

Abstract
In this issue of Cell Stem Cell, Jia et al. (2020) identify residual cancer stem cells (CSCs) as a mechanism of immunotherapy resistance in head and neck squamous cell carcinoma (HNSCC). Remarkably, targeting this population of CSCs can be exploited to potentiate immunotherapy and reduce tumor recurrence and metastasis.
Every year more than 600,000 new cases of head and neck squamous cell carcinoma (HNSCC) are diagnosed worldwide, ranking sixth in incidence. HNSCC has a poor 5-year survival rate at 63%, due to limited responses to currently available therapies and frequent development of recurrent disease. Immunotherapy has recently revolutionized HNSCC treatment, but unfortunately <20% of patients respond to immune check point blockade (ICB), performed primarily by blocking PD-1 and often not leading to durable responses. There is an urgent need to develop new approaches to achieve durable cure and relapse-free survival. In this regard, HNSCCs are heterogeneous cancer lesions. Cancer stem cells (CSCs), also known as tumor-initiating cells, are considered a population of poorly differentiated cells responsible for maintaining long-term tumor growth that mediate therapeutic resistance (Prager et al., 2019). Due to these characteristics, eliminating CSCs may provide an opportunity to help increase immunotherapy efficacy and achieve durable responses (cure). In a study published in this issue of Cell Stem Cell, Jia et al. (2020) explore the link between immunotherapy and CSC targeting in HNSCC (Figure 1).
Figure 1. Multimodal Precision Immunotherapy for HNSCC.
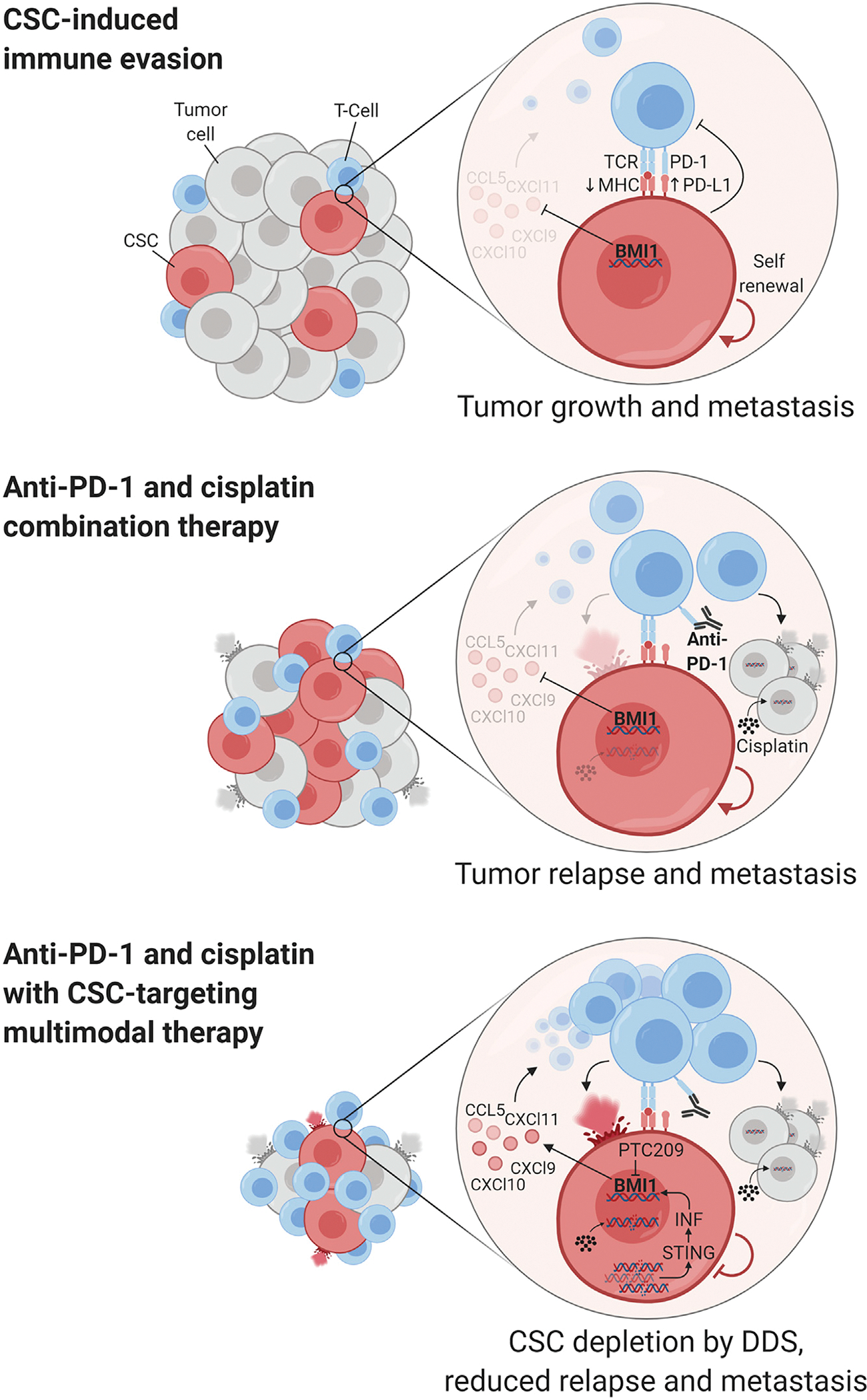
Jia and coworkers describe that BMI1+ CSCs in HNSCC contribute to immune evasion and tumor relapse and metastasis. Combining cisplatin and immunotherapy with concomitant inhibition of CSC self-renewal triggers an increased recruitment of intratumoral immune cells, enhancing ICB. BMI1 targeting can also lead to the activation of senescent pathways in CSCs that contribute to stem cell depletion (the DDS response). See text for more details. Figure created with BioRender.com.
Using elegant cell tracing approaches, HNSCC CSCs were shown to represent a distinct cell population that can be characterized by the expression of BMI1 (Chen et al., 2017). Jia and coworkers assessed the effect of combination immunotherapy on this BMI1+ CSC population taking advantage of the 4 nitroquinoline-1 oxide (4NQO) model of chemically induced HNSCC, which recapitulates the mutational and immune complexity of tobacco-related HNSCC (Wang et al., 2019). Following treatment of tumors with cisplatin and anti-PD-1 they found that, although this combination therapy reduces tongue HNSCC lesions and metastasis, apoptosis is induced preferentially in BMI1− tumor cells, thus enriching the tumors for BMI1+ CSCs that may ultimately mediate cancer relapse. This raised the possibility that eliminating BMI1+ CSCs could increase the efficacy of combination immunotherapy. Indeed, knocking out BMI1 genetically within HNSCC lesions or using a small-molecule inhibitor of BMI1 (PTC209) increased tumor immune cell infiltration and effectively increased the cancer cell killing effect of cisplatin and anti-PD-1, further reducing tumor growth and halting metastasis. Moreover, targeting BMI1+ CSCs resulted in reduced HNSCC relapse compared with combination immunotherapy alone.
Interestingly, BMI1 inhibition per se resulted in increased CD8+ T cell infiltration in tumors, suggesting that CSCs could be responsible for immune evasion in HNSCC. Mechanistically, BMI1 is part of the polycomb repressive complex 1 (PRC1), which participates in gene silencing. By performing transcriptional analysis of HNSCC cells treated with PTC209, Jia and coworkers found that BMI1 inhibition results in the accumulation of cytosolic double stranded DNA damage. This in turn causes the stimulation of the STING pathway and the consequent activation of interferon signaling and increased expression of interferon-regulated chemokines, including CCL5, CXCL9, CXCL10, and CXCL11, which are known to promote CD8+ T cell recruitment. BMI1 was found bound to the promoters of these chemokines, suggesting their direct repression by PRC1 that is relieved by BMI1 blockade (Figure 1). These findings provide exciting avenues for increasing the effectiveness of immunotherapy for HNSCC, particularly by combining ICB with CSC-specific targeted therapies.
BMI1 is part of a larger group of DNA and histone modifying proteins that participate in epigenetic silencing of genes involved in the activation of senescence and differentiation pathways. These chromatin modifications interplay with the expression and function of transcription factors and signaling circuitries in the determination of stem cell fate. Tumor suppressive mechanisms triggered by oncogenic stimulation often converge on an epigenetic regulatory network triggering the expression of the p16/Ink4a and p19/Arf cell-cycle inhibitors, thus promoting the demise of epithelial stem cells by death, differentiation, or senescence (the DDS response) (Iglesias-Bartolome and Gutkind, 2011). CSCs must override these epigenetic protective mechanisms for cancer initiation, which otherwise would lead to the loss of stem cell self-renewal capacity and depletion. Aberrant expression and function of BMI1 is known to circumvent these tumor suppressive pathways (Heffner and Fearon, 2007; Jacobs et al., 1999); thus their reactivation may provide a mechanistic framework for the anti-tumor activity of BMI1 inhibition.
The study from Jia and coworkers also raises the possibility that the repression of cell senescence programs by BMI1 may contribute to cancer immune evasion. In this case, BMI1 inhibitors may act as single agents or in combination with cisplatin to trigger DNA damage, the activation of the cGAS-STING cytosolic DNA-sensing pathway, and the consequent release of senescent associated secretome, including the release of T cell chemoattractants, thus contributing to immune cell recruitment (Figure 1). The combination of triggering CSC epigenetic senescence and differentiation programs with the reestablishment of immune surveillance might ultimately result in enhanced CSC depletion and tumor regression. While this is beneficial to increase immunotherapy success, the effects of BMI1 in other tissues have to be considered. Loss of BMI1 has been associated with T cell senescence (Heffner and Fearon, 2007), a process closely related to T cell exhaustion and immunotherapy failure, indicating that while short-term BMI1 suppression may expose an HNSCC CSC vulnerability, BMI1 inhibitors could have long-term detrimental effects on T cells. Indeed, Bmi1 knockout mice show reduced frequency of CD4+CD8+ thymocytes (Liu et al., 2009), which should be considered for translating these findings to the clinic.
Since the realization of the existence of CSCs, numerous markers and mechanisms have been postulated to label and target this population with mixed results. Preclinical models showed promising outcomes for some interventions; however, most of these new therapies have not come to fruition in the clinic. Several aspects have been attributed to CSC targeting failure, including the difficulty to precisely identify and isolate these cells and the increasingly recognized property of stem cell plasticity, where stemness can be gained by non-CSCs and restart tumor growth. Despite these hiccups, targeting of CSCs provides a general understanding of how to approach cancer treatment, particularly for tumors with high incidence of recurrence (Saygin et al., 2019). The findings from Jia and coworkers demonstrating that CSCs participate in immune evasion can bring new uses for CSC-targeting agents. Nevertheless, better tracing and markers for CSCs would greatly improve our ability to find precision therapeutic agents that can differentiate between normal and cancer cells. Current efforts in the application of single-cell techniques to tumor biology (Ji et al., 2020) will greatly improve the identification and tracking of CSCs and aid in finding new avenues to target this specific cell population for cancer therapy.
Footnotes
DECLARATION OF INTERESTS
J.S.G. is a Member of the Scientific Advisory Board of Oncoceutics, Vividion Therapeutics, and Domain Therapeutics.
REFERENCES
- Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M, Deng P, Yu B, Yu Y, Dong J, et al. (2017). Targeting BMI1(+) Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell 20, 621–634 e626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner M, and Fearon DT (2007). Loss of T cell receptor-induced Bmi-1 in the KLRG1(+) senescent CD8(+) T lymphocyte. Proc. Natl. Acad. Sci. USA 104, 13414–13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias-Bartolome R, and Gutkind JS (2011). Signaling circuitries controlling stem cell fate: to be or not to be. Curr. Opin. Cell Biol. 23, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, and van Lohuizen M (1999). The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168. [DOI] [PubMed] [Google Scholar]
- Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstråhle J, et al. (2020). Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell, in press. Published online June 23, 2020. 10.1016/j.cell.2020.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Zhang W, and Wang C-Y (2020). BMI1 Inhibition Eliminates Residual Cancer Stem Cells after PD1 Blockade and Activates Antitumor Immunity to Prevent Metastasis and Relapse. Cell Stem Cell 27, this issue, 238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, et al. (2009). Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459, 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager BC, Xie Q, Bao S, and Rich JN (2019). Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 24, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saygin C, Matei D, Majeti R, Reizes O, and Lathia JD (2019). Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 24, 25–40. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu VH, Allevato MM, Gilardi M, He Y, Luis Callejas-Valera J, Vitale-Cross L, Martin D, Amornphimoltham P, Mcdermott J, et al. (2019). Syngeneic animal models of tobacco-associated oral cancer reveal the activity of in situ antiCTLA-4. Nat. Commun. 10, 5546. [DOI] [PMC free article] [PubMed] [Google Scholar]