescue the phenotype, with α5-SNP neurons displaying a similar increased response to ACh during nicotine exposure and early stages of withdrawal. These results demonstrate the importance of VTA α5-nAChRs in the response to nicotine and implicate them in the time course of withdrawal.
Keywords: α5 nicotinic subunit, nicotine, withdrawal, VTA, dopamine, CHRNA5, rs16969968 SNP
Introduction
Tobacco use is a major public health problem, leading to hundreds of thousands of preventable deaths each year in the United States (United States. Public Health Service. Office of the Surgeon General., 2014). Establishing and maintaining abstinence from tobacco is difficult. In the United States, nearly 70% of active smokers express interest in quitting, but less than 10% report past-year success (Babb et al., 2017). Moreover, those who successfully quit typically require multiple attempts (Chaiton et al., 2016). This phenomenon seems to arise, at least in part, from the withdrawal syndrome that accompanies cessation, which includes physical, affective, and cognitive symptoms (McLaughlin et al., 2015; Paolini and De Biasi, 2011). Intriguingly, the risk of relapse remains high even after symptoms of withdrawal have abated (Garcia-Rodriguez et al., 2013), suggesting long-term adaptations that increase the difficulty of smoking cessation.
Nicotine, the principal addictive component of tobacco, acutely increases dopamine (DA) neuron activity in the ventral tegmental area (VTA) (Forget et al., 2018; Grenhoff et al., 1986; Pidoplichko et al., 1997) and—like other addictive drugs—increases DA release in the nucleus accumbens (NAc) (Di Chiara and Imperato, 1988) to exert its rewarding and reinforcing effects (Dani and Heinemann, 1996; De Biasi and Dani, 2011). In addition to reinforcement, DA in the NAc is also associated with nicotine withdrawal. Animals show significantly reduced NAc DA concentrations during the first few days of abstinence (Hildebrand et al., 1998; Rada et al., 2001; Zhang et al., 2012), and enhancing NAc DA activity has been suggested to ameliorate affective signs and symptoms of nicotine withdrawal (Paterson et al., 2007; Radke and Gewirtz, 2012). Nicotine modulates DA release in the brain through complex interactions with nicotinic acetylcholine receptors (nAChRs) (Dani and Bertrand, 2007; Dani and Heinemann, 1996). These pentameric receptors comprise five α subunits, alone or in combination with β subunits, with the subunit composition and position determining receptor pharmacology and function (see (Dani, 2015; Dani and Bertrand, 2007) for reviews).
Receptors containing the α5 subunit are expressed on neurons within the VTA and on DA terminals in the dorsal striatum (Azam et al., 2002; Exley et al., 2012; Grady et al., 2007; Klink et al., 2001), contributing to the importance of α5-containing receptors during the study of nicotine dependence. Distinguished by its role as an obligate accessory subunit, the α5 subunit does not contribute to the agonist binding site (Dani, 2015). However, several studies have consistently shown that its genetic deletion significantly reduces nAChR function (Chatterjee et al., 2013; Morel et al., 2014; Sciaccaluga et al., 2015). Moreover, the non-synonymous single nucleotide polymorphism (SNP) rs16969968 in CHRNA5 encodes a change in amino acid (Asp398Asn) in the α5 subunit that results in a partial loss of function (Bierut et al., 2008; Kuryatov et al., 2011; Sciaccaluga et al., 2015). Inclusion of the α5 subunit modulates nicotine-induced nAChR desensitization, which depends upon the broader receptor composition (Bailey et al., 2010; Gerzanich et al., 1998; Grady et al., 2012 Marks 2012) and prevents the subsequent upregulation of high-affinity α4β2-nAChRs (Mao et al., 2008).
Expression of α5-containing nAChRs in VTA DA neurons powerfully modulates nicotine intake in rodents (Morel et al., 2014). Additionally, a highly replicated genetic association of habitual smoking and nicotine dependence links the rs16969968 SNP with heavy smoking in humans (Bierut et al., 2008; Lips et al., 2010; Sarginson et al., 2011; Sherva et al., 2008; Stevens et al., 2008). Fewer studies have addressed the role of the α5 subunit in nicotine withdrawal and relapse. However, mice lacking the α5 subunit do not display physical signs of nicotine withdrawal (Salas et al., 2009) or withdrawal-induced hyperalgesia (Jackson et al., 2008), suggesting that the α5 subunit plays an important role during withdrawal from chronic nicotine intake. Overall, the contribution of the α5 subunit to the cellular adaptations associated with long-term nicotine exposure remain largely unknown. To shed light on the mechanisms behind the influence of α5-containing nAChRs and the rs16969968 SNP on nicotine use and dependence, we examined changes in nAChR function arising from chronic nicotine exposure and during multiple stages of nicotine withdrawal.
Methods
Subjects
We studied 149 α5-null mice and 162 wild-type (WT) littermate controls (male and female, aged 4-8 months). We also crossed α5-null mice with mice expressing Cre recombinase under the control of the DA transporter (DAT-Cre) to be able to express the rs16969968 SNP exclusively in midbrain DA neurons (see below). All mice were on a C57BL/6J background and were maintained in a 12h light:dark cycle, temperature- and humidity-controlled vivarium. Experiments were performed during the light cycle and were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania and in accordance with the guidelines provided by the National Institutes of Health Guide for care and use of laboratory animals. Our animal care facility is approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Mesolimbic α5-SNP re-expression
Fifty-one adult α5-null x DAT-Cre mice (age ~2 months), which from here on are referred to as SNP mice, were deeply anesthetized with isoflurane (1-2% in O2) and mounted in a stereotactic apparatus. The α5-SNP virus (AAVDJ-DIO-α5SNP-copGFP) was infused bilaterally into the VTA (AP: −3.2 to −3.25 cm; ML: ± 0.05 to 0.12; DV: −4.2 to −4.35) at a rate of 0.01 μL/min for a total of 1.7-2.0 μL using a 10 μL syringe (Hamilton, Reno, Nevada) coupled to a microinfusion pump (KdScientific, Hollistin, MA). Meloxicam was administered to minimize post-operative pain. The vector was designed to express cop-GFP to aid identification of infected neurons expressing the α5-SNP.
Drugs
Nicotine hydrogen tartrate salt was purchased from Glentham Life Sciences (Corsham, United Kingdom). Neurobiotin Tracer was purchased from Vector Laboratories (Burlingame, California, USA). All other chemicals were procured from Sigma-Aldrich (St. Louis, MO, USA).
Nicotine Treatment and Withdrawal
Mice were randomly assigned to one of five treatment groups: saccharin control (SAC), chronic nicotine (NIC), short-term/early withdrawal (1-4 days), intermediate withdrawal (2 weeks), or long-term/late withdrawal (5-10 weeks). Mice received 200 mg/L nicotine + 0.2 % saccharin or 0.2% saccharin alone in the drinking water for at least 8 weeks. Mice used to evaluate the effect of nicotine vs. saccharin were studied during nicotine exposure, while those used to evaluate the effects of withdrawal were provided with plain drinking water until reaching their assigned endpoint.
Slice Preparation
Mice were deeply anesthetized using intraperitoneal ketamine/xylazine followed by exsanguination via transcardial perfusion, performed as previously described (Broussard et al., 2016; Yang et al., 2017) using ice-cold N-methyl-D-glucamine (NMDG) based artificial cerebrospinal fluid (ACSF, in mM): 92 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, and 10 MgSO4, pH 7.3-7.4 with concentrated HCl (Ting et al., 2014). After perfusion, the brain was rapidly removed and placed in ice-cold, oxygenated NMDG solution. Horizontal slices containing the VTA (230 μm) were obtained using a Leica VT1200S vibratome and allowed to recover in 32°C NMDG for 13 min. Slices were then transferred to a HEPES-based holding ACSF solution (in mM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2, and 2 MgSO4. Slices were kept in the ACSF holding solution at room temperature for at least 1 hour prior to recording.
Electrophysiological Recordings
Slices were placed in a home-made recording chamber and were continuously bathed in well-oxygenated standard recording ACSF (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 glucose, 2 CaCl2, and 2 MgSO4, maintained at 32–34 °C using an inline heater system (TC-324B, Warner Instrument Corp, Hamden, CT). Responses were recorded using glass recording electrodes (~2-3 MΩ), which were pulled from borosilicate glass capillaries (TW 150-4, World Precision Instruments, Inc, Sarasota, FL) using a micropipette puller (Narishige PC-10, Tokyo, Japan) and were filled with a K-gluconate-based intracellular solution (in mM): 140 K-gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP, and 10 Na2-phosphocreatine, pH 7.3 with KOH. The tight-seal patch-clamp recording configuration was first achieved by applying a brief gentle suction. Under this configuration, spontaneous action potential firing was observed in most recorded neurons which could be blocked by puff-applied quinpirole (2 μM), a D2-receptor agonist (Fig 1A). Whole-cell mode configuration was then achieved by briefly applying strong suction. Only access resistance (Ra) < 10 MΩ was accepted, and the Ra was monitored throughout the experiment. Putative DA neurons were then identified by the presence of hyperpolarization-activated cationic currents (H-current, Fig 1C), a typical electrophysiological property of DA neurons of the lateral VTA (Zhang et al., 2010). A subset of recorded putative DA neurons was labeled with neurobiotin during patch-clamp recording to confirm that they were dopaminergic (as verified by positivity to tyrosine hydroxylase (TH) staining) and were located in the VTA (Fig 1B & D). To fully activate nAChRs in the recorded neurons, 1 mM acetylcholine (ACh) was pressure-applied (100 ms at 20 psi) by Picospritzer II (Parker Instrumentation, Fairfield, NJ) every 2 min via a puffer pipette. The puffer pipette was identical to those used for electrical recording. During the ACh puff, the recorded neurons were clamped at −60 mV (VH = −60 mV) under voltage-clamp mode. Atropine (1 μM) was added to recording ACSF to block muscarinic receptors, ensuring that the recorded response to ACh was mediated entirely by nAChRs (Yang et al., 2011; Yang et al., 2009). The data presented were collected from a minimum of 6 mice for each genotype within each experimental condition.
Figure 1.
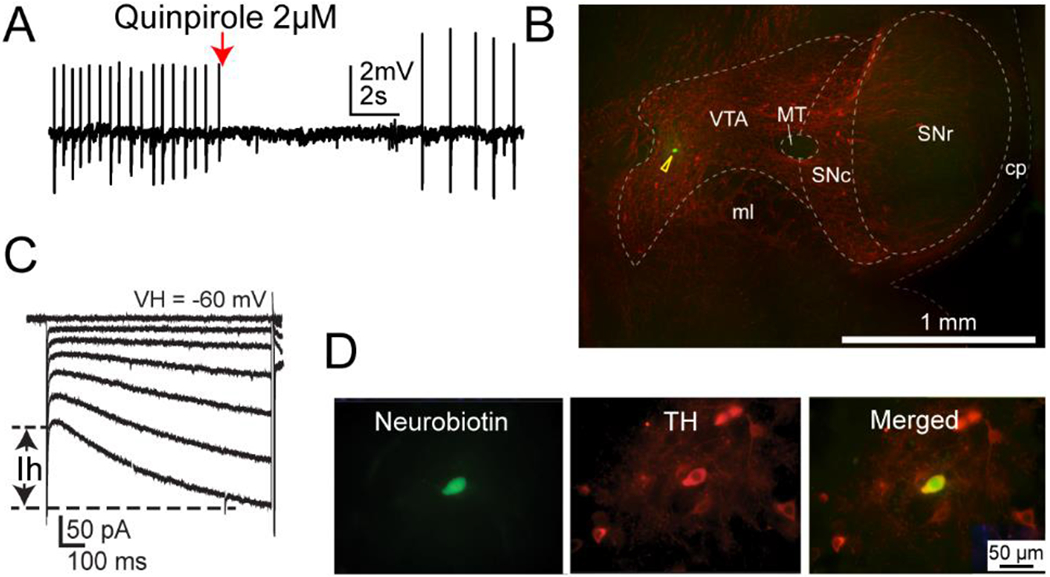
Identification of VTA DAergic neurons. Cell DAergic identity was confirmed by several approaches. DAergic cells displayed inhibited response to quinpirole (A), were anatomically localized to the VTA region (B), and were defined by the presence of hyperpolarization-activated cationic currents (H-currents, C). In addition, recorded cells were back-filled with neurobiotin and confirmed as DAergic neurons using TH immunohistochemical staining (D).
Immunohistochemistry
Slices used for TH staining were fixed overnight and washed with PBS before incubation in a blocking solution comprising 3% normal goat serum (NGS) and 0.3% Triton-X in PBS for 2 hours. Slices were then incubated in primary mouse anti-TH (1:200) at 4°C overnight, and in secondary goat anti-mouse Alexa Fluor 594 (1:200) and streptavidin DyLight 488 (1:1000) for 7 hours. Slices were imaged on an epifluorescence microscope to confirm TH expression in filled neurons.
Statistical Analyses
All values are expressed as mean ± SEM, and the number of cells analyzed is denoted by “n”. Variance in the peak ACh-induced currents in WT SAC mice was >30 times higher than in both α5-SNP and α5-null mice (see Fig. 2B). The baseline current differences between drug-naïve/SAC-treated groups were therefore determined using one-way ANOVA with Welch’s correction for unequal variances and multiple comparisons were performed using Games-Howell post-hoc. Planned within-group comparisons were then conducted using one-way Welch’s ANOVA with Games-Howell post-hoc for multiple comparisons between timepoints. Statistical tests were performed using SPSS v27 (IBM, Armonk, NY).
Figure 2.
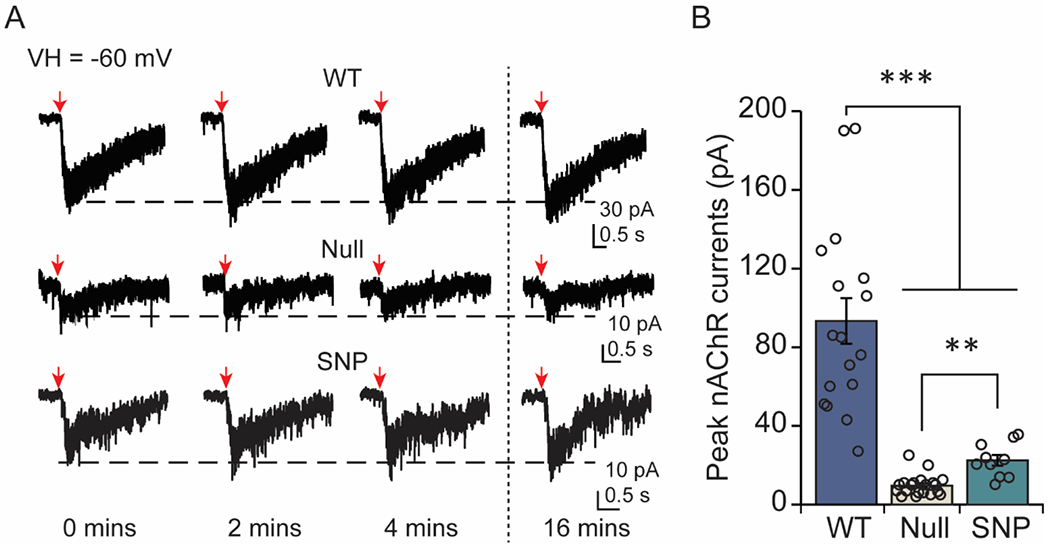
The α5 subunit regulates baseline response to ACh in DA neurons. (A) Example traces are shown from WT, α5-null, and α5-SNP neurons. Downward arrows indicate ACh puffs, which were applied once every 2 mins. ACh induced stable inward currents in DA neurons that were stable and persistent. Recordings were taken at the indicated times (in mins). (B) Currents (mean ± SEM) in response to the ACh puff. The amplitude of the response to ACh was significantly higher in WT neurons than neurons from animals without α5 or with the α5 SNP. **p<0.01, ***p<0.001
Results
The α5 subunit is necessary for baseline nAChR currents in DA neurons
To better understand the role of the α5 subunit in nAChR modulation of DA neuron activity, we first evaluated the response to ACh in drug-naive/saccharin-treated WT, α5-null, and SNP mice. Whole-cell currents were elicited through pressure puff-application of 1 mM ACh every two minutes, evoking stable and repeatable responses in all groups (Fig 2A). Disruption of the α5 subunit resulted in significant differences in the response to ACh [Welch’s F(2, 18.663)=32.591, p<0.001] (Fig 2B). Recordings from α5-null mice showed very limited function, demonstrated by the small peak amplitude of inward-currents (9.66 ± 1.17 pA, n=20 from 16 mice) in VTA DA neurons relative to recordings from WT mice (99.34 ± 11.59 pA, n=17 from 13 mice) [p<0.001]. These results corroborate previous work demonstrating that α5-containing nAChRs are prominent contributors to baseline nAChR currents in VTA DA neurons in both juvenile (Chatterjee et al., 2013; Sciaccaluga et al., 2015) and adult (Morel et al., 2014) mice. A reduction in nAChR function was also observed when the α5 subunit encoding the rs16969968 polymorphism was expressed in VTA DA neurons (22.51 ± 2.76 pA, n=10 from 6 mice; p<0.001). The response to ACh was significantly larger in SNP-expressing neurons compared to α5-null cells [p<0.005], but the SNP-currents were still significantly smaller than WT, suggesting partial diminution of nAChR function when the α5 subunit is present but mutated.
Dramatic, long-lasting reduction in nAChR currents after withdrawal in WT mice
There are very few patch-clamp studies in VTA DA neurons of adult animals that examine the dynamic changes in nAChR-expressing VTA DA neurons following prolonged nicotine exposure (≥ 2 months). We first measured nAChR-mediated whole-cell currents in VTA DA neurons in WT mice to determine the effects of long-term nicotine exposure and withdrawal on nAChR function (Fig 3A). One-way Welch’s ANOVA revealed a significant difference between time points associated with withdrawal [ F(4,26.441)=5.969, p<0.01] in WT animals. ACh-induced whole-cell currents were comparable between saccharine-treated (99.34± 11.59 pA, n=17) and nicotine-treated (89.47 ± 10.15 pA, n=16 from 12 mice) groups [p=0.999], consistent with previous findings indicating that chronic nicotine does not functionally alter nAChRs in midbrain DA neurons (Nashmi et al., 2007; Xiao et al., 2009) (Fig 3B).
Figure 3.
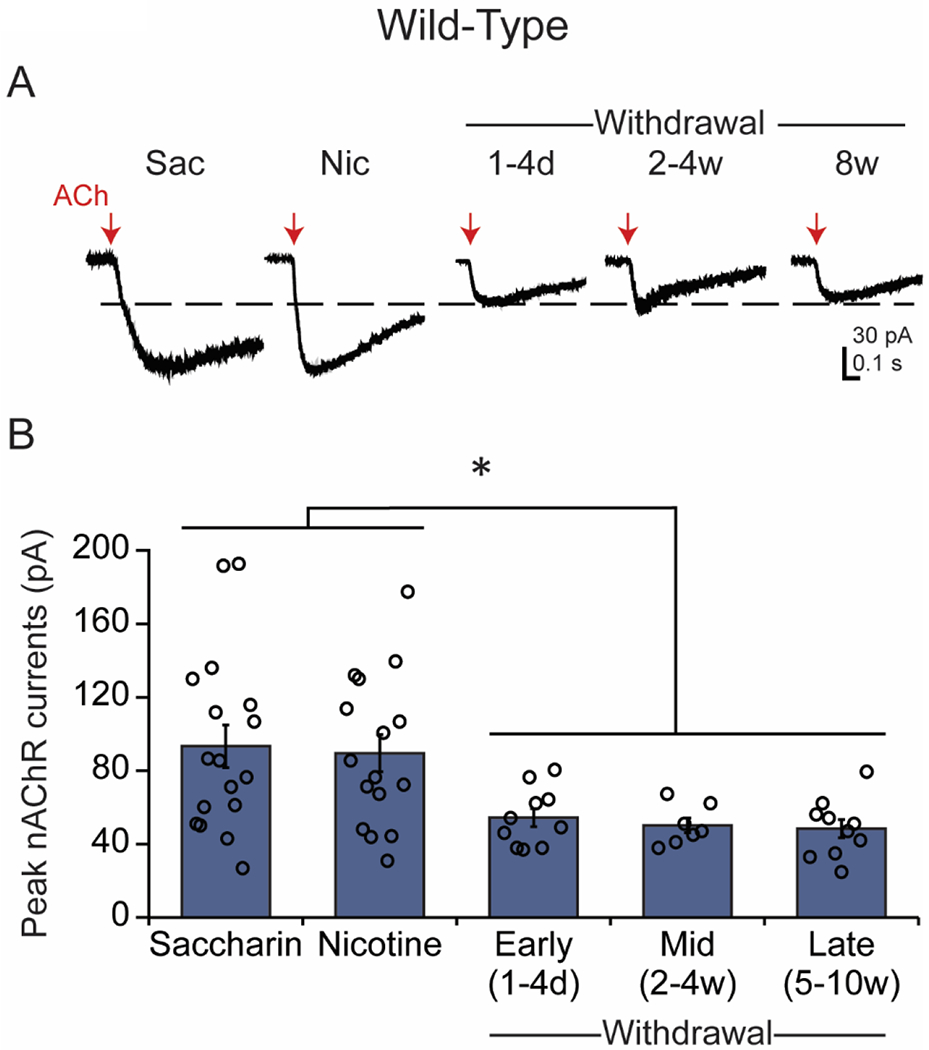
Long-term withdrawal depresses the response to ACh in WT DA neurons. (A) Example traces are shown from WT DA neurons under saccharin/drug-naïve and chronic nicotine conditions, and in different stages of withdrawal. Red arrows indicate the ACh puffs. (B) Current (mean ± SEM) responses to the ACh puff. The amplitude of the response to ACh was significantly reduced during withdrawal compared to saccharin/drug-naïve and nicotine-treated conditions. *p<0.05
However, ACh-induced whole-cell currents became significantly smaller following 1-4 days of nicotine withdrawal [WD 1-4 days: 54.40 ± 4.96 pA, n=10 from 8 mice, p<0.05 vs SAC/NIC], indicating reduced nAChR function resulting from nicotine withdrawal. The suppressed ACh-mediated currents did not recover after 2-4 weeks of withdrawal [WD 2-4 weeks: 50.14 ± 4.06 pA, n=7 from 6 mice, p<0.05 vs SAC/NIC, p=0.96 vs WD 1-4 days] and, surprisingly, remained similarly depressed even after 10 weeks of withdrawal [WD 5-10 weeks: 48.40 ± 4.97 pA, n=10 from 9 mice]. ACh-induced currents from mice following 5-10 weeks of withdrawal were significantly lower than saccharin only- or nicotine-drinking mice [p<0.05] but did not significantly differ from earlier time points during withdrawal [p=0.91 vs WD 1-4 days; p=0.99 vs WD 2-4 weeks]. These results suggest that chronic nicotine does not change nAChR function in WT mice, but that nicotine withdrawal results in long-lasting down-regulation of nAChR function (Gould et al., 2012).
Nicotine-induced increase in nAChR currents in α5-null mice is long-lasting
We evaluated the effect of chronic nicotine exposure and withdrawal in mice lacking the α5 subunit. One-way ANOVA revealed a significant difference across treatment conditions [Welch’s F(4,26.165)=14.262, p<0.001] in α5-null animals (Fig 4 A,B). In contrast to the observations in WT mice, VTA DA neurons in α5-null mice chronically treated with nicotine were significantly more responsive to ACh [26.34 ± 3.39 pA, n=15 from 12 mice; p<0.005 vs SAC] than those from SAC-treated mice [9.66 ± 1.17pA, n=20], implying that chronic nicotine treatment leads to enhanced nAChR function in the absence of the α5 subunit. This increase in nAChR function persisted beyond cessation of nicotine intake. The peak amplitude of ACh-induced currents in α5-null mice in the first 1-4 days of withdrawal (42.65 ± 7.9, n=13 from 12 mice) remained significantly higher than SAC-treated controls [p<0.01] but did not significantly differ from α5-nulls drinking nicotine [p=0.355]. The enhanced response to ACh persisted after 2-4 weeks [36.91 ± 6.67 pA, n=13 from 11 mice, p<0.05 vs SAC] and 5-10 weeks [37.08 ± 7.33 pA n=12 from 10 mice, p<0.05 vs SAC] of withdrawal from chronic nicotine treatment.
Figure 4.
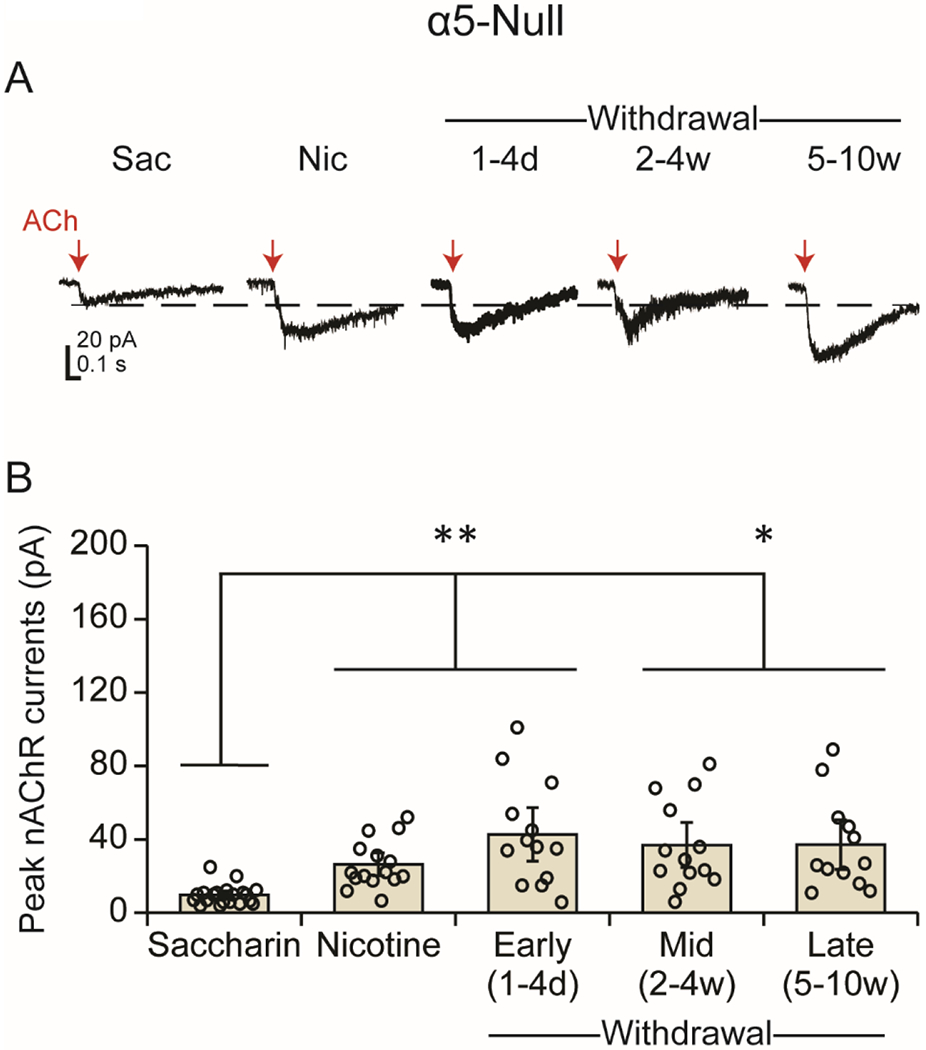
In the absence of the α5 subunit, the ACh-induced currents are smaller than WT (Fig 2), but chronic nicotine leads to an increase in the ACh response that continues into withdrawal in VTA DA neurons. (A) Example traces are shown from α5-null DA neurons under saccharin/drug-naïve and chronic nicotine conditions and in different stages of withdrawal. Red arrows indicate the ACh puffs. (B) Current (mean ± SEM) responses to the ACh puff. The amplitude of the response to ACh was significantly enhanced under chronic nicotine treatment and throughout withdrawal relative to saccharin/drug-naïve conditions. *p<0.05, **p<0.01
The effects of nicotine in α5-SNP mice are complex, with some similarities to α5-null mice
Neurons from α5-SNP mice displayed an interesting response to nicotine and withdrawal. As in WT and α5-null mice, one-way ANOVA revealed a significant effect of treatment condition on ACh-induced currents [Welch’s F(4, 21.883)=6.335, p<0.005] in α5-SNP mice (Fig 5A,B). The peak nAChR current amplitude in the α5-SNP mice was significantly higher during nicotine-drinking (60.03 ± 10.22 pA, n=11 from 8 mice) than SAC-treated controls (22.51 ±2.8 pA, n=10; p<0.05 NIC vs SAC), demonstrating that chronic nicotine exposure enhances nAChR function in mice expressing the rs16969968 SNP in VTA DA neurons. Although the currents in SAC-treated controls are smaller in the α5-SNP mice than the WT mice, the ACh-induced currents in α5-SNP mice approached amplitudes closer to those observed in WT mice before and during nicotine treatment. Enhanced nAChR function in α5-SNP mice persisted in the days immediately following cessation of nicotine treatment (WD 1-4 days: 72.59 ± 12.22 pA, n=12 from 9 mice; p<0.05 vs SAC). However, unlike the WT and α5 null mice, the peak amplitude of ACh-induced currents was no longer significantly different from SAC controls at either 2-4 weeks (35.19 ± 9.65 pA, n= 8 from 5 mice; p=0.718 vs. SAC) or 5-10 weeks after nicotine cessation (28.15 ± 3.24 pA, n= 14 from 11 mice; p=0.68 vs. SAC), with a trend towards reduced function compared to nicotine-drinking α5-SNP mice [p=0.072, WD 5-10 weeks vs. NIC].
Figure 5.
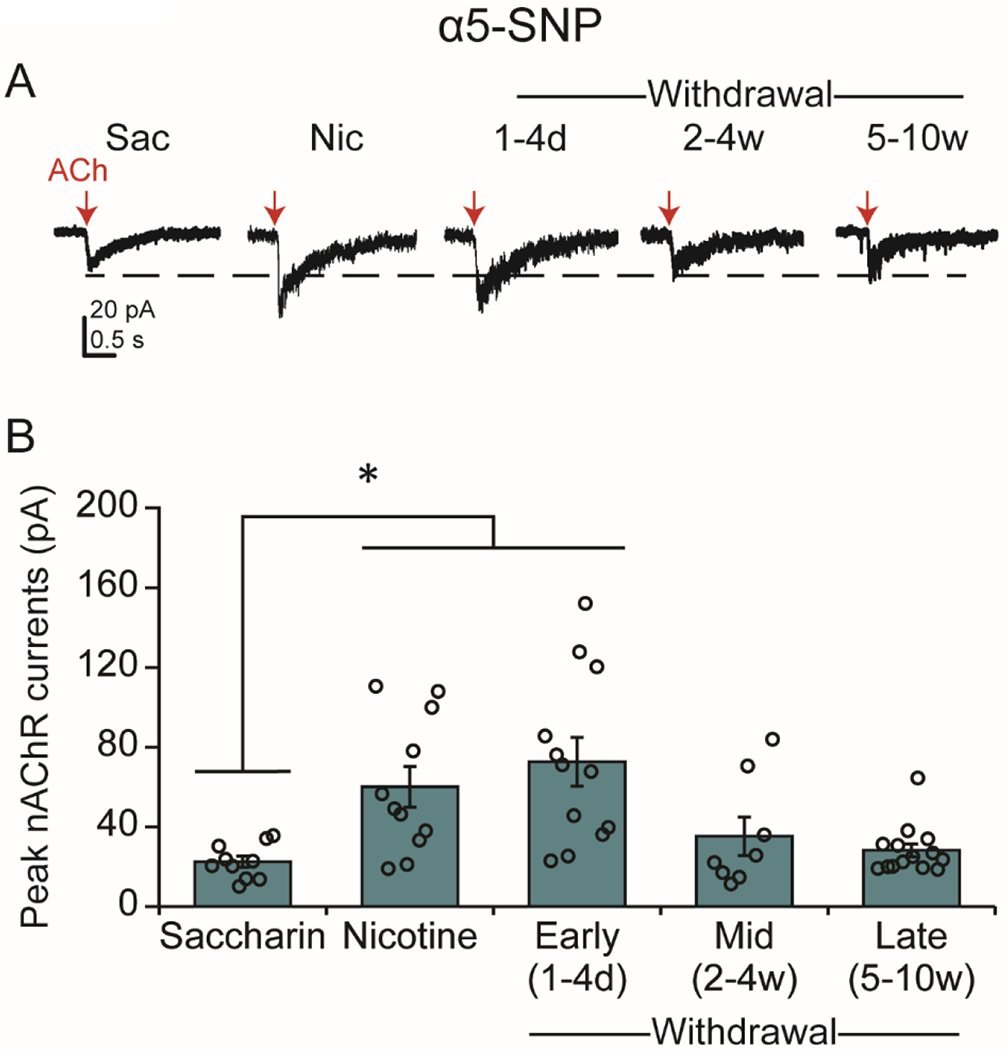
Re-expression of the α5-SNP in VTA DA neurons of α5-null mice produces a complex response to nicotine and withdrawal. (A) Example traces from α5-SNP DA neurons under saccharin/drug-naïve and chronic nicotine conditions and in different stages of withdrawal. Red arrows indicate the ACh puffs. (B) Current (mean ± SEM) responses to the ACh puffs. The amplitude of the response to ACh was significantly enhanced under chronic nicotine treatment and the initial stage of withdrawal, but currents returned to saccharin/drug-naïve levels at later stages of withdrawal. *p<0.05
Discussion
Our study addressed the long-term changes in nAChR function that take place in DA neurons both during and after prolonged exposure to nicotine. We demonstrated that α5-containing nAChRs are essential for the adaptations in nAChR function that occur during nicotine exposure and withdrawal. We also showed that expression of the hypofunctional rs16969968 CHRNA5 variant in VTA neurons leads to abnormal nAChR responses during both long-term nicotine use and the early phases of withdrawal, shedding light on the role of this human polymorphism in nicotine use and abuse.
The α5 subunit governs basal nAChR responsivity in VTA DA Neurons
Studies using α5-null animals have consistently demonstrated reduced nAChR functionality when the α5 subunit is absent (Chatterjee et al., 2013; Morel et al., 2014; Sciaccaluga et al., 2015). Corroborating these observations, we found that ACh-induced whole-cell currents were significantly reduced in DA neurons from α5-null mice.
Recent studies suggest that, under baseline conditions, observed differences in the neuronal response to ACh between WT neurons and neurons with disrupted α5 subunits are balanced by other factors, resulting in a similar level of functionality at a systems level. For example, although the absence of the α5 subunit decreases nAChR function (Chatterjee et al., 2013; Morel et al., 2014; Sciaccaluga et al., 2015), baseline firing frequency and bursting in VTA DA neurons of α5-null and α5-SNP mice and rats did not significantly differ from WT controls (Forget et al., 2018; Morel et al., 2014). Similarly, basal levels of DA in the NAc—a primary target of the VTA—did not differ between α5-null and WT mice (Besson et al., 2016). When challenged with acute nicotine, however, disruptions in DA neuron function are unmasked. α5-null mice show blunted DA release at nicotine doses that reliably increase DA in WT mice (Besson et al., 2016), and VTA DA neurons lacking α5 fail to respond to doses that increase firing in WT cells (Forget et al., 2018; Morel et al., 2014).
Cholinergic inputs to VTA DA neurons are thought to promote fast, “phasic” firing (Floresco et al., 2003), which is believed to be a critical component of reward and reinforcement (Dautan et al., 2016; Grace et al., 2007). Because α5-null mice demonstrate a hypofunctional response to ACh, it seems reasonable to hypothesize that initial exposure to nicotine might be less rewarding/reinforcing when mutated CHRNA5 is expressed in VTA neurons. Indeed, both α5 null and α5 SNP mice required a dose of nicotine at least twice as high as WT mice to self-administer intravenous nicotine in a model of initiation of drug-taking behavior (Morel et al., 2014). This result suggests that the dose of nicotine required to elicit a sufficient DA response to support reinforcement during early use is higher when CHRNA5 is mutated.
α5 regulates nAChR changes in response to chronic nicotine exposure
Adaptations to chronic nicotine are different when α5 is absent or mutated. Consistent with previous reports suggesting that homeostatic regulation masked differences in nAChR number and function in WT animals (Besson et al., 2007), we found that responsivity to ACh in cells from WT mice undergoing chronic nicotine consumption was similar to that of cells from drug-naïve mice. In contrast to neurons from WT mice, we observed a significant increase in the response to ACh in α5-null VTA DA neurons from mice undergoing chronic nicotine treatment compared to drug-naïve mice. Therefore, chronic nicotine exposure enhanced nAChR function, thereby shifting nAChR responsivity closer to the higher response level observed in drug-naïve WT mice.
The effects of chronic nicotine on nAChR expression and function are complex, with regional and subunit-specific variability. Chronic nicotine exposure is believed to trigger a homeostatic response to nAChR desensitization, including subunit-specific increases in nAChR binding that are primarily attributable to an upregulation of β2-containing nAChRs (Besson et al., 2007; Dani and Heinemann, 1996; Mao et al., 2008)—particularly in mesolimbic areas (Nashmi et al., 2007; Nguyen et al., 2003; Renda and Nashmi, 2014)—in both rodents (Nashmi et al., 2007; Renda and Nashmi, 2014) and humans (Staley et al., 2006). Nicotine-induced upregulation of α4β2-nAChRs accounts for a large proportion of the increase in nAChRs (Marks et al., 2014; Nguyen et al., 2003), whereas evidence suggests that α6β2-nAChRs are downregulated (Perez et al., 2008; Perry et al., 2007). The presence of the α5 subunit modulates these dynamics: expression of α5 in α4α5β2-nAChRs reduces desensitization and speeds subsequent recovery in response to nicotine (Grady et al., 2012) and prevents upregulation following chronic nicotine administration (Mao et al., 2008). α4β2-nAChR upregulation in the VTA has been primarily reported in GABA neurons in WT animals, where α5 is found in fewer than 20% of those neurons compared to approximately 80% in VTA DA neurons (Klink et al., 2001). Because α5-null mice do not express any α4α5β2 nAChRs, the observed increased responsivity to ACh in VTA DA neurons may be the result of an overall upregulation of high-affinity α4β2-nAChRs that, under normal circumstances, is selective to GABA neurons.
As previously mentioned, α5-null and α5-SNP mice require nicotine doses at least twice as high as WT mice to reach the same levels of self-administration (Morel et al., 2014). Furthermore, while WT mice titrate the amount of nicotine they self-administer—maintaining a preferred level of intake despite increasing doses—α5-null and α5-SNP mice increase the amount of nicotine taken (Fowler et al., 2011; Morel et al., 2014) when receiving doses that would normally be aversive (Fowler et al., 2011) or even toxic (Salas et al., 2003) to WT mice. This ‘loss of control’ over nicotine consumption at high doses was corrected by re-expression of α5 in VTA DA cells (Morel et al., 2014), suggesting a direct involvement of the VTA in this behavior.
Although the mesolimbic DA system plays a role in the dose-dependent effects of nicotine on both reinforcement and aversion (Morel et al., 2014; Wills et al., 2022), other brain areas expressing CHRNA5 might also contribute to these effects. Besides the VTA, α5-containing nAChRs are highly expressed in few other brain areas, with the interpeduncular nucleus (IPN) representing the region most enriched in those receptors (Beiranvand et al., 2014; Forget et al., 2018; Salas et al., 2003; Salas et al., 2009). Although stimulation of α5-expressing IPN GABAergic neurons does not induce withdrawal, it does trigger aversion-like behavior in animals exposed to nicotine (Morton et al., 2018). Interestingly, a subpopulation of IPN GABA neurons implicated in nicotine aversion sends projections to the laterodorsal tegmentum (LDTg) (Wolfman et al., 2018), which in turn mediates reward (Lammel et al., 2012) and promotes DA-neuron burst firing (Lodge and Grace, 2006; Omelchenko and Sesack, 2005) via projections to the VTA. Therefore, altered nAChR function in both VTA DA neurons and IPN GABA neurons could contribute to the escalation of nicotine consumption observed in α5-null and α5-SNP mice.
α5 regulates nAChR changes in response during withdrawal from chronic nicotine
Nicotinic responses during withdrawal from nicotine differ from WT mice when α5 is absent or mutated. Although WT nAChR peak currents were unchanged during nicotine exposure, we observed a dramatic reduction in nAChR function during nicotine withdrawal that persisted for at least 10 weeks in tested mice (Fig 3B). The plastic changes that maintain homeostasis during chronic nicotine intake become inappropriate during abstinence, leading to a withdrawal syndrome that includes physical, affective, and cognitive symptoms (McLaughlin et al., 2015; Shiffman et al., 2004). Withdrawal severity is believed to contribute to the risk of relapse (Zhou et al., 2009) and relies on multiple areas of the brain in addition to the VTA, particularly the medial habenula and interpeduncular nucleus, where α5-containing nAChRs are fundamental to nicotine withdrawal (Salas et al., 2009). The mechanisms underlying the dramatic, long-lasting reduction in nAChR function observed in DA neurons from WT mice during nicotine withdrawal are unclear but appear to rely on the α5 subunit. In stark contrast to WT mice, the increased nAChR function observed in neurons from nicotine-treated α5-null mice persisted throughout withdrawal, remaining significantly elevated above ACh-induced currents in drug-naive controls. Because α5-null mice do not display physical signs of withdrawal (Salas et al., 2009), it is tempting to speculate that the effects we describe in this study might contribute to the behavioral phenotype of α5-null mice.
Nicotine withdrawal is associated with a reduction in tonic and phasic DA release in the nucleus accumbens shell (Zhang et al., 2012). There are many factors controlling VTA cell function and DA release (Kramer et al., 2022; Liu and Kaeser, 2019), and nAChRs are particularly important contributors. Nicotine exposure enhances DA release through a dynamic balance of activation and desensitization of nAChRs in the VTA and the striatum (Mansvelder and McGehee, 2000; Picciotto et al., 2008; Pidoplichko et al., 2004). That nAChR function does not decrease during withdrawal in α5 null mice, remaining elevated above drug-naïve levels, might result in persistent changes in phasic and tonic DA signaling (Zhang et al., 2012). Furthermore, nicotine withdrawal is associated with increased activity of IPN GABAergic neurons (Klenowski et al., 2022), many of which contain α5 nAChRs. Therefore, mutated α5 might alter VTA and IPN cellular and circuit-level adaptations that normally lead to the manifestation of nicotine withdrawal.
Similar effect of the rs16969968 SNP and the a5 null mutation.
The human polymorphism rs16969968 produces nAChRs with hypofunctional α5 subunits, resulting in significantly lower calcium permeability and diminished α4β2α5-nAChR function in heterologous expression systems and neurons expressing the α5 SNP in an α5 null background (Bierut et al., 2008; Sciaccaluga et al., 2015). We used a viral vector to re-express the SNP variant of the α5 subunit in DA neurons within the VTA of α5-null mice to determine how the SNP modifies nAChR function at baseline and across nicotine use and withdrawal. Similar to nicotine-naive α5-null mice, VTA nAChR currents from nicotine-naïve α5-SNP mice are significantly smaller than those recorded from WT neurons, but nAChR currents from α5-SNP neurons were over ≈33% larger than those from α5-null neurons. We also showed that, similar to the α5-null mutation, α5-SNP nAChR currents increase after chronic nicotine exposure.
Functional alterations produced by CHRNA5 mutations—such as the rs16969968—are relevant when considering VTA neuronal adaptations to chronic nicotine exposure. Smokers with the risk allele encoding the D398N CHRNA5 mutation alter their smoking behavior to obtain more nicotine (Macqueen et al., 2014) and display lower nicotine aversion than subjects carrying the common allele (Jensen et al., 2015). Interestingly, smokers with high-risk genetic variants in the CHRNA5-CHRNA3-CHRNB4 region, including rs16969968, are at increased risk of nicotine cessation failure, a phenomenon that may reflect the fact that, in subjects carrying the mutation, continued nicotine exposure might partially ameliorate the decreased nAChR function, making it more difficult to quit (Chen et al., 2012).
Neither α5-null nor α5-SNP mice demonstrated the dramatic and persistent reduction in ACh-induced currents observed in WT mice during nicotine withdrawal. During the early stages of withdrawal, neurons from α5-SNP mice most resembled those from α5-null mice, in that ACh-induced currents did not fall below levels observed in nicotine-naïve controls and were instead elevated in the first few days of withdrawal, the time of highest risk for relapse (Hughes et al., 2004). Zhang and colleagues (Zhang et al., 2012) showed that, during withdrawal, phasic DA release is increased relative to tonic DA release in WT animals, thereby increasing DA’s “signal-to-noise” ratio. Although the effect of α5 disruption on this phenomenon is unknown, it is possible that increased α4ß2 nAChR function in DA neurons from α5-null and α5-SNP mice results in a higher propensity for fast, “phasic” firing than WT mice. Therefore, the DA signal produced by an acute nicotine re-exposure would be especially elevated in the α5-SNP mice, producing a DA response that may alter the vulnerability to some forms of relapse. To that end, a recent preclinical study demonstrated an enhanced propensity to nicotine-primed reinstatement in α5-SNP rats (Forget et al., 2018), whereas another study suggests that smokers expressing rs16969968 are less responsive to cigarette cues than those with fully functional α5-containing nAChRs (Janes et al., 2012). These individuals have a three-fold increased likelihood of responding to pharmacologic cessation treatments that include nicotine replacement therapy or DA and norepinephrine reuptake inhibition (bupropion), compared to smokers with low-risk genetic variants in the same gene cluster (Chen et al., 2012). Our findings might provide a mechanism by which drugs that enhance/maintain DA activity are more effective during nicotine cessation in individuals carrying the CHRNA5 polymorphism.
Summary
Our data demonstrate the significant role of α5-nAChRs in the effects of chronic nicotine exposure on VTA DA neurons. Lack of α5 subunits or the expression of the rs16969968 SNP leads to long-lived alterations of VTA nAChR receptors. Further research is needed to investigate how these DA neuron adaptations to nicotine influence the broader mesolimbic DA system to impact nicotine use and abuse.
Highlights.
CHRNA5 mutation significantly reduces VTA nAChR function at baseline.
Chronic nicotine exposure enhances nAChR function when CHRNA5 is mutated.
nAChR currents decrease during extended withdrawal in control mice.
nAChR currents remain enhanced during nicotine withdrawal when CHRNA5 is mutated.
ACKNOWLEDGEMENTS
We would like to thank Dr. Olivia Swanson for her expert assistance in the finalization of the manuscript.
FUNDING AND DISCLOSURES
This work was supported in part by NIH Bench to Bedside supplement for NIDA DA 036572 and NIAAA U01 AA025931 to MDB, and NIAAA R01 AA026267 and NIDA R01 DA053296 to JAD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest: All authors declare no conflict of interest
Contributor Roles Taxonomy (CRediT) author statement
Kechun Yang: Conceptualization, Methodology, Formal analysis, Investigation, Writing – Original Draft, Writing – Reviewing & Editing. Ian McLaughlin: Conceptualization, Methodology, Investigation. Jessica Shaw: Conceptualization, Formal analysis, Writing – Original Draft, Writing – Reviewing & Editing. Natalia Quijano Carde’: Conceptualization, methodology. John A. Dani: Conceptualization, Methodology, Writing – Reviewing & Editing, Supervision, Funding acquisition.
Mariella De Biasi: Conceptualization, Methodology, Writing – Reviewing & Editing, Supervision, Funding acquisition.
References
- Azam L, Winzer-Serhan UH, Chen Y, & Leslie FM (2002). Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol, 444(3), 260–274. [DOI] [PubMed] [Google Scholar]
- Babb S, Malarcher A, Schauer G, Asman K, & Jamal A (2017). Quitting Smoking Among Adults - United States, 2000-2015. MMWR Morb Mortal Wkly Rep, 65(52), 1457–1464. [DOI] [PubMed] [Google Scholar]
- Bailey CD, De Biasi M, Fletcher PJ, & Lambe EK (2010). The nicotinic acetylcholine receptor alpha5 subunit plays a key role in attention circuitry and accuracy. J Neurosci, 30(27), 9241–9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiranvand F, Zlabinger C, Orr-Urtreger A, Ristl R, Huck S, & Scholze P (2014). Nicotinic acetylcholine receptors control acetylcholine and noradrenaline release in the rodent habenulo-interpeduncular complex. Br J Pharmacol, 171(23), 5209–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson M, Granon S, Mameli-Engvall M, Cloez-Tayarani I, Maubourguet N, Cormier A, Cazala P, David V, Changeux JP, Faure P (2007). Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A, 104(19), 8155–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson M, Guiducci S, Granon S, Guilloux JP, Guiard B, Reperant C, Faure P, Pons S, Cannazza G, Zolie M, Gardier AM, Maskos U (2016). Alterations in alpha5* nicotinic acetylcholine receptors result in midbrain- and hippocampus-dependent behavioural and neural impairments. Psychopharmacology (Berl), 233(18), 3297–3314. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J Jr., Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice JP, Goate AM (2008). Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry, 165(9), 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard JI, Yang K, Levine AT, Tsetsenis T, Jenson D, Cao F, Garcia I, Arenkiel BR, Zhou FM, De Biasi M, Dani JA (2016). Dopamine Regulates Aversive Contextual Learning and Associated In Vivo Synaptic Plasticity in the Hippocampus. Cell Rep, 14(8), 1930–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaiton M, Diemert L, Cohen JE, Bondy SJ, Selby P, Philipneri A, & Schwartz R (2016). Estimating the number of quit attempts it takes to quit smoking successfully in a longitudinal cohort of smokers. BMJ Open, 6(6), e011045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Santos N, Holgate J, Haass-Koffler CL, Hopf FW, Kharazia V, Lester H, Bonci A, Bartlett SE (2013). The alpha5 subunit regulates the expression and function of alpha4*-containing neuronal nicotinic acetylcholine receptors in the ventral-tegmental area. PLoS One, 8(7), e68300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LS, Baker TB, Piper ME, Breslau N, Cannon DS, Doheny KF, Gogarten SM, Johnson EO, Saccone NL, Wang JC, Weiss RB, Goate AM, Bierut LJ (2012). Interplay of genetic risk factors (CHRNA5-CHRNA3-CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry, 169(7), 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA (2015). Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int Rev Neurobiol, 124, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, & Bertrand D (2007). Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol, 47, 699–729. [DOI] [PubMed] [Google Scholar]
- Dani JA, & Heinemann S (1996). Molecular and cellular aspects of nicotine abuse. Neuron, 16(5), 905–908. [DOI] [PubMed] [Google Scholar]
- Dautan D, Souza AS, Huerta-Ocampo I, Valencia M, Assous M, Witten IB, Deisseroth K, Tepper JM, Bolam JP, Gerdjikov TV, Mena-Segovia J (2016). Segregated cholinergic transmission modulates dopamine neurons integrated in distinct functional circuits. Nat Neurosci, 19(8), 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi M, & Dani JA (2011). Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci, 34, 105–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, & Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A, 85(14), 5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, McIntosh JM, Marks MJ, Maskos U, & Cragg SJ (2012). Striatal alpha5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J Neurosci, 32(7), 2352–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, & Grace AA (2003). Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci, 6(9), 968–973. [DOI] [PubMed] [Google Scholar]
- Forget B, Scholze P, Langa F, Morel C, Pons S, Mondoloni S, Besson M, Durand-de Cuttoli R, Hay A, Tricoire L, Lambolez B, Mourot A, Faure P, Maskos U (2018). A Human Polymorphism in CHRNA5 Is Linked to Relapse to Nicotine Seeking in Transgenic Rats. Curr Biol, 28(20), 3244–3253 e3247. [DOI] [PubMed] [Google Scholar]
- Fowler CD, Lu Q, Johnson PM, Marks MJ, & Kenny PJ (2011). Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature, 471(7340), 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez O, Secades-Villa R, Florez-Salamanca L, Okuda M, Liu SM, & Blanco C (2013). Probability and predictors of relapse to smoking: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Drug Alcohol Depend, 132(3), 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerzanich V, Wang F, Kuryatov A, & Lindstrom J (1998). alpha 5 Subunit alters desensitization, pharmacology, Ca++ permeability and Ca++ modulation of human neuronal alpha 3 nicotinic receptors. J Pharmacol Exp Ther, 286(1), 311–320. [PubMed] [Google Scholar]
- Gould TJ, Portugal GS, Andre JM, Tadman MP, Marks MJ, Kenney JW, Yildirim E, Adoff M (2012). The duration of nicotine withdrawal-associated deficits in contextual fear conditioning parallels changes in hippocampal high affinity nicotinic acetylcholine receptor upregulation. Neuropharmacology, 62(5-6), 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Floresco SB, Goto Y, & Lodge DJ (2007). Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci, 30(5), 220–227. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, & Marks MJ (2007). The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol, 74(8), 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Wageman CR, Patzlaff NE, & Marks MJ (2012). Low concentrations of nicotine differentially desensitize nicotinic acetylcholine receptors that include alpha5 or alpha6 subunits and that mediate synaptosomal neurotransmitter release. Neuropharmacology, 62(5-6), 1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, & Svensson TH (1986). Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand, 128(3), 351–358. [DOI] [PubMed] [Google Scholar]
- Hildebrand BE, Nomikos GG, Hertel P, Schilstrom B, & Svensson TH (1998). Reduced dopamine output in the nucleus accumbens but not in the medial prefrontal cortex in rats displaying a mecamylamine-precipitated nicotine withdrawal syndrome. Brain Res, 779(1-2), 214–225. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Keely J, & Naud S (2004). Shape of the relapse curve and long-term abstinence among untreated smokers. Addiction, 99(1), 29–38. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Martin BR, Changeux JP, & Damaj MI (2008). Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Ther, 325(1), 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes AC, Smoller JW, David SP, Frederick BD, Haddad S, Basu A, Fava M, Evins AE, Kaufman MJ (2012). Association between CHRNA5 genetic variation at rs16969968 and brain reactivity to smoking images in nicotine dependent women. Drug Alcohol Depend, 120(1-3), 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KP, DeVito EE, Herman AI, Valentine GW, Gelernter J, & Sofuoglu M (2015). A CHRNA5 Smoking Risk Variant Decreases the Aversive Effects of Nicotine in Humans. Neuropsychopharmacology, 40(12), 2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenowski PM, Zhao-Shea R, Freels TG, Molas S, & Tapper AR (2022). Dynamic activity of interpeduncular nucleus GABAergic neurons controls expression of nicotine withdrawal in male mice. Neuropsychopharmacology, 47(3), 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink R, de Kerchove d’Exaerde A, Zoli M, & Changeux JP (2001). Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci, 21(5), 1452–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PF, Brill-Weil SG, Cummins AC, Zhang R, Camacho-Hernandez GA, Newman AH, Eldridge MAG, Averbeck BB, Khaliq ZM (2022). Synaptic-like axo-axonal transmission from striatal cholinergic interneurons onto dopaminergic fibers. Neuron, 110(18), 2949–2960 e2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Berrettini W, & Lindstrom J (2011). Acetylcholine receptor (AChR) alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)(2)alpha5 AChR function. Mol Pharmacol, 79(1), 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC (2012). Input-specific control of reward and aversion in the ventral tegmental area. Nature, 491(7423), 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips EH, Gaborieau V, McKay JD, Chabrier A, Hung RJ, Boffetta P, Hashibe M, Zaridze D, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Field JK, Liloglou T, Xinarianos G, McLaughlin J, Liu G, Skorpen F, Elvestad MB, Hveem K, Vatten L, Study E, Benhamou S, Lagiou P, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Castellsague X, Macfarlane TV, Barzan L, Canova C, Lowry R, Conway DI, Znaor A, Healy C, Curado MP, Koifman S, Eluf-Neto J, Matos E, Menezes A, Fernandez L, Metspalu A, Heath S, Lathrop M, Brennan P (2010). Association between a 15q25 gene variant, smoking quantity and tobacco-related cancers among 17 000 individuals. Int J Epidemiol, 39(2), 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, & Kaeser PS (2019). Mechanisms and regulation of dopamine release. Curr Opin Neurobiol, 57, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, & Grace AA (2006). The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc Natl Acad Sci U S A, 103(13), 5167–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macqueen DA, Heckman BW, Blank MD, Janse Van Rensburg K, Park JY, Drobes DJ, & Evans DE (2014). Variation in the alpha 5 nicotinic acetylcholine receptor subunit gene predicts cigarette smoking intensity as a function of nicotine content. Pharmacogenomics J, 14(1), 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, & McGehee DS (2000). Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron, 27(2), 349–357. [DOI] [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, & Kellar KJ (2008). The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem, 104(2), 446–456. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Grady SR, Salminen O, Paley MA, Wageman CR, McIntosh JM, & Whiteaker P (2014). alpha6beta2*-subtype nicotinic acetylcholine receptors are more sensitive than alpha4beta2*-subtype receptors to regulation by chronic nicotine administration. J Neurochem, 130(2), 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin I, Dani JA, & De Biasi M (2015). Nicotine withdrawal. Curr Top Behav Neurosci, 24, 99–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel C, Fattore L, Pons S, Hay YA, Marti F, Lambolez B, De Biasi M, Lathrop M, Fratta W, Maskos U, Faure P (2014). Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol Psychiatry, 19(8), 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton G, Nasirova N, Sparks DW, Brodsky M, Sivakumaran S, Lambe EK, & Turner EE (2018). Chrna5-Expressing Neurons in the Interpeduncular Nucleus Mediate Aversion Primed by Prior Stimulation or Nicotine Exposure. J Neurosci, 38(31), 6900–6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ , Lester HA (2007). Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci, 27(31), 8202–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, & Perry DC (2003). Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther, 307(3), 1090–1097. [DOI] [PubMed] [Google Scholar]
- Omelchenko N, & Sesack SR (2005). Laterodorsal tegmental projections to identified cell populations in the rat ventral tegmental area. J Comp Neurol, 483(2), 217–235. [DOI] [PubMed] [Google Scholar]
- Paolini M, & De Biasi M (2011). Mechanistic insights into nicotine withdrawal. Biochem Pharmacol, 82(8), 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NE, Balfour DJ, & Markou A (2007). Chronic bupropion attenuated the anhedonic component of nicotine withdrawal in rats via inhibition of dopamine reuptake in the nucleus accumbens shell. Eur J Neurosci, 25(10), 3099–3108. [DOI] [PubMed] [Google Scholar]
- Perez XA, Bordia T, McIntosh JM, Grady SR, & Quik M (2008). Long-term nicotine treatment differentially regulates striatal alpha6alpha4beta2* and alpha6(nonalpha4)beta2* nAChR expression and function. Mol Pharmacol, 74(3), 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Mao D, Gold AB, McIntosh JM, Pezzullo JC, & Kellar KJ (2007). Chronic nicotine differentially regulates alpha6-and beta3-containing nicotinic cholinergic receptors in rat brain. J Pharmacol Exp Ther, 322(1), 306–315. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, & Brunzell DH (2008). It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol, 84(4), 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, & Dani JA (1997). Nicotine activates and desensitizes midbrain dopamine neurons. Nature, 390(6658), 401–404. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, & Dani JA (2004). Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem, 11(1), 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada P, Jensen K, & Hoebel BG (2001). Effects of nicotine and mecamylamine-induced withdrawal on extracellular dopamine and acetylcholine in the rat nucleus accumbens. Psychopharmacology (Berl), 157(1), 105–110. [DOI] [PubMed] [Google Scholar]
- Radke AK, & Gewirtz JC (2012). Increased dopamine receptor activity in the nucleus accumbens shell ameliorates anxiety during drug withdrawal. Neuropsychopharmacology, 37(11), 2405–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renda A, & Nashmi R (2014). Chronic nicotine pretreatment is sufficient to upregulate alpha4* nicotinic receptors and increase oral nicotine self-administration in mice. BMC Neurosci, 15, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, & De Biasi M (2003). The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol, 63(5), 1059–1066. [DOI] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J, & De Biasi M (2009). Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci, 29(10), 3014–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarginson JE, Killen JD, Lazzeroni LC, Fortmann SP, Ryan HS, Schatzberg AF, & Murphy GM Jr. (2011). Markers in the 15q24 nicotinic receptor subunit gene cluster (CHRNA5-A3-B4) predict severity of nicotine addiction and response to smoking cessation therapy. Am J Med Genet B Neuropsychiatr Genet, 156B(3), 275–284. [DOI] [PubMed] [Google Scholar]
- Sciaccaluga M, Moriconi C, Martinello K, Catalano M, Bermudez I, Stitzel JA, Maskos U, Fucile S (2015). Crucial role of nicotinic alpha5 subunit variants for Ca2+ fluxes in ventral midbrain neurons. FASEB J, 29(8), 3389–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherva R, Wilhelmsen K, Pomerleau CS, Chasse SA, Rice JP, Snedecor SM, Bierut LJ, Neuman RJ, Pomerleau OF (2008). Association of a single nucleotide polymorphism in neuronal acetylcholine receptor subunit alpha 5 (CHRNA5) with smoking status and with ‘pleasurable buzz’ during early experimentation with smoking. Addiction, 103(9), 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiffman S, West R, Gilbert D, Craving S. W. G. o. t. A. o., & Withdrawal in Clinical, T. (2004). Recommendation for the assessment of tobacco craving and withdrawal in smoking cessation trials. Nicotine Tob Res, 6(4), 599–614. [DOI] [PubMed] [Google Scholar]
- Staley JK, Krishnan-Sarin S, Cosgrove KP, Krantzler E, Frohlich E, Perry E, Dubin JA, Estok K, Brenner E, Baldwin RM, Tamagnan GD, Seibyl JP, Jatlow P, Picciotto MR, London ED, O’Malley S, van Dyck CH (2006). Human tobacco smokers in early abstinence have higher levels of beta2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci, 26(34), 8707–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens VL, Bierut LJ, Talbot JT, Wang JC, Sun J, Hinrichs AL, Thun MJ, Goate A, Calle EE (2008). Nicotinic receptor gene variants influence susceptibility to heavy smoking. Cancer Epidemiol Biomarkers Prev, 17(12), 3517–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, & Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol, 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States. Public Health Service. Office of the Surgeon General. (2014). The health consequences of smoking--50 years of progress : a report of the surgeon general. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Office of the Surgeon General. [Google Scholar]
- Wills L, Ables JL, Braunscheidel KM, Caligiuri SPB, Elayouby KS, Fillinger C, Ishikawa M, Moen JK, Kenny PJ (2022). Neurobiological Mechanisms of Nicotine Reward and Aversion. Pharmacol Rev, 74(1), 271–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfman SL, Gill DF, Bogdanic F, Long K, Al-Hasani R, McCall JG, Bruchas MR, McGehee DS (2018). Nicotine aversion is mediated by GABAergic interpeduncular nucleus inputs to laterodorsal tegmentum. Nat Commun, 9(1), 2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, & Lester HA (2009). Chronic nicotine selectively enhances alpha4beta2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci, 29(40), 12428–12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Broussard JI, Levine AT, Jenson D, Arenkiel BR, & Dani JA (2017). Dopamine receptor activity participates in hippocampal synaptic plasticity associated with novel object recognition. Eur J Neurosci, 45(1), 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Buhlman L, Khan GM, Nichols RA, Jin G, McIntosh JM, Whiteaker P, Lukas RJ, Wu J (2011). Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J Neurosci, 31(7), 2537–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Hu J, Lucero L, Liu Q, Zheng C, Zhen X, Jin G, Lukas RJ, Wu J (2009). Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol, 587(2), 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Dong Y, Doyon WM, & Dani JA (2012). Withdrawal from chronic nicotine exposure alters dopamine signaling dynamics in the nucleus accumbens. Biol Psychiatry, 71(3), 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TA, Placzek AN, & Dani JA (2010). In vitro identification and electrophysiological characterization of dopamine neurons in the ventral tegmental area. Neuropharmacology, 59(6), 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Nonnemaker J, Sherrill B, Gilsenan AW, Coste F, & West R (2009). Attempts to quit smoking and relapse: factors associated with success or failure from the ATTEMPT cohort study. Addict Behav, 34(4), 365–373. [DOI] [PubMed] [Google Scholar]