

SUMMARY
Schwann cells (SCs) are the primary glia of the peripheral nervous system. SCs are involved in many debilitating disorders including diabetic peripheral neuropathy (DPN). Here we present a strategy for deriving SCs from human pluripotent stem cells (hPSCs) that enables comprehensive studies of SC development, physiology and disease. hPSC-derived SCs recapitulate the molecular features of primary SCs and are capable of in vitro and in vivo myelination. We established a model of DPN that revealed the selective vulnerability of SCs to high glucose. We performed a high-throughput screen and found that an antidepressant drug, bupropion, counteracts glucotoxicity in SCs. Treatment of hyperglycemic mice with bupropion prevents their sensory dysfunction, SC death and myelin damage. Further, our retrospective analysis of health records revealed that bupropion treatment is associated with a lower incidence of neuropathy among diabetic patients. These results highlight the power of this approach for identifying therapeutic candidates for DPN.
Keywords: Metabolic stress, Myelination disorders, Demyelination, Drug repurposing, Nerve repair
eTOC blurb
hPSC-derived Schwann cells recapitulate the molecular and functional features of primary Schwan cells and enable the discovery of disease mechanisms and therapeutic candidates to prevent and treat nerve damage in diabetes.
Bupropion decreases diabetic nerve damage in mice and reduces neuropathy risk in patients.
Graphical Abstract
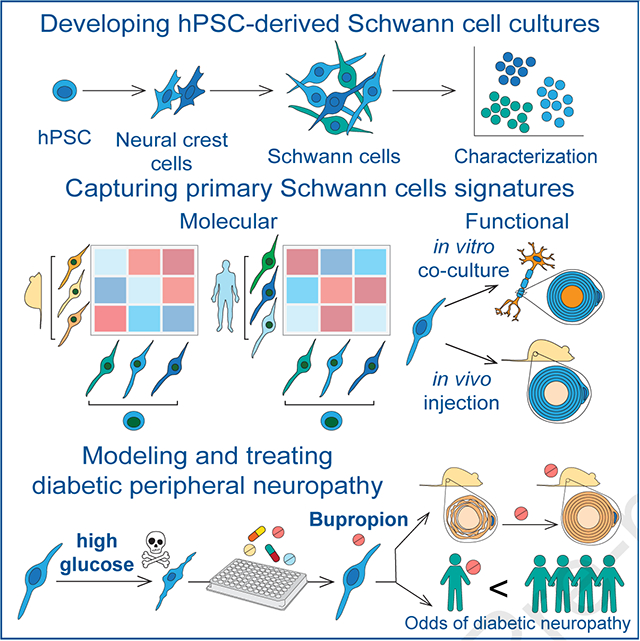
INTRODUCTION
Schwann cells (SCs) are vital components of the peripheral nervous system (PNS). They are crucial for the development, structural maintenance and function of the nerves and exhibit a remarkable ability to promote neural repair following injury1,2. SCs support axons by forming insulating myelin sheaths and Remak bundles, and provide essential neurotrophic factors. SCs develop from the neural crest (NC) via a SC precursor (SCP) intermediate that is highly proliferative and migratory. SCPs further differentiate into immature SCs that ultimately give rise to mature myelinating or non-myelinating SCs. In addition to SCs, SCPs can give rise to other derivatives (SCPDs) such as melanocytes3–5. SC defects are involved in genetic and acquired PNS disorders such as Charcot-Marie-Tooth disease, Schwannomatosis, Guillain-Barre Syndrome and diabetic peripheral neuropathy (DPN) for which there are currently no faithful disease models or effective therapies.
Understanding the development and function of SCs and their roles in PNS health and disease has broad basic and translational implications. However, access to authentic models of human SCs at large scale has been a major challenge. Here, we developed hPSC differentiation strategies for efficient derivation of SCs that recapitulate molecular features of primary SCs and are capable of myelination. We characterized the diversity of cell types in our cultures using a combination of imaging and high-resolution transcriptomic profiling and identified markers and molecular signatures for human SC subtypes. We further validated the engraftment potential of these cells upon transplantation into a rat model of peripheral neuropathy. Finally, we leveraged hPSC-derived SCs to model the most common cause of peripheral neuropathy, i.e. diabetic peripheral neuropathy (DPN) that affects 30–60% of diabetic patients6 and is the leading cause of diabetes-related hospital admissions and nontraumatic lower-extremity amputations7. The pathogenesis of DPN is complex involving vascular disease, hyperglycemia, hypoxia and oxidative stress that result in cytotoxicity and progressive degeneration of peripheral nerves8. While symptoms arise from neuronal dysfunction, it is unclear whether sensory neuron damage is the primary event in DPN, and there is evidence that SC degeneration and peripheral demyelination may be contributing factors9. Dissecting cell type specific mechanisms is challenging using current animal models of DPN given the involvement of various non-cell autonomous factors including systemic vascular abnormalities. We utilized hPSC-derived SCs and sensory neurons to determine cell type specific vulnerabilities to high glucose, establish an alternative human-based model of DPN and identify potential therapeutic candidates.
RESULTS
Derivation and prospective isolation of SC lineages from hPSCs
We previously established hPSC differentiation protocols to access various NC lineages including enteric and sensory neurons10–13. However, reliable methods for derivation of SCs from hPSCs have not been available. Previous reports on the derivation of SC-like cells from hPSCs did not show molecular authenticity through gene expression profiling and failed to demonstrate functional myelination14–17. While the mechanisms of SC specification during human development remain to be elucidated, SCs are known to arise from SOX10+ NC cells. Based on studies in the mouse and chick embryos, NC first gives rise to SC precursors that associate with developing neuronal fibers. These neurons produce NRG1 which promotes the survival and further differentiation of SC precursors (SCPs) by activating ERBB3 receptors18. By E13.5 of mouse development, SCPs give rise to immature SCs which express markers such as GFAP, S100 and POU3F1 while maintaining the expression of SOX10. Terminal differentiation of SCs into myelinating and non-myelinating fates continues for extended time periods and concludes only after birth19.
Initial NC differentiation protocols relied on the delamination of putative NC cells from neuroepithelial lineages combined with the prospective isolation of p75+ and/or HNK1+ NC precursors 20,21. While those protocols yield various NC-derived lineages, the levels of SOX10 expression are generally low. In contrast, more directed NC induction protocols based on timed exposure to activators of WNT signaling show robust induction of SOX10 in the majority of cells by day 11 of differentiation10,12,13,22,23. Upon further culture, those NC cells can be directed into SOX10+ melanocytes23 but also give rise to SOX10− mesenchymal and neuronal precursors12,13,21,23. Since SOX10 is a key marker in the SC lineage24, we first screened for conditions that maintained its expression in cultured NC precursors. After screening several factors including EGF, FGF, WNT, Notch, TGFβ, BMP and endothelin 3, we observed that activation of WNT signaling through CHIR99021 exposure and treatment with FGF2 resulted in the maintenance of SOX10 expression in 3D aggregates that we refer to as developing precursors (Figure 1 A and B).
Figure 1: Deriving Schwann cells from hPSCs.
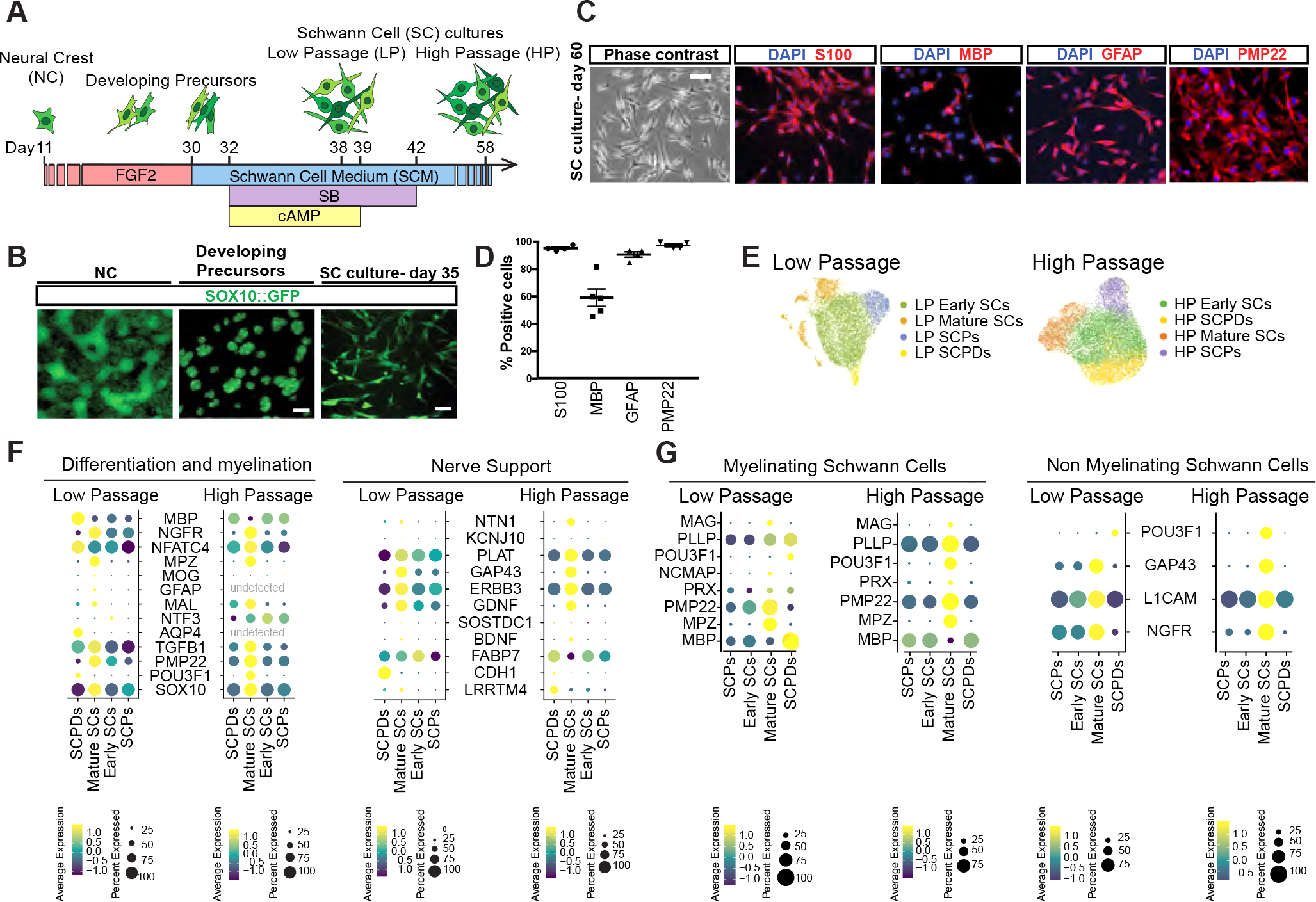
A) Schematic of the protocol for deriving developing precursors and SC cultures from hPSCs-derived neural crests (NCs).
B) SOX10::GFP expression at days 11, 25 and 35 of differentiation. Scale bars = 100 μm (left and middle) and 25 μm (right).
C, D) Representative (C) and quantification (D) of immunofluorescence images of hPSC-derived SCs for lineage markers (D60). Scale bar= 25 μm.
E) UMAP visualization of scRNA-seq data for low passage (LP, Day 38) and high passage (HP, Day 58) SC cultures. SCPD: SC precursor derived; SCP: SC precursor.
F, G) Dot plot of the scaled average expression of SC differentiation and myelination (left) and nerve support (right), myelinating (mySC) and non-myelinating (nmSC) SC markers in scRNA-seq data of LP and HP SCs.
Further treatment of developing precursors with SC media (SCM) containing FGF2, SB431542 and dbcAMP enabled the induction of additional SC markers and upregulation of genes involved in glial-neuronal interactions and support (Figure S1 A and B). These cultures can be passaged and maintained for several weeks while retaining the expression of key SC markers S100, MBP, GFAP and PMP22 (Figure 1 C and D).
In order to determine the cellular diversity of our hPSC-derived SC cultures, we performed single cell RNA-sequencing (scRNA-seq) at two differentiation time points; low passage (LP, day 38) and high passage (HP, day 58). Unbiased clustering of both LP and HP datasets revealed four transcriptionally distinct cell types namely SCPs, early SCs, mature SCs and SCP derivatives (SCPDs) (Figure 1E, S1C). These cell types differentially expressed canonical markers of SC differentiation and function (Figure 1F). For example, mature SCs in both datasets expressed higher levels of myelinating and non-myelinating SC markers such as PMP22 and NGFR. Nerve support markers and neurotrophic factors including ERBB3, GDNF NGF, BDNF and GAP43 were also enriched in mature SCs particularly in the HP cultures (Figure 1F, S1D). We module scored a number of SC functional gene sets including neurotrophic factors, neurotransmitter receptors and transcription factors (Table S1) and detected differential expression of many neurotransmitter receptors and postsynaptic signal transmission genes by our SC types (Figure S1E). We also identified transcription factors that were specifically expressed by each population in LP and HP cultures. For example, early SCs were enriched for E2F7 and E2F8 while mature SCs expressed SOX10 and FOXO1, POU3F2, TBX19 at higher levels. POU6F2 was the common enriched transcription factor in LP and HP SCPDs (Figure S1F).
To validate the authenticity of our hPSC-derived SCs, we assessed the expression of top DEG in glial precursor, early SC, myelinating and non-myelinating SC and satellite glial genes derived from a mouse single cell transcriptomics dataset previously published by Segal and colleagues25 (Figure 1G, S2 A–C). We detected the expression of these markers in both our LP and HP cultures with cell type specific expression patterns (Figure 1G, S2A–C). The highest expression of almost all mouse glial precursor markers was detected in our SCP clusters (Figure S2A). We also observed the expression of a number of mouse immature SC markers such as DLX1, DLX2, GAP43, SLC39A8 and PCSK6 in our cultures (Figure S2C), however, some of these markers are also expressed by mature non-myelinating SCs (Figure 1G). Some markers such as MPZ and MATN2 were specifically expressed by mature SCs. However, the majority of other genes showed differential transcript levels between cell types while not exclusively expressed in a single population (Figure 1G). Interestingly, our LP and HP mature SC populations were highly enriched for myelinating and non-myelinating markers indicating that our hPSC-derived SC cultures are stable over time and resemble mature primary SCs (Figure 1G).
For a more in-depth assessment of the authenticity of the hPSC-derived SC cultures, we compared their gene expression profile with tissue-derived primary SCs using the SingleCellNet machine learning pipeline26 that enables classification of query scRNA-seq data in comparison to reference datasets. Remarkably, when the model was trained on the human cell atlas datasets composed of a diverse range of cell types from adult human tissue27(Figure 2A, S2D), a large proportion of our hPSC-derived cells, particularly the mature SCs, were classified as primary human SCs (Figure 2B). In addition, while all clusters contained cells classified as melanocyte the highest proportion of melanocyte classification was observed in the SCPD clusters (Figure 2B). As the primary human dataset used for training the SingleCellNet algorithm contained numerous different cell types, it is noteworthy that the unbiased classifications validate the authenticity of our hPSC-derived SCs and their precursor derivatives. Next, we performed a similar analysis by generating a model trained on a primary mouse SC dataset25 (Figure 2C and S2E) because the human cell atlas27 does not specify SC subtypes, and SingleCellNet26 is also capable of cross-species classification. The majority of our cells were classified as g1 and g9 which were clusters originally annotated by the authors as support glia and glial precursors respectively (Figure 2D). We also observed many cells classified as g5 and g3 identities which were described as non-myelinating and immature SCs respectively (Figure 2D). These data confirm that our hPSC-derived SCs recapitulates the molecular features of bona-fide SCs and capture the diversity of SC lineages in vivo.
Figure 2: hPSC-derived Schwann cells are transcriptionally similar to primary Schwann cells.
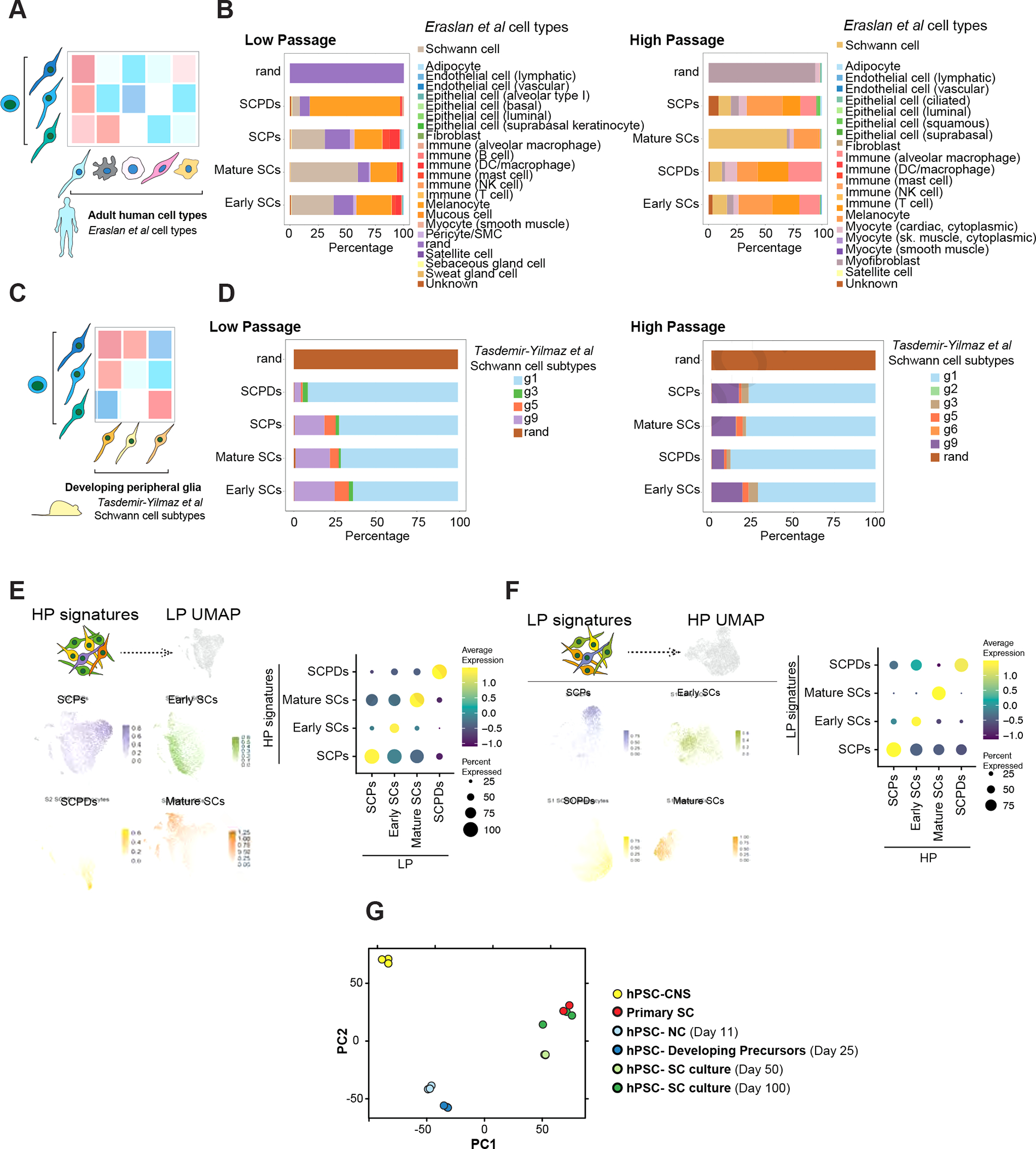
A) Schematic of comparing SCs with primary human adult cell types.
B) SingleCellNet26 classification of LP (left) and HP (right) SC clusters using the human cell atlas dataset27 as a reference.
C) Schematic of comparing SCs with primary mouse developing peripheral glia.
D) SingleCellNet26 classification of HP (left) and HP (right) SC clusters using a single cell transcriptomics mouse peripheral glia25 dataset as reference.
E, F) Module scoring of top 100 HP SC type specific differentially expressed (DE) marker genes in LP SC types (left). Feature (left) and dot plot (right) visualizations are depicted. Panel E shows the HP dataset module scored in LP dataset and panel F shows the other way comparison.
G) Principal component analysis (PCA) of NC cells, developing precursors, human primary SCs, and hPSC-derived SC cultures at D50 and D100 of differentiation in comparison with central nervous system (CNS) precursors.
The proliferative capacity of our SC cultures enables their expansion and scalability. To characterize the proliferation potential of our cell types, we determined the proportional distribution of cell cycle phases within individual populations (Figure S2 F and G). As cultures transition from low to high passage, all cell types progressively exit the cell cycle. This is particularly evident in SCPs and early SCs that are predominantly cycling in LP (Figure S2 F and G). This is in agreement with the lower expression of the proliferation marker MKI67 in HP populations (Figure S2H)
To assess the stability of SC identity after long-term expansion, we compared the LP and HP datasets by module scoring the transcriptional signature of LP cell types in HP cell types and vice versa (Figure 2 E and F). Notably, each cell type signature was most similar to its corresponding cell type in the other dataset (Figure 2 E and F). The similarity between corresponding LP and HP clusters was further demonstrated when we performed clustering on a merged dataset and obtained the same cell populations (Figure S2 I and J). Bulk transcriptomics analysis of cultures at different time points demonstrated that hPSC-derived developing precursors were closely related to early NC cells while SC cultures, particularly in higher passages, showed a gene expression pattern closely matching primary adult human SCs (Figure 2G).
To characterize the diversity in these cultures in higher resolution, we performed further sub-clustering and revealed two early SCs and two SCPDs populations (Figure S2K). While early SCs 1 and 2 separated solely based on their cell cycle phase distributions (Figure S2L), SCPDs sub-clusters were predominantly LP or HP specific (Figure S2M). To determine if LP and HP SCPDs were functionally distinct, we performed gene ontology (GO) enrichment analysis of their top 250 differentially expressed genes (DEGs). Interestingly, despite high level expression of canonical melanocytic genes such as MITF, MLANA and PMEL, LP SCPDs were enriched for myelin production terms, such as cholesterol and lipid metabolism pointing to a dual melanocyte-SC identity (Figure S1C, S2N). On the other hand, HP SCPDs displayed enrichment for melanin synthesis and pigmentation (Figure S2N) indicating that as cultures age SCPDs become more melanocytic.
Strategies for prospective isolation enables the purification of SC populations from heterogeneous cultures. To enable fluorescence-activated cell sorting (FACS) based purification of hPSC-SCs, we analyzed surface marker expression in our scRNA-seq datasets and identified transcripts that were specifically enriched in each cell type (Figure S3A). While this analysis revealed a number of surface markers that were almost exclusively expressed by one cluster, for example CDH1 in LP SCPDs, and CD24 and IL10RA in HP mature SCs, these markers were not expressed by all cells in these clusters. Also, the majority of surface markers that were highly expressed in each cluster were also expressed at lower levels by cells of the other clusters. To evaluate the expression of surface markers at the protein level, we next screened a library of 242 antibodies for human surface antigens that specifically mark GFAP+ SCs (Figure S3 B and C). We identified 11 surface antibodies that stained >20% of the GFAP+ SCs of which CD44, CD49e, CD81 and CD98 labeled most of the target population (Figure S3C). Interestingly, 9 of the 11 surface marker hits identified by the antibody screening were among the proteins identified in the scRNA-seq data analysis (Figure S3A). Among these, CD46, CD146, CD147 and CD166 were enriched in mature SCs and CD44 was highly expressed in SCPDs in both LP and HP cultures (Figure S3A). Further validations revealed that CD98 was the only marker specifically expressed in SCs but not in NCs or SCPs (Figure S3D).
Collectively these data demonstrate that our hPSC differentiation system generates scalable and proliferative human SC cultures that can be further enriched using FACS.
hPSC-SCs promote neuronal maturation and myelination in vitro and engraft into injured sciatic nerves in rats
SCs play fundamental roles in maintaining and protecting the structure and function of the peripheral nerves. Premyelinating SCs interact with axons and receive signals from neurons that lead to the formation of lipid rich myelin sheaths, enabling fast neuronal signal propagation in the PNS. Since our hPSC-derived mature SCs express genes involved in myelination and lipid metabolism in high levels (Figure 1 F and G, S2N), we set out to identify and characterize premyelinating SCs in our cultures. We module scored a curated list of premyelinating SC markers and canonical myelination genes (Table S1) 28 in our LP and HP mature SCs (Figure 3A). We found that more than 25% of mature SCs in each dataset show high expression of myelination genes (Figure 3A). These cells, which we termed mySCs, were specifically enriched for a number of neurotrophic factors and neurotransmitter and postsynaptic transmission genes (Figure S4 A and B). To define the unique functional properties of these cells we performed pathway enrichment analysis using GO BP, KEGG and Reactome gene sets on the significantly upregulated genes in mySCs (Figure 3B). Interestingly, of the top 50 significantly enriched pathways, we identified multiple pathways related to axon development, myelination, neuronal development, synapse assembly, cell adhesion and cell motility. HP mySCs upregulated pathways related to extracellular matrix organization, cell adhesion and cell motility among others. This is intriguing given the known contribution of SCs in depositing and organizing ECM components, formation of lamellipodia and cytoplasmic protrusions and forming contact and recognizing axons during radial sorting and myelination. These indicate our mySCs are equipped with the molecular programs that enable myelinating SCs to perform their function.
Figure 3: hPSC-derived Schwann cells myelinate hPSC-derived sensory neurons and engraft in injured rat sciatic nerves.
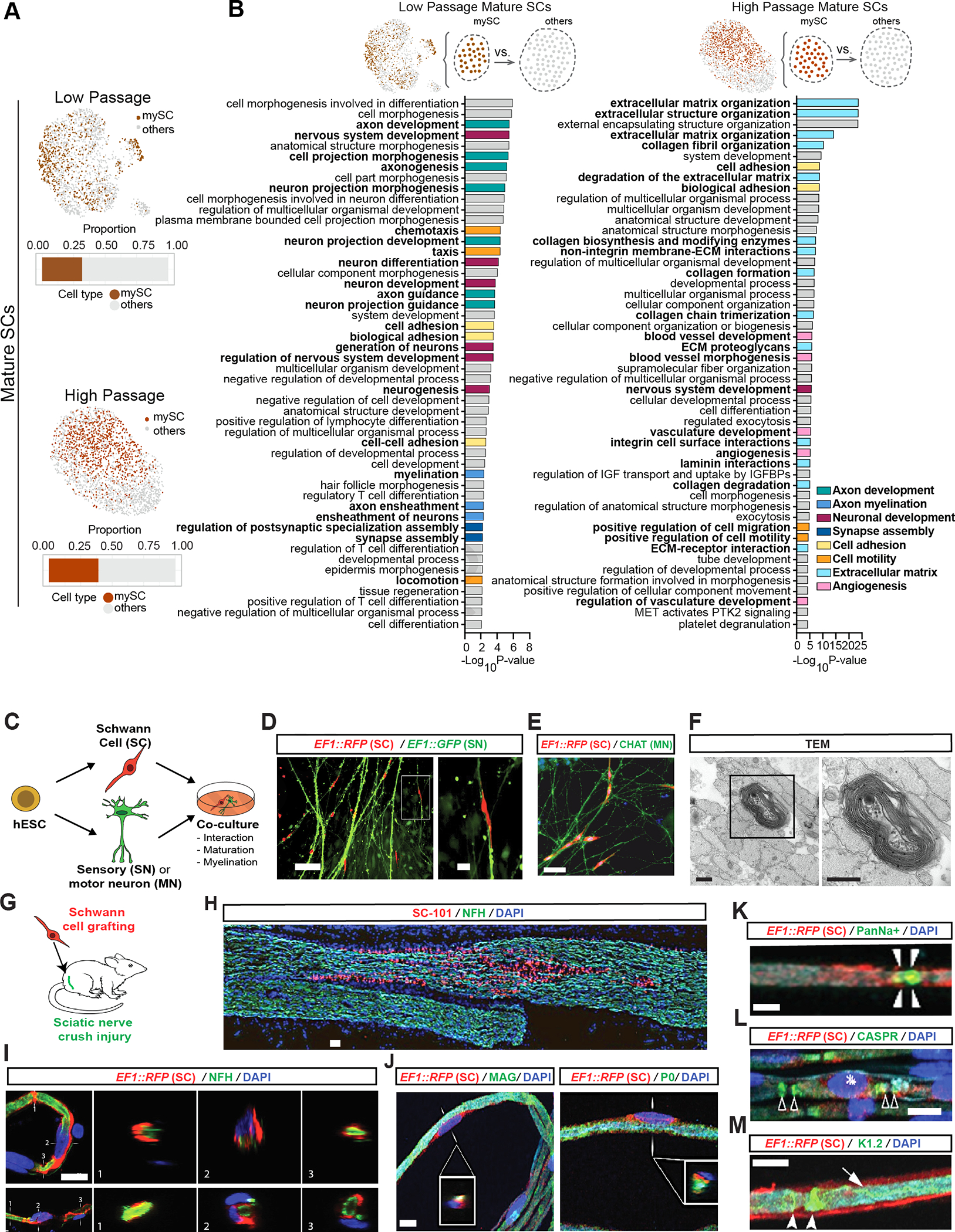
A) Feature plots of mature SC clusters isolated from LP (top) and HP (bottom) scRNA-seq data. dark colors: myelinating (mySC), bar plots: relative population of mySCs.
B) Pathway enrichment analysis of top 250 DEGs of myelinating mature SCs in LP (left) and HP (right). Top 50 pathways from combined gene ontology biological process (GOBP), Reactome and KEGG analysis.
C) Schematic of the SC co-cultures with sensory or motor neurons.
D, E) Immunofluorescence imaging of SCs (EF1::RFP) co-cultured with sensory (EF1::GFP,D) or motor neurons (stained for CHAT,E).
F) TEM of myelin in sensory neuron and SC co-cultures. Images were taken at 80 kV. Scale bars= 500 nm.
G) Schematic of SC transplantation in adult rat sciatic nerves. RFP+ SCs were injected following nerve crush at the site of injury.
H) Immunofluorescence staining of grafted sciatic nerves for human specific nuclear marker SC101 at 8 weeks post transplantation.
I and J) Confocal analysis of teased sciatic nerve fibers for RFP (grafted human cells), axonal marker (NFH, I), myelin markers (MAG and P0, J), node markers Pan-Na+ (sodium channel, arrow heads, K), CASPR (arrow heads, L) and Kv1.2 (K+ channel, arrow heads, M) and DAPI. Scale bars= 100 μm in D left, E and B 20 μm in D right, I and J, 10 μm in K-M.
Since cell adhesion molecules (CAMs) play an important role in SC association with axons and cell adhesion was enriched in mySCs (Figure 3B), we module scored a list of CAMs combining cell-cell and cell-matrix gene sets in mature SCs and identified many CAMs that were specifically depleted and enriched in mySCs relative to the other mature SCs (Figure S4C). For example, HLA-DR that was also a hit in our SC antibody screen (Figure S3B, S4C) was enriched in mySCs. The proinflammatory interleukin IL18 was depleted while MAG and KIT were enriched in mySCs (Figure S4C). Members of the contactin protein family (CNTN1, 4 and 6) that are axon-associated CAMs and play roles in the formation of axon connections in the developing nervous system were specifically enriched in HP mySCs (Figure S4C). Similarly, HP mySCs were highly enriched for PLXNB3 which is important for axon guidance and cell migration (Figure S4C).
High level expression of these CAMs prompted us to evaluate the ability of our SCs to physically interact with axons. We first determined whether our hPSC-derived SC cultures contain any neurons by measuring the expression of the pan neuronal marker TUBB3 by flow cytometry and did not detect a substantial neuronal population (Figure S4D). Therefore, to assess the interaction of SCs with neurons, we established co-cultures with hPSC-derived sensory11 and motor neurons28 (Figure 3C). SCs associated closely with both sensory neurons and motor neuros (Figure 3 D and E). To determine the ability of the SCs to produce myelin, we performed transmission electron microscopy (TEM) on co-cultures and observed wrapping of numerous axons by SCs and formation of myelin sheaths (Figure 3F).
The protracted process of neuronal maturation in hPSC-derived cultures is a major hurdle in the field. Glial cells such as astrocytes have been shown to promote the functional maturation of hPSC-derived CNS neurons29. To assess the impact of SCs on neuronal maturation, we performed calcium imaging in hPSC-derived motoneurons co-cultured with SCs. Interestingly, there was a marked increase in the calcium response of stage-matched motoneurons co-cultured with SCs (Figure S4E). By day 70, the responsiveness to glutamate and KCl stimulations further improved and remained distinct from cultures containing motor neurons only (Figure S4F). Our findings demonstrate the capability of hPSC-SCs to modulate neuronal function in vitro. In order to determine whether hPSC-SCs are functional in vivo and are capable of producing myelin, we asked whether they could survive and engraft in a rat model of sciatic nerve injury. We depleted endogenous SCs through a mechanical crush of the nerve and injected RFP-labeled hPSC-SC cultures at the site of injury (Figure 3G). The transplanted SCs could be readily detected at eight weeks after nerve injection using the human-specific nuclear marker SC101 (Figure 3H). Transplanted hPSC-SCs wrapped the host neurons (Figure 3I) and expressed the myelin markers MAG and P0 (Figure 3J). In mature myelinated fibers, sodium channels are localized at nodes of Ranvier: the site of action potential electrogenesis. This area is flanked by a CASPR-expressing domain (juxtaparanodal region) where the axon membrane is in close contact to myelin membrane. Adjacent to the CASPR+ region is an axon membrane domain marked by the expression of potassium channels. Remarkably, we observed appropriate localization of both sodium and potassium channels in axons that were wrapped by RFP-labelled hPSC-SCs (Figure 3K–M). These studies demonstrate the ability of hPSC-SCs to engraft and produce myelin that is appropriately associated with nerve fibers and the nodes of Ranvier in injured adult peripheral nerves.
These results demonstrate that our hPSC-derived cultures of functional SCs offer a framework for modeling pathologies in which SCs play central roles in disease initiation and progression. For example, a large subset of Charcot-Marie-Tooth (CMT) patients suffer from debilitating myelin defects. Importantly, genes associated with CMT including de-myelinating CMT1, axonal CMT2, and intermediate CMT are expressed by our SC cultures confirming their applicability for modeling CMT in future studies (Figure S4G).
hPSC-derived SCs enable modeling, mechanistic understanding and treating diabetic peripheral neuropathy
In addition to rare inherited defects such as CMT, SCs are associated with a broad range of other neuropathies. The most prominent form of an acquired neuropathy is diabetic peripheral neuropathy (DPN) which results from the progressive degeneration of peripheral nerves 8. While symptoms arise from neuronal dysfunction, it is unclear whether sensory neuron damage is the primary event in DPN, and there is evidence that SC degeneration and peripheral demyelination may be contributing factors 9. As a proof of concept, we set out to leverage our human hPSC differentiation system to model DPN by investigating the effect of high glucose on hPSC-derived sensory neurons11,13 and SCs (Figure 4A).
Figure 4: Schwann cells are selectively vulnerable to high glucose exposure.
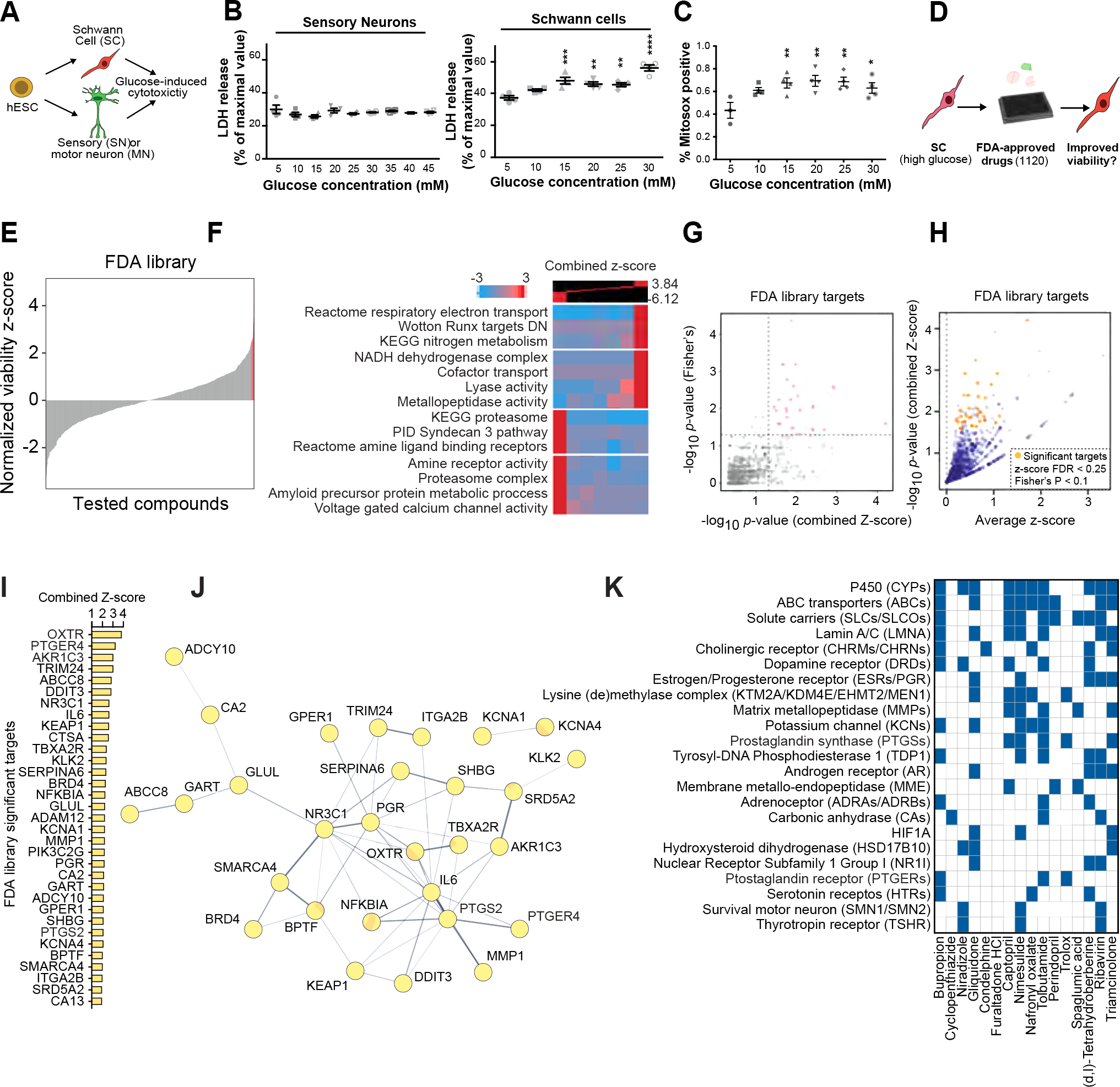
A) Schematic of the experimental paradigm for modeling diabetic nerve damage.
B) Cytotoxicity analysis of SCs and sensory neurons at different glucose concentrations.
C) Oxidative stress measurement of SCs exposed to increasing concentration of glucose. Statistical analysis (one-way ANOVA) comparing to low glucose (5 mM) condition. ns, not significant. * p<0.05; ** p<0.01.
D) Schematic of high-throughput drug screening for identification of compounds that enhance the viability of high glucose-treated hPSC-SCs.
E) Presentation of the distribution of library compounds by their corresponding normalized viability z-score.
F) Gene-set enrichment analysis using iPAGE for the library compounds’ targets identifies GO terms associated with hits improving and worsening SC viability.
G) p-value correlation plot to identify significant drug targets. See methods for details.
H) One-sided volcano plot for all genes with positive z-scores. Gold: thresholds of combined z-score FDR<0.25 and Fisher’s p-value <0.1.
I, J) Identified target genes (gold in H) and their protein-protein interaction network (J).
K) Predicted targets of hits compiled from the following databases: BindingDB51, Carlsbad52, Dinies53, PubChem bioassays54, SEA32, Superdrug 255 and SwisTargetPrediction56.
Measurement of apoptosis using cleaved caspase-3 flow cytometry and the LDH release cell death assay showed no overt toxicity in sensory neurons at glucose levels of up to 45 mM (Figure 4B, S5A). In contrast, LDH and Annexin V assays revealed that hPSC-derived SC cultures were exquisitely sensitive to even moderately increased glucose levels (Figure 4B, S5B). Treatment with high glucose induced oxidative stress in SC cultures (Figure 6C).
Figure 6: Bupropion treatment prevents diabetic nerve damage in vivo.
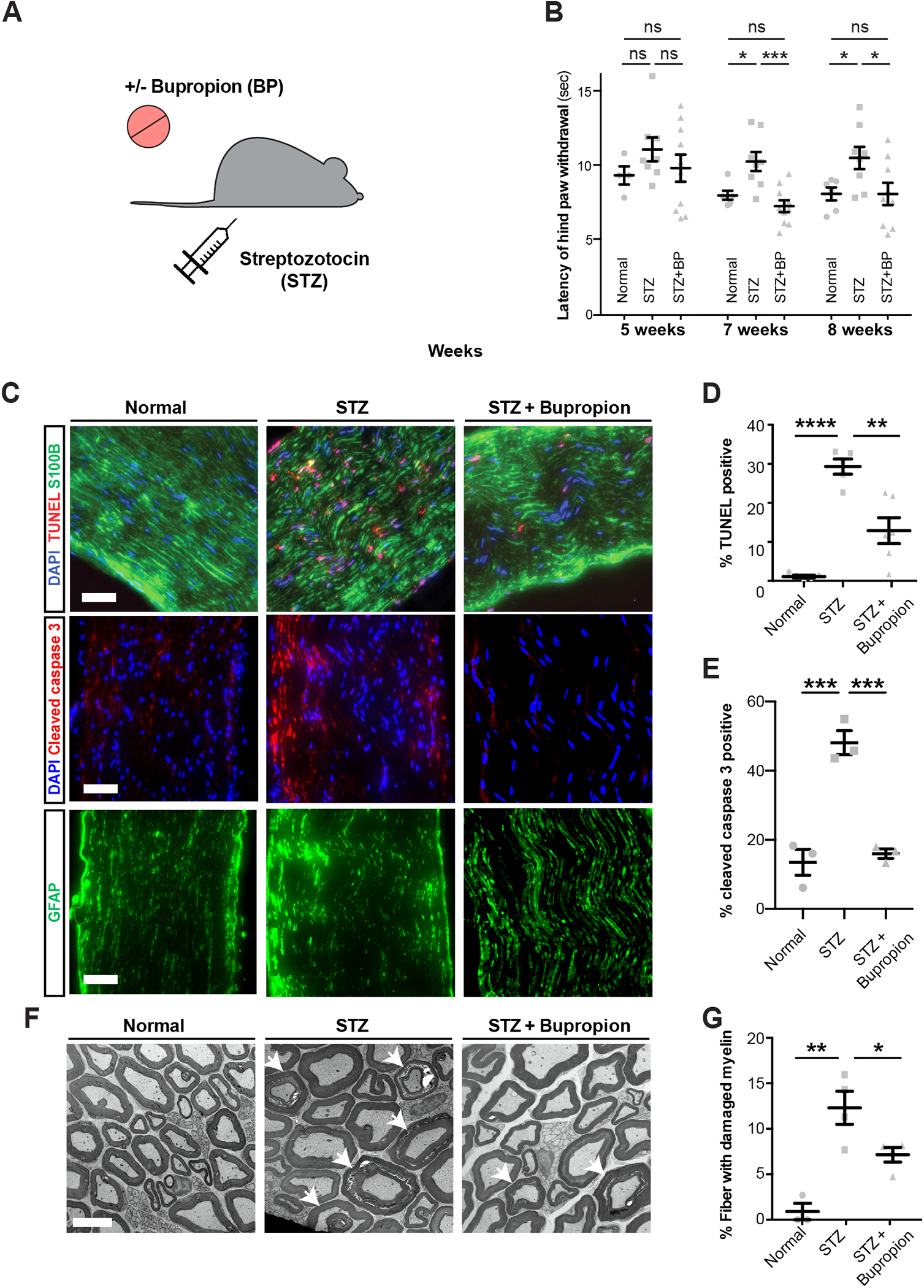
A) Schematic of modeling diabetes and bupropion treatment in mice.
B) Thermal sensitivity test measuring the latency of hind paw withdrawal in untreated mice or treated with STZ and bupropion.
C-E) TUNEL and cleaved caspase-3 staining (C) and quantification (D, E) in sciatic nerves of untreated mice or treated with STZ and bupropion. Scale bar= 100 μm.
F, G) Transmission electron microscopy and quantification of damaged myelin structures in sciatic nerves of untreated mice or treated with STZ and bupropion. Error bars: standard errors. Scale bar= 5 μm. * p<0.05; ** p<0.01; *** p<0.001; **** p < 0.0001.
Given the selective glucotoxicity in SCs, strategies that prevent glucose-mediated cellular damage in SCs may represent novel therapeutic opportunities for treating DPN. We established a high-throughput screen (HTS) to measure the viability of hPSC-SCs in the presence of high glucose. We screened the Prestwick library containing 1,120 small molecules of FDA approved drugs (Figure 4D, Figure S5C) and identified several hit compounds that significantly increased SC viability under high glucose conditions (Figure 4E, Table S2). Given that the library compounds target many different cellular pathways, we sought to determine the shared pathways among candidate drugs that improved SC viability under the high glucose condition.
We first predicted drug protein interactions for the entire Prestwick library using our previously established HTS analysis pipeline30–32. Among the GO terms associated with the SC protecting drug candidates we identified oxidative phosphorylation (OXPHOS), nitrogen metabolism and metallopeptidases (Figure 4F). We validated the expression of these candidate pathways in our SC cultures using module scoring analysis (Figure S5D). To determine the degree to which positive z-scores were enriched among the drugs targeting each protein, we performed a Fisher’s exact test. Through this analysis, we identified 33 proteins as significant drug targets filtered based on average combined z-score>0, false discovery rate (FDR)<0.25 and Fisher’s p<0.1 (Figure 4 G–I).
We performed a protein-protein interaction network analysis to identify interactions between our 33 significant drug targets using the STRING database33. In the resulting network, IL6, NR3C1, PGR and PTGS2 had the highest degree centrality (Figure 3J). In addition, for a more comprehensive target prediction, we generated a list of potential targets for the top hits derived from the HTS dataset (Figure S5E) or computational predictions that use network-based and similarity-based algorithms (Figure 4K, Table S3). Intriguingly, many of the potential target proteins were shared between multiple hits; including potassium channels (KCNs), estrogen and progesterone receptors (ESRs and PGRs), prostaglandin synthases (PTGSs) and prostaglandin receptors (PTGERs). Notably, the predicted target pattern of tolbutamide resembled that of our top hit, bupropion (BP) (Figure 4K).
BP is a widely used antidepressant marketed as Wellbutrin®. BP treatment increased the viability of high glucose treated SC cultures in a dose dependent manner (Figure S5A, Figure S5 F and G) but led to toxicity at concentrations higher than 2 μM (Figure S5G). In line with previous reports on glucose-mediated activation of oxidative stress response34,35, we observed the activation of cellular inflammatory response via NF-kB p65 localization to the nucleus in SCs exposed to high glucose and BP treatment was able to counteract this phenotype (Figure S5 H and I).
To understand the mechanism of glucotoxicity in SC cultures and its rescue with BP, we performed transcriptional and metabolite profiling (Figure 5A). We compared gene expression profiles of SCs treated with low glucose and high glucose with or without BP treatment followed by pathway enrichment analysis of DEGs (Figure 5E, Figure S5J). Of the 1,559 SC transcripts that were enriched or depleted in response to high glucose, 66 showed a reversed expression pattern in cells treated with BP (Figure 5E). Interestingly, of these, PTGER4 was also a predicted targets of BP (Figure 4 I–K, Table S3). The affected processes in SCs exposed to high glucose included several metabolic and cell cycle signaling pathways and BP treatment modulated pathways related to DNA replication and transcription as well as cell cycle and stress response (Figure S5J). Module scoring of top DEGs suggested different clusters might be differentially responsive to high glucose and/or BP treatments. Transcripts upregulated in high glucose showed higher expression in Mature SCs and early SCs (Figure S5K), while the module containing transcripts downregulated in high glucose were highly expressed in all clusters except SCPs (Figure S5K). Differentially upregulated and downregulated gene modules upon BP treatment were almost exclusively scored in matures SCs and SCPDs respectively (Figure S5K). We also evaluated the expression of the BP predicted targets as well as the targets of other HTS hits (Figure 3I) and observed varying expression levels of these transcripts among different clusters (Figure S5L).
Figure 5: Bupropion treatment counteracts high-glucose induced molecular changes in Schwann cells.
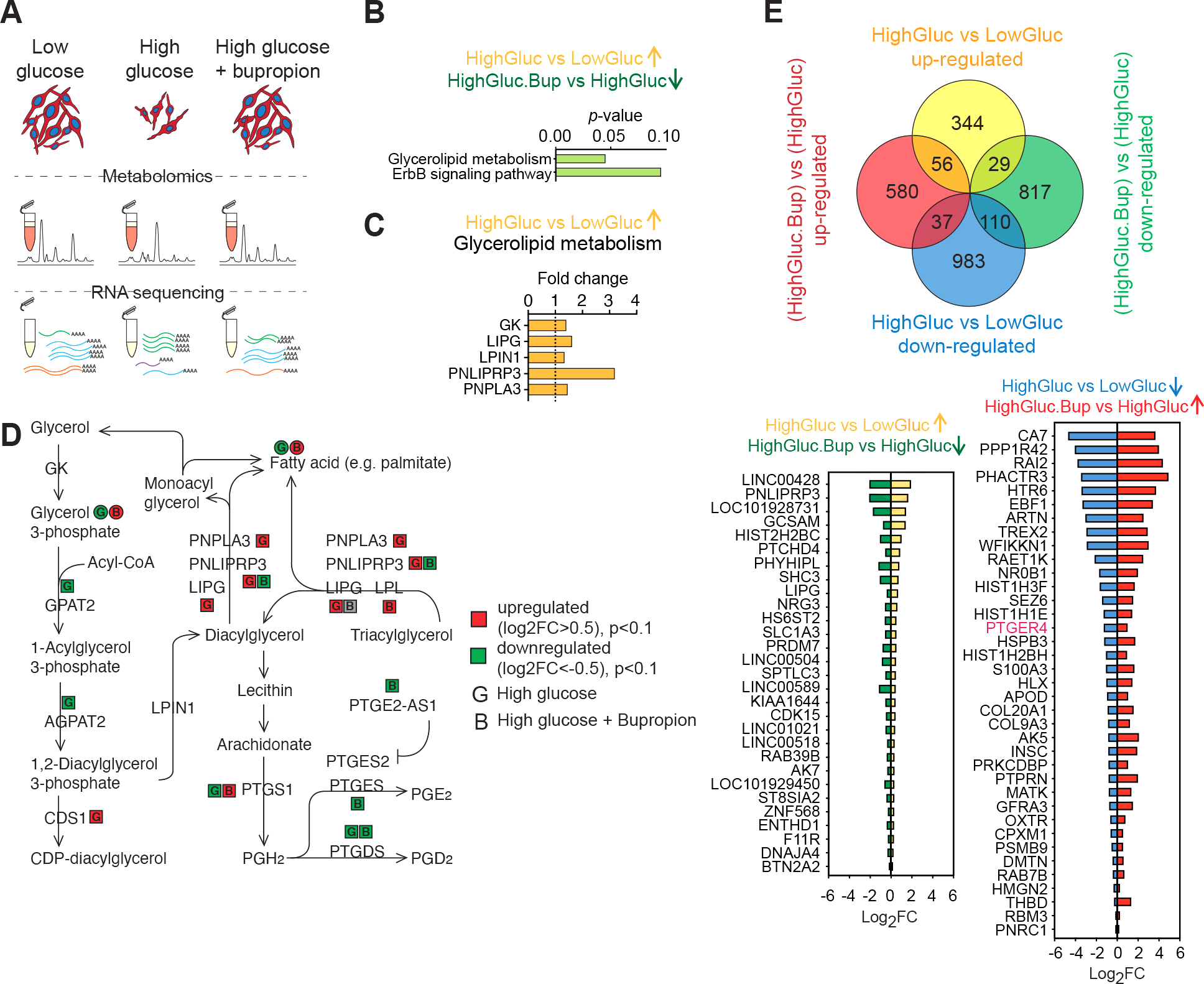
A) Schematic of the unbiased metabolite and transcriptional profiling of differentially treated SCs.
B) Pathway enrichment analysis of genes upregulated in high glucose and downregulated upon BP treatment.
C) Glycerolipid metabolism enzymes upregulated in high glucose condition.
D) Glycerolipid metabolism schematic adopted from KEGG shows changes in the enzymes and metabolites in response to high glucose and BP treatments in SCs.
E) Venn diagram showing DEGs in SCs treated with high glucose, with or without BP (top). DEGs in hyperglycemia that show a reversed pattern of expression when treated with BP (bottom).
Next, we performed metabolomics and integrated the data with our bulk RNA sequencing results (Figure S6 A–C). The detected primary metabolites fell into seven categories based on their pattern of abundance in response to glucose and BP (Figure S6 A and B). BP treatment led to a reversed response in a subset of these metabolites in particular groups 4 and 7. Metabolic pathway enrichment analysis suggested modulations in citric acid cycle, urea cycle, amino acid metabolism, glycolysis and gluconeogenesis (Figure S6 A and B). In parallel, BP treatment led to an increase in TCA cycle metabolites succinate, fumarate, citrate, α-ketoglutarate and malate (Figure S6 A–C). Citrate was reduced in response to high glucose and was reversed by BP treatment. Citrate transporter SLC13A2 expression followed the same trend (Figure S6 A–C). High glucose exposure resulted in elevation of cellular urea accompanied by increased transcript levels of the urea transporter SLC14A2 (Figure S6 A–C). Both urea concentration and its transporter mRNA level were decreased by BP (Figure S6 A–C). We observed an increase in cellular pyruvate in high glucose that was lowered by BP treatment (Figure S6 A–C). BP treatment led to reduced lactate and downregulation of membrane monocarboxylate transporters SLC16A3, SLC16A14, SLC5A2 RNA levels (Figure S6 A–C). It has been previously reported that in some cell types elevated glucose levels can activate the polyol pathway36. This pathway metabolizes excess intracellular glucose into sorbitol and subsequently into fructose via two enzymatic steps catalyzed by aldose reductase (AR) and sorbitol dehydrogenase (SDH), respectively. Osmotic and oxidative stress triggered by the polyol pathway have been proposed as mediators of high glucose-induced tissue damage in the lens more than 50 years ago37. Sorbitol accumulation has been implicated in peripheral nerve damage in multiple animal models of diabetes36,38. We asked whether hPSC-SCs show increased sorbitol levels in response to high glucose as a potential mechanism of their selective vulnerability. In agreement with studies in the mouse39,40, we observed a higher AR to SDH ratio in hPSC-derived SCs compared to sensory neurons (Figure S6D). Furthermore, SCs but not sensory neurons showed increased levels of sorbitol when exposed to high glucose (Figure S6E). BP treatment reduced sorbitol accumulation in SCs in a dose-dependent manner (Figure S6F). Similarly, we observed an increase in the cellular level of fructose in SCs treated with high glucose and BP treatment countered this effect (Figure S6G). This is in agreement with the protective effect of aldose reductase inhibitors in diabetic SCs41. Collectively, these results point to a global metabolic shift in high glucose treated SCs that is reversed by BP treatment.
Pathway enrichment analysis of transcripts that were upregulated in response to high glucose and reversed in response to BP revealed glycerolipid metabolism and ErbB signaling pathways (Figure 5B). Transcriptional changes in the glycerolipid metabolism pathway (Figure 5C) were accompanied by corresponding changes in pathway metabolites measured in the metabolomics dataset (Figure 5D). Increased tri- and diacylglycerol degradation into free fatty acid and glycerol was suggested by upregulation of LIPG and PNLIPRP3 transcripts in response to high glucose treatment (Figure 5C). This is interesting given the crucial role of lipid metabolism in myelin production and SC physiology (Figure S2N). Further, through the action of prostaglandin synthases, the glycerolipid metabolic pathway is directly linked to the prostaglandin metabolism. This piqued our interest for multiple additional reasons. For example, prostaglandin E2 receptor (PTGER4) and prostaglandin-endoperoxide synthase 2 (PTGS2) were among the top significant protein targets identified by our high-throughput drug screening (Figure 4I). Remarkably, they were both part of the protein-protein interaction network with PTGS2 showing a high degree network centrality (Figure 4J) and PTGSs and PTGERs were part of the protein families shared between our top drug hits (Figure 4K). Moreover, PTGER4 transcript had a reversed pattern of expression in response to high glucose in presence and absence of BP (Figure 5E).
Bupropion treatment prevents DPN in vivo
Given the ability of BP in rescuing the viability of hPSC-SCs in vitro, we next assessed the therapeutic potential of BP in a mouse model of DPN. We treated wild type mice with streptozotocin (STZ) which leads to beta cell death, impaired insulin production and hyperglycemia in mice42. In DPN, sensory nerve damage commonly leads to loss of sensation in the extremities. We evaluated the impact of BP treatment in STZ-treated mice by measuring thermal sensation as a readout of sensory nerve function and by histological analysis of the sciatic nerve to assess structural damage (Figure 6A). STZ-treated mice showed a dramatic increase in blood glucose levels independent of BP treatment as compared to non-diabetic control animals (Figure S7A) indicating that BP treatment does not affect glucose levels. Hyperglycemic mice maintained without BP treatment showed a delayed response to thermal stimulation at seven and eight weeks post-STZ treatment. Remarkably, BP treated hyperglycemic mice showed a significantly shorter response time compared to untreated hyperglycemic mice and no significant difference compared to normal animals (Figure 6B). Histological analysis revealed a marked increase in the percentage of TUNEL+ and cleaved caspase-3+ apoptotic cells in the sciatic nerves of STZ mice. BP+STZ treated animals showed significantly fewer apoptotic cells than animals with vehicle + STZ treatment (Figure 6 C–E). Finally, we evaluated the impact of STZ and BP treatment on peripheral myelin using transmission electron microscopy. We observed a large percentage of fibers with damaged myelin in the sciatic nerves of STZ treated animals which was significantly reduced in BP+STZ treated animals (Figure 6 F and G). These data indicate that BP can partially prevent DPN in STZ treated mice. To assess whether BP is capable of reversing sensory defects and has therapeutic potential in more advanced disease states, we started BP treatment 8 weeks post-STZ in a separate cohort of mice. These mice already showed a delayed response time to thermal stimulation before treatment with BP but showed no significant difference after 4–8 weeks of BP treatment compared to normal mice (Figure S7B). These studies demonstrate robust therapeutic effects of BP in the STZ-model of DPN.
We next aimed to determine if the potential of BP for alleviating DPN is supported by real-world data. We extracted a cohort of diabetic individuals from the UCSF de-identified health record database consisting of 71499 cases with 11% prevalence of neuropathy. A number of medications are commonly prescribed for management of neuropathic pain including gabapentin and pregabalin, BP and a subset of antidepressant drugs such as tricyclic antidepressants (TCAs). We first fitted a logistic regression model on the filtered cohort (age >40 years, duration of diabetes >10 years) to confirm this prescription pattern in our cohort (Figure S7 C and D). The result confirmed the positive association between prescription of these drugs and presence of neuropathy (Figure S7E). Interestingly, aside from BP, the high throughput screen results did not show a protective effect of anti-psychotics and anti-depressive medications present in our drug library against glucotoxicity in high glucose treated SCs (Figure S7F). Next, to examine the neuroprotective effect of BP versus other prescribed drugs while controlling for the potential prescription confounder effect, we selected individuals who had at least two episodes of prescription of these drugs in their health records (Figure 7 A and B, S7G, Table S4). A treatment score was then calculated based on the number of instances of prescription for each drug and each patient. Neuropathy was then evaluated with a logistic regression model taking BP score and other drugs as predictors, as well as smoking status intensity score, sex and duration of diabetes as other covariates (Figure 7C). BP prescription score was negatively associated with diabetic neuropathy suggesting a potential protective effect of BP in comparison to other drugs (Figure 7C). Collectively these results highlight the promise of hPSC-derived cultures for modeling SC disorders, understanding disease mechanisms and identifying potential therapeutic strategies.
Figure 7: Bupropion treatment is associated with lower odds of neuropathy in diabetic patients.
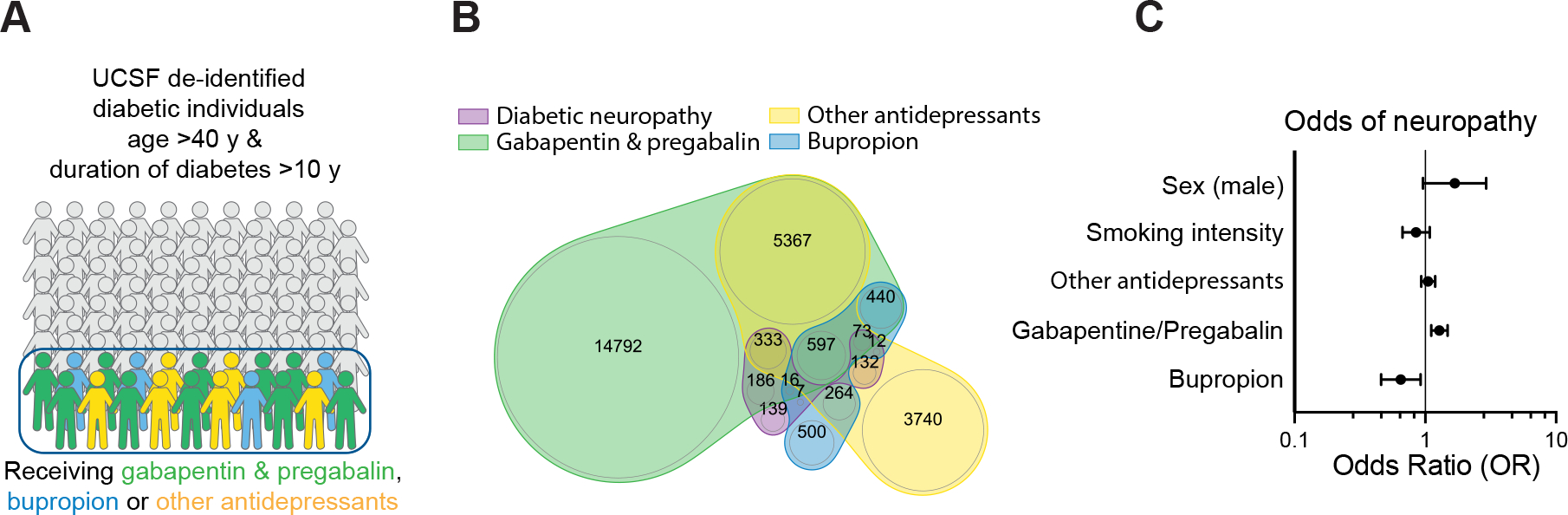
A, B) Schematic (A) and Venn diagram (B) of the cohort of diabetes individuals derived from health records.
C) Association of Bupropion with neuropathy in diabetic patients in multivariate logistic models adjusted for age, duration of diabetes, sex, smoking, and antidepressant drug treatment.
DISCUSSION
Our study reports on the highly efficient derivation of SCs from hPSCs which overcomes the limitations of previous studies such as low yield, protracted differentiation, limited SC maturation and lack of myelination data14–16,16,17,21. An important feature of our hPSC-based model is the scalability and purity of the resulting SCs and the ability to culture cells for extended periods without losing SC properties. Important developmental questions that are now accessible using this differentiation technology include the mechanisms controlling the transition from a multipotent NC stem cell to committed SCs and the study of human SC plasticity given data in the mouse suggesting that both melanocytes and parasympathetic neurons can be derived from early SC-lineages3–5,43. Our stepwise SC differentiation protocol generates SCs following the developmental processes thought to occur in vivo. It hence provides a powerful framework for lineage tracing studies that would shed light on how NC and SCP fate specification mechanisms lead to the emergence of SCs and other SCPDs such as melanocytes. SCPDs in LP cultures display molecular features of both melanocytes and SCs pointing to a potential bipotent precursor that transitions to a more melanogenic identity in HP cultures (Figure 2B, S1C, S2N). Future experiments are necessary to provide further insights into the lineage relationship between these subtypes.
Our transcriptomics profiling of hPSC-derived SCs provides a comprehensive molecular characterization of human SC lineage. Using single cell transcriptomics datasets of adult human SC as a reference, we validated the bona fide identity of the hPSC-derived SCs using machine learning-based unbiased classification. A surprising feature of the cultured hPSC-derived SC is their gene expression pattern that not only confirms SC identity but suggests that hPSC-derived cells match the expression pattern of adult SC. This is in contrast to most other hPSC differentiations that lead to fetal-like cells44.
Based on the proof-of-concept data presented here, studies on SC-mediated neuronal maturation and myelination should be other areas of focus. The modeling of PNS pathologies and the development of a drug screening platform for compounds modulating peripheral myelination could be of particular interest. Autologous SCs are currently being tested for applications in regenerative medicine targeting both PNS and CNS disorders2,45. Our transplantation data demonstrate robust engraftment of hPSC-SCs in a model of traumatic nerve injury. While future studies are required to assess the cellular composition of grafts and their therapeutic potential in models of PNS and CNS injury, our results set a solid foundation for the application of hPSC-SCs in regenerative medicine including spinal cord injury.
We present an hPSC-based DPN model that revealed a selective susceptibility of SCs to glucotoxicity and enabled the identification of candidate drugs that counteract this toxicity. For validation studies, we focused on our top candidate BP. Transcriptional and metabolic profiling identified candidate pathways involved in SC vulnerability. For example, glycerolipid metabolism was identified as a pathway significantly altered in high glucose and BP treatments (Figure 5B–D). This is of significant functional relevance considering the role of lipid metabolism and myelin synthesis in SC function. Glycerolipid pathway products can generate arachidonate, which is converted by prostaglandin synthases (PTGS1 and PTGS2) to prostaglandin H2 (PGH2). PGH2 is the precursor for other prostaglandins including PGD2 and PGF2 and PGE2. These molecules bind to prostaglandin receptor PTGER4 which is predicted target of BP and showed a reversed pattern of expression under high glucose and BP treatment conditions (Figure 5E). In addition to BP, PTGER4 and PTGS2 are among the significant targets of other hit compounds (Figure 4 I–K). Intriguingly, prostaglandin analogs have shown moderate efficacy in a randomized clinical trial for DPN7.
The ability of BP to counteract glucose-mediated SC toxicity correlated with a decrease in intracellular glucose and sucrose levels. BP treatment resulted in reduced cellular lactate concentration and its membrane transporters that might be potentially important given the importance of lactate as a fuel in neuron-SC metabolic coupling46,47. Our transcriptional and metabolomics profiling provide evidence on upregulation of glycolysis and downregulation of mitochondrial respiration in SCs in response to high glucose that were rescued by BP treatment. iPAGE gene set enrichment analysis also identified oxidative phosphorylation as pathways by which HTS hits may mediate their protective effects. Interestingly, BP appears to be the only antidepressant drug commonly associated with moderate weight loss in patients rather than a weight gain48. It is tempting to speculate whether BP mediated changes in glucose metabolism could be related to those systemic effects.
Importantly, our in vivo studies demonstrate that BP treatment can rescue DPN-related behavioral deficits and nerve damage. Interestingly, BP and other antidepressant have shown some benefit in the treatment of patients suffering from neuropathic pain49,50 raising the question whether their primary effects are on pain perception or on the underlying mechanism of SC vulnerability. Although the histological improvements in myelin and nerves in BP treated hyperglycemic mice support our in vitro findings in which BP directly impacts SC health and viability, it remains to be addressed whether BP provided additional protective mechanisms through alterations in physical activity or pain sensation. Remarkably, retrospective analysis of diabetic patients’ health records supported a higher protective effect of BP compared to other antidepressants (Figure 7C). Further prospective studies and clinical trials are necessary to demonstrate the relationship between BP, pain and neuropathy in diabetes.
In addition to BP, we identified several additional compounds capable or rescuing SC vulnerability including gliquidone among our top hits that is currently prescribed for diabetic patients (Figure 4K). It will be interesting to determine whether these compounds act via a common or distinct mechanism and whether they show in vivo activity in STZ mice comparable to BP.
In conclusion, our findings facilitate human SC-based studies for applications in regenerative medicine and human disease modeling. The work further implicates SC defects in the pathogenesis of DPN and presents BP as an FDA-approved drug that can treat DPN-related damage in vitro and in vivo.
Limitations of the study
While we demonstrated SC glucotoxicity, and rescue and restoration of multiple cellular phenotypes by candidate drugs, further experiments are required to understand the relative contribution of various mechanisms. We did not determine differential impact of diabetic stress on SC subtypes. Additional studies are needed to validate the findings in female animal models. Future prospective clinical studies will be necessary to define the protective effect of drug candidates in diabetic neuropathy.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Faranak Fattahi (faranak.fattahi@ucsf.edu).
Materials availability
Research reagents generated by the authors will be distributed upon request to other researchers via MTA.
Data and Code Availability
The raw and processed datasets from bulk and scRNA-seq of hESC-derived SC are available on GEO under accession number GSE195730. Metabolomics raw and processed data are accessible at NIH NMDR with study ID: ST002540
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal experiments were done in accordance with approved protocols by the Institutional Animal Care and Use Committee (IACUC) and Research Animal Resource Center (RARC) at Weill Cornell Medicine and Yale University. The mice were handled in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. We used wildtype male C57BL/6J acquired at 5–8 weeks old from JAX (RRID:IMSR_JAX:000664) and 10 weeks old wildtype male Sprague Dawley rats (RRID:RRRC_00239). Animals were handled and euthanized according to procedures approved by RARC at Weill Cornell Medicine and Yale University. Mice weighted at 25–35g at the beginning of their treatment, housed with littermates at 4–5 mice/cage under standard conditions and fed with standard chow diet. Rats were transplated at 10–12 weeks old and weighed 185–215g at the time of transplantation. They were housed individually under standard conditions and fed with standard chow diet. Animals were randomly assigned to experimental groups and compared with littermate controls.
Cell lines
We used the female human embryonic stem cell (hESC) line H9 (WA-09; RRID: CVCL_1240) obtained from WiCell, or reporter lines derived from H9. We also used the male human induced pluripotent stem cell (iPSCs) line WTC11 (RRID: CVCL_Y803) to repeat critical experiments. Cells were cultured in chemically defined E8 medium and grown on Geltrex™ Growth Factor Reduced (GFR) Basement Membrane Matrix (GIBCO) at 37 C with 5% CO2. Cells were fed every other day and passaged every 5–6 days using 0.5mM EDTA (Thermo Fisher).
hPSC lines were quality controlled with karyotyping, STR profiled to confirm cell identity at the initiation of the study and were subjected to routine mycoplasma testing.
METHODS DETAILS
Culture of human pluripotent stem cells (hPSCs)
hPSC line H9 (WA-09) and derivatives (SOX10::GFP; EF1::RFP were maintained on mouse embryonic fibroblasts (MEF, Global Stem, Rockville, MD) in KSR (Life Technologies, 10828–028) containing hPSC medium as described previously 57 or were plated on geltrex™-coated (Thermo Fisher Scientific, A1413302) plates and maintained in chemically-defined Essential 8 (E8) medium as described previously10. WTC11 cells were also maintained in E8 medium.
Neural crest (NC) and Schwann cell (SC) differentiation
To induce neural crest-derived Schwann cells we used two different methods. In option 1, we performed neural crest induction using KnockOut serum replacement (KSR, Life Technologies, 10828028) containing media and SC induction media supplemented with NRG1 (R&D 378-SM-025). In option 2, we used Essential 6 (E6, Life Technologies, A1516401) medium for neural crest induction and SC induction media without additional NRG1 supplement.
NC induction option 1
hESCs were plated on Matrigel (BD Biosciences, 354234) coated dishes (105 cells/cm2) in hESC medium containing 10 ng/ml FGF2 (R&D Systems, 233-FB-001MG/CF). NC differentiation was initiated in knockout serum replacement (KSR) medium (KO DMEM+15% KSR, L-glutamine (Life Technologies, 25030–081), NEAA (Life Technologies, 11140–050) containing LDN193189 (100 nM, Stemgent, Cambridge, MA) and SB431542 (10 μM, Tocris, Ellisville, MI). For NC induction, cells are treated with 3 μM CHIR99021 (Tocris Bioscience, 4423) in addition to LDN and SB from day 2 through day 11. The KSR medium was gradually replaced with increasing amounts of N2 medium from day 4 through day 10 as described previously (Chambers et al., 2009). The differentiated cells are sorted for CD49D at day 11. CNS precursor control cells were generated by treatment with LDN and SB from day 0 through day 11 as previously described (Chambers et al., 2009). Throughout the manuscript, day 0 is the day the medium is switched from hESC medium to differentiation medium. Days of differentiation in text and figures refer to the number of days since the pluripotent stage (day 0).
Induction and expansion of Schwann cells option 1
At day 11, NC cells were aggregated into 3D spheroids (5 million cells/well) in Ultra Low Attachment 6-well culture plates (Fisher Scientific, 3471) and cultured in Neurobasal (NB) medium supplemented with L-Glutamine (Gibco, 25030–164), N2 (Stem Cell Technologies, 07156) and B27 (Life Technologies, 17504044) containing CHIR (3 μM, Tocris Bioscience, 4423) and FGF2 (10 ng/ml, R&D Systems, 233-FB-001MG/CF) and NRG1 (10 ng/ml, R&D 378-SM-025). After 14 days of suspension culture, the spheroids were plated on Poly Ornithine/Laminin/ Fibronectin (PO/LM/FN) coated dishes (prepared as described previously (17)) in Neurobasal (NB) medium supplemented with L-Glutamine (Gibco, 25030–164), N2 (Stem Cell Technologies, 07156) and B27 (Life Technologies, 17504044) containing NRG1 (20 ng/ml, R&D 378-SM-025), FGF2 (10 ng/ml, R&D Systems, 233-FB-001MG/CF) and cAMP (100 mM, Sigma, D0260). The SC precursors migrate out of the plated spheroids and differentiate into SCs within 10 days. For long-term expansion, cells were cultured in Schwann cell medium (Sciencell, 1701 without FBS) on PO/LM/FN coated dishes. Cells were fixed for immunostaining or harvested for gene expression analysis at Day 25, Day 35, Day 50 and Day 100 of differentiation.
NC induction option 2
Differentiations of hPSCs towards NC were carried out following previously established methods using the chemically defined Essential 6 (E6, Life Technologies, A1516401) medium13. Briefly, when the monolayer culture of hPSCs reached about 70% confluency, NC induction protocol was initiated (D0) by aspirating the maintenance medium (E8, Life Technologies, A2858501) and replacing it with NC induction medium A [BMP4 (1 ng ml−1, R&D Systems, 314-BP), SB431542 (10 μM, R&D Systems, 1614), and CHIR 99021 (600 nM, Tocris Bioscience, 4423) in Essential 6 medium (Life Technologies, A1516401)]. Subsequently, on D2-D12 cultures were fed with NC induction medium B [SB431542 (10 μM) and CHIR 99021 (1.5 μM) in Essential 6 medium].
Induction and expansion of Schwann cells option 2
On D12, NC are dissociated to form 3D spheroids and maintained until D30 to facilitate the emergence of glial progenitors. To do so, we removed medium B on D12 and detached the NC monolayers using Accutase (Innovative Cell Technologies, AT104) for 30 min at 37°C, 5% CO2. After centrifuging the samples at 290 × g for 1 min, we re-suspended the cells in NC-C medium [FGF2 (10 ng ml−1, R&D Systems, 233-FB-001MG/CF), CHIR 99021 (3 μM), N2 supplement (10 μl ml−1, CTS, A1370701), B27 supplement (20 μl ml−1, Life Technologies, 17504044), glutagro (10 μl ml−1, Corning, 25–015-CI), and MEM NEAAs (10 μl ml−1, Corning, 25–025-CI) in neurobasal medium (Life Technologies, 21103049)] and transferred them to ultra-low-attachment plates (Fisher Scientific, 3471) to form free-floating 3D developing precursors. Two days later, when the free-floating developing precursors could be observed, we gently gathered them in the center of each well using a swirling motion. Then, the old media was carefully aspirated from the circumference of each well without removing developing precursors. After addition of the fresh NC-C medium, the cultures were incubated for 48 h (37 °C and 5% CO2) prior to passaging using Accutase. Similarly, cultures were fed with fresh medium every other day and passaged every four days until D30. On D30, the free-floating developing precursors were dissociated using Accutase and cultured in Schwann cell medium (Sciencell, 1701) without FBS, plated on dishes coated with Poly Ornithine/Laminin/ Fibronectin (PO/LM/FN, Sigma, P3655, Cultrex 3400–10 and Corning, 356008 at 15 μg/ml, 2 μg/ml, 2 μg/ml respectively). Typically, starting with one 6-well plate on D0 should yield three 10-cm PO/LM/FN coated dishes on D30. Cells were treated with Schwann Cell medium without FBS with 10 μM SB431542 at D32-D42 and 100 μM cAMP (Sigma, D0260) at D32-D39 to improve induction efficiency and purity. Cultures were fed with fresh medium every day and passaging was done using 0.05% trypsin (Fisher Scientific, MT25052CI) when the cultures expanded and became confluent (>80%). Long-term maintenance and expansion of cells were in Schwann cell medium without FBS.
FACS and Immunofluorescence (IF) analysis
For IF, the cells were fixed with 4% paraformaldehyde (PFA, SCBT, sc-281692) for 20 minutes, then blocked and permeabilized using 1% Bovine Serum Albumin (BSA, Thermo Scientific, 23209) and 0.3% triton X-100 (Sigma, T8787). The cells were then incubated in primary antibody solutions overnight at 4°C and stained with fluorophore conjugated secondary antibodies at RT for 1 hour, the stained cells were then incubated with DAPI (1 ng/ml, Sigma, D9542–5MG) and washed several times before imaging. For Flow Cytometry analysis, the cells were dissociated with Accutase (Innovative Cell Technologies, AT104) and fixed and permeabilized using BD Cytofix/Cytoperm (BD Bioscience, 554722) solution, then washed, blocked and permeabilized using BD Perm/Wash buffer (BD Bioscience, 554723) according to the manufacturer’s instructions. The cells were then stained with primary (overnight at 4°C) and secondary (30 min at room temperature) antibodies and analyzed using a flow cytometer (FlowJo software). The antibodies and the dilutions used are as follows: CD49D (Biolegend, 304301, 1:800), CHAT (Proteintech, 20747–1-AP, 1:1000), CHAT (Sigma, AB144P, 1:1000), GFAP (Abcam, ab4674, 1:1000), MAG (Millipore, LS-C279052–200, 1:200), MBP (Millipore, MAB386, 1:200), MPZ (abcam, ab39375, 1:500), NFH (Encor, RPCA-NF-H, 1:1000), NFkB p65 (Invitrogen, 710048, 1:500), PMP22 (Novus Biologicals, NB110–59086, 1:100), S100 (Thermo Scientific, RB-9018-P0, 1:500), SC101 (Takara, Y40400, 1:1000), TUBB3 (Millipore Sigma, ab9354, 1:350), TUBB3 (Biolegend, 801202, 1:1500). CASPR, Kv1.2 and PanNa were generous gifts from Dr. Joel Black.
Single cell RNA-sequencing data analysis
Processing
FASTq files were aligned using the 10X Genomics CellRanger 6.0.0 pipeline 58 to the human GRCh38 reference transcriptome to generate gene-expression counts matrices using the “include introns” option.
Quality control and cell filtration
Datasets were analyzed in R v4.1.0 with Seurat v4 59. The number of reads mapped to mitochondrial and ribosomal transcripts per cell were derived using the “PercentageFeatureSet” function. We identified cells of poor quality and subsequently removed them independently for each dataset based on the number of unique features captured per cell, the number of unique molecular identifiers (UMI) captured per cell and the percentage of mitochondrial gene transcripts per cell. Datasets were filtered based on the following quality control metrics: nFeatures>200, nFeatures<7000, nCounts< 40000 and percent mitochondrial reads < 20%.
Clustering and annotation
Transcript count matrices were log normalized applying a scaling factor of 10,000 with 2,000 variable features identified using the “vst” method. Cell cycle phase distribution was predicted using the “CellCycleScoring” function with Seurat’s S and G2M features available in “cc.genes”. We first filtered the dataset for cells with nFeatures>200 and <7000, nCounts< 40000 and mitochondrial gene percentage <20%, and then selected the top 2000 features to scale and center, and regressed residual variability from cell cycle, mitochondrial and ribosomal genes and difference in feature count. Principal Components Analysis (PCA) was run using default settings. Number of PCs used in downstream analysis for was 46 for LP SCs and 35 for HP SCs. PCA reduction was used to perform Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction using UMAP minimum distance of 0.3, cosine as UMAP metric and uwot as umap method with UMAP n.neighbors and clustering resolution customized for each dataset (LP SC n.neighbors = 46, HP SC n.neighbors = 35 and LP SC resolution = 0.15, HP SC resolution = 0.35). The shared nearest neighbors (SNN) graph was computed using default settings followed by cell clustering achieved using the default Louvain algorithm. Quality control metrics were visualized per cluster to identify and remove clusters of low-quality cells (see Quality Control and Cell Filtration). The above pipeline was performed again on each dataset after the removal of low-quality cell clusters. Cluster markers were derived using the Wilcoxon Rank Sum test. Cluster annotation was based on the expression of known cell type marker genes. Following cell type annotation, gene dropout values were imputed using adaptively-thresholded low rank approximation (ALRA) 60. The rank-k approximation was automatically chosen for each dataset with default values selected for all other parameters. The imputed gene expression is depicted in all plots and used as default in all downstream analyses unless otherwise specified.
Analysis of published datasets
Primary tissue derived Schwann cell type markers for Schwann cell precursor, myelinating and non-myelinating Schwann cells were obtained from Tasdemir-Yilmaz et.al.25 using interactive webpage Pagoda 2 by performing differential feature expression analysis of cell type cluster of interest against the entirety of dataset cells. Differentially expressed (DE) genes were sorted by Z-score and converted to human gene names with “biomaRt” 61 package using human and mouse genome databases available at ensembl.
Gene group expression characterization
Gene lists were compiled for genes belonging to transcription factor, surface marker, cell adhesion, neurotransmitter receptor and neurotrophic factor functional groups from Molecular Signatures Database (MSigDB) 62. For each dataset, the gene lists were filtered to remove low abundance genes (detected in less than 25% of cells of each cell type cluster). Genes were then determined to be exclusively expressed by a cluster if greater than 25% of cells within that cluster only expressed the gene.
To further selectively filter transcription factor and surface marker gene sets we derived genes shared by transcription factor and surface marker gene sets and cell type specific differentially expressed (DE) gene lists.
Cell type transcriptional signature scoring
To find transcriptionally similar cell populations between two datasets, first the differentially expressed (DE) genes of the reference dataset were calculated from the imputed gene counts with the “FindAllMarkers” function using the Wilcoxon Rank Sum test and only genes with a fold change (FC) above 0.25 were returned. The reference DE gene lists were filtered to remove genes not present in the query dataset. Then for each cell cluster in the reference dataset, a transcriptional signature gene list was made from the top 100 DE genes filtered for p value below 0.05 and sorted by decreasing fold change (FC). The query dataset is then scored for the transcriptional signature gene lists of each reference dataset cell cluster using the “AddModuleScore” function based on the query dataset’s imputed feature counts.
Myelinating SC identity specification
Myelinating Schwann cell (mySC) specific marker gene set was curated by combining well known lineage markers1 and canonical myelination associated markers for a total of 21 marker genes (Table S1). LP and HP specific mature Schwann cell clusters were subset from entire LP and HP datasets and scored for mySC gene set using the “AddModuleScore” function. Cells with positive mySC score were isolated and identified as mySC in further analyses.
Gene ontology analysis
Cell type of interest specific DE genes with positive fold change (FC) were calculated from the imputed gene counts with the “FindAllMarkers” function. Each gene set was filtered to include genes with p value<0.05 and sorted by decreasing fold change. Where possible up to 250 genes from each cell type specific dataset were used in gene functional profiling analysis by using g:Profiler 63 online tool and selecting pathways from GO biological process, KEGG and Reactome databases. Term enrichment was ranked by decreasing value of negative log10 transformed p values.
Unbiased scRNA-seq annotation
We used SingleCellNet26 (SCN) for unbiased classification of our hPSC-derived SC subtypes. Feature expression matrices and associated metadata object were derived from LP and HP SC datasets and used as query datasets. Reference datasets included the human cell atlas27 and a primary mouse tissue derived peripheral glia dataset 25. For classification, model training custom parameters were used for each reference dataset. Model was trained on a subset of 500 randomly selected cells for each cell cluster present in the Eraslan et.al. reference dataset, selecting top 20 DE genes and top 50 gene pairs for training. A subset of 2000 cells per cluster were used from the Tasdemir-Yilmaz et.al. reference dataset selecting top 40 DE genes and top 100 gene pairs for training.
Module scoring of bulk RNA-seq data
Gene sets derived from bulk RNA-seq of treated Schwann cell cultured were filtered based on log fold change (logFC) values to include upregulated (logFC >1) and downregulated (logFC <−1) genes for each treatment group (Table S5). Gene sets were filtered to only include genes expressed in scRNA-seq dataset and used for module scoring and visualization in the HP SC dataset.
Hit target expression in scRNA-seq data
Bupropion predicted targets and high throughput screen (HTS) hit compound significant target gene sets (Table S6) were filtered to include only genes expressed in scRNA-seq dataset and used for relative expression visualization in high passage HP SC dataset.
Myelination assessment
For IF imaging, RFP-labeled SCs (day 60) were mixed with GFP-labeled sensory neurons (day 50) and analyzed at 72 hours of co-culture. Similarly, co-cultures of SCs and hPSC-derived motor neurons (day 25) showed robust interaction along neuronal fibers with IF. For transmission electron microscopy, hPSC-derived Schwann cells (D40-D50) were added to hPSC-derived sensory neurons (D20-D30) and were co-cultured for 8 weeks in Neurobasal medium (no glucose, no pyruvate, Gibco, A2477501), glucose (5 mM, Sigma, 5767)] containing GDNF (10 ng ml−1, Peprotech, 450–10), ascorbic acid (100 μM, Sigma, A4034), N2 supplement (10 μl ml−1, CTS, A1370701), B27 supplement (20 μl ml−1, Life Technologies, 17504044), glutagro (10 μl ml−1, Corning, 25–015-CI), and MEM NEAAs (10 μl ml−1, Corning, 25–025-CI) on 8-well chamber slides (Permanox plastic, Thermo Fisher, 12–565-22) were fixed with Karnovsky’s Fixative EM Grade (EMS, 15732–10) containing 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium phosphate buffer overnight and were washed with 0.1 M sodium phosphate. Then 1% osmium tetroxide was used for secondary fixation. Samples were rinsed with distilled water and were dehydrated with a graded ethanol series from 30–100%. Then, they were suspended in propylene oxide twice for 15 min and then pre-infiltrated overnight in 1:1 propylene oxide: resin (Dodecenyl Succinic Anhydride, Araldite 6005, Epon 812, Dibutyl Phthalate, Benzyldimethylamine). Then samples were infiltrated for 5 h in 100% resin. The samples were embedded in fresh resin and polymerized for 24 h at 70°C. 100 nm sections were cut using a Leica EM UC6 ultramicrotome and collected on copper grids. The sections were stained with 4% aqueous uranyl acetate followed by 0.15% lead citrate in 0.1 N sodium hydroxide and imaged at 80 kV with a FEI Talos L120C transmission electron microscope equipped with CETA 16 MP camera at (Thermo Fisher).
Surface marker screening
Screening for specific surface antigens was performed using BD Lyoplate library® (BD, 560747) on hPSC-SCs at day 80 of differentiation. Cells were plated in 96 well plates (10,000 cells/well) and stained with primary and secondary antibodies according to manufacturer’s instructions. The stained wells were fixed for total plate imaging and quantification. The percentage of double positive cells out of total GFAP was quantified for each antibody. Top hits (>60% double positive) were validated further using flow cytometry.
Gene expression analysis
For RNA sequencing, total RNA was extracted using RNeasy RNA purification kit (Qiagen, 74106). For qRT-PCR assay, total RNA samples were reverse transcribed to cDNA using Superscript II Reverse Transcriptase (Life Technologies, 18064–014). qRT-PCR reactions were set up using QuantiTect SYBR Green PCR mix (Qiagen, 204148). Each data point represents three independent biological replicates. RNA-seq reads were mapped to the human reference genome (hg19) using TopHat v2.0. TopHat was run with default parameters with exception to the coverage search. Alignments were then quantified using HTSeq and differential gene expression was calculated using DESeq normalized to the cranial neural crest sample.
Viability assay
To monitor the viability of SCs, cells were assayed for LDH activity using CytoTox 96 cytotoxicity assay kit (Promega, G1780). Briefly, the cells are plated in 96 well plates at 30,000 cells/cm2. The supernatant and the cell lysate was harvested 24 hours later and assayed for LDH activity using a plate reader (490 nm absorbance). Cytotoxicity is calculated by dividing the LDH signal of the supernatant by total LDH signal (from lysate plus supernatant). The cells were cultured in Schwann cell medium (Sciencell, 1701) on PO/LM/FN coated dishes during the assay.
Oxidative stress measurement
We used MitoSOX™ Mitochondrial Superoxide Indicator (Invitrogen, M36008) according to the manufacturer’s instructions.
Calcium imaging
MN-only cultures and MN-SC co-cultures were subjected to calcium imaging at days 40 and 70 post-co-culture as previously described 64. Briefly, The cells were loaded with 2 μmol/L Fluo-4 AM dissolved in 1:1 (v/v) amount of 20% Pluronic®-F127 and DMSO with stock concentration of 1 mmol/L for 45 min at RT in Tyrode solution consisting of (mmol/L): 140 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 10 glucose and 10 HEPES at pH 7.4. For activation, cells were spiked with a solution containing glutamate (50 mM) or KCl (300 mM). Time lapse images were acquired using an Axiovert Inverted Microscope (Zeiss) on a heated stage. Ratiometric analysis was performed using Metamorph Software (Molecular Devices).
SC transplantation in rat sciatic nerves
All procedures were performed following NIH guidelines, and were approved by the local Institutional Animal Care and Use Committee (IACUC). Rats were placed under isoflurane gas anesthesia and both sciatic nerves were exposed below the sciatic notch and crushed using Dumont #5 forceps for 30 seconds twice in the same location. Immediately afterwards, a suspension of hPSC-derived HP SCs at 3×104 cells/μl was transplanted via injection of ~3–4 μl at proximal and distal locations to the crush site with a glass micropipette. Differentiated cultures were not purified for any specific subtype marker. Rats were immunosuppressed with cyclosporine (15 mg/kg, S.C.) daily beginning 1 day prior to cell transplantation continuing until the day of sacrifice and treated with Baytril (0.005 mg/ml in drinking water) to prevent infection. For immunohistochemistry, tissue was fixed through intracardiac perfusion of 4% PFA in 0.1 M PBS. Sciatic nerves were dissected from rats at 2, 3, 4, 8 weeks after crush lesion and transplantation. After dissection, sciatic nerves where prepared by placing them in 30% sucrose in 0.1 M PBS overnight and embedding them in OCT blocks for cryosectioning, or, by removing the perineurium and teasing them in cold 0.1 M Phosphate Buffer (pH 7.4). Some nerves were teased after perfusion and immunostained to examine individual axons. Regenerated axons distal to the crush site were analyzed.
High-throughput drug screening
The chemical compound screening was performed using the Prestwick Chemical Library®. RFP-labeled HP hPSC-SC cultures were generated from multiple independent differentiations and pooled for the screen. Cells were plated in 384 well plates (1,000 cells/well) and treated with 30 mM glucose after 24 hours immediately before addition of the compounds. The compounds were added at 1 μM concentration. After 72 hours, the plates were treated with DAPI for ten minutes, washed twice and fixed for total plate imaging. The number of viable cells was quantified for each well by counting the number of DAPI negative, RFP positive cells. HP cultures are slow to proliferate (Figure S2 G and H) so we did not anticipate a significant increase in cell numbers over the course of the experiment. z-scores were calculated for each 384 well plate separately as Z= (x−μ)/σ where x is the measured value for each condition, μ is the mean value for all test conditions and negative controls within each plate, and σ is the standard deviation for all tests and negative controls. The average and standard deviation of each plate was as follows: plate 1 (average of all wells: 187.62, SD: 83.08; average of ctrl DMSO wells: 163.25, SD: 55.22), plate 2 (average of all wells: 147.22, SD: 58.14; average of ctrl DMSO wells: 163.25, SD: 55.22), plate 3 (average of all wells: 185.03, SD: 54.96; average of ctrl DMSO wells: 189.84, SD: 38.95), plate 4 (average of all wells: 138.54, SD: 86.29; average of ctrl DMSO wells: 93.5, SD: 34.87). The full primary screen dataset was used for the computational target analysis. The HTS was performed once on mixed SC cultures pooled from five independent hPSC differentiations. For validation of the selected hit compound Bupropion (BP) (Sigma, B102), the cells were treated with various concentrations of the compound for dose response analysis (Figure S5G and S5A). To calculate the EC50 we used non-linear curve fitting (Hill slope=1). The EC50 calculated using these methods was 0.083 μM but we chose the highest non-toxic dose of 1 μM (Figure S5 A and G) for follow-up experiments.
Drug target prediction
Z-scores for primary hit compounds were calculated as Z= (x−μ)/σ. X is the number of viable cells. μ is the mean number of viable cells and σ is the standard deviation for all compounds and DMSO controls. The normalized z-score values reported for all the compounds were first transformed to N(0,1) using the bestNormalize package (v1.4.0) in R (v3.5.1). The treatments with transformed z-scores greater than 2 were selected, which resulted in 16 hit compounds.
To identify the proteins that are most likely targeted by the SC protecting drugs against glucotoxicity, we employed two independent tests. First, we calculated combined z-score in which we integrated normalized z-score from all of the treatments associated with a particular protein. Second, we performed a Fisher’s exact test to see if a target protein is enriched among the targets of treatments with positive z-scores. We report the correlation between the p--values calculated by these two independent tests. For every compound, possible target proteins were identified as above. Weighted combined z-scores were then calculated for each protein by combining normalized z-scores across all treatments 65. The p-values were then calculated based on the combined z-scores and adjusted using p.adjust (method=FDR). As an orthogonal approach, for each protein, we recorded the number of treatments with positive normalized z-scores as well as the total number of compounds predicted to target that protein. Using the sum of counts for all other proteins and drugs, we performed a Fisher’s exact test to evaluate the degree to which positive z-scores were enrichment among the treatments likely to affect a protein of interest. As expected, the two p-values, i.e. combined z-score and Fisher’s, are generally correlated.
Protein-protein interaction network construction
Protein-protein interaction network analysis was performed using the Search Tool for the Retrieval of Interacting Genes (STRING) database. The minimum required interaction score was set to 0.4 corresponding to medium confidence. The edge thickness indicates the degree of data support from the following active interaction sources: textmining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence.
iPAGE pathway enrichment analysis
The iPAGE algorithm was used for gene set and pathway enrichment analysis 31. iPAGE first quantize continuous input data into equally populated bins and then, it calculates the Mutual Information (MI) between a vector of values for genes within each cluster bin and a binary vector of gene set memberships. The significance of the calculated MI values is then assessed through a randomization-based statistical test. Finally, it uses hypergeometric distribution to determine the level with which the significantly informative pathways are overrepresented (red) or underrepresented (blue) in each cluster bin. The resulting p-values asses to draw heatmap visualization, in which rows represent significant pathways and columns correspond to cluster bins. We first ordered target genes identified from FDA approved drug library screening with positive z-score (see drug target prediction above) based on their combined z-score from left to right and followed by dividing them into seven equally populated bins. We then assessed the enrichment (red boxes) and depletion (blue boxes) of various gene sets across the spectrum (MSigDB v6.0). Gene sets provided were from the Molecular Signatures Database (MSigDB v6.0) database 66. Here, we report enriched gene sets from cluster 2 (C2, curated gene sets), and cluster 5 (C5, ontology gene sets).
NF-kB signal quantification
Image analysis was performed using FIJI 67. Watershedding was applied to separate nuclei that were grouped together. Nuclear ROIs were overlayed onto the NF-κB image to calculate nuclear co-localization. To analyze NF-κB in each condition, several measures were taken. Because the cytoplasm could not be isolated to represent the cell to which it belongs and because the average of the mean intensity of the cytoplasmic ROIs would not account for the distribution of intensities per ROI area, the integrated densities of the cytoplasmic ROIs were summed together and divided by the sum of the cytoplasmic ROIs’ area to give the mean intensity of NF-κB across all cytoplasms in each image. The mean intensities of each nuclei were divided by the mean intensity of the cytoplasm to give a ratio of nuclear to cytoplasmic NF-κB expression.
Metabolomics
hPSC-Scs were treated with 5mM and 30mM glucose for 72 h and harvested for metabolomics analysis. Frozen total cell pellets from at least three biological repeats were submitted to the West Coast Metabolomics Center at the University of California, Davis that used Agilent 7890C gas chromatographer and Pegasus III TOF mass spectrometer for an untargeted primary metabolomics analysis. Metaboanalyst 68 was used to generate the heat maps and perform path analysis.
Targeted metabolite measurements
Cellular levels of sorbitol (Abcam, ab118968) and fructose (Abcam, ab83380) were measured by colorimetric assays following the instructions provided by the manufacturers. Data were normalized according to cell numbers and averaged across 3–6 biological replicates.
Drug treatment of diabetic mice
All procedures were performed following NIH guidelines, and were approved by the local Institutional Animal Care and Use Committee (IACUC). 3–8 weeks old male C57BL6 mice were treated with one dose IP injection of STZ (180 mg/kg, sigma, 85882) to induce pancreatic beta cell death. Blood glucose levels were measured using a standard Glucometer (Freestyle Lite) in weekly intervals starting at one week post-treatment, by drawing a drop of blood from tail tip. BP treatment was initiated one week post-STZ treatment. BP was mixed with standard chow at 1.63 mg/g of chow to be administered orally at ~300 mg/kg daily. The dose was calculated based on average daily food intake (5.5 g/day) and initial body weight (30 g).
Mouse thermal sensitivity test
Thermal nociception was assessed using the hot plate test. The hot plate (Ugo Basile 35100) consisted of a metal surface (55 °C) with a transparent Plexiglas cylinder to contain the mouse. The subject is placed upon a constant temperature hot plate and the latency necessary to demonstrate discomfort, assessed by either licking/shaking the hind paw or jumping, is determined. Typical baseline latencies are 5–10 seconds, with maximal latencies of 30 seconds. Any animal that does not demonstrate discomfort behavior will be removed after 30 seconds, the maximal latency, to avoid tissue damage.
Retrospective clinical data analysis
We used the UCSF Information Commons computational research platform to extract the patient cohorts (UCSF DeID CDW-OMOP. 2022-September.) ICD codes used to identify Diabetes and diabetes-associated neuropathy can be found in Table S7. We filtered the cohort to include only those between the ages of 40 and 90 with a diabetes duration of more than 10 years. We calculated a smoking intensity score by ranking “Never Smoker” as 0, “Light Tobacco Smoker” and “Passive Smoke Exposure - Never Smoker” as 1, “Former Smoker” and “Current Some day Smoker” as 2, “Current Every day Smoker” as 3 and “Heavy Tobacco Smoker” as 4. The list of drugs that are included in the bupropion, gabapentin, pregabalin, and other drugs group can be found in Table S4. To conduct the second regression analysis, we filtered the cohort to include individuals who had at least two episodes of gabapentin, pregabalin, or the “other drugs” prescribed (Table S4). Prescription scores are determined by the number of episodes prescribed for each drug. Analysis and visualization of data was performed in Python (version 3.8.8) using Pandas (version 1.4.1), NumPy (version 1.22.2) and Seaborn (version 0.11.2) as well as in R (version 4.2.0) using nVennR (version 0.2.3).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM and were derived from at least 3 independent experiments. Data on replicates (n) is shown in figures. Statistical analysis was performed using the Student t-test (comparing 2 groups) or ANOVA with Dunnett test (comparing multiple groups against control). Distribution of the raw data approximated normal distribution (Kolmogorov Smirnov normality test) for data with sufficient number of replicates to test for normality.
Supplementary Material
Table S1: List of gene categories used for characterization of SC lineages, Related to Figure 1
Table S2: List of normalized z-scores from the high-throughput screen, Related to Figure 4
Table S3: List of all predicted targets for hit compounds, Related to Figure 4
Table S4: List of drugs used for de-identified clinical data analysis, Related to Figure 7
Table S5: Differentially expressed transcripts in high glucose and BP treated SCs, Related to STAR Methods
Table S6: Predicted targets of BP and HTS hits, Related to STAR Methods
Table S7: ICD codes for diabetes and diabetic neuropathy used for de-identified clinical data analysis, Related to STAR Methods
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CASPR | Generous gift from Dr. Joel Black | N/A |
| CD49D | Biolegend | Cat#304301; RRID:AB_314427 |
| CHAT | Proteintech | Cat#20747–1-AP; RRID:AB_10898169 |
| CHAT | Sigma | Cat#AB144P; RRID:AB_2079751 |
| GFAP | Abcam | Cat#ab4674; RRID:AB_304558 |
| Kv1.2 | Generous gift from Dr. Joel Black | N/A |
| MAG | Millipore | Cat#LS-C279052-200 |
| MBP | Millipore | Cat#MAB386; RRID:AB_94975 |
| MPZ | Abcam | Cat# ab39375; RRID:AB_881430 |
| NFH | Encor | Cat#RPCA-NF-H; RRID:AB_2572360 |
| NFkB p65 | Invitrogen | Cat#710048; RRID:AB_2532539 |
| PanNa | Generous gift from Dr. Joel Black | N/A |
| PMP22 | Novus Biologicals | Cat#NB110–59086; RRID:AB_892372 |
| S100B | Thermo Scientific | Cat#RB-9018-P0; RRID:AB_149782 |
| SC101 | Takara | Cat#Y40400; RRID:AB_2895096 |
| TUBB3 | Millipore Sigma | Cat#ab9354; RRID:AB_570918 |
| TUBB3 | Biolegend | Cat#801202; RRID:AB_10063408 |
| donkey a-goat AF488 | Invitrogen | Cat#A11055; RRID:AB_2534102 |
| donkey a-goat AF568 | Invitrogen | Cat#A11057; RRID:AB_142581 |
| donkey a-goat AF647 | Invitrogen | Cat#A21447; RRID:AB_141844 |
| donkey a-mouse AF488 | Invitrogen | Cat#A21202; RRID:AB_141607 |
| donkey a-mouse AF568 | Invitrogen | Cat#A10037; RRID:AB_2534013 |
| donkey a-mouse AF647 | Invitrogen | Cat#A31571; RRID:AB_162542 |
| donkey a-rabbit AF488 | Invitrogen | Cat#A11008; RRID:AB_143165 |
| donkey a-rabbit AF568 | Invitrogen | Cat#A10042; RRID:AB_2534017 |
| donkey a-rabbit AF647 | Invitrogen | Cat#A31573; RRID:AB_2536183 |
| goat a-chicken AF488 | Invitrogen | Cat#A11039; RRID:AB_142924 |
| Chemicals, peptides, and recombinant proteins | ||
| Accutase | Innovative Cell Technologies | Cat# AT104 |
| Araldite 6005 | EMS | Cat#10920 |
| Ascorbic acid | Sigma-Aldrich Inc | Cat#A4034 |
| B27 | Life Technologies | Cat#17504044 |
| Cytofix/Cytoperm buffer | BD Bioscience | Cat#554722 |
| Perm/Wash buffer | BD Bioscience | Cat#554723 |
| Benzyldimethylamine | EMS | Cat#11400 |
| BMP4 | R&D Systems | Cat#314-BP |
| BSA | Thermo Scientific | Cat#23209 |
| Bupropion, HCl | Sigma-Aldrich Inc | Cat#B102 |
| cAMP | Sigma-Aldrich Inc | Cat#D0627 |
| CHIR99021 | Tocris Bioscience | Cat#4423 |
| DAPI | Sigma-Aldrich Inc | Cat#D9542–5MG |
| Dibutyl Phthalate | Ted Pella | Cat#18231 |
| DMEM/F12 | Thermo Fisher Scientific | Cat#21331020 |
| Dodecenyl Succinic Anhydride | EMS | Cat#13700 |
| Essential 6 (E6) medium | Thermo Fisher Scientific | Cat#A1516401 |
| Essential 8 (E8) Flex medium | Thermo Fisher Scientific | Cat#A2858501 |
| 0.5 M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat#15575020 |
| FGF2 | R&D Systems | Cat#233-FB-001MG/CF |
| Fibronectin | Corning | Cat#356008 |
| Fluo-4 AM | Life Technologies | Cat#F14201 |
| GDNF | Peprotech | Cat#450–10 |
| Geltrex | Thermo Fisher Scientific | Cat# A1413302 |
| glucose | Sigma-Aldrich Inc | Cat#5767 |
| L-Glutamine | Gibco | Cat#25030–164 |
| Glutagro | Corning | Cat#25–015-CI |
| Karnovsky's Fixative EM Grade | EMS | Cat#15732–10 |
| KSR | Life Technologies | Cat#10828–028 |
| Laminin | Cultrex | 3400–10 |
| LDN193189 | Stemgent | 04–0074-base |
| Lead Citrate | Eastman | Cat#11534 |
| Matrigel | BD Biosciences | Cat#354234 |
| MEM non-essential amino acids (NEAA) | Corning | Cat#25–025-CI |
| Mouse embryonic fibroblast (MEF) | Gibco | Cat#A34959 |
| Neurobasal medium | Gibco | Cat#21103049 |
| NNeurobasal medium (no glucose, no pyruvate) | Gibco | Cat#A2477501 |
| N2 | CTS | Cat#A1370701 |
| NRG1 | R&D Systems | Cat#378-SM-025 |
| Osmium tetroxide | Electron Microscopy Sciences (EMS) | Cat#19150 |
| Paraformaldehyde, 4% solution | SCBT | Cat#:sc-281692 |
| Pluronic®-F127 | Life Technologies | Cat#P3000MP |
| Poly/Bed 812 | Polysciences | Cat#08791–500 |
| Poly-L-ornithine hydrobromide | Sigma | Cat#P3655 |
| Prestwick Library of FDA approved drugs | Prestwick Chemical Libraries | N/A |
| Propylene oxide | EMS | Cat#20401 |
| SB431542 | R&D Systems | Cat#:1614 |
| Schwann Cell Medium | Sciencell | Cat#1701 |
| STZ | Sigma-Aldrich Inc | Cat#85882 |
| triton X-100 | Sigma-Aldrich Inc | Cat#T8787 |
| 0.05% Trypsin/0.53 mM EDTA | Fisher Scientific | Cat#MT25052CI |
| Uranyl Acetate | Ted Pella | Cat#19481 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 2c Kit v3.1 | 10X Genomics | Cat#1000121 |
| CytoTox 96 cytotoxicity assay kit | Promega | Cat#G1780 |
| D-sorbitol Assay kit | Abcam | Cat#ab118968 |
| eBioscience™ Permeabilization Buffer (10X) | Invitrogen | Cat#00–8333-56 |
| Fructose assay kit | Abcam | Cat# ab83380 |
| Human cell surface marker screening panel | BD | Cat#560747 |
| QuantiTect SYBR Green PCR mix | Qiagen | Cat#204148 |
| MitoSOX™ Mitochondrial Superoxide Indicators | Invitrogen | Cat# M36008 |
| RNeasy RNA purification kit | Qiagen | Cat#74106 |
| Superscript II Reverse Transcriptase | Life Technologies | Cat#18064–014 |
| Deposited data | ||
| Bulk and single cell RNA sequencing data on hPSC-derived Schwann Cells | This study | GEO: GSE195730 |
| Targeted metabolomics data | This study | NMDR: ST002540 |
| Experimental models: Cell lines | ||
| Human embryonic stem cells, H9 | WiCell | RRID: CVCL_1240 |
| H9-derived SOX10::GFP reporter line | Chambers et al.11 | N/A |
| H9 RFP line | Fattahi et al.12 | N/A |
| Human induced pluripotent stem cells, WTC11 | Coriell Institute | RRID: CVCL_Y803 |
| Experimental models: Organisms/strains | ||
| 8 weeks old male C57BL6 mice | The Jackson Laboratory | RRID:IMSR_JAX:00 0664 |
| 10 weeks old male Sprague Dawley rats | Charles River | RRID:RRRC_00239 |
| Oligonucleotides | ||
| Oct6 | Qiagen | QuantiTect Primer Assays |
| Egr2 | Qiagen | QuantiTect Primer Assays |
| Gfap | Qiagen | QuantiTect Primer Assays |
| Pax3 | Qiagen | QuantiTect Primer Assays |
| S100b | Qiagen | QuantiTect Primer Assays |
| Plp1 | Qiagen | QuantiTect Primer Assays |
| Pmp22 | Qiagen | QuantiTect Primer Assays |
| Erbb3 | Qiagen | QuantiTect Primer Assays |
| Sox10 | Qiagen | QuantiTect Primer Assays |
| Mbp | Qiagen | QuantiTect Primer Assays |
| Software and algorithms | ||
| iPAGE gene ontology analysis | Goodarzi et al.31 | https://tavazoielab.c2b2.columbia.edu/iPAGE/ |
| SEA computational tool | Keiser et al. 32 | https://bkslab.org/ |
| STRING v11 protein-protein interaction (PPI) network analysis | Szklarczyk et al.33 | https://string-db.org/ |
| Flow Jo | FlowJo LLC | https://www.flowjo.com/ |
| Prism 9 | GraphPad Software | https://graphpad.com |
| Python (version 3.8.8) | Python | https://www.python.org/downloads/ |
| Pandas (version 1.4.1) | Python Package Index | https://pypi.org/project/pandas/ |
| NumPy (version 1.22.2) | Python Package Index | https://pypi.org/project/numpy/1.22.2/ |
| Seaborn (version 0.11.2) | Python Package Index | https://pypi.org/project/seaborn/ |
| Metamorph Software | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy |
| Tophat v.2.1.1 | Johns Hopkins University Center for Computational Biology | https://ccb.jhu.edu/software/tophat/index.shtml |
| R package: HTSeq v0.12.4 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: DESeq2 v1.30.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: bestNormalize (v1.4.0) | CRAN package repository | https://cran.r-project.org/web/packages/bestNormalize/index.html |
| R (v4.1.3) | The R Project forStatisticalComputing | https://www.r-project.org/ |
| R (v4.2.0) | The R Project forStatisticalComputing | https://www.r-project.org/ |
| R package: nVennR (version 0.2.3) | Github Inc | https://github.com/cran/nVennR |
| R package: Seurat v4.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: MSigDB v1.6.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: BiomaRt v2.54.0 | CRAN package repository | https://cran.r-project.org/web/packages/ |
| R package: SingleCellNet v0.4.1 | Tan and Cahan et al.26 | https://github.com/ |
| g:Profiler | Raudvere et al.63 | https://biit.cs.ut.ee/gprofiler/gost |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator/free-trial-download.html |
| Other | ||
| Molecular Signatures Database | MSigDB v6.0 | https://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/MSigDB_v6.0_Release_Notes |
| Previously published bulk RNA-seq dataset | Tasdemir-Yilmaz et al.25 | GEO: GSE172110 |
| Previously published bulk RNA-seq dataset | Eraslan et al.27 | https://singlecell.broadinstitute.org/single_cell/study/SCP1479/single-nucleus-cross-tissue-molecular-reference-maps |
| Previously published bulk RNA-seq dataset | Fattahi et al.12 | GEO/GSE66148 |
Highlights.
Differentiation of hPSCs yields authentic myelinating Schwann cells.
Schwann cells are selectively vulnerable to glucotoxicity.
Drug screen identifies candidates that protect Schwann cells against glucotoxicity.
Bupropion reduces diabetic nerve damage in mice and risk of neuropathy in patients.
ACKNOWLEDGEMENTS
We acknowledge the UCSF Information Commons supported by UCSF Bakar Computational Health Sciences Institute. We are very grateful for technical support provided by Harold S. Ralph (Weill Cornell Cell Screening Core) and Leona Cohen-Gould (Weill Cornell histology and electron microscopy Core). The work was supported by grants from UCSF Program for Breakthrough Biomedical Research and Sandler Foundation, the NIH Director’s New Innovator Award (DP2NS116769) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK121169) to F.F., the Starr Foundation and by NYSTEM contract C026446 to L.S. and supported by the NIH Cancer Center support grant P30 CA008748; by TRI-SCI 2014–030 to L.S., and S.C., and by the New York Stem Cell Foundation (R-103), NIDDK (DP2 DK098093–01) to S.C. S.C. is a New York Stem Cell Foundation – Robertson Investigator. H.G. is supported by NIH (R01CA240984). H.M. is supported by Larry L. Hillblom Foundation postdoctoral fellowship, NIH diabetes, Endocrinology, and Metabolic Diseases (DEM) T32 fellowship and UCSF Program for Breakthrough Biomedical Research independent postdoctoral fellowship.
Footnotes
DECLARATION OF INTERESTS
F.F., L.S., S.C. and Z.G. are inventors on several patent applications owned by UCSF, MSKCC and Weill Cornell Medicine related to hPSC-differentiation technologies including the technologies reported in this manuscript. L.S. is a scientific co-founder and paid consultant of BlueRock Therapeutics and DaCapo BrainScience.
INCLUSION AND DIVERSITY STATEMENT
We support inclusive, diverse, and equitable conduct of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Jessen KR, and Mirsky R (2005). The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682. 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- 2.Lavdas AA, Papastefanaki F, Thomaidou D, and Matsas R (2008). Schwann cell transplantation for CNS repair. Curr. Med. Chem. 15, 151–160. [DOI] [PubMed] [Google Scholar]
- 3.Adameyko I, Lallemend F, Aquino JB, Pereira JA, Topilko P, Müller T, Fritz N, Beljajeva A, Mochii M, Liste I, et al. (2009). Schwann Cell Precursors from Nerve Innervation Are a Cellular Origin of Melanocytes in Skin. Cell 139, 366–379. 10.1016/j.cell.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 4.Bonnamour G, Soret R, and Pilon N (2021). Dhh-expressing Schwann cell precursors contribute to skin and cochlear melanocytes, but not to vestibular melanocytes. Pigment Cell Melanoma Res. 34, 648–654. 10.1111/pcmr.12938. [DOI] [PubMed] [Google Scholar]
- 5.Nitzan E, Pfaltzgraff ER, Labosky PA, and Kalcheim C (2013). Neural crest and Schwann cell progenitor-derived melanocytes are two spatially segregated populations similarly regulated by Foxd3. Proc. Natl. Acad. Sci. U. S. A. 110, 12709–12714. 10.1073/pnas.1306287110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callaghan BC, Cheng HT, Stables CL, Smith AL, and Feldman EL (2012). Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol. 11, 521–534. 10.1016/S1474-4422(12)70065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulton AJM, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D, et al. (2005). Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 28, 956–962. 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 8.Simmons Z, and Feldman EL (2002). Update on diabetic neuropathy. Curr. Opin. Neurol. 15, 595–603. [DOI] [PubMed] [Google Scholar]
- 9.Eckersley L (2002). Role of the Schwann cell in diabetic neuropathy. Int. Rev. Neurobiol. 50, 293–321. [DOI] [PubMed] [Google Scholar]
- 10.Barber K, Studer L, and Fattahi F (2019). Derivation of enteric neuron lineages from human pluripotent stem cells. Nat. Protoc. 14, 1261–1279. 10.1038/s41596-019-0141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chambers SM, Qi Y, Mica Y, Lee G, Zhang X-J, Niu L, Bilsland J, Cao L, Stevens E, Whiting P, et al. (2012). Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 30, 715–720. 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fattahi F, Steinbeck JA, Kriks S, Tchieu J, Zimmer B, Kishinevsky S, Zeltner N, Mica Y, El-Nachef W, Zhao H, et al. (2016). Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 531, 105–109. 10.1038/nature16951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tchieu J, Zimmer B, Fattahi F, Amin S, Zeltner N, Chen S, and Studer L (2017). A Modular Platform for Differentiation of Human PSCs into All Major Ectodermal Lineages. Cell Stem Cell 21, 399–410.e7. 10.1016/j.stem.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C-W, Huang W-C, Qiu X, Fernandes Ferreira da Silva F, Wang A, Patel S, Nesti LJ, Poo M-M, and Li S. (2017). The Differentiation Stage of Transplanted Stem Cells Modulates Nerve Regeneration. Sci. Rep. 7, 17401. 10.1038/s41598-017-17043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H-S, Lee J, Lee DY, Kim Y-D, Kim JY, Lim HJ, Lim S, and Cho YS (2017). Schwann Cell Precursors from Human Pluripotent Stem Cells as a Potential Therapeutic Target for Myelin Repair. Stem Cell Rep. 8, 1714–1726. 10.1016/j.stemcr.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Q, Spusta SC, Mi R, Lassiter RNT, Stark MR, Höke A, Rao MS, and Zeng X (2012). Human neural crest stem cells derived from human ESCs and induced pluripotent stem cells: induction, maintenance, and differentiation into functional schwann cells. Stem Cells Transl. Med. 1, 266–278. 10.5966/sctm.2011-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ziegler L, Grigoryan S, Yang IH, Thakor NV, and Goldstein RS (2011). Efficient generation of schwann cells from human embryonic stem cell-derived neurospheres. Stem Cell Rev. 7, 394–403. 10.1007/s12015-010-9198-2. [DOI] [PubMed] [Google Scholar]
- 18.Newbern J, and Birchmeier C (2010). Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin. Cell Dev. Biol. 21, 922–928. 10.1016/j.semcdb.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jessen KR, Mirsky R, and Lloyd AC (2015). Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb. Perspect. Biol. 7, a020487. 10.1101/cshperspect.a020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang C-P, Zhao Y, Swigut T, and Wysocka J (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, Tabar V, and Studer L (2007). Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol. 25, 1468–1475. 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- 22.Menendez L, Yatskievych TA, Antin PB, and Dalton S (2011). Wnt signaling and a Smad pathway blockade direct the differentiation of human pluripotent stem cells to multipotent neural crest cells. Proc. Natl. Acad. Sci. 108, 19240–19245. 10.1073/pnas.1113746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mica Y, Lee G, Chambers SM, Tomishima MJ, and Studer L (2013). Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 3, 1140–1152. 10.1016/j.celrep.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finzsch M, Schreiner S, Kichko T, Reeh P, Tamm ER, Bösl MR, Meijer D, and Wegner M (2010). Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J. Cell Biol. 189, 701–712. 10.1083/jcb.200912142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tasdemir-Yilmaz OE, Druckenbrod NR, Olukoya OO, Dong W, Yung AR, Bastille I, Pazyra-Murphy MF, Sitko AA, Hale EB, Vigneau S, et al. (2021). Diversity of developing peripheral glia revealed by single-cell RNA sequencing. Dev. Cell 56, 2516–2535.e8. 10.1016/j.devcel.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan Y, and Cahan P (2019). SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Syst. 9, 207–213.e2. 10.1016/j.cels.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eraslan G, Drokhlyansky E, Anand S, Fiskin E, Subramanian A, Slyper M, Wang J, Van Wittenberghe N, Rouhana JM, Waldman J, et al. (2022). Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290. 10.1126/science.abl4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calder EL, Tchieu J, Steinbeck JA, Tu E, Keros S, Ying S-W, Jaiswal MK, Cornacchia D, Goldstein PA, Tabar V, et al. (2015). Retinoic Acid-Mediated Regulation of GLI3 Enables Efficient Motoneuron Derivation from Human ESCs in the Absence of Extrinsic SHH Activation. J. Neurosci. Off. J. Soc. Neurosci. 35, 11462–11481. 10.1523/JNEUROSCI.3046-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang X, Zhou L, Wagner AM, Marchetto MCN, Muotri AR, Gage FH, and Chen G (2013). Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem Cell Res. 11, 743–757. 10.1016/j.scr.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samuel RM, Majd H, Richter MN, Ghazizadeh Z, Zekavat SM, Navickas A, Ramirez JT, Asgharian H, Simoneau CR, Bonser LR, et al. (2020). Androgen Signaling Regulates SARS-CoV-2 Receptor Levels and Is Associated with Severe COVID-19 Symptoms in Men. Cell Stem Cell 27, 876–889.e12. 10.1016/j.stem.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodarzi H, Elemento O, and Tavazoie S (2009). Revealing global regulatory perturbations across human cancers. Mol. Cell 36, 900–911. 10.1016/j.molcel.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, and Shoichet BK (2007). Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197–206. 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 33.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corrêa-Silva S, Alencar AP, Moreli JB, Borbely AU, de S Lima L, Scavone C, Damasceno DC, Rudge MVC, Bevilacqua E, and Calderon IMP (2018). Hyperglycemia induces inflammatory mediators in the human chorionic villous. Cytokine 111, 41–48. 10.1016/j.cyto.2018.07.020. [DOI] [PubMed] [Google Scholar]
- 35.Kowluru RA, Koppolu P, Chakrabarti S, and Chen S (2003). Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic. Res. 37, 1169–1180. 10.1080/10715760310001604189. [DOI] [PubMed] [Google Scholar]
- 36.Oates PJ (2002). Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 50, 325–392. [DOI] [PubMed] [Google Scholar]
- 37.Van Heyningen R (1959). Formation of Polyols by the Lens of the Rat with ‘Sugar’ Cataract. Nature 184, 194–195. 10.1038/184194b0. [DOI] [Google Scholar]
- 38.Mizisin AP (2014). Mechanisms of diabetic neuropathy: Schwann cells. Handb. Clin. Neurol. 126, 401–428. 10.1016/B978-0-444-53480-4.00029-1. [DOI] [PubMed] [Google Scholar]
- 39.Maekawa K, Tanimoto T, Okada S, Suzuki T, Suzuki T, and Yabe-Nishimura C (2001). Expression of aldose reductase and sorbitol dehydrogenase genes in Schwann cells isolated from rat: effects of high glucose and osmotic stress. Brain Res. Mol. Brain Res. 87, 251–256. [DOI] [PubMed] [Google Scholar]
- 40.Mizisin AP, and Powell HC (1993). Schwann cell injury is attenuated by aldose reductase inhibition in galactose intoxication. J. Neuropathol. Exp. Neurol. 52, 78–86. [DOI] [PubMed] [Google Scholar]
- 41.Hao W, Tashiro S, Hasegawa T, Sato Y, Kobayashi T, Tando T, Katsuyama E, Fujie A, Watanabe R, Morita M, et al. (2015). Hyperglycemia Promotes Schwann Cell De-differentiation and De-myelination via Sorbitol Accumulation and Igf1 Protein Down-regulation. J. Biol. Chem. 290, 17106–17115. 10.1074/jbc.M114.631291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu KK, and Huan Y (2001). Streptozotocin-Induced Diabetic Models in Mice and Rats. In Current Protocols in Pharmacology (John Wiley & Sons, Inc.). [DOI] [PubMed] [Google Scholar]
- 43.Espinosa-Medina I, Outin E, Picard CA, Chettouh Z, Dymecki S, Consalez GG, Coppola E, and Brunet J-F (2014). Neurodevelopment. Parasympathetic ganglia derive from Schwann cell precursors. Science 345, 87–90. 10.1126/science.1253286. [DOI] [PubMed] [Google Scholar]
- 44.Studer L, Vera E, and Cornacchia D (2015). Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell Stem Cell 16, 591–600. 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodrigues MCO, Rodrigues AA, Glover LE, Voltarelli J, and Borlongan CV (2012). Peripheral Nerve Repair with Cultured Schwann Cells: Getting Closer to the Clinics. Sci. World J. 2012, e413091. 10.1100/2012/413091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babetto E, Wong KM, and Beirowski B (2020). A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 23, 1215–1228. 10.1038/s41593-020-0689-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domènech-Estévez E, Baloui H, Repond C, Rosafio K, Médard J-J, Tricaud N, Pellerin L, and Chrast R (2015). Distribution of monocarboxylate transporters in the peripheral nervous system suggests putative roles in lactate shuttling and myelination. J. Neurosci. Off. J. Soc. Neurosci. 35, 4151–4156. 10.1523/JNEUROSCI.3534-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arterburn D, Sofer T, Boudreau DM, Bogart A, Westbrook EO, Theis MK, Simon G, and Haneuse S (2016). Long-Term Weight Change after Initiating Second-Generation Antidepressants. J. Clin. Med. 5, E48. 10.3390/jcm5040048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Semenchuk MR, Sherman S, and Davis B (2001). Double-blind, randomized trial of bupropion SR for the treatment of neuropathic pain. Neurology 57, 1583–1588. [DOI] [PubMed] [Google Scholar]
- 50.Cayley WE (2006). Antidepressants for the treatment of neuropathic pain. Am. Fam. Physician 73, 1933–1934. [PubMed] [Google Scholar]
- 51.Liu T, Lin Y, Wen X, Jorissen RN, and Gilson MK (2007). BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 35, D198–201. 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathias SL, Hines-Kay J, Yang JJ, Zahoransky-Kohalmi G, Bologa CG, Ursu O, and Oprea TI (2013). The CARLSBAD database: a confederated database of chemical bioactivities. Database J. Biol. Databases Curation 2013, bat044. 10.1093/database/bat044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamanishi Y, Kotera M, Moriya Y, Sawada R, Kanehisa M, and Goto S (2014). DINIES: drug-target interaction network inference engine based on supervised analysis. Nucleic Acids Res. 42, W39–45. 10.1093/nar/gku337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, et al. (2019). PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siramshetty VB, Eckert OA, Gohlke B-O, Goede A, Chen Q, Devarakonda P, Preissner S, and Preissner R (2018). SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res. 46, D1137–D1143. 10.1093/nar/gkx1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, and Zoete V (2014). SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 42, W32–38. 10.1093/nar/gku293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049. 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linderman GC, Zhao J, and Kluger Y (2018). Zero-preserving imputation of scRNA-seq data using low-rank approximation 10.1101/397588. [DOI] [Google Scholar]
- 61.Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191. 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinforma. Oxf. Engl. 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198. 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barreto-Chang OL, and Dolmetsch RE (2009). Calcium imaging of cortical neurons using Fura-2 AM. J. Vis. Exp. JoVE, 1067. 10.3791/1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaykin DV (2011). Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J. Evol. Biol. 24, 1836–1841. 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia J, Psychogios N, Young N, and Wishart DS (2009). MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 37, W652–660. 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of gene categories used for characterization of SC lineages, Related to Figure 1
Table S2: List of normalized z-scores from the high-throughput screen, Related to Figure 4
Table S3: List of all predicted targets for hit compounds, Related to Figure 4
Table S4: List of drugs used for de-identified clinical data analysis, Related to Figure 7
Table S5: Differentially expressed transcripts in high glucose and BP treated SCs, Related to STAR Methods
Table S6: Predicted targets of BP and HTS hits, Related to STAR Methods
Table S7: ICD codes for diabetes and diabetic neuropathy used for de-identified clinical data analysis, Related to STAR Methods
Data Availability Statement
The raw and processed datasets from bulk and scRNA-seq of hESC-derived SC are available on GEO under accession number GSE195730. Metabolomics raw and processed data are accessible at NIH NMDR with study ID: ST002540
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.