

Abstract
Disease Overview:
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma with immunoglobulin M (IgM) monoclonal protein. Clinical features include anemia, thrombocytopenia, hepatosplenomegaly, lymphadenopathy, and rarely hyperviscosity.
Diagnosis:
Presence of IgM monoclonal protein associated with ≥10% clonal lymphoplasmacytic cells in bone marrow confirms the diagnosis. The L265P mutation in MYD88 is detectable in more than 90% of patients and is found in most IgM MGUS patients. MYD88 is not required for the diagnosis.
Risk Stratification:
Age, hemoglobin level, platelet count, β2 microglobulin, LDH, and monoclonal IgM concentrations are characteristics that are predictive of outcomes.
Risk-Adapted Therapy:
Not all patients who fulfill WM criteria require therapy; these patients can be observed until symptoms develop. Rituximab-monotherapy is inferior to regimens that combine it with bendamustine, an alkylating agent, a proteosome inhibitor, or a BTK inhibitor. The preferred Mayo Clinic induction is either rituximab and bendamustine (without rituximab maintenance) or zanubrutinib.
Management of Refractory Disease:
Bortezomib, cyclophosphamide, fludarabine, thalidomide, everolimus, Bruton Tyrosine Kinase inhibitors, carfilzomib, lenalidomide, bendamustine, and venetoclax have all been shown to have activity in relapsed WM. Given WM’s natural history, the reduction of therapy toxicity is an important part of treatment selection.
1 |. PATIENT
A 55-year-old male was found to have an IgM monoclonal gammopathy in January 2005. He was observed until November 2010 when his IgM level climbed to 9135 mg/dl. He was treated with rituximab, bortezomib, and dexamethasone for 4 months. Treatment was interrupted due to neuropathy, but the IgM level fell to 1150. He was observed until April 2015. At the time of progression his IgM was 11 500 mg/dl and he was found to have acquired von Willebrand disease.1 He was treated with single-agent rituximab which failed to produce a minor response. He received bendamustine, lenalidomide, and rituximab with a response of <1 year. In 2018 he was placed on an experimental trial of oprozomib with a response of 5 years duration. At relapse in August of 2022, he was placed on a trial of ixazomib and ibrutinib. Ixazomib was poorly tolerated due to diarrhea, and he continued ibrutinib alone and has achieved a very good partial response.
Comment: This patient illustrates many common features of the disease. A smoldering phase lasted 5 years, multiple therapeutic interventions resulted in responses more than 4 years. Failure of single-agent rituximab is typical. Bortezomib neuropathy was severe but resolved completely.
2 |. DISEASE OVERVIEW
The World Health Organization defines Waldenström macroglobulinemia (WM) as a lymphoplasmacytic lymphoma associated with a monoclonal immunoglobulin M (IgM) protein.2 The physical manifestations of the disorder are hepatomegaly (20%), splenomegaly (15%), and lymphadenopathy (15%). The most common presenting symptom is fatigue related to a normocytic anemia. The median hemoglobin value at diagnosis is 10 g/dl. Many patients who fulfill the criteria of WM do not require immediate therapy because they are asymptomatic (smoldering).3 Virtually all patients have a preceding phase of IgM MGUS, but the clonal MGUS B cells already contain the molecular signature of a malignant clone.4 Patients under the age of 70 have a median survival in excess of 10 years; those 70–79, approximately 7 years; and those 80 or older, approximately 4 years. In patients with Waldenstrom over age 65 at diagnosis, the most common cause of death is not cancer related.5 Clonal hematopoiesis is present in 14% of patients with macroglobulinemia. These patients are more likely to progress from IgM MGUS or smoldering macroglobulinemia to symptomatic disease.6 The key clinical features of the disease are represented in the graphical abstract.
The median age at the time of diagnosis is 71 years. The age-adjusted incidence rate is .42/100 000 person-years with an age- and sex-adjusted incidence of 0.57 per 100 000 person-years with a male-to-female ratio of 3.2:1. There is no evidence of a change in the incidence of WM over the past 50 years.7
WM incidence is higher in whites (4.1 per million per year) than in blacks (1.8 per million per year). Waldenström patients had a positive family history of lymphoplasmacytic lymphoma or WM in 4.3%, and a family history was associated with poorer survival than the non-familial forms. A study of monoclonal immunoglobulins (MGUS) showed that the M protein isotype was IgM in 2% and 16% of black and white patients, respectively. The median M protein concentration for blacks was 0.44 g/dl, whereas it was 1.2 g/dl in whites. Black patients less commonly have IgM monoclonal gammopathy compared with white patients. Median age at diagnosis is 63 years for blacks and 73 for whites, with blacks having a shorter survival than whites.8
Survival of WM is improving. The SEER database contained 5784 patients with WM. Median OS from 1991 to 2000 and 2001 to 2010 improved from 6 to 8 years, respectively. Deaths in the 2001 to 2010 cohorts were reduced both from WM related and non-WM-related causes. Age at diagnosis continues to have the greatest impact on survival. The hazard ratio for death for WM patients’ age 80 or greater was 6.99, compared with a reference group less than age 50.9
The presence of a monoclonal IgM protein adds a unique dimension to the disorder because it can result in hyperviscosity syndrome, peripheral neuropathy, hemolytic anemia, and immune complex vasculitis. The 10-year survival rate is now 66%. In a population-based study of Latin American patients with WM and lymphoplasmacytic lymphoma, 5-year relative survival was 81%. Survival improvements were seen in all age groups, although increasing age was associated with inferior survival.10
The management of peripheral neuropathy associated with IgM monoclonal protein remains frustrating for clinicians. The mechanism of the neuropathy is thought to be demyelination due to direct binding of the antibody to myelin-associated glycoprotein. The treatment of IgM-associated peripheral neuropathy can be like that of WM. Overall improvement following rituximab treatment in IgM associated neuropathy was seen in 54.5%. Six patients who were unchanged after the 1st treatment with rituximab improved after another rituximab cycle. Rituximab monotherapy retains a role in IgM-mediated neuropathy.11 In a double blind, placebo-controlled trial, 54 patients with anti-MAG IgM chronic demyelinating neuropathy were randomized to receive either placebo or rituximab. The primary outcome of absolute improvement in ISS (INCAT sensory score) from baseline at 12 months was not achieved in the study as no significant difference in the change in ISS was seen between rituximab and placebo groups. Ibrutinib was administered to 3 patients with anti-myelin associated glycoprotein immunoglobulin M neuropathy all reported an improvement pointing to possible efficacy of ibrutinib in this setting.12 The therapy of IgM-associated neuropathy remains inadequate.13
2.1 |. Diagnosis
In the original description of WM, Jan Gösta Waldenström described two patients with oronasal bleeding (hyperviscosity), lymphadenopathy, anemia, thrombocytopenia, and an elevated sedimentation rate. The disorder is a lymphoplasmacytic lymphoma with a monoclonal pentameric IgM protein. Bone marrow and lymph nodes are infiltrated with pleomorphic B-lineage cells at different stages of maturation. The bone marrow pattern is predominantly intertrabecular.14 Many patients who fulfill all other criteria for the diagnosis have a presymptomatic phase and may not require therapy.3 The cells express pan B-cell markers (e.g., CD19, CD20) and typically test negative for CD3 and CD103.15 A recurring sequence variant at position 38 182 641 in chromosome 3p22.2 has been identified. A single-nucleotide change from T to C in the MYD88 gene resulted in a leucine-to-proline change at amino acid position 265. This mutation is seen in 93% of patients. CXCR4 mutations are seen in 29%. 53% of patients with hyperviscosity have mutations of CXCR4.16 Together, these studies demonstrate an important somatic variant in the malignant cells of WM.17 MYD88 status does not predict survival 10.2 (mutant) versus 13.9 years (wild type) in patients not treated with BTK inhibitors.18 Others have reported a survival difference between those with mutant and those with wild-type MYD88. The estimated 10-year survival was 73% for MYD88WT versus 90% for mutated MYD88MUT. Median cause specific survival in cyclophosphamide treated patients was 166 months.19
MYD88 can be detected by a polymerase chain reaction in the peripheral blood of patients with WM.20 CXCR4 is mutated in 30% of patients with WM. In animal models, this mutation predicts resistance to ibrutinib and everolimus. CXCR4 mutation is associated with a shorter treatment free survival.21
Distinguishing between WM and marginal zone lymphoma can be challenging. MYD88 mutation L265P is specifically associated with WM and IgM monoclonal gammopathy of undetermined significance. MYD88 L265P also is seen in splenic marginal zone lymphoma (4%), IgM amyloidosis (71%), mucosa-associated lymphatic tissue lymphoma (7%), and WM (67%–90%). MYD88 L265P cannot be used to differentiate between WM and IgM MGUS. The mutation is not found in IgM multiple myeloma, and mutation expression is concordant with the extent of bone marrow involvement. Responses after chemotherapy are associated with declines in mutation expression.22
Patients can present with markedly elevated IgM levels and infiltration of the bone marrow more than 30% yet still not require therapy because they have no symptoms. Conversely, patients can have low levels of monoclonal IgM protein and minimal clonal marrow infiltration and still require therapy for complications associated with the IgM protein, including moderate or severe peripheral neuropathy, amyloid deposition, cold agglutinin hemolytic anemia, and type II mixed cryoglobulinemia—all a consequence of the antibody-binding specificity and protein folding of the IgM protein. A classification scheme for WM is provided in Table 1. Symptoms can be produced by the tumor mass or the monoclonal protein. The disease is incurable with current therapies.23
IgM multiple myeloma is a distinct entity; although constituting only 1% of all multiple myeloma cases, it must be distinguished from WM. MYD88 is not mutated in IgM myeloma. Useful clues to the diagnosis of multiple myeloma include the presence of lytic bone lesions (rare in WM) and a translocation at chromosome 14 (does not occur in WM). Patients with IgM multiple myeloma tend to have plasmacytic differentiation with high expression of CD138 and cytoplasmic immunoglobulin, whereas WM expresses CD20.24
Monoclonal IgM proteins are found in 1 of 600 persons older than 50 years. More patients have IgM MGUS than have WM. However, all patients with IgM MGUS require lifelong monitoring. Among patients with IgM MGUS, the presence of two adverse risk factors—namely, an abnormal serum free light-chain ratio (ratio of kappa to lambda free light chains) and a high serum monoclonal protein (M protein) level (≥1.5 g per deciliter)—was associated with a risk of progression at 20 years of 55%. In a study of 176 IgM MGUS patients a monoclonal protein peak of >1 gram/deciliter and the presence of an MYD88 mutation successfully predicted progression to symptomatic macroglobulinemia with hazard ratios of over 20 for both variables. The cumulative incidence of progression at 10 years was 38%.25 MYD88 wild type is also an independent predictor of transformation to large-cell lymphoma which carries an inferior overall survival.26
Patients with IgM values greater than 3000 mg/dl may have no symptoms, a normal hemoglobin value, and no clinically important increase in serum viscosity. In these instances, observation continues to be an appropriate option. Symptomatic hyperviscosity was only seen in 13% of Mayo Clinic patients with WM. Even among patients presenting with an IgM greater than 6000 mg/dl the median time to initial therapy was 6.9 years.27 In patients with smoldering macroglobulinemia independent predictors of disease progression to symptomatic disease included immunoglobulin M level greater than 4500 mg/dl, bone marrow infiltration with 70% or greater lymphoplasmacytic lymphoma, beta 2 microglobulin >4 mg/L or greater and albumin <3.5 g/dl.28 Wild type MYD 88 is also an independent predictor of progression to symptomatic disease.29 The Box 1 lists the recommended diagnostic tests for a new patient with suspected WM. Imaging plays a minor role since the majority of patients have modest lymphadenopathy, however, there is a suggestion that the results from FDG PET are prognostic in patients with macroglobulinemia.30
BOX 1. Diagnostic approach to suspected Waldenström macroglobulinemia.
Serum protein electrophoresis.
Serum immunofixation to validate the immunoglobulin M (IgM) heavy chain and the type of light chain.
Quantitative test for immunoglobulin G, immunoglobulin A, and IgM.
24-Hour urine collection for protein electrophoresis; monoclonal light chains are detected in the urine of 40%–80% of patients tested.
Immunoglobulin free light chain assay (long-term value not established).
Serum β2 microglobulin and LDH evaluation for prognosis; part of the international staging system for Waldenström macroglobulinemia.
Bone marrow biopsy; intertrabecular monoclonal lymphoplasmacytic infiltrate ranges from predominantly lymphocytic cells to overt plasma cells.
Perform MYD88L265P mutational analysis on bone marrow sample in all cases of WM by allele-specific polymerase-chain-reaction (AS-PCR) assay.
CXCR4 mutational analysis, if available.
Computed tomography of abdomen and pelvis to detect organomegaly and lymphadenopathy or a combined 18F-FDG positron emission tomography (PET)/CT scan (a skeletal survey and radiographic imaging of the bones are unnecessary in the absence of symptoms; lytic bone lesions are unusual).
Serum viscosity required when signs and symptoms of hyperviscosity syndrome are present or when IgM >4000 mg/dl.
Ophthalmologic evaluation for hyperviscosity.
Based on clinical presentation, analysis involves Coombs test (cold autoantibody), cryoglobulin, Von Willebrand Ag, Factor VIII C or tissue stains for amyloid deposits.
Of myeloma patients, 1% have IgM, and their disorder behaves like other multiple myeloma.
Hepatitis B and C screening is necessary if rituximab therapy is planned.
Response in WM is defined by reduction in the M protein. A minor response is an M-spike reduction of at least 25%. A partial response is defined as a 50% or greater reduction in M protein. A very good partial response is a 90% reduction in M protein, and a complete response is immunofixation negativity in the serum. There may be discrepancies between IgM levels and bone marrow response. The involved serum free light chain is a useful marker of tumor burden and acts as a leading indicator of response and progression before the intact IgM. The immunoglobulin free light chain assay, which is quite valuable in myeloma, has not been well established in WM. It is not required for serial monitoring of patients with WM.31
2.2 |. Risk stratification
As WM is a distinct lymphoproliferative process with unique cell surface and genetic characteristics, the International Prognostic Index and the Follicular Lymphoma International Prognostic Index are not used to determine prognosis. Table 2 gives the currently accepted international staging system for WM.
The five criteria shown in Table 2 are not weighted equally. Age has the greatest impact on prognosis. Patients older than 65 years cannot be in a low-risk category. Although IgM protein levels are important prognostically, they do not enter the staging system until the IgM level exceeds 7000 mg/dl. In the largest study of single-agent rituximab therapy for WM, the IgM level did not affect response rate.32 Lactate dehydrogenase is absent from the International Prognostic Scoring System for Waldenström Macroglobulinemia. In a revision of the prognostic scoring system age (≤65 vs 66–75 vs ≥76 years), β2-microglobulin ≥4 mg/L, serum albumin <3.5 g/dl, and LDH ≥250 IU/L (ULN < 225) was able to stratify patients in five different prognostic groups and identify a very-low risk as well as a very-high risk group with a 3-year WM-related death rate of 0, 10, 14, 38, and 48% (p < 0.001) and 10-year survival rate of 84, 59, 37, 19, and 9% (p < 0.001). LDH is a predictor of early mortality in this disease. Age is the most powerful predictor of outcome. The 10 year survival of patients age 45 years or younger is 86%.33
The International Prognostic Scoring System for Waldenström Macroglobulinemia is to be used only for patients who require treatment. The system should not be used to determine whether a patient requires intervention; this determination continues to be a clinical decision. Serial measurements of β2 microglobulin are not useful in monitoring therapy. Both hemoglobin and beta 2 microglobulin levels at diagnosis are independent predictors of progression to active macroglobulinemia.34
Because most patients with WM have an indolent disease course and often are elderly, nearly half of all patients succumb to diseases unrelated to WM.35 The impact of age on OS was investigated in 238 patients with WM. The poorest survival of patients older than 65 years at diagnosis was attributable to the higher number of non-WM-related deaths.7 Cause-specific survival has been introduced as an important outcome measure.36 This statistical technique censors patients who die of causes unrelated to the malignancy and accounts for the competing risks of death that these patients face. In a competing risk survival analysis, 23% of deaths were unrelated to WM, and 40% of patients >75 years do not die of WM. Patients with WM have greater overall risk of a second malignancy that is 1.69 times higher than expected (p = 0.002). Compared with the general population, patients with WM appear to have a higher risk of large cell lymphoma, myelodysplasia, and brain cancer. Therapy related myeloid neoplasms occur in 2.7% of patients.
3 |. MANAGEMENT
3.1 |. Hyperviscosity syndrome
Hyperviscosity syndrome is seen in a decreasing proportion of patients with WM because WM is being diagnosed earlier. Symptomatic hyperviscosity is rare in patients with an IgM concentration less than 4000 mg/dl, and viscosity measurements are not required in patients whose IgM levels are below that threshold. The symptoms of hyperviscosity are primarily due to shear forces that rupture unsupported venous channels. Therefore, the presentation generally includes epistaxis, gingival bleeding, and visual changes due to retinal hemorrhage.37 Central nervous system findings, including dizziness, light-headedness, and generalized fatigue, are nonspecific and must be confirmed with measures of serum viscosity. Reference serum viscosity is 1.8; water has a viscosity of 1. Hyperviscosity syndrome is not likely unless the serum viscosity exceeds 4. When hyperviscosity is present, plasma exchange should be considered a temporizing measure until systemic therapy successfully lowers the tumor mass and thereby reduces the IgM protein concentration in the serum. A single plasma exchange is often sufficient to relieve symptoms and allow initiation of systemic therapy.27
3.2 |. Systemic chemotherapy to reduce tumor mass
3.2.1 |. Rituximab cyclophosphamide
Rituximab is a widely available treatment for the management of WM. Its lack of long-term toxicity and nonmyelosuppressive treatment profile has led to its incorporation in most therapeutic regimens for this disorder. However, rituximab alone is generally a poor choice for patients in need of therapy. Including both minor (25%–50% reduction of M protein) and objective (>50% reduction of IgM protein) responses, the response rate to rituximab (<55%) is inferior to every other reported combination regimen. A meta-analysis confirmed that a greater response was produced with combination therapy of 2+ drugs than with rituximab monotherapy (73% vs. 44%). In a double-blind randomized placebo-controlled phase 3 trial rituximab was the control arm. The median progression-free survival with rituximab was 20.3 months partial response or better was observed in only 31%. Median time to next treatment was 18 months. Hemoglobin improvement was seen in only 43%.38 Rituximab monotherapy is generally only suited for IgM-associated symptoms such as type II cryoglobulinemia, MGUS-associated neuropathy and cold agglutinin disease.
Use of rituximab is associated with the risk of “flare” for many patients. In this phenomenon, the initiation of rituximab treatment results in a transient rise in the level of IgM, which can produce an increase of serum viscosity. This flare is seen less frequently when rituximab is combined with cytotoxic chemotherapy. In some trials, rituximab is delayed until the second cycle to allow cytotoxic therapy to reduce IgM levels and reduce the risk of hyperviscosity associated with the introduction of rituximab.39
The use of maintenance rituximab therapy has been controversial. In a trial of patients treated with bendamustine and rituximab followed by randomization to observation or rituximab, an improvement in progression-free survival was not seen.40
Rituximab is not the only monoclonal antibody that has been used in WM. Ofatumumab, an anti-CD20 monoclonal human anti-body, has shown activity in WM. In a trial of 37 Waldenstrom patients receiving ofatumumab 15 (41%) achieved a partial response, seven (19%) a minor response. All 37 patients had at least one adverse event.41
3.3 |. Rituximab cyclophosphamide dexamethasone RCd
Rituximab treatment combined with cyclophosphamide (orally) and dexamethasone has been reported, with a response rate of 83% and minimal toxicity. Two-year PFS was 67%; 2-year disease-specific survival was 90%. In an updated final analysis in 72 patients treated with rituximab cyclophosphamide and dexamethasone, the response rate on an intent-to-treat basis was 83%. Median PFS was 35 months. Median OS was 95 months. This three-agent combination is currently an alternative regimen for first-line therapy if the disease burden is low, based on Mayo Clinic mSMART guidelines.
Selected outcomes with RCd.
3.4 |. Proteosome inhibition
Bortezomib has been shown to have high levels of activity in the management of relapsed WM in schedules of twice weekly, 2 of 3 weeks, with response rates ranging from 81% to 96%.45 CXCR4 mutation does not lower the response rate to bortezomib. In newly diagnosed patients, weekly treatment with bortezomib and rituximab resulted in a better-than-minimal response in 23 of 26 patients and a 1-year event-free survival rate of 79%. Most importantly, no grade 3 or 4 neuropathy was seen with the weekly bortezomib schedule. The European Myeloma Network reported outcomes of bortezomib, rituximab, and dexamethasone in previously untreated symptomatic patients. Rituximab was delayed to cycles 2 and 5 to reduce the risk of flare. No patient required plasma exchange for flare. The response rate was 85%, the median PFS was 42 months, and the 3-year OS was 81%. Peripheral neuropathy was seen in 46%. Bortezomib-rituximab-dexamethasone is a reasonable choice for front-line therapy, but attention to early neurotoxicity is required. Bortezomib also has reported activity in cold agglutinin disease with an overall response rate of 32%.
In view of the high neuropathy rates, the less neurotoxic proteosome inhibitor, carfilzomib, was combined with rituximab and dexamethasone in patients not previously treated with the combination of rituximab and bortezomib. The overall response rate was 87%, with 36% having at least a very good partial response. At 2 years, 65% were progression-free. The peripheral neuropathy rate and cardiomyopathy rates were both 3%. The oral proteosome inhibitor, ixazomib, was combined with dexamethasone and rituximab, 26 patients were enrolled with an overall response rate of 96%, and a major response rate of 77%. The median time to response was 8 weeks. Fifty-nine previously treated patients received ixazomib subcutaneous rituximab and dexamethasone. After 8 cycles overall response rate was 71% with 14% very good partial response. Median response duration was 36 months. At 2 years progression-free and overall survival were 56 and 88% respectively.46 Proteosome Inhibitiors are an important treatment option for the management of macroglobulinemia.
Selected Outcomes with Proteosome Inhibitors.
| Regimen | ND/RR | Response Rate > MR, % | Response Rate > PR, % | Response rate > VGPR, % | PFS, mo | Reference |
|---|---|---|---|---|---|---|
| Vd | 10/0 | 80 | 20 | 39 | 47 | |
| RituximabVd | 0/34 | 59 | 32 | 3 | 15.3 | 48 |
| VCd | 4/11 | 93 | 53 | 7 | 18.6/7.3 (TTP) | 49 |
| Carfilzomib rituximab dex | 28/3 | 86 | 67 | 35 | 75%@1 year | 41 |
| Ixazomib rituximab dexamethasone | 26/0 | 96 | 77 | 15 | 75%@22 months | 50 |
3.5 |. Bendamustine
In a prospective randomized study of bendamustine plus rituximab compared with R-CHOP in low-grade lymphoma, a subset analysis identified 41 patients with WM, of whom 22 received bendamustine and rituximab, and 19 received R-CHOP. In both groups, the response rate was 95%, but median PFS was significantly prolonged with bendamustine. The median PFS for R-CHOP was 36 months in contrast to not being reached with bendamustine and rituximab (p < 0.001). At the time of analysis, 4 relapses were identified (18%) in the bendamustine and rituximab group and 11 relapses (58%) in the R-CHOP group. Bendamustine and rituximab treatment was better tolerated, with no alopecia, less hematotoxicity, lower frequency of infection, lower incidence of neuropathy, and reduced stomatitis.51 Twenty-four previously treated patients received Bendamustine (90 mg/m2) plus rituximab on two consecutive days. Each cycle was 4 weeks, with a median of five treatment cycles. The overall response rate was 83% (20/24). The median PFS was 13.2 months. Prolonged myelosuppression was more common in patients who previously had received fludarabine or cladribine.52 In a cohort of 71 patients, with a median age of 72, all with relapsed/refractory WM (median two prior lines of therapy), R-bendamustine produced a PR of 74.6% and PR + MR of 80.2%. One- and 3-year PFS was approximately 80% and 60%, respectively. The risk of progression appears to be lower in patients treated with bendamustine-rituximab or bortezomib-dexamethasone-rituximab when compared to cyclophosphamide-dexamethasone-rituximab.53 At Mayo Clinic 60 patients receiving rituximab bendamustine were compared to100 patients receiving rituximab cyclophosphamide dexamethasone. Two-year PFS was 88 vs. 61% favoring bendamustine, outcomes independent of MYD88 status. A total of 69 patients enrolled in a study of bendamustine plus rituximab. The overall response rate at 18 months was 97%. The progression-free survival at 2 years was 87%. MYD 88 and CXCR4 mutations had no impact on response rate or PFS. One patient developed myelodysplastic syndrome 6 months after bendamustine initiation (1.4%).52
In an East German lymphoma Study group trial 293 patients received bendamustine rituximab. The overall response rate was 91.4% The 5 year survival is estimated to be 78%, two therapy related myeloid neoplasms were seen (0.7%). Median progression-free survival was 65.3 months, There was no difference between those receiving maintenance rituximab and those that did not. As a result of this large trial Mayo clinic does not recommend rituximab maintenance therapy.54
Rituximab-bendamustine is one of the Mayo Clinic preferred induction regimen for newly diagnosed WM due to its ease of use and low rates of non-hematologic adverse events. It also has the advantage of time limited therapy usually less than 6 months and there is no risk of IgM flare. However, in the presence of central nervous system infiltration so-called Bing Neel syndrome ibrutinib is preferred as it is known to cross the blood–brain barrier. Ibrutinib shows rapid and durable symptomatic and radiologic responses in patients with central nervous system infiltration with lymphoplasmacytic lymphoma.55 A patient with Bing Neel syndrome was treated with tirabrutinib and within 2 months of treatment lower extremity muscle strength had normalized and T2 weighted magnetic resonance imaging showed improvement in contrast enhancement in the spinal cord.56
Bendamustine based therapies.
3.6 |. BTK inhibitors
Sixty-three previously treated patients received 420 mg of the BTK inhibitor ibrutinib. The major response rate was 73.0%, including minor responses of 90.5%. Two-year PFS and OS survival rates were 69.1% and 95.2%, respectively. Neutropenia and thrombocytopenia were the most common adverse events. The median time to response was 4 weeks. Median IgM fell from 3610 to 1340. Median Hb rose from 10.5 to 12.6. Diarrhea, bleeding, and atrial fibrillation (10.7%) were seen as non-hematologic toxicities.61 Ibrutinib should be given indefinitely as rapid IgM increases have been reported on its cessation.62 Unmutated MYD 88 patients have a lower response rate to ibrutinib.
In an open-label sub-study of Ibrutinib that was multi-center and phase 3, 31 patients, all of whom were rituximab refractory with a median age of 67, were enrolled. Overall response rate was 71%, progression-free survival at 18 months was 86%, and overall survival was 97%.63 A trial of 30 patients who were newly diagnosed and received ibrutinib was reported. The major response rate was 80% with no difference between patients with wild-type or mutated MYD 88. Atrial arrhythmias were seen in 10%. In treatment naïve patients overall (minor or more than minor) and major (partial or greater than partial) responses for all patients were 100% and 83%, respectively.64 It does not appear that a deep response is critical with ibrutinib. Comparing patients with a greater than very good partial response at 6 months versus others no significant differences in progression-free survival were observed.65
Outside of a clinical trial setting (“real world”) 80 patients were reported receiving ibrutinib therapy, achieving an overall response rate of 91% with an 18 month progression free survival of 82%; 21% of patients discontinued therapy due to treatment related toxicity. Atrial fibrillation was seen in 11%. IgM rebound was seen in 36% of patients following ibrutinib discontinuation.66
In newly diagnosed and relapsed Waldenstrom a phase 3 trial randomized patients to ibrutinib with rituximab vs. rituximab placebo. At 30 months, the progression-free survival rate was 82% with ibrutinib–rituximab versus 28% with placebo–rituximab (hazard ratio for progression or death, 0.20; p < 0.001). The benefit in the ibrutinib–rituximab group over that in the placebo–rituximab group was independent of the MYD88 or CXCR4 genotype. More patients had sustained increases in hemoglobin level with ibrutinib–rituximab than with placebo–rituximab (73% vs. 41%, p < 0.001). Events of grade 3 or higher that occurred more frequently with ibrutinib–rituximab than with placebo–rituximab included atrial fibrillation (12% vs. 1%) and hypertension (13% vs. 4%).38When treating macroglobulinemia with ibrutinib, administered dose is important. Patients with a dose intensity lower than 97% had a shorter progression-free survival.67 Holding ibrutinib for longer than 1 week is associated with a 4-fold increased risk of progression. When initiated, ibrutinib therapy should be considered indefinite and compliance should be emphasized to optimize outcomes. Following discontinuation of ibrutinib rapid increase in serum immunoglobulin M level was observed in 60% of patients. Ten acutely developed symptomatic hyperviscosity.68 Adverse events associated with stopping ibrutinib include fever, body aches, night sweats, arthralgias, chills and headache.
Acalabrutinib was developed to be more potent and selective than ibrutinib. Acalabrutinib is rapidly absorbed, has a short half-life, and lacks irreversible targeting to alternative kinases including the epidermal growth factor receptor, interleukin-2-inducible T-cell kinase, and T cell X chromosome kinase. Acalabrutinib is approved for the treatment of chronic lymphatic leukemia and mantle cell lymphoma. In a trial of 106 patients with WM a response was achieved in 93%. Grade 3/4 atrial fibrillation occurred in only 1 patient.69
Zanubrutinib is a second-generation Bruton tyrosine kinase inhibitor. At a dose of 160 milligrams twice daily 77 patients achieved an overall response rate of 95.9% with a ≥ VGPR rate at 24 months of 43.8%. Three year progression-free survival was 80.5%. However, the incidence and severity of toxicities was lower with zanubrutinib. Atrial fibrillation was seen in only 2% of zanubrutinib patients compared with 15% of those receiving ibrutinib. In both arms, 84 and 85% of patients were progression-free at 18 months with ibrutinib and zanubrutinib, respectively.69–71
Tirabrutinib is a 2nd generation irreversible BTK inhibitor. Of 27 enrolled patients the major response rate was 93%. Including 1 complete and 5 very good partial responses. The progression-free and overall survival rates at 24 months were 92.6 and 100%. One patient experienced grade 2 atrial fibrillation. Treatment related skin adverse events were observed in 14 patients (52%).72
Selected outocimes with BTKi.
| Regimen | ND/RR | ≥25%↓ IgM;% | ≥50%↓ IgM;% | ≥90%↓ IgM;% | PFS, mo | Reference |
|---|---|---|---|---|---|---|
| ibrutinib | 0/63 | 90.5 | 79.4 | 30.2 | 54%@5 years | 61 |
| ibrutinib | 30/0 | 100 | 87 | 30 | 76%@48mo | 64 |
| Ibrutinib-R | 34/41 | 93 | 73 | 26 | 82%@30mo | 38 |
| acalabrutinib | 14/92 | 93/94 | 79/81 | 0/9 | 90/82@2 years | 69 |
| zanubrutinib | 19/83 | 94 | 77 | 28 | 85%@18mo | 71 |
| Tirabrutinib | 27 | 93 | 22 | 81% on therapy @ 24.8 | 72 |
A multi Center global case series of 347 symptomatic patients compared the use of rituximab bendamustine to ibrutinib. Deeper responses were obtained with rituximab bendamustine. Overall survival and progression-free survival were not different between the 2 groups. Both are rational options for the treatment of macroglobulinemia. Often the decision rests on the patient’s desire for oral versus parenteral therapy or indefinite versus time-limited therapy.73
Since macroglobulinemia cells highly express Bcl-2 venetoclax is a logical therapy for this disorder. Waldenstrom macroglobulinemia cells devoid of BTKC481S or CXCR4WHIM-like mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability toward venetoclax treatment. In a phase 2 clinical trial, venetoclax (at a maximum target dose of 800 mg daily) demonstrated an ORR and MRR of 87% and 80%, respectively.74 CD19 directed car T therapy is currently being explored in macroglobulinemia. Three patients were treated all responded but all 3 developed recurrent disease from 3 to 26 months after infusion.75
Figure 1 shows the Mayo Clinic algorithm for the recommended management of patients with newly diagnosed WM. Figure 2 illustrates treatment recommendations for patients with relapsing WM, based on consensus criteria developed by the WM treatment and research group at Mayo Clinic. Unlike other low-grade lymphomas, early disease progression within 24 months does not impact mortality. Survival after progression is not influenced by time to progression regardless of treatment.76 The National Comprehensive Cancer Network has recently published its consensus recommendations on diagnosis and therapy of WM. (NCCN Guidelines for Waldenström Macroglobulinemia/Lymphoplasmacytic Lymphoma V.1.2022) Recent consensus reviews on WM are available.77
FIGURE 1.
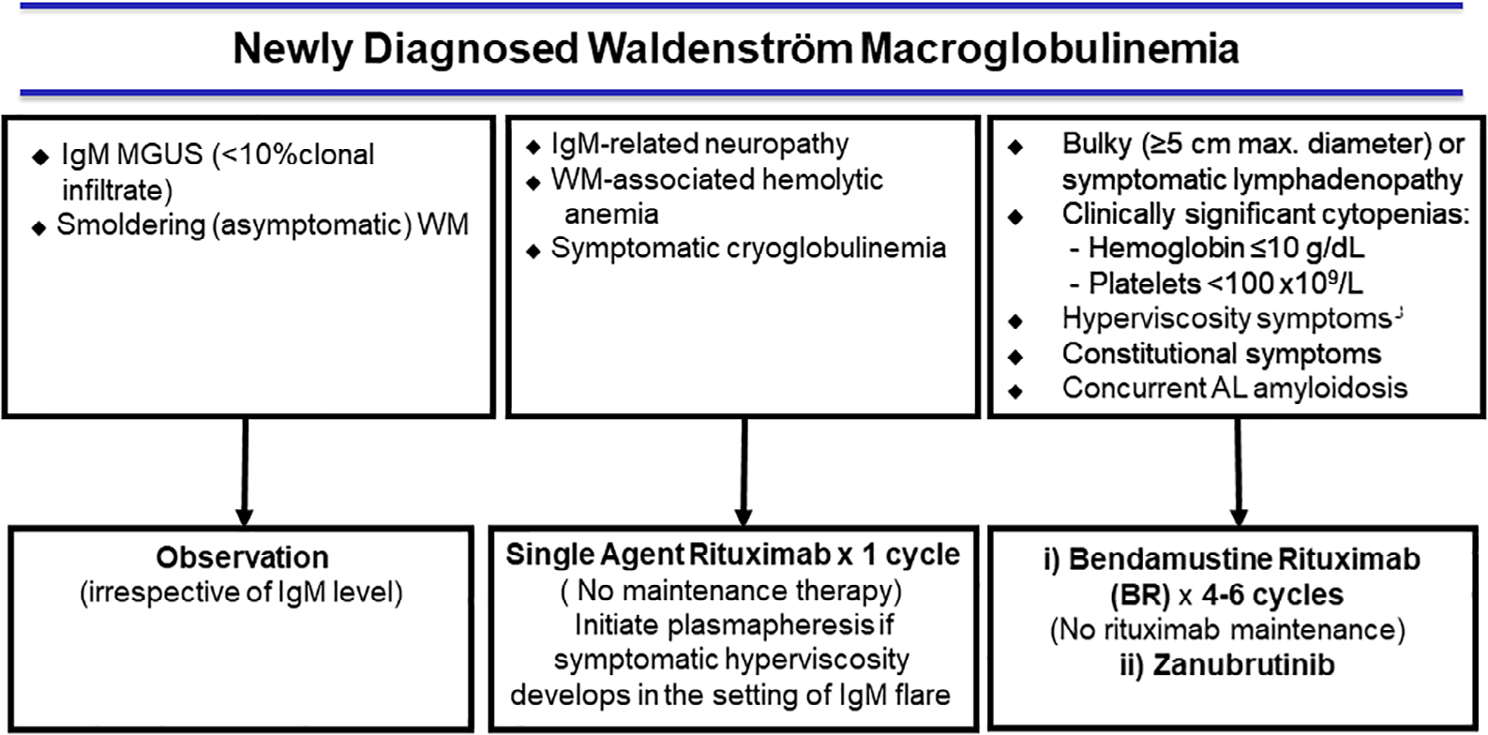
Mayo Clinic Consensus for Newly Diagnosed Waldenström Macroglobulinemia (WM). Hb indicates hemoglobin; IgM, immunoglobulin M; MGUS, monoclonal gammopathy of undetermined significance; RCD, rituximab, cyclophosphamide, and dexamethasone. (https://www.msmart.org/wm-treatment-guidelines)
FIGURE 2.
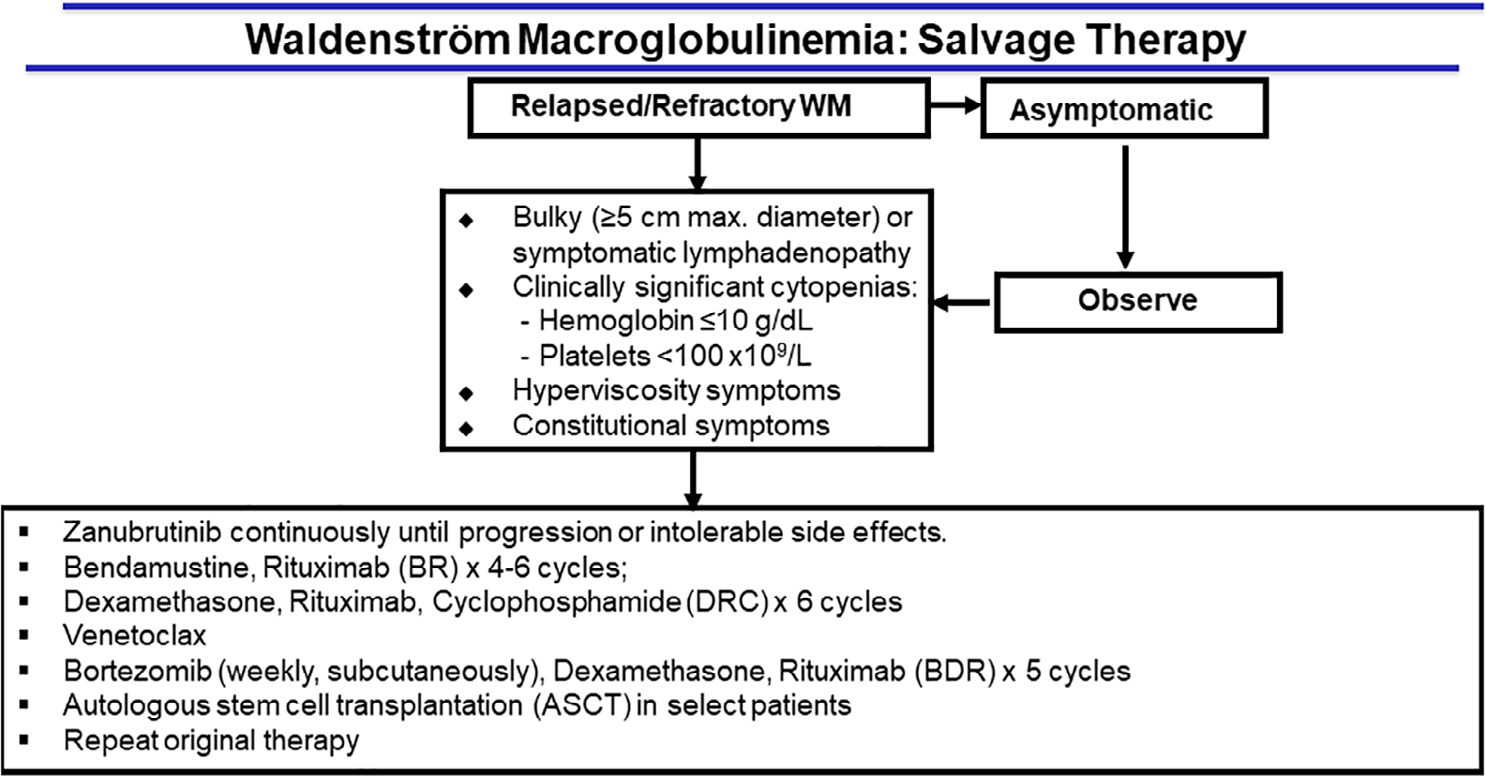
Mayo Clinic Consensus for Salvage Therapy in Waldenström Macroglobulinemia. (https://www.msmart.org/wm-treatment-guidelines)
4 |. CONCLUSION
When WM is diagnosed before the development of symptoms, patients may be safely observed and monitored. However, patients with symptoms require chemotherapy. Nonstudy Mayo Clinic–preferred options are rituximab and bendamustine or zanubrutinib. The clinician should focus on methods to minimize the toxicity associated with therapy and avoid late complications.
TABLE 1.
Definitions of IgM-related phenomenon in macroglobulinemia
| IgM Monoclonal Component | Symptoms of Tumor Mass/Infiltration (Adenopathy Anemia) | Marrow Infiltration > 10% | IgM-Mediated Symptoms | |
|---|---|---|---|---|
| MGUS | + | − | − | − |
| Smoldering macroglobulinemia | + | − | + | − |
| IgM-related disorder (eg, cold agglutinin hemolytic anemia, type II cryoglobulin, neuropathy, amyloidosis) | + | − | ± | + |
| Macroglobulinemia | + | + | + | ± |
Abbreviations: IgM, immunoglobulin M; MGUS, monoclonal gammopathy of undetermined significance; +, positive; −, negative; ±, equivocal.
TABLE 2.
International prognostic scoring system for Waldenström macroglobulinemia
| Factor associated with prognosis | Value | |
| Age, year | >65 | |
| Hemoglobin, g/dl | ≤11.5 | |
| Platelet count, No./mcL | ≤100 000 | |
| β2-Microglobulin, mg/L | >3 | |
| Monoclonal IgM, g/dl | >7 | |
| Risk stratum and survival | ||
|
| ||
| Risk category | Score a | Median survival, month |
| Low | 0 or 1 (except age) | 142.5 |
| Intermediate | 2 or age > 65 years | 98.6 |
| High | >2 | 43.5 |
Abbreviation: IgM, immunoglobulin M.
One point is assigned for each positive factor and the risk score is the sum of points.
Funding information
National Cancer Institute, Grant/Award Number: 5P50 CA186781-04
Abbreviations:
- BTK
Bruton tyrosine kinase
- IgM
immunoglobulin M
- MGUS
monoclonal gammopathy of undetermined significance
- mTOR
mammalian target of rapamycin
- OS
overall survival
- PFS
progression-free survival
- R-CHOP
cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab
- WM
Waldenström macroglobulinemia
Footnotes
CONFLICT OF INTEREST
Dr Gertz has received honoraria from Celgene Corporation (Summit, NJ), Millennium: The Takeda Oncology Company (Cambridge, MA), The Binding Site Group Ltd (Birmingham, United Kingdom), Onyx (San Francisco, CA), Novartis (Basel, Switzerland), Ionis (Carlsbad, CA), Amgen (Thousand Oaks, CA); Prothena (San Francisco, CA) Sandoz (Princeton, NJ), AbbVie (North Chicago, IL), Alnylam (Cambridge, MA), Prothena (South San Francisco, CA), Janssen (Beerse, Belgium), Spectrum (Henderson, NV), Apellis (Louisville, KY), Medscape(New York, NY), Physicians Education Resource(Cranbury NJ), Research to Practice (Miami, FL), Teva (Petah Tikva, Israel), Astra Zeneca.
REFERENCES
- 1.Brysland SA, Maqbool MG, Talaulikar D, Gardiner EE. Bleeding propensity in Waldenstrom macroglobulinaemia: potential causes and evaluation. Thromb Haemost. 2022;122:1843–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castillo JJ, Advani RH, Branagan AR, et al. Consensus treatment recommendations from the tenth international workshop for Waldenstrom macroglobulinaemia. Lancet Haematol. 2020;7(11):e827–e837. [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA, Ansell SM, Kapoor P. Prognostic factors and indications for treatment of Waldenstrom’s macroglobulinemia. Best Pract Res Clin Haematol. 2016;29(2):179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trojani A, Di Camillo B, Bossi LE, et al. Identification of a candidate gene set signature for the risk of progression in IgM MGUS to smoldering/symptomatic Waldenstrom macroglobulinemia (WM) by a comparative transcriptome analysis of B cells and plasma cells. Cancers. 2021;13(8):1837. doi: 10.3390/cancers13081837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amaador K, Kersten MJ, Visser O, et al. Primary therapy and relative survival in patients with lymphoplasmacytic lymphoma/Waldenstrom macroglobulinaemia: a population-based study in The Netherlands, 1989–2018. Br J Haematol. 2022;196(3):660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tahri S, Mouhieddine TH, Redd R, et al. Clonal hematopoiesis is associated with increased risk of progression of asymptomatic Waldenström macroglobulinemia. Blood Adv. 2022;6(7):2230–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeong S, Kong SG, Kim DJ, Lee S, Lee HS. Incidence, prevalence, mortality, and causes of death in Waldenstrom macroglobulinemia: a nationwide, population-based cohort study. BMC Cancer. 2020;20(1):623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas SK. SOHO state of the art updates and next questions: Waldenstrom macroglobulinemia - 2021 update on management and future directions. Clin Lymphoma Myeloma Leuk. 2022;22(6):347–355. [DOI] [PubMed] [Google Scholar]
- 9.Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenström macroglobulinaemia: an analysis of the surveillance, epidemiology and end results database. Br J Haematol. 2015;169(1):81–89. [DOI] [PubMed] [Google Scholar]
- 10.Riva E, Duarte PJ, Valcarcel B, et al. Treatment and survival outcomes of Waldenstrom macroglobulinemia in Latin American patients: a multinational retrospective cohort study. JCO Glob Oncol. 2022;8:e2100380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menon D, Katzberg HD, Bril V. Treatment approaches for atypical CIDP. Front Neurol. 2021;12:653734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castellani F, Visentin A, Campagnolo M, et al. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol Neuroimmunol Neuroinflamm. 2020;7(4):e720. doi: 10.1212/NXI.0000000000000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visentin A, Pravato S, Castellani F, et al. From biology to treatment of monoclonal gammopathies of neurological significance. Cancers. 2022;14(6):1562. doi: 10.3390/cancers14061562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kyle RA, Anderson KC. A tribute to Jan Gosta Waldenström. Blood. 1997;89(12):4245–4247. [PubMed] [Google Scholar]
- 15.Paulus A, Ailawadhi S, Chanan-Khan A. Novel therapeutic targets in Waldenstrom macroglobulinemia. Best Pract Res Clin Haematol. 2016;29(2):216–228. [DOI] [PubMed] [Google Scholar]
- 16.Krzisch D, Guedes N, Boccon-Gibod C, et al. Cytogenetic and molecular abnormalities in Waldenström’s macroglobulinemia patients: correlations and prognostic impact. Am J Hematol. 2021;96(12):1569–1579. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez S, Celay J, Goicoechea I, et al. Preneoplastic somatic mutations including MYD88(L265P) in lymphoplasmacytic lymphoma. Sci Adv. 2022;8(3):eabl4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abeykoon JP, Paludo J, King RL, et al. MYD88 mutation status does not impact overall survival in Waldenstrom macroglobulinemia. Am J Hematol. 2018;93(2):187–194. [DOI] [PubMed] [Google Scholar]
- 19.Zanwar S, Abeykoon JP, Ansell SM, et al. Characteristics and outcome of patients with MYD88 wild-type Waldenstrom macroglobulinemia. J Clin Oncol. 2020;38(15):8550. [Google Scholar]
- 20.Demos MG, Hunter ZR, Xu L, et al. Cell-free DNA analysis for detection of MYD88(L265P) and CXCR4(S338X) mutations in Waldenstrom macroglobulinemia. Am J Hematol. 2021;96(7):E250–E253. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in Waldenstrom’s Macroglobulinema: chances and challenges. Leukemia. 2021;35(2):333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juarez-Salcedo LM, Castillo JJ. Lymphoplasmacytic lymphoma and marginal zone lymphoma. Hematol Oncol Clin North Am. 2019;33(4):639–656. [DOI] [PubMed] [Google Scholar]
- 23.Maqbool MG, Tam CS, Morison IM, et al. A practical guide to laboratory investigations at diagnosis and follow up in Waldenstrom macroglobulinaemia: recommendations from the medical and scientific advisory group, myeloma Australia, the pathology sub-committee of the lymphoma and related diseases registry and the Australasian association of clinical biochemists monoclonal gammopathy working group. Pathology. 2020;52(2):167–178. [DOI] [PubMed] [Google Scholar]
- 24.Grunenberg A, Buske C. Monoclonal IgM gammopathy and Waldenstrom’s macroglobulinemia. Dtsch Arztebl Int. 2017;114(44):745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kyle RA, Rajkumar SV, Therneau TM, Larson DR, Plevak MF, Melton LJ 3rd. Prognostic factors and predictors of outcome of immunoglobulin M monoclonal gammopathy of undetermined significance. Clin Lymphoma. 2005;5(4):257–260. [DOI] [PubMed] [Google Scholar]
- 26.Zanwar S, Abeykoon JP, Durot E, et al. Impact of MYD88(L265P) mutation status on histological transformation of Waldenstrom macroglobulinemia. Am J Hematol. 2020;95(3):274–281. [DOI] [PubMed] [Google Scholar]
- 27.Abeykoon JP, Zanwar S, Ansell SM, et al. Predictors of symptomatic hyperviscosity in Waldenstrom macroglobulinemia. Am J Hematol. 2018;93(11):1384–1393. [DOI] [PubMed] [Google Scholar]
- 28.Cesana C, Miqueleiz S, Bernuzzi P, et al. Smouldering Waldenstrom’s macroglobulinemia: factors predicting evolution to symptomatic disease. Semin Oncol. 2003;30(2):231–235. [DOI] [PubMed] [Google Scholar]
- 29.Zanwar S, Abeykoon JP, Ansell SM, et al. Disease outcomes and biomarkers of progression in smouldering Waldenstrom macroglobulinaemia. Br J Haematol. 2021;195(2):210–216. [DOI] [PubMed] [Google Scholar]
- 30.Pan Q, Cao X, Luo Y, Li J, Li F. Baseline 18 F-FDG PET/CT may portend the prognosis of patients with Waldenström macroglobulinemia/lymphoplasmacytic lymphoma after first-line treatment. Clin Nucl Med. 2022;47(11):954–960. [DOI] [PubMed] [Google Scholar]
- 31.Leleu X, Moreau AS, Weller E, et al. Serum immunoglobulin free light chain correlates with tumor burden markers in Waldenstrom macroglobulinemia. Leuk Lymphoma. 2008;49(6):1104–1107. [DOI] [PubMed] [Google Scholar]
- 32.Gertz MA, Rue M, Blood E, Kaminer LS, Vesole DH, Greipp PR. Multicenter phase 2 trial of rituximab for Waldenström macroglobulinemia (WM): an eastern cooperative oncology group study (E3A98). Leuk Lymphoma. 2004;45(10):2047–2055. [DOI] [PubMed] [Google Scholar]
- 33.Kastritis E, Morel P, Duhamel A, et al. A revised international prognostic score system for Waldenström’s macroglobulinemia. Leukemia. 2019;33(11):2654–2661. [DOI] [PubMed] [Google Scholar]
- 34.Zanwar S, Abeykoon JP, Ansell SM, et al. Disease outcomes and biomarkers of progression in smouldering Waldenstrom macroglobulinaemia. Brit J Haematol. 2021;195(2):210–216. [DOI] [PubMed] [Google Scholar]
- 35.Sarosiek S, Treon SP, Castillo JJ. Reducing treatment toxicity in Waldenstrom macroglobulinemia. Expert Opin Drug Saf. 2021;20(6):669–676. [DOI] [PubMed] [Google Scholar]
- 36.Dalal NH, Dores GM, Curtis RE, Linet MS, Morton LM. Cause-specific mortality in individuals with lymphoplasmacytic lymphoma/Waldenstrom macroglobulinaemia, 2000–2016. Br J Haematol. 2020;189(6):1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez Rogers A, Estes M. Hyperviscosity Syndrome: StatPearls. StatPearls Publishing LLC; 2022. [PubMed] [Google Scholar]
- 38.Buske C, Tedeschi A, Trotman J, et al. Ibrutinib plus rituximab versus placebo plus rituximab for Waldenstrom’s macroglobulinemia: final analysis from the randomized phase III iNNOVATE study. J Clin Oncol. 2022;40(1):52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amaador K, Kersten MJ, Minnema MC, Vos JMI. Dutch Physician’s perspectives on diagnosis and treatment of Waldenstrom’s macroglobulinemia before and after the implementation of a national guideline. Hema. 2022;6(7):e746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenstrom macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncologia. 2017;3(9):1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castillo JJ, Treon SP. Management of Waldenstrom macroglobulinemia in 2020. Hematology Am Soc Hematol Educ Program. 2020;2020(1):372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kastritis E, Gavriatopoulou M, Kyrtsonis MC, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: final analysis of a phase 2 study. Blood. 2015;126(11):1392–1394. [DOI] [PubMed] [Google Scholar]
- 43.Paludo J, Abeykoon JP, Gertz MA, et al. Dexamethasone, rituximab and cyclophosphamide (DRC) in relapsed/refractory (R/R) and treatment naïve (TN) Waldenström macroglobulinemia (WM). J Clin Oncol. 2016;34(15_suppl):7552. [Google Scholar]
- 44.Paludo J, Abeykoon JP, Kumar S, et al. Dexamethasone, rituximab and cyclophosphamide for relapsed and/or refractory and treatment-naive patients with Waldenstrom macroglobulinemia. Brit J Haematol. 2017;179(1):98–105. [DOI] [PubMed] [Google Scholar]
- 45.Pratt G, El-Sharkawi D, Kothari J, et al. Diagnosis and management of Waldenstrom macroglobulinaemia-a British Society for Haematology guideline. Br J Haematol. 2022;197(2):171–187. [DOI] [PubMed] [Google Scholar]
- 46.Kersten MJ, Amaador K, Minnema MC, et al. Combining Ixazomib with subcutaneous rituximab and dexamethasone in relapsed or refractory Waldenstrom’s macroglobulinemia: final analysis of the phase I/II HOVON124/ECWM-R2 study. J Clin Oncol. 2022;40(1):40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang YP, Yang X, Lin ZH, et al. Low-dose bortezomib and dexamethasone as primary therapy in elderly patients with Waldenstrӧm macroglobulinemia. Eur J Haematol. 2017;99(6):489–494. [DOI] [PubMed] [Google Scholar]
- 48.Leblond V, Morel P, Dilhuidy M-S, et al. A phase II Bayesian sequential clinical trial in advanced Waldenström macroglobulinemia patients treated with bortezomib: interest of addition of dexamethasone. Leukemia Lymphoma. 2017;58(11):2615–2623. [DOI] [PubMed] [Google Scholar]
- 49.Leblebjian H, Noonan K, Paba-Prada C, Treon SP, Castillo JJ, Ghobrial IM. Cyclophosphamide, bortezomib, and dexamethasone combination in waldenstrom macroglobulinemia. Am J Hematol. 2015;90(6):E122–E123. [DOI] [PubMed] [Google Scholar]
- 50.Castillo JJ, Meid K, Flynn CA, et al. Ixazomib, dexamethasone, and rituximab in treatment-naive patients with Waldenstrom macroglobulinemia: long-term follow-up. Blood Adv. 2020;4(16):3952–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benevolo G, Nicolosi M, Santambrogio E, Vitolo U. Current options to manage Waldenstrom’s macroglobulinemia. Expert Rev Hematol. 2017;10(7):637–647. [DOI] [PubMed] [Google Scholar]
- 52.Paludo J, Abeykoon JP, Shreders A, et al. Bendamustine and rituximab (BR) versus dexamethasone, rituximab, and cyclophosphamide (DRC) in patients with Waldenstrom macroglobulinemia. Ann Hematol. 2018;97(8):1417–1425. [DOI] [PubMed] [Google Scholar]
- 53.Abeykoon JP, Zanwar S, Ansell SM, et al. Outcomes with rituximab plus bendamustine (R-Benda), dexamethasone, rituximab, cyclophosphamide (DRC), and bortezomib, dexamethasone, rituximab (BDR) as primary therapy in patients with Waldenstrom macroglobulinemia (WM). J Clin Oncol. 2019;37(15):7509. [Google Scholar]
- 54.Rummel M, Lerchenmüller C, Hensel M, et al. Two years rituximab maintenance vs. observation after first line treatment with Bendamustine plus rituximab (B-R) in patients with Waldenström’s macroglobulinemia (MW): results of a prospective, randomized, multicenter phase 3 study (the StiL NHL7-2008 MAINTAIN trial). Blood. 2019;134:343. [Google Scholar]
- 55.Nanah A, Al HS. Bing-Neel syndrome: update on the diagnosis and treatment. Clin Lymphoma Myeloma Leuk. 2022;22(3):e213–e219. [DOI] [PubMed] [Google Scholar]
- 56.Saburi M, Saburi Y, Kawano K, Sato R, Urabe S, Ohtsuka E. Successful treatment with tirabrutinib for relapsed lymphoplasmacytic lymphoma complicated by Bing-Neel syndrome. Int J Hematol. 2022;115(4):585–589. [DOI] [PubMed] [Google Scholar]
- 57.Treon SP, Hanzis C, Tripsas C, et al. Bendamustine therapy in patients with relapsed or refractory Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011;11(1):133–135. [DOI] [PubMed] [Google Scholar]
- 58.Tedeschi A, Picardi P, Ferrero S, et al. Bendamustine and rituximab combination is safe and effective as salvage regimen in Waldenström macroglobulinemia. Leuk Lymphoma. 2015;56(9):2637–2642. [DOI] [PubMed] [Google Scholar]
- 59.Laribi K, Poulain S, Willems L, et al. Bendamustine plus rituximab in newly-diagnosed Waldenström macroglobulinaemia patients. A study on behalf of the French innovative Leukaemia organization (FILO). Br J Haematol. 2019;186(1):146–149. [DOI] [PubMed] [Google Scholar]
- 60.Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. [DOI] [PubMed] [Google Scholar]
- 61.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. [DOI] [PubMed] [Google Scholar]
- 62.Gustine JN, Meid K, Dubeau T, et al. Ibrutinib discontinuation in Waldenström macroglobulinemia: etiologies, outcomes, and IgM rebound. Am J Hematol. 2018;93(4):511–517. [DOI] [PubMed] [Google Scholar]
- 63.Trotman J, Buske C, Tedeschi A, et al. Single-agent ibrutinib for rituximab-refractory Waldenstrom macroglobulinemia: final analysis of the substudy of the phase III innovate(TM) trial. Clin Cancer Res. 2021;27(21):5793–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castillo JJ, Meid K, Gustine JN, et al. Long-term follow-up of ibrutinib monotherapy in treatment-naive patients with Waldenstrom macroglobulinemia. Leukemia. 2022;36(2):532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vitolo U, Novo M, Santambrogio E. Partial responses to ibrutinib in Waldenstrom macroglobulinaemia - good enough? Br J Haematol. 2021;192(3):423–424. [DOI] [PubMed] [Google Scholar]
- 66.Abeykoon JP, Zanwar S, Ansell SM, et al. Ibrutinib monotherapy outside of clinical trial setting in Waldenstrom macroglobulinaemia: practice patterns, toxicities and outcomes. Br J Haematol. 2020;188(3):394–403. [DOI] [PubMed] [Google Scholar]
- 67.Castillo JJ, Gustine JN, Meid K, et al. Impact of ibrutinib dose intensity on patient outcomes in previously treated Waldenström macroglobulinemia. Haematologica. 2018;103(10):e466–e468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gustine JN, Sarosiek S, Flynn CA, et al. Natural history of Waldenstrom macroglobulinemia following acquired resistance to ibrutinib monotherapy. Haematologica. 2022;107(5):1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenstrom macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112–e121. [DOI] [PubMed] [Google Scholar]
- 70.Buske C Bruton tyrosine-kinase inhibitor on the rise: acalabrutinib in Waldenstrom macroglobulinemia. Lancet Haematol. 2020;7(2):e85–e86. [DOI] [PubMed] [Google Scholar]
- 71.Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sekiguchi N, Rai S, Munakata W, et al. Two-year outcomes of tirabrutinib monotherapy in Waldenstrom’s macroglobulinemia. Cancer Sci. 2022;113(6):2085–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abeykoon JP, Kumar S, Castillo JJ, et al. Bendamustine rituximab (BR) versus ibrutinib (Ibr) as primary therapy for Waldenström macroglobulinemia (WM): an international collaborative study. J Clin Oncol. 2022;40(16_suppl):7566. [Google Scholar]
- 74.Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in previously treated Waldenstrom macroglobulinemia. J Clin Oncol. 2022;40(1):63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palomba ML, Qualls D, Monette S, et al. CD19-directed chimeric antigen receptor T cell therapy in Waldenstrom macroglobulinemia: a preclinical model and initial clinical experience. J Immunother Cancer. 2022;10(2):e004128. doi: 10.1136/jitc-2021-004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Labreuche J, Assouan D, Durot E, et al. Does early disease progression predict survival after first line-treatment of Waldenström macroglobulinemia? Hematol Oncol. 2022;40(3):400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abushukair H, Syaj S, Ababneh O, et al. First- versus second-generation Bruton tyrosine kinase inhibitors in Waldenstrom’s macroglobulinemia: a systematic review and meta-analysis. Am J Hematol. 2022;97(7):942–950. [DOI] [PubMed] [Google Scholar]