

Abstract
Post-stroke secondary brain damage is significantly influenced by the induction and accumulation of α-Synuclein (α-Syn). α-Syn positive inclusions are often present in tauopathies and elevated tau levels and phosphorylation promotes neurodegeneration. Glycogen synthase kinase 3β (GSK-3β) is a known promoter of tau phosphorylation. We currently evaluated the interaction of α-Syn with GSK-3β and tau in post-ischemic mouse brain. Transient focal ischemia led to increased cerebral protein-protein interaction of α-Syn with both GSK-3β and tau and elevated tau phosphorylation. Treatment with a GSK-3β inhibitor prevented post-ischemic tau phosphorylation. Furthermore, α-Syn interaction was observed to be crucial for post-ischemic GSK-3β-dependent tau hyperphosphorylation as it was not seen in α-Syn knockout mice. Furthermore, tau knockout mice show significantly smaller brain damage after transient focal ischemia. Overall, the present study indicates that GSK-3β catalyzes the α-Syn-dependent tau phosphorylation and preventing this interaction is crucial to limit post-ischemic secondary brain damage.
Keywords: Focal ischemia, interaction, hyperphosphorylation, brain damage, neuroprotection
INTRODUCTION
α-Synuclein (α-Syn) is one of the abundant proteins in the brain that is known to cause synucleinopathies and chronic neurodegeneration (Lashuel et al. 2013; Clinton et al. 2010; Savica et al. 2013). It is highly induced in both human and rodent brains following stroke (Kim et al. 2016; Kim et al. 2018). The accumulation and aggregation of α-Syn promote ischemic brain damage, whereas its deletion, silencing, or inhibition decreases infarction, mitochondrial fragmentation, oxidative stress, apoptosis, and autophagy and better sensorimotor and cognitive recovery after transient focal ischemia in rodents (Kim et al. 2016; Kim et al. 2018; Unal-Cevik et al. 2011). α-Syn positive inclusions are frequently observed in tauopathies and increased tau levels and phosphorylation are known to occur in Parkinson’s disease (Moussaud et al. 2014). This indicates that α-Syn and tau promote each other’s fibrillization and solubility indicating a synergistic mechanism of secondary brain damage (Clinton et al. 2010; Postina 2008). In CNS, both α-Syn and tau are predominately induced in neurons (Kim et al. 2016; Pluta et al. 2018).
Tau plays a crucial role in protecting microtubule integrity that is essential for vesicular transport, polarity, and signal transduction in axons and dendrites (Chen and Jiang 2019). However, tau phosphorylation leads to its aggregation and formation of neurofibrillary tangles that promote apoptosis after cerebral ischemia (Wen et al. 2007; Morioka et al. 2006; Gordon-Krajcer et al. 2007). Tau phosphorylation exacerbates and its deficiency protects the post-ischemic rodent brain (Bi et al. 2017). Glycogen synthase kinase 3β (GSK-3β) promotes tau hyperphosphorylation and its inhibition reduces brain damage and improves neurologic function after focal ischemia (Toral-Rios et al. 2020; Wang et al. 2017; Wang et al. 2016). We presently evaluated if α-Syn and GSK-3β interactively modulate tau phosphorylation and the downstream brain damage after transient focal ischemia in adult mice.
MATERIALS AND METHODS
All the procedures using animals were approved by the University of Wisconsin Research Animal Resources and Care Committee, conducted in compliance with the “Animal Research: Reporting of In Vivo Experiments (ARRIVE)” guidelines and cared in accordance with the Guide for the Care and Use of Laboratory Animals, U.S. Department of Health and Human Services Publication Number. 86-23 (revised) (Percie du Sert et al. 2020). Animals were randomly divided into experimental groups. The outcome measures were evaluated by an investigator blinded to study groups.
Focal ischemia:
Adult, male, C57BL/6J, C57BL/6N-Snca tm1Mjff/J (SNCA knockout; α-Syn−/−) and B6.129X1-Mapttm1Hnd/J (Tau knockout; Tau−/−) and corresponding wild-type controls (12 weeks, 27±2 g,) were obtained from Jackson Labs USA. Transient focal ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO) using silicon-coated monofilament (6-0, Doccol Corporation USA) under isoflurane anesthesia followed by 12h or 24h of reperfusion (Chelluboina et al. 2021; Chokkalla et al. 2019; Kim et al. 2016; Kim et al. 2018; Mehta et al. 2017; Mehta et al. 2022; Kim et al. 2021). Sham-operated animals were used as control. The body temperature during surgery was maintained at 37.0 ± 0.5°C with a heating blanket. Regional cerebral blood flow (rCBF) and physiological parameters (pH, PaO2, PaCO2, hemoglobin, and blood glucose) were monitored. For the inclusion and exclusion criteria, mice subjected to transient MCAO that showed no signs of neurologic deficits during reperfusion or those that showed signs of hemorrhage during imaging or after euthanasia were excluded (Kim et al. 2018).
GSK-3β inhibitor administration:
A cell-permeable GSK-3β Inhibitor VIII, CAS 487021-52-3 (Cat # 361549; Calbiochem USA) that regulates the biological activity of GSK-3β by phosphorylation and dephosphorylation at different residues and prevents tau phosphorylation at a GSK-3β-specific site was injected (4 mg/kg dissolved in 5 mg/ml of DMSO and supplemented with 0.9% sterile NaCl; IV) at 30 min of reperfusion following transient MCAO. Mice in the control group received an equal volume of vehicle (Venna et al. 2015).
Co-immunoprecipitation (Co-IP):
The protein-protein interaction between α-Syn, tau, and GSK-3β was analyzed using Co-IP Assay (Cat. # ab206996; Abcam USA) as described previously (Kim et al. 2021). Briefly, the ipsilateral peri-infarct cortex was homogenized in a non-denaturing lysis buffer containing a cocktail of protease and phosphatase inhibitors. The homogenate was centrifuged (10,000g; 5 min at 4°C) and 300 μg protein equivalent of the supernatant was incubated overnight at 4°C with antibodies against α-Syn or GSK-3β (each at 1:50; Cell Signaling Technology USA) and with 25 μl of protein A/G Sepharose beads slurry (1h at 4°C). The precipitated protein complex was eluted, washed in buffer, incubated (5 min at 80°C) with NuPAGE LDS Sample Buffer (Cat. # NP0008; Life Technologies USA) and immunoblotted to evaluate the protein-protein interaction. Rabbit IgG was used as a control.
Western blotting:
Ipsilateral peri-infarct cortex was homogenized in T-PER Tissue Protein Extraction Reagent (Cat. # 78510; Life Technologies USA) supplemented with a cocktail of protease and phosphatase inhibitors. Homogenates were centrifuged (10,000g for 10 min at 4°C) and 40 μg protein equivalents were electrophoresed, transferred to nitrocellulose membranes, and probed with antibodies against α-Syn, tau, p-tau, GSK-3β, p-GSK-3β (S9) and GAPDH (each at 1:1,000; Cell Signaling Technology USA), followed by HRP-conjugated (1:3,000) or IRDye Infrared Fluorescent Dye-conjugated (1:20,000; LI-COR) anti-rabbit or anti-mouse antibodies. Protein bands were detected using enhanced chemiluminescence or scanned with a NIR spectrum (between 680 and 800 nm) using a Li-COR Odyssey analyzer and the band intensity was quantitated with Image Studio software (LI-COR Biotechnology USA).
Infarct volume assessment:
As infarct size after transient MCAO in mice is known to reach ~90% to 95% by 24h of reperfusion, it was assessed with T2-MRI (4.7T small animal system scanner; Agilent Technologies USA) at that time. Briefly, mice were anesthetized with isoflurane and 8-10 coronal brain slices at 1.0 mm thickness and 20 × 20 mm2 fields were scanned under isoflurane anesthesia as described previously (Chelluboina et al. 2021). The respiration rate was monitored during the imaging. The Infarct size was computed blindly, and the total infarct volume was configured by numeric integration of data from serial coronal sections factoring-in sectional intervals and corrected for edema using NIH ImageJ software with an FDF plugin as described before (Chelluboina et al. 2021; Kim et al. 2018; Mehta et al. 2022; Mehta et al. 2015).
Results
Transient focal ischemia induced phosphorylation of GSK-3β and tau:
The p-tau levels increased (by 2 to 2.5 fold; p<0.05) and p-GSK-3β levels decreased (by 3 to 4 fold; p<0.05) in the ipsilateral peri-infarct cortex at 6h, 12h and 24h of reperfusion following transient MCAO in adult mice compared with sham (Fig. 1A and B). Whereas protein levels of total tau and total GSK-3β were unaltered at any reperfusion period tested compared with sham (Fig. 1A and B).
Fig. 1: Transient focal ischemia induced tau phosphorylation but prevented GSK3β S9 phosphorylation.
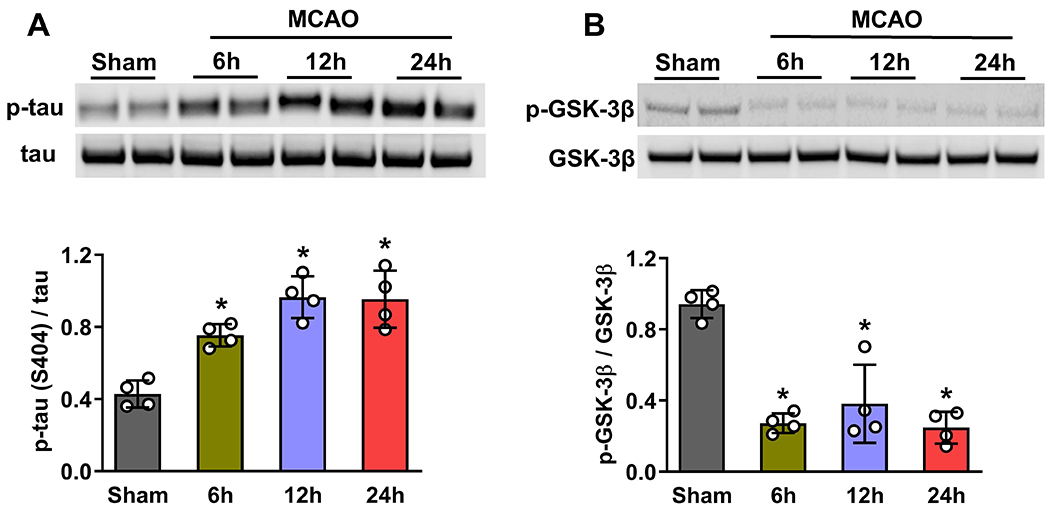
Protein abundance of p-tau increased (A) and p-GSK-3β decreased (B) in the peri-infarct cortex of mice at 6h, 12h, and 24h of reperfusion following transient MCAO. Total tau and GSK3β protein levels were unaltered after focal ischemia. Each band in the blot is the biological representation. Values are mean ± of SD (n=4/group); *p<0.05 compared with sham by Mann-Whitney U test.
α-Syn forms a complex with p-tau and p-GSK-3β after focal ischemia:
We previously demonstrated that induction of α-Syn after focal ischemia in rodents increases DRP1 phosphorylation that promotes mitochondrial fragmentation (Kim et al. 2016). To understand if α-Syn promotes GSK-3β phosphorylation and the further downstream tau hyperphosphorylation that modulates DRP1 and mitochondrial damage after focal ischemia, we immunoprecipitated protein complex with α-Syn antibodies and evaluated the interaction of α-Syn with p-tau, and p-GSK-3β at 12h reperfusion following transient MCAO in the ipsilateral cerebral cortex. As expected, α-Syn protein levels increased significantly in the post-ischemic brain (Fig. 2A). α-Syn binding to p-tau increased and to p-GSK-3β decreased significantly after transient MCAO compared to sham (Fig. 2A). Whereas α-Syn binding to total tau and total GSK-3β were not altered after transient MCAO (Fig. 2A).
Fig. 2: α-Syn interacted with p-tau and p-GSK-3β after transient focal ischemia.
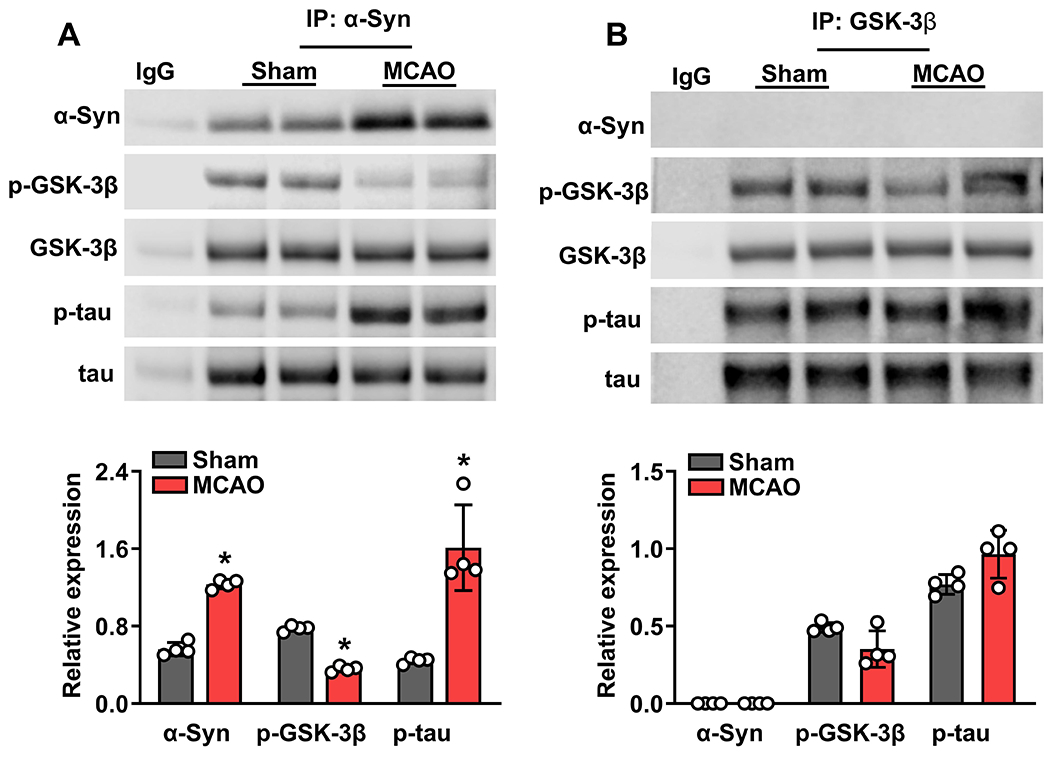
The protein lysates from peri-infarct cortical tissue of mice subjected to transient MCAO and 12h reperfusion were immunoprecipitated with α-Syn antibody. Immunoprecipitated proteins were subjected to western blotting and probed with antibodies against tau, p-tau, GSK-3β and p-GSK-3β. Following transient MCAO, α-Syn/p-tau interaction increased while α-Syn/p-GSK-3β interaction decreased compared with sham (A). We then repeated the experiment with protein lysates from α-Syn−/− mice subjected to transient MCAO and 12h reperfusion (B). As α-Syn protein is absent in α-Syn−/− mice, we used GSK-3β antibodies for immunoprecipitation. In α-Syn−/− mice, there was no interaction of p-GSK-3β and p-tau in sham or ischemic mice, indicating the essential nature of α-Syn in this process. Each band in the blot is the biological representation. Values are mean ± SD (n=4/group); *p<0.05 compared with sham by Mann-Whitney U test.
Using α-Syn−/− mice, we investigated if post-ischemic GSK-3β activation and tau hyperphosphorylation are α-Syn dependent. In α-Syn−/− mouse brain, total GSK-3β and total tau protein levels didn’t change following transient MCAO and 12h of reperfusion in the ipsilateral cortex compared to sham (Fig. 2B). This is similar to what we observed in wild-type mice. However, α-Syn deletion prevented post-ischemic tau hyperphosphorylation seen in wild-type mice (Fig. 2B). We further evaluated the interaction of α-Syn, tau, and GSK-3β following immunoprecipitation with GSK-3β antibody. In α-Syn−/− mice subjected to transient MCAO and 12h reperfusion, both GSK-3β phosphorylation and tau phosphorylation were similar to sham (Fig. 2B), unlike in wild-type control. This indicates that GSK-3β bound to tau is S9 phosphorylated (inactive), which can’t prevent tau phosphorylation in the wild-type mice when it was decreased following transient MCAO. However, in α-Syn−/− mice, the reversal of GSK-3β phosphorylation prevents tau hyperphosphorylation. This suggests that α-Syn availability is essential for GSK-3β suppression and the resulting tau hyperphosphorylation in the post-ischemic brain.
Inhibition of GSK-3β prevented tau hyperphosphorylation in the ischemic brain:
We injected mice with a potent GSK-3 inhibitor VIII that prevents tau phosphorylation at a GSK-3-specific site to confirm the role of GSK-3β in controlling tau hyperphosphorylation (Venna et al. 2015). GSK-3β inhibitor given IV at 30 min of reperfusion significantly reversed the decrease in GSK-3β S9 phosphorylation and subsequently decreased tau phosphorylation at 12h of reperfusion following transient MCAO compared to the vehicle control group (Fig. 3). This confirms that GSK-3β mediates tau phosphorylation after focal ischemia.
Fig. 3: GSK-3β inhibitor VIII prevented GSK-3β S9 dephosphorylation, leading to reduced tau phosphorylation after transient focal ischemia.
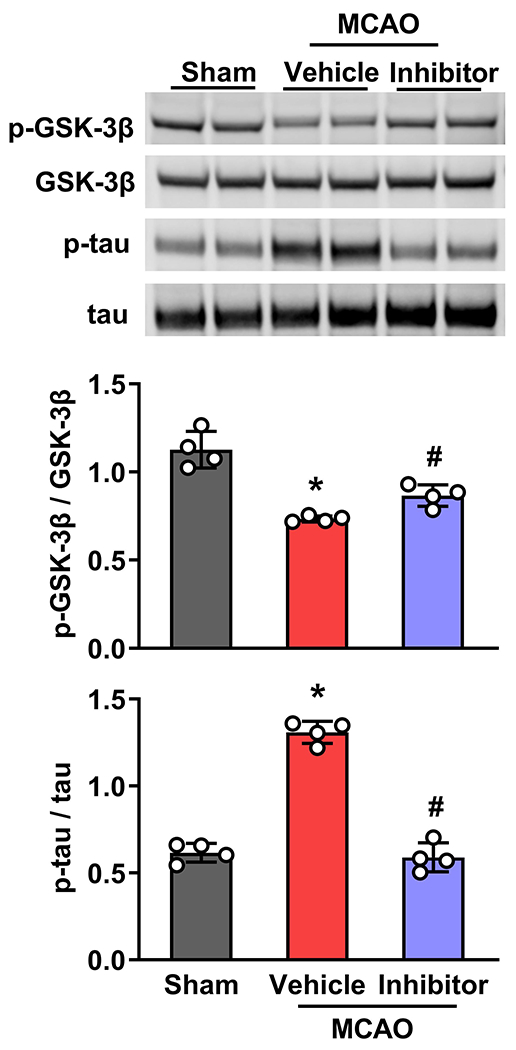
Treatment with GSK-3β Inhibitor VIII at 30 min of reperfusion following transient MCAO reversed post-ischemic changes in both GSK-3β phosphorylation and tau phosphorylation compared to vehicle control. GSK-3β Inhibitor VIII did not change the protein levels of total tau and total GSK-3β. Each band in the blot is the biological representation. Values are mean ± SD (n=4/group); *p<0.05 compared with vehicle control by Mann-Whitney U test.
Tau deletion decreased brain damage after transient focal ischemia:
We previously demonstrated that mice lacking α-Syn suffered less brain damage and recovered motor functions after focal ischemia better than wild-type mice. As shown above, α-Syn seems to influence GSK-3β and its downstream tau signaling. As tau hyperphosphorylation and its aggregation are known to promote neurodegeneration (Ballatore et al. 2007), we tested the effect of focal ischemia in Tau−/− mice. Tau−/− mice showed no total tau or p-tau protein after transient MCAO or sham surgery (Fig. 4A). Tau−/− mice showed significantly smaller infarct volume (by 38%; p<0.05) at 1 day of reperfusion following 1h of transient MCAO compared with Tau+/+ mice (Fig. 4B).
Fig. 4: Tau deletion decreased post-ischemic secondary brain damage.
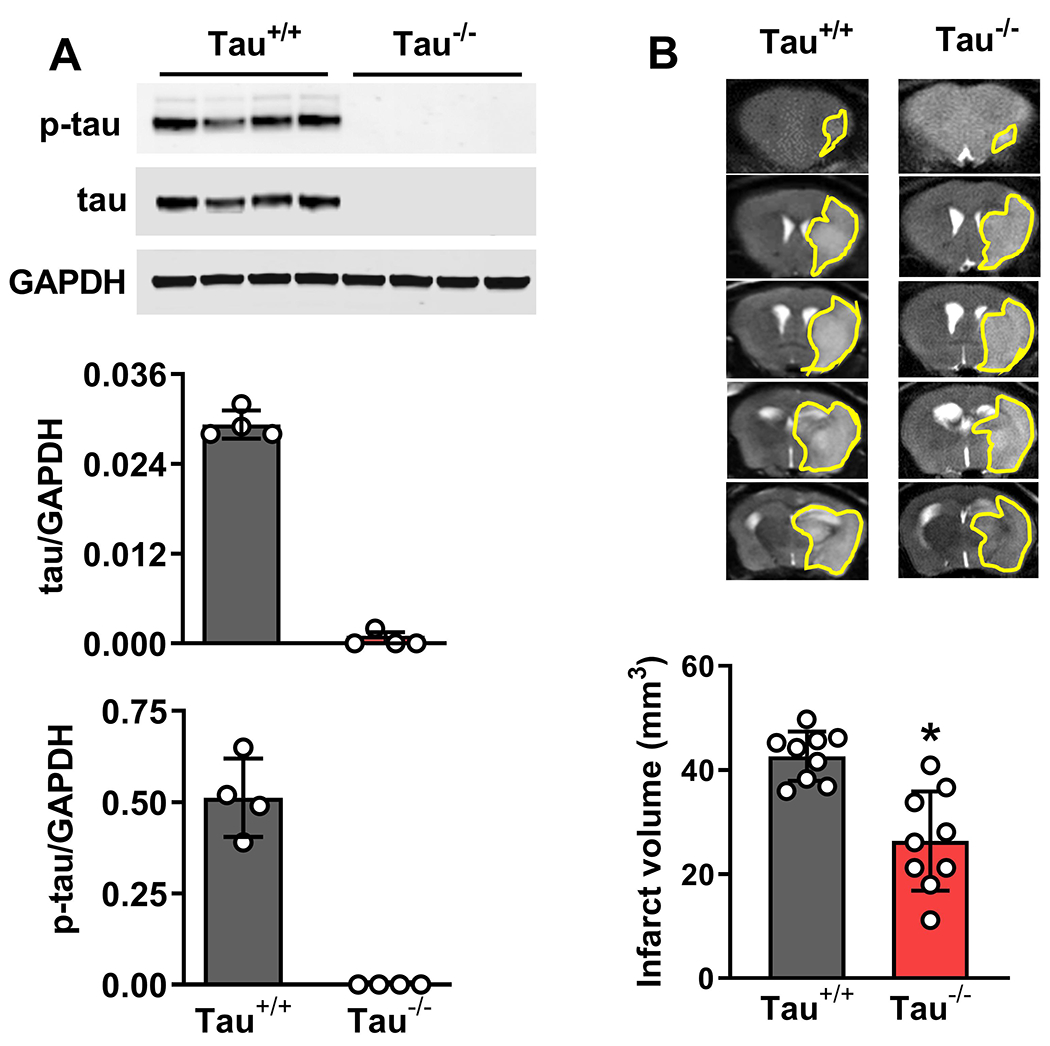
Tau−/− mice showed no tau or p-tau protein expression (A) in the peri-infarct cortex of mice subjected to 1h of transient MCAO and 1 day of reperfusion compared with Tau+/+ mice (A). T2-MRI showed reduced infarct volume in Tau−/− mice compared to Tau+/+ mice on day 1 of reperfusion (B). Each band in the blot is the biological representation. Values are mean ± SD (n = 4/group in A and 8/group in B); *p<0.05 by the Mann-Whitney U test.
DISCUSSION
We previously reported that induction and accumulation of α-Syn significantly contribute to post-ischemic brain damage (Kim et al. 2016). Previous studies showed that tau and GSK-3β are also promoters of secondary brain damage after focal ischemia (Bi et al. 2017; Venna et al. 2015). Both α-Syn and tau cause synucleinopathies and tauopathies associated with neurodegenerative disorders (Ballatore et al. 2007; Galpern and Lang 2006; Savica et al. 2013). Clinical studies indicate a substantial correlation between these abnormalities with the co-occurrence of α-Syn and tau inclusions (Savica et al. 2013; Galpern and Lang 2006; Forman et al. 2002).
GSK-3β is a major kinase that hyperphosphorylates tau leading to its accumulation and tauopathies (Credle et al. 2015; Duka et al. 2009; Lei et al. 2011). Transgenic mice overexpressing GSK-3β showed tau hyperphosphorylation and hippocampal neuronal death (Wang et al. 2017). Whereas inhibiting GSK-3β enhanced synaptic and cognitive capabilities in Alzheimer’s disease mouse models and reduced infarct volume and BBB disruption and enhanced cognitive recovery after stroke (Farr et al. 2014; Zhou et al. 2022; Wang et al. 2017; Venna et al. 2015).
The current studies show that the basal levels of GSK-3β and tau are unaltered after focal ischemia, but their phosphorylation status changes significantly. Importantly, GSK-3β S9 phosphorylation is reduced, leading to its activation and downstream tau phosphorylation. The key component that enables this seems to be the increased post-ischemic expression of α-Syn, which binds to both GSK-3β and tau. Previous studies showed that α-Syn is directly involved in tau hyperphosphorylation (Duka et al. 2006). Our studies confirm this by showing that post-ischemic tau phosphorylation does not change in α-Syn knockout mice (known to be protected after focal ischemia) compared to sham, indicating that α-Syn binding is critical. Following transient MCAO, tau phosphorylation peaked at 12h of reperfusion and remained elevated up to 24h of reperfusion. GSK-3β is the major kinase that phosphorylates tau, and we observed reduced GSK-3β S9 phosphorylation between 6h and 24h of reperfusion. GSK-3β activity is inhibited when the S9 residue is phosphorylated (Duka et al. 2009), leading to GSK-3β activation and subsequent downstream tau phosphorylation. Thus, further interaction and inhibitor studies were conducted at 12h of reperfusion following transient MCAO. Our studies also show that the GSK-3β inhibitor promoted S9 phosphorylation and prevented downstream tau phosphorylation. This indicates the essentiality of GSK-3β in Tau phosphorylation and an association between α-Syn, GSK-3β and tau in mediating post-ischemic brain damage.
Tau is a neuron-specific cytoskeletal protein involved in the assembly and stabilization of microtubules regulating axonal transport and maintaining neuronal morphology (Guo et al. 2017). Progressive hyperphosphorylation leads to its aggregation/deposition, impairing normal cellular functions and leading to excitotoxicity, whereas lowering tau is neuroprotective (Roberson et al. 2007). Tau can be phosphorylated at various serine, threonine, and tyrosine residues, including S199-202, Thr205, and S396-404, which are linked to abnormal tau processing in tauopathies. Studies have revealed that tau phosphorylation at S396-404 is one of the earliest events in neurodegenerative diseases, where nearly half of all structures harboring p-S396-404 form early aggregates with a well-preserved neuronal soma (Mondragón-Rodríguez et al. 2014). Additionally, it has been demonstrated that oxidative damage and mitochondrial dysfunction accompany an increase in tau phosphorylation (S396/404) in the hippocampus of aged mice (Torres et al. 2021). GSK-3β can directly phosphorylate tau at S396, S400, and S404 without the involvement of other kinases (Morfini et al. 2002), and this favors the interaction of tau with α-Syn and GSK3β. Consistent with these reports, we observed a significant increase in p-tau (S404) levels and interaction with α-Syn and GSK-3β following transient MCAO. This suggests that tau S404 phosphorylation is an indicator of neuronal damage after ischemic stroke. We previously reported that post-ischemic mitochondrial dysfunction (increase in Drp1 and p-Drp1 protein levels) and oxidative stress could be curtailed by silencing or repressing α-Syn (Kim et al. 2016; Kim et al. 2018). The oxidative damage and mitochondrial dysfunction accompany an increase in tau phosphorylation (S396/404) in the hippocampus of aged mice (Torres et al. 2021), suggesting that tau and α-Syn are linked in aggravating post-ischemic secondary brain damage. Consistent with this, the present study shows that tau knockout mice that lack total tau as well as p-tau show significantly lower infarction following transient focal ischemia. Since the tau protein regulates synaptic function, tau deletion or knockdown in mice could lower brain-derived neurotrophic factor, leading to deficiencies in long-term depression and long-term hippocampal potentiation in aging (Gonçalves et al. 2020; Velazquez et al. 2018). Our studies also indicate a progression from α-Syn to GSK-3β to tau in post-ischemic brain damage.
Overall, the current study demonstrates that a complex formed by α-Syn, tau, and GSK-3β is necessary for GSK-3β-catalyzed tau hyperphosphorylation in the post-ischemic brain and that α-Syn serves as a key initiator of tau-mediated ischemic brain damage. Disrupting these interactions might be a novel strategy to prevent post-ischemic brain damage.
ACKNOWLEDGMENTS
Partially supported by NIH RO1 NS101960 and UW Department of Neurological Surgery. Dr. Vemuganti is the recipient of a Research Career Scientist award (# IK6BX005690) from the US Department of Veterans Affairs. We thank Mr. Shreyas Kumar for proofreading the text.
Footnotes
COMPETING INTEREST
The authors declare that they have no competing interests.
DATA AND MATERIALS AVAILABILITY
All data needed to evaluate the conclusions in the paper are present in the main document.
REFERENCES
- Ballatore C, Lee VM, & Trojanowski JQ (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci, 8(9), 663–672, doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, et al. (2017). Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun, 8(1), 473, doi: 10.1038/s41467-017-00618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelluboina B, Chokkalla AK, Mehta SL, Morris-Blanco KC, Bathula S, Sankar S, et al. (2021). Tenascin-C induction exacerbates post-stroke brain damage. J Cereb Blood Flow Metab, 271678x211056392, doi: 10.1177/0271678x211056392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, & Jiang H (2019). Tau as a potential therapeutic target for ischemic stroke. Aging (Albany NY), 11(24), 12827–12843, doi: 10.18632/aging.102547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokkalla AK, Mehta SL, Kim T, Chelluboina B, Kim J, & Vemuganti R (2019). Transient Focal Ischemia Significantly Alters the m(6)A Epitranscriptomic Tagging of RNAs in the Brain. Stroke, 50(10), 2912–2921, doi: 10.1161/strokeaha.119.026433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, & LaFerla FM (2010). Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci, 30(21), 7281–7289, doi: 10.1523/jneurosci.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle JJ, George JL, Wills J, Duka V, Shah K, Lee YC, et al. (2015). GSK-3β dysregulation contributes to parkinson’s-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ, 22(5), 838–851, doi: 10.1038/cdd.2014.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duka T, Duka V, Joyce JN, & Sidhu A (2009). Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J, 23(9), 2820–2830, doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, et al. (2006). Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J, 20(13), 2302–2312, doi: 10.1096/fj.06-6092com. [DOI] [PubMed] [Google Scholar]
- Farr SA, Ripley JL, Sultana R, Zhang Z, Niehoff ML, Platt TL, et al. (2014). Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic Biol Med, 67, 387–395, doi: 10.1016/j.freeradbiomed.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM, & Trojanowski JQ (2002). Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol, 160(5), 1725–1731, doi: 10.1016/s0002-9440(10)61119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galpern WR, & Lang AE (2006). Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol, 59(3), 449–458, doi: 10.1002/ana.20819. [DOI] [PubMed] [Google Scholar]
- Gonçalves RA, Wijesekara N, Fraser PE, & De Felice FG (2020). Behavioral Abnormalities in Knockout and Humanized Tau Mice. Front Endocrinol (Lausanne), 11, 124, doi: 10.3389/fendo.2020.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Krajcer W, Kozniewska E, Lazarewicz JW, & Ksiezak-Reding H (2007). Differential changes in phosphorylation of tau at PHF-1 and 12E8 epitopes during brain ischemia and reperfusion in gerbils. Neurochem Res, 32(4–5), 729–737, doi: 10.1007/s11064-006-9199-3. [DOI] [PubMed] [Google Scholar]
- Guo T, Noble W, & Hanger DP (2017). Roles of tau protein in health and disease. Acta Neuropathol, 133(5), 665–704, doi: 10.1007/s00401-017-1707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Chokkalla AK, & Vemuganti R (2021). Deletion of ubiquitin ligase Nedd4l exacerbates ischemic brain damage. J Cereb Blood Flow Metab, 41(5), 1058–1066, doi: 10.1177/0271678x20943804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Mehta SL, Kaimal B, Lyons K, Dempsey RJ, & Vemuganti R (2016). Poststroke Induction of alpha-Synuclein Mediates Ischemic Brain Damage. J Neurosci, 36(26), 7055–7065, doi: 10.1523/jneurosci.1241-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Mehta SL, Morris-Blanco KC, Chokkalla AK, Chelluboina B, Lopez M, et al. (2018). The microRNA miR-7a-5p ameliorates ischemic brain damage by repressing alpha-synuclein. Sci Signal, 11(560), doi: 10.1126/scisignal.aat4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, & Masliah E (2013). The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci, 14(1), 38–48, doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Ayton S, Bush AI, & Adlard PA (2011). GSK-3 in Neurodegenerative Diseases. Int J Alzheimers Dis, 2011, 189246, doi: 10.4061/2011/189246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Chokkalla AK, Bathula S, & Vemuganti R (2022). MicroRNA miR-7 Is Essential for Post-stroke Functional Recovery. Transl Stroke Res, doi: 10.1007/s12975-021-00981-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Kim T, & Vemuganti R (2015). Long Noncoding RNA FosDT Promotes Ischemic Brain Injury by Interacting with REST-Associated Chromatin-Modifying Proteins. J Neurosci, 35(50), 16443–16449, doi: 10.1523/jneurosci.2943-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Pandi G, & Vemuganti R (2017). Circular RNA Expression Profiles Alter Significantly in Mouse Brain After Transient Focal Ischemia. Stroke, 48(9), 2541–2548, doi: 10.1161/strokeaha.117.017469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragón-Rodríguez S, Perry G, Luna-Muñoz J, Acevedo-Aquino MC, & Williams S (2014). Phosphorylation of tau protein at sites Ser(396–404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol Appl Neurobiol, 40(2), 121–135, doi: 10.1111/nan.12084. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, & Brady ST (2002). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J, 21(3), 281–293, doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka M, Kawano T, Yano S, Kai Y, Tsuiki H, Yoshinaga Y, et al. (2006). Hyperphosphorylation at serine 199/202 of tau factor in the gerbil hippocampus after transient forebrain ischemia. Biochem Biophys Res Commun, 347(1), 273–278, doi: 10.1016/j.bbrc.2006.06.096. [DOI] [PubMed] [Google Scholar]
- Moussaud S, Jones DR, Moussaud-Lamodière EL, Delenclos M, Ross OA, & McLean PJ (2014). Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener, 9, 43, doi: 10.1186/1750-1326-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol, 18(7), e3000410, doi: 10.1371/journal.pbio.3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta R, Ułamek-Kozioł M, Januszewski S, & Czuczwar SJ (2018). Tau Protein Dysfunction after Brain Ischemia. J Alzheimers Dis, 66(2), 429–437, doi: 10.3233/jad-180772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postina R (2008). A closer look at alpha-secretase. Curr Alzheimer Res, 5(2), 179–186, doi: 10.2174/156720508783954668. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science, 316(5825), 750–754, doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Savica R, Grossardt BR, Bower JH, Ahlskog JE, & Rocca WA (2013). Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism. JAMA Neurol, 70(7), 859–866, doi: 10.1001/jamaneurol.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toral-Rios D, Pichardo-Rojas PS, Alonso-Vanegas M, & Campos-Peña V (2020). GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front Cell Neurosci, 14, 19, doi: 10.3389/fncel.2020.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AK, Jara C, Olesen MA, & Tapia-Rojas C (2021). Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci Rep, 11(1), 4448, doi: 10.1038/s41598-021-83910-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal-Cevik I, Gursoy-Ozdemir Y, Yemisci M, Lule S, Gurer G, Can A, et al. (2011). Alpha-synuclein aggregation induced by brief ischemia negatively impacts neuronal survival in vivo: a study in [A30P]alpha-synuclein transgenic mouse. J Cereb Blood Flow Metab, 31(3), 913–923, doi: 10.1038/jcbfm.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez R, Ferreira E, Tran A, Turner EC, Belfiore R, Branca C, et al. (2018). Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell, 17(4), e12775, doi: 10.1111/acel.12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venna VR, Benashski SE, Chauhan A, & McCullough LD (2015). Inhibition of glycogen synthase kinase-3β enhances cognitive recovery after stroke: the role of TAK1. Learn Mem, 22(7), 336–343, doi: 10.1101/lm.038083.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li M, Wang Y, Li Q, Deng G, Wan J, et al. (2016). GSK-3β inhibitor TWS119 attenuates rtPA-induced hemorrhagic transformation and activates the Wnt/β-catenin signaling pathway after acute ischemic stroke in rats. Mol Neurobiol, 53(10), 7028–7036, doi: 10.1007/s12035-015-9607-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li M, Wang Y, Wang Z, Zhang W, Guan F, et al. (2017). GSK-3β as a target for protection against transient cerebral ischemia. Int J Med Sci, 14(4), 333–339, doi: 10.7150/ijms.17514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, et al. (2007). Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta, 1772(4), 473–483, doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Li S, Li M, Ke D, Wang Q, Yang Y, et al. (2022). Human tau accumulation promotes glycogen synthase kinase-3β acetylation and thus upregulates the kinase: A vicious cycle in Alzheimer neurodegeneration. EBioMedicine, 78, 103970, doi: 10.1016/j.ebiom.2022.103970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the main document.