

Abstract
Background:
Fat mass and obesity-associated protein (FTO) demethylates N6-methyladenosine (m6A), which is a critical epitranscriptomic regulator of neuronal function. We previously reported that ischemic stroke induces m6A hypermethylation with a simultaneous decrease in FTO expression in neurons. Currently, we evaluated the functional significance of restoring FTO with an adeno-associated virus 9 (AAV9), and thus reducing m6A methylation in post-stroke brain damage.
Methods:
Adult male and female C57BL/6J mice were injected with FTO AAV9 (intracerebral) at 21 days prior to inducing transient middle cerebral artery occlusion. Post-stroke brain damage (infarction, atrophy and white matter integrity) and neurobehavioral deficits (motor function, cognition, depression and anxiety-like behaviors) were evaluated between days 1 and 28 of reperfusion.
Results:
FTO overexpression significantly decreased the post-stroke m6A hypermethylation. More importantly, exogenous FTO substantially decreased post-stroke grey and white matter damage and improved motor function recovery, cognition and depression-like behavior in both sexes.
Conclusions:
These results demonstrate that FTO-dependent m6A demethylation minimizes long-term sequelae of stroke independent of sex.
Keywords: Neuroprotection, Epitranscriptomics, m6A demethylation, Acute brain injury
Graphical Abstract
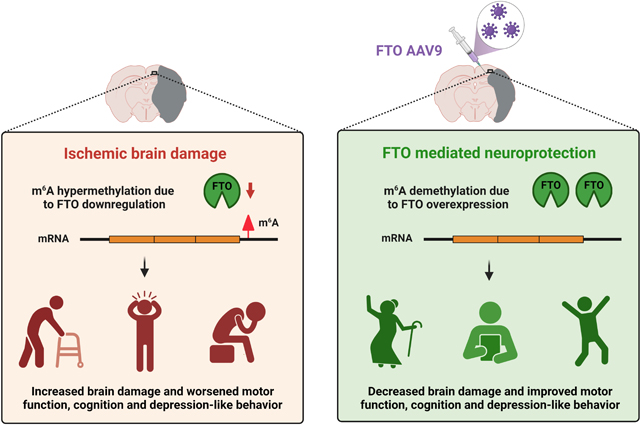
INTRODUCTION
Epitranscriptomic modifications rapidly fine-tune gene expression after an acute CNS injury.1 Particularly, N6-methyladenosine (m6A) is at least twice more abundant in the brain than in other vital organs and is highly sensitive to various types of injuries, including stroke, traumatic brain injury, spinal cord injury and peripheral nerve injury.2 Dynamic alterations in the activity of m6A methylase complex or demethylases were thought to shape post-injury m6A epitranscriptome.2 We and others have previously reported that experimental stroke in rodents triggers m6A hypermethylation and a concomitant downregulation of m6A demethylase fat mass and obesity-associated protein (FTO) in the brain.3,4
Among the two mammalian m6A demethylases, FTO is brain-enriched, specifically in neurons.5,6 FTO is thought to demethylate ~1,500 neuronal transcripts in the healthy brain.7 Notable mRNA substrates of FTO include Bdnf, Grin1, Myc, Jun and Olig2.8 FTO-dependent m6A demethylation regulates a plethora of developmental and physiological processes like neurogenesis, gliogenesis, axonal growth, synaptic plasticity, and stress response.8 Thus, FTO deficiency causes severe brain deficits like microcephaly, postnatal growth retardation, aberrant dopaminergic neurotransmission, cognitive dysfunction and anxiety-associated behavior in mice and humans.8 In the neurons, FTO is also detected in the extra-somatic regions such as axons and dendrites, where it mediates dynamic erasure of m6A.9 For example, FTO localized in axons during neuronal development demethylated the axon elongation factor Gap43 mRNA leading to its translation.10 Overall, the pleiotropic activity of FTO is crucial for normal brain function.
FTO requires alpha-ketoglutarate, non-heme iron and oxygen as co-factors to catalyze m6A demethylation.5 Given that FTO is an oxygen-dependent enzyme, its demethylase activity is severely compromised after ischemic injury to various organs, including the brain, heart, liver and kidney.11 Mounting evidence suggests that FTO minimizes post-ischemic cell death. 4, 12 Notably, FTO overexpression prevented apoptosis in the primary neurons subjected to oxygen-glucose deprivation.4 We presently investigated the role of the FTO/m6A axis in modulating the post-stroke grey and white matter damage, motor function, cognitive function, anxiety- and depression-like behavior.
METHODS
Data availability:
The authors declare that all supporting data are available in the article and online-only Supplementary material.
Focal Ischemia:
Animal procedures were approved by the Research Animal Resources and Care Committee of the University of Wisconsin-Madison. Mice were cared for in accordance with the Guide for the Care and Use of Laboratory Animals [U.S. Department of Health and Human Services Publication Number. 86–23 (revised)]. Surgical procedures were conducted in compliance with the “Animal Research: Reporting of In Vivo Experiments” guidelines.13 Focal ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO) for 1 h using a 6–0 silicon-coated monofilament (Doccol Corporation) in adult C57BL/6J male and female mice (12 weeks, The Jackson Laboratory) under isoflurane anesthesia as described earlier.14 The regional cerebral blood flow and physiological parameters were monitored (Supplementary Table 1), and rectal temperature was maintained at 37°C during the surgery. Mice were randomly assigned to study groups using GraphPad random number generator tool, and the neurological deficits were assessed by modified neurological severity scoring with the following criteria: 0-no neurological deficit, 1-failure to fully extend right forepaw, 2-turning to the right, 3-circling to the right, 4-unable to spontaneously walk, and 5-death from a stroke. The mice that showed no signs of neurological deficits during the acute phase after MCAO (8 males and 6 females) or showed subarachnoid (1 male) or intracerebral hemorrhage (2 males and 1 female) during postmortem analyses were excluded from the study. The outcome parameters were evaluated by a person blinded to the study groups. Sham-operated mice underwent a similar surgical procedure except for MCAO. Cohorts of mice were euthanized at 1 day, 3 days or 28 days after MCAO as needed.
FTO overexpression:
A recombinant AAV9 (Vector Biolabs) carrying a full-length FTO coding sequence or a control (null) AAV9 was injected intracerebrally. Briefly, mice were anesthetized and a burr hole was drilled in the pericranium (2.5 mm lateral to the sagittal suture and 0.25 mm posterior to the coronal suture) and 2 μl of AAV9 (viral load of ~1011 genome copies) was injected into the cortex (0.2 μl/min) using a Hamilton syringe positioned 1.5 mm below the cranium at an angle of 370 with sagittal plane. The needle was withdrawn after 10 min, and the wound was closed.
Dot blot:
Total RNA was extracted from the peri-infarct cortex using mirVana RNA Isolation Kit (Thermo Scientific) and purified with Dynabeads mRNA Purification Kit (Thermo Scientific). Bulk m6A abundance in the mRNA was measured by dot blot analysis as described previously.3 Briefly, 40 ng of denatured mRNA was spotted onto Hybond N+ membrane (GE Healthcare), UV cross-linked, blocked with SuperBlock Blocking Buffer (Thermo Scientific) and probed with rabbit anti-m6A antibody (1:250; Synaptic Systems) followed by horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:3,000; Cell Signaling Technology). Blots were visualized with enhanced chemiluminescence (ECL) and quantified using Image Studio software (LI-COR Biotechnology).
Real-time PCR:
One μg of total RNA was reverse transcribed to cDNA with High Capacity RNA to cDNA Kit (Applied Biosystems). The mRNA levels of FTO, METTL3, METTL14, WTAP and ALKBH5 were estimated by real-time PCR with the SYBR-green chemistry using gene-specific primers (Supplementary Table 2). 18S rRNA was used as housekeeping control, and relative gene expression was calculated by the comparative Ct method (2-ΔΔCt). Real-time PCR assays were conducted in triplicates.
Western blot:
Protein samples (15 μg) were electrophoresed, transferred to a nitrocellulose membrane, blocked and probed with antibodies against FTO (1:1,000; PhosphoSolutions) followed by HRP-conjugated anti-mouse IgG (1:3,000; Cell Signaling Technology). Blots were stripped and reprobed with an antibody against GAPDH (1:1,000, Santa Cruz Biotechnology) followed by HRP-conjugated anti-mouse IgG. Blots were visualized with ECL and quantified using Image Studio software (LI-COR Biotechnology).
Immunostaining:
Mice were euthanized by transcardiac 4% paraformaldehyde perfusion, brains were post-fixed, cryoprotected and sectioned (coronal, 30 μm). Brain sections (+0.5 mm from bregma) were immunostained with antibodies against FTO (1:100; Abcam), GFAP (1:200; EMD Millipore) and TMEM119 (1:400; Synaptic Systems) followed by donkey Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies (1:300; Thermo Scientific) as described earlier.3
Lesion, atrophy volume and white matter damage estimation:
On day 7 of reperfusion following transient MCAO, mice were anesthetized with isoflurane and subjected to T2-weighted MRI (4.7-T small animal system with 205/120/HD/S gradient 210 mm bore varian magnet; Agilent Technologies, USA). Respiration rate was monitored during the imaging. 8–10 equidistant coronal slices/mice were acquired with a slice thickness of 1.0 mm. MRI scans were analyzed using NIH ImageJ software with an FDF plugin by a person blinded to the study groups. On day 28, mice were euthanized by transcardiac 4% paraformaldehyde perfusion. Six serial sections (coronal, 40 μm between +2.28 to −5.64 mm from bregma) from each brain were stained with 0.1% cresyl violet, scanned and analyzed using the NIH ImageJ software. The brain atrophy volume was calculated as the difference in the volume of the contralateral and ipsilateral hemispheres. For white matter damage evaluation, sections near the coordinate +0.5 mm from bregma were stained with 0.1% luxol fast blue (56°C for 12 h) followed by counterstaining with 0.1% cresyl violet (room temperature for 5 min). Stained sections were scanned, and corpus callosum thickness was computed by NIH ImageJ software.
Neurobehavioral assessment:
Post-stroke motor function was evaluated on days 1, 3, 5 and 7 of reperfusion by rotarod test (cylinder rotating at 8 rpm for 4 min) and beam walk test (foot faults while crossing 120 cm long beam) as described earlier.14 Mice were trained for 3 days before testing. Mice were subjected to Morris water maze test (eight blocks of 4 sequential trials/block of training over 4 days followed by a probe trial) to assess spatial learning and memory. For each trial, a mouse was placed in a circular pool surrounded by visual cues at a different start location and was allowed to swim until it located the hidden platform or reached the end of the 60 sec trial. After the completion of eight blocks of training, mice were placed in the pool for a probe trial without the platform, and the swimming behavior was tracked for 60 sec. The two measures of cognitive function, escape latency (duration to reach the platform) for the training blocks and the time spent in the platform quadrant during the probe trial, were retrieved from ANY-maze software. Post-stroke depression-like behavior was assessed by the tail suspension test, in which a mouse was suspended by tail and movements were recorded for 6 min. The duration of immobility was used as a measure of depression-like behavior. Post-stroke anxiety-like behavior was assessed by an open field test in which the mice were placed in a closed arena, and the spontaneous locomotor activity was tracked for 30 mins using Fusion 6.5 SuperFlex system. The time spent in the center zone was measured as an indicator of the less anxious behavior.
Statistics:
Statistical significance between two groups was estimated by the Mann-Whitney U test. For analyzing data that was collected repeatedly from the same set of mice at different time points, a nonparametric two-way repeated-measures ANOVA (Sidak post hoc correction) was used. All these analyses were performed in the GraphPad Prism 9 software.
RESULTS
FTO modulated post-stroke m6A abundance
We have previously shown a significant downregulation of FTO expression and concomitant increase in m6A levels after transient MCAO in mice.3 To determine if FTO directly modulates post-ischemic m6A levels, we overexpressed FTO in adult mouse brain by injecting an FTO AAV9, which showed widespread distribution in the ipsilateral cortex, striatum and hippocampus by 21 days (Fig. 1A). FTO AAV9 treatment significantly increased the FTO mRNA (by ~30-fold) (Fig. 1B) and protein (by ~4.7-fold) levels (Fig. 1C and 1D) in the peri-infarct cortex at 1 day of reperfusion following transient MCAO compared to sham, reversing the post-ischemic loss of FTO seen in the control AAV9 treated group. Importantly, FTO AAV9 treatment partially reduced the post-ischemic m6A hypermethylation (by ~32%) relative to the control AAV9 group (Fig. 1E). As expected, both FTO AAV9 and control AAV9 treatment did not alter the mRNA expression of the m6A methylase complex subunits METTL3, METTL14 and WTAP (Supplementary Fig. 1A to 1C) and m6A demethylase ALKBH5 (Supplementary Fig. 1D) in the peri-infarct cortex at 1 day of reperfusion following transient MCAO compared to sham. This indicates that post-stroke m6A hypermethylation is FTO-dependent.
Fig. 1: FTO overexpression reversed post-stroke m6A hypermethylation.
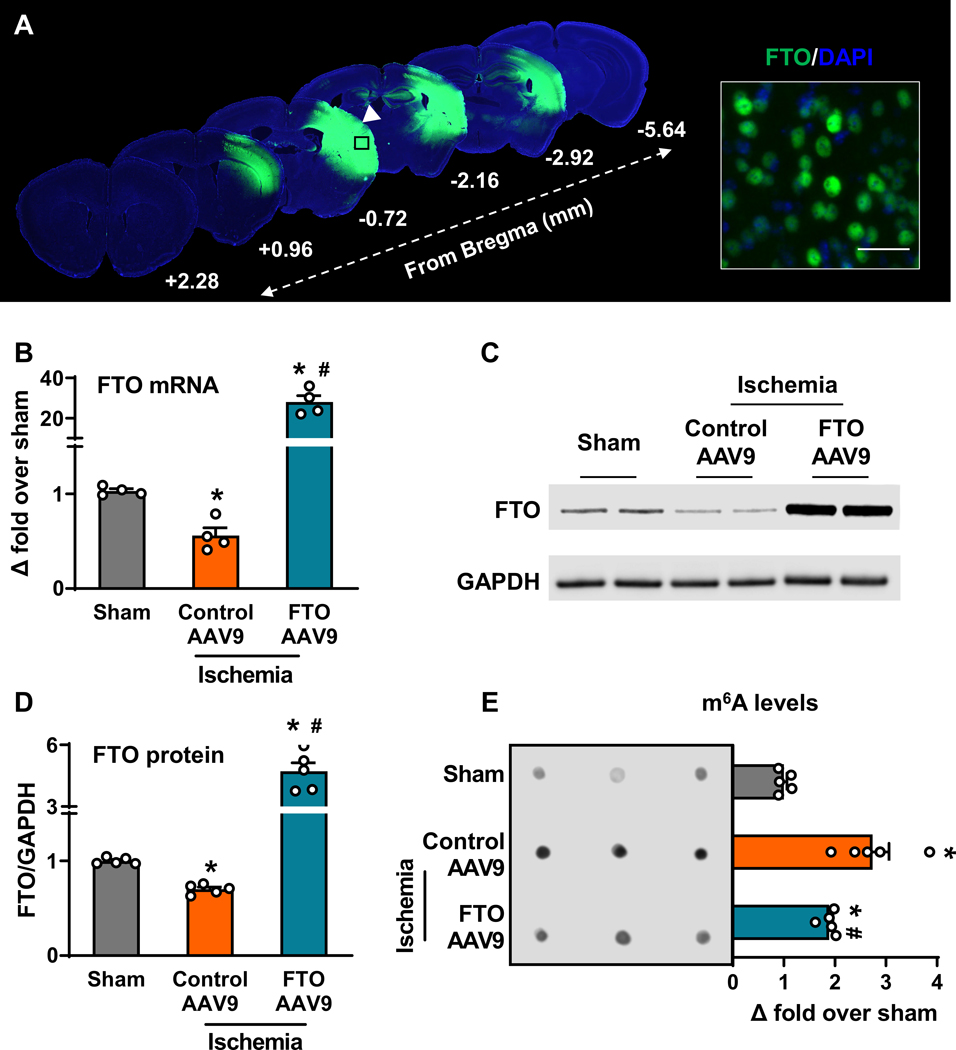
Immunostaining of the transgenic FTO AAV9 in mouse brain 21 days after intracerebral injection (n=4/group) (A). Injection site is indicated by a white arrowhead and the magnified image shows the cellular localization of FTO. DAPI, 4’,6-diamidino-2-phenylindole. Scale bar, 30 μm. Quantification of FTO mRNA (B), FTO protein (C and D) and m6A levels (E) in the cortical peri-infarct region of control AAV9 and FTO AAV9 treated mice at 1 day of reperfusion following transient MCAO compared to sham. Data are mean±SEM (n=4–5/group). *p<0.05 compared with sham and #p<0.05 compared with control AAV9 ischemia group, by Mann-Whitney U test.
Exogenous FTO decreased post-ischemic secondary brain damage in both sexes
To determine whether exogenous expression of FTO modulates post-stroke functional recovery, we injected a cohort of adult male and female mice with either FTO AAV9 or control AAV9 (intracerebral), and after 21 days, they were subjected to 1 h transient MCAO (Fig. 2A). Mice were then subjected to a battery of neurobehavioral tests between day 1 and 28 of reperfusion to assess motor function, cognition, anxiety and depression, MRI on day 7 to estimate infarct volume (Fig. 2A). Mice were euthanized on day 28 of reperfusion and histopathological analysis was performed to estimate grey and white matter damage (Fig. 2A). FTO AAV9 treated mice showed significantly smaller infarcts (by ~55% in males and by ~29% in females) at 7 days of reperfusion compared with the sex-matched control AAV9 treated mice (Fig. 2B). Brain atrophy was also significantly decreased in the FTO AAV9 treated male (by ~41%) and female mice (by ~44%) at 28 days of reperfusion compared with the sex-matched control AAV9 treated mice (Fig. 2C). FTO AAV9 treated male, but not female, mice showed significant protection of white matter integrity estimated as corpus callosum thickness (by ~39%), at 28 days of reperfusion compared with the sex-matched control AAV9 treated mice (Fig. 2D). The in vivo laser speckle imaging revealed that the cerebroprotection offered by FTO AAV9 treatment was not due to changes in the regional cerebral blood flow either before or during MCAO, or during reperfusion (Supplementary Fig. 2A and 2B). FTO AAV9 treatment had no gross effect on post-stroke astrocyte and microglial activation at 3 days of reperfusion compared with the control AAV9 treated group (Supplementary Fig. 3).
Fig. 2: FTO overexpression curtailed post-stroke brain damage in mice of both sexes.
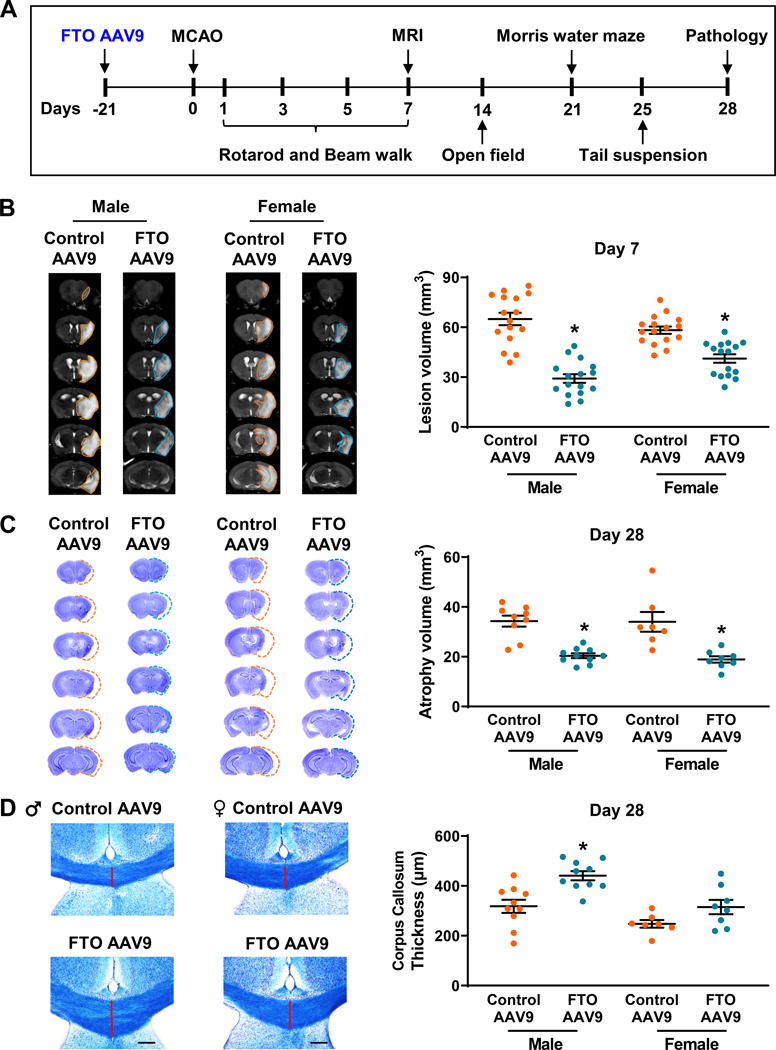
Experimental paradigm for testing the role of FTO overexpression on long-term functional recovery after stroke (A). Representative MRI scans and infarct volume quantification of control AAV9 and FTO AAV9 treated male and female mice (B). Infarct volume was measured at 7 days of reperfusion after 1 h transient MCAO. Data are mean±SEM (n=16/group). *p<0.05 compared with the control AAV9 group by Mann-Whitney U test. Representative cresyl violet-stained coronal brain sections (between +2.28 to −5.64 mm from bregma) and atrophy volume quantification of control AAV9 and FTO AAV9 treated male and female mice (C). Atrophy was estimated at 28 days of reperfusion after 1 h transient MCAO. Data are mean±SEM (n=9–10/group for males and n=7–8/group for females). *p<0.05 compared with the control AAV9 group by Mann-Whitney U test. Representative luxol fast blue-stained images (+0.5 mm from bregma) and white-matter damage quantification of control AAV9 and FTO AAV9 treated male and female mice (D). Corpus callosum thickness (medial) was measured at 28 days of reperfusion after 1 h transient MCAO to identify white-matter integrity. Data are mean±SEM (n=10/group for males and n=7–8/group for females). *p<0.05 compared with the control AAV9 group by Mann-Whitney U test.
Exogenous FTO accelerated functional recovery after stroke in both sexes
Post-stroke motor function recovery assessed by rotarod (Fig. 3A and 3B) and beam walk (Fig. 3C and 3D) tests was significantly improved in the FTO AAV9 treated male and female mice between days 3 to 7 of reperfusion compared with sex-matched control AAV9 treated mice. There was no significant difference in the anxiety-like behavior between FTO AAV9 or control AAV9 treated mice of both sexes assessed at 14 days of reperfusion by open field test (Supplementary Fig. 4A and 4B). However, FTO AAV9 treatment significantly reduced post-stroke depression-like behavior estimated at 25 days of reperfusion by tail suspension test in both male (by ~31%) and female mice (by ~37%) compared with sex-matched control AAV9 treated mice (Fig. 3E and 3F). Both male and female mice treated with FTO AAV9 showed improved memory retention compared with sex-matched control AAV9 treated mice (Fig. 4A and 4B). Specifically, FTO AAV9 treated mice of both sexes located the platform faster during the training trials (Fig. 4C and 4D) and stayed in the platform quadrant significantly longer during the probe trial in the Morris water maze test, compared with sex-matched control AAV9 treated mice (Fig. 4E and 4F).
Fig. 3: FTO overexpression ameliorated post-stroke motor dysfunction and depression-like behavior in mice of both sexes.
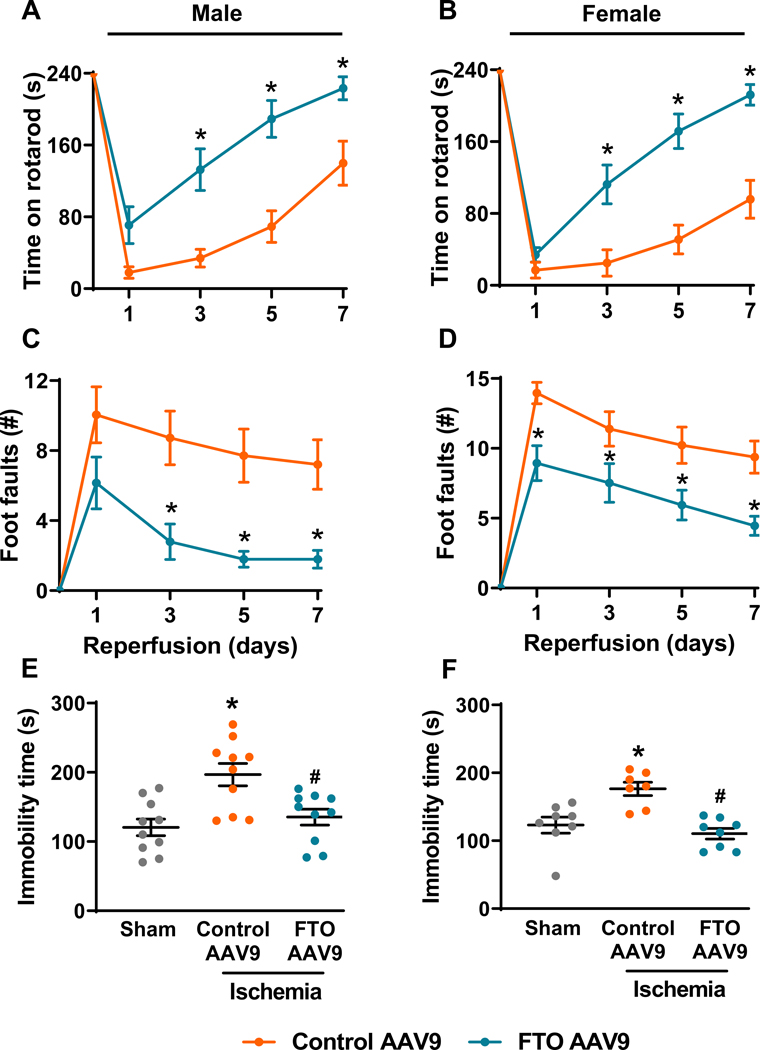
Motor function was assessed by rotarod (A and B) and beam walk (C and D) tests in control AAV9 and FTO AAV9 treated mice at 1 day before occlusion and on days 1, 3, 5, and 7 of reperfusion after 1 h transient MCAO. Data are mean±SEM (n=16/group). *p<0.05 compared with the control AAV9 treated group by two-way repeated-measures ANOVA (Sidak post hoc correction). Depression-like behavioral assessment in sham, control AAV9 and FTO AAV9 treated male (E) and female mice (F) by tail-suspension test at 25 days of reperfusion after 1 h transient MCAO. Data are mean±SEM (n=10/group for males and n=7–8/group for females). *p<0.05 compared with sham and #p<0.05 compared with control AAV9 ischemia group, by Mann-Whitney U test.
Fig. 4: FTO overexpression improved post-stroke cognitive function in mice of both sexes.
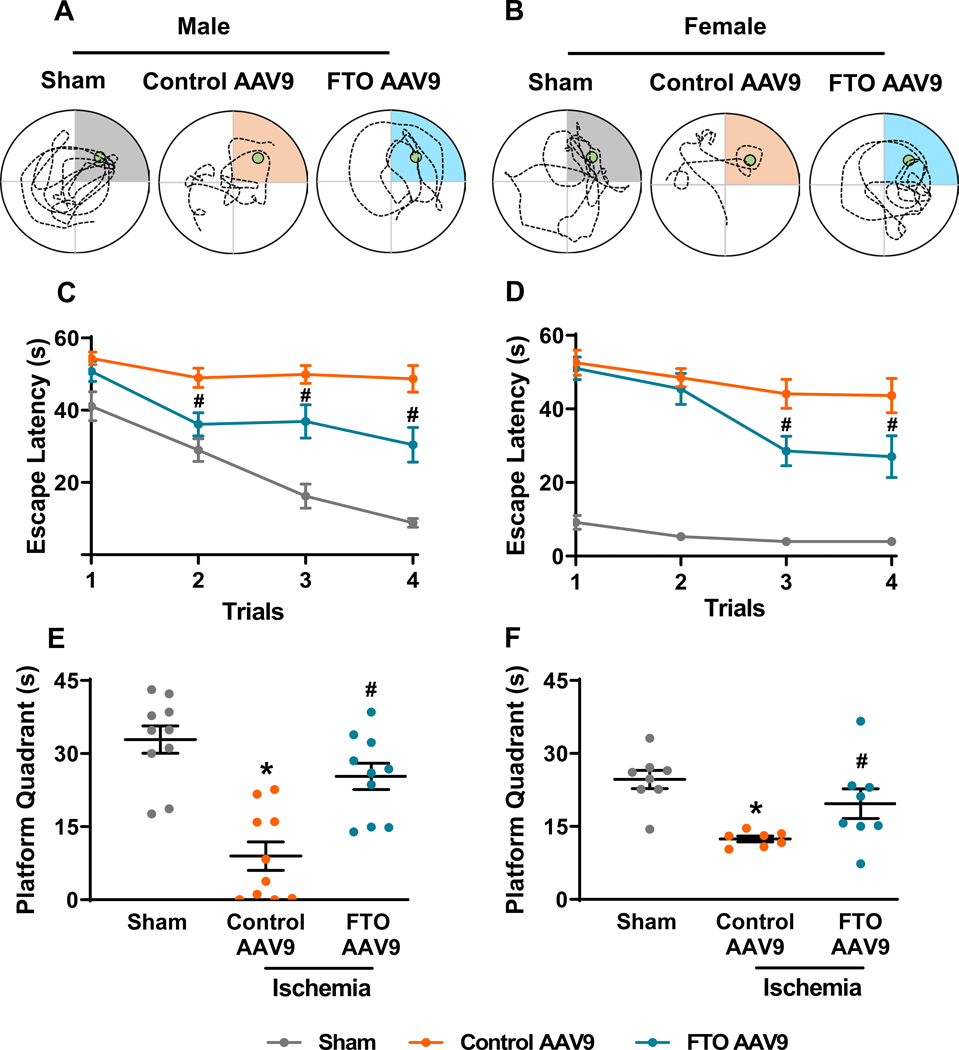
Representative swimming tracks of the sham, control AAV9 and FTO AAV9 treated male (A) and female mice (B) during the probe trial of the Morris water maze test. Learning curves based on the escape latency for the sham, control AAV9 and FTO AAV9 treated male (C) and female mice (D) during the training phase of the Morris water maze test. Data are mean±SEM (n=10/group for males and n=7–8/group for females). #p<0.05 compared with the control AAV9 treated group by two-way repeated-measures ANOVA (Sidak post hoc correction). Time spent in the platform quadrant by the sham, control AAV9 and FTO AAV9 treated male (E) and female mice (F) during the probe trial phase of the Morris water maze test. Data are mean±SEM (n=10/group for males and n=7–8/group for females). *p<0.05 compared with sham and #p<0.05 compared with control AAV9 ischemia group, by Mann-Whitney U test.
DISCUSSION
Reversible chemical tuning of mRNAs by m6A methylation facilitates neurons to respond rapidly to acute stress.8 We and others previously demonstrated an inverse correlation between the abundance of m6A and the expression/activity of its demethylase FTO during the acute phase of ischemic stroke.3,15,4 However, if these alterations serve as disease drivers or merely disease manifestations is not clear. The present study is the first to show that FTO-dependent m6A methylation contributes to post-stroke secondary brain damage. This indicates FTO as a potential therapeutic target for ischemic stroke.
Despite robust overexpression of FTO near the MCA territory, only a partial reversal of post-stroke m6A abundance was observed in FTO AAV9 treated mice. Previous studies also reported a limited demethylation efficiency of exogenous FTO in mammalian cells in vitro.12,16,17 This raises the possibility that FTO might require additional interacting partners to assist in its function. Along those lines, a recent study showed that the RNA binding protein, splicing factor proline- and glutamine-rich (SFPQ), interacts and recruits FTO to promote m6A demethylation.18 However, the role of SFPQ in modulating the FTO-dependent post-stroke m6A methylation is not known at this time.
FTO overexpression alleviated post-stroke secondary brain damage, enhanced motor and cognitive functional recovery and decreased depression-like behavior in both sexes. These findings unequivocally suggest that post-stroke loss of FTO is detrimental. Interestingly, FTO overexpression did not alter post-stroke anxiety in either sex. Consistently, others also showed that FTO does not affect anxiety in mice after acute or chronic stress.19,20 Emerging evidence indicates the protective role of FTO in various neuropathological conditions.20, 21, 22 For example, FTO inhibition exacerbated the neurologic dysfunction after traumatic brain injury in rats21 and FTO activation conferred resilience to depression-like behavior in mice.20, 22 Furthermore, loss of FTO triggered cognitive dysfunction and depression-like behavior in mice.23,24,25 These impairments were attributed to the degradation of key transcripts involved in synaptic plasticity (Kcnj6, Grin1 and Drd3) and neurogenesis (Bdnf, Pi3k and Akt1) due to a lack of m6A erasure by FTO.7,23 Therefore, it is plausible that FTO-mediated long-term functional recovery after stroke might be secondary to enhanced neural plasticity and neurogenesis apart from reduced infarction. Future studies are needed to evaluate this possibility.
Sex differences due to hormones and chromosomes significantly impact post-stroke functional outcomes.26 Several crucial post-stroke pathological processes, such as excitotoxicity, oxidative stress, mitochondrial dysfunction, autophagy, apoptosis and inflammation, exhibit sexual dimorphism.3, 27, 28 A recent study showed that m6A methylase differentially modulates the fate of lipogenic mRNAs in the liver, thereby affecting the fasting triglyceride levels in a sex-specific manner.29 To date, studies investigating sex-specific differences in m6A methylation during brain development and injuries are lacking. The present study showed that FTO is cerebroprotective after stroke in both sexes. Nonetheless, we observed robust protection of the white matter in the FTO AAV9-treated male but not female mice after stroke. Previous studies also showed the prevalence of sex-specific differences in white matter architecture during neurodevelopmental and other neurological conditions.30,31,32,33 Further analysis using more sensitive techniques such as diffusion tensor imaging is necessary to understand these differences after stroke.34 Additionally, profiling studies are necessary to uncover the sex-biased m6A targets involved in white matter injury after stroke.
Over the past few decades, hundreds of neuroprotective agents have been tested and proven effective against ischemic stroke in preclinical animal models.35 However, none of the candidate neuroprotectants has entered the clinic. This translational failure is thought to be due to their nature of action against a single mechanism.36 Given the clinical heterogeneity, acute onset and complex sequelae, it is suggested to test pleiotropic agents that target multiple mechanisms of the ischemic cascade.36 Epitranscriptomic enzymes like FTO possess immense pleiotropic potential as they can modulate the expression of numerous transcripts in m6A dependent manner. Overall, we showed that reversing post-stroke m6A hypermethylation by exogenous FTO reduces secondary brain damage and rescues mice of both sexes from behavioral deficits, including motor dysfunction, cognitive dysfunction and depression-like phenotype. Pre-stroke treatment with AAV9 and a lack of a mechanism to turn off exogenous FTO limit our findings. The development of specific small molecule activators of FTO is essential to overcome these challenges. This also awaits further validation of the cerebroprotective role of FTO in aged mice and comorbid mice (diabetic and hypertensive) using FTO activators.
Supplementary Material
Acknowledgments:
Dr. Chokkalla and Dr. Vemuganti contributed to the conception and design of the study. Dr. Chokkalla, Ms. Jeong, Dr. Mehta, Dr. Davis, Dr. Morris-Blanco, Mr. Bathula and Ms. Qureshi contributed to the data acquisition and analysis. Dr. Chokkalla and Dr. Vemganti drafted and edited the article. Graphical abstract is created using Biorender.
Funding:
These studies were supported in part by the Department of Neurological Surgery, University of Wisconsin, National Institute of Health and American Heart Association (20PRE3512023). Dr. Vemuganti is the recipient of a Research Career Scientist Award (IK6BX005690) from the US Department of Veterans Affairs.
Non-standard Abbreviations and Acronyms:
- AAV9
adeno-associated virus 9
- FTO
fat mass and obesity-associated protein
- m6A
N6-methyladenosine
- MCAO
middle cerebral artery occlusion
Footnotes
Disclosures: None
REFERENCES
- 1.Chokkalla AK, Mehta SL, Vemuganti R. Epitranscriptomic modifications modulate normal and pathological functions in cns. Transl Stroke Res. 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, Liang Y, Luo X, Liu Y, Zhang X, Gao L. N6-methyladenosine rna modification: A promising regulator in central nervous system injury. Exp Neurol. 2021;345:113829. [DOI] [PubMed] [Google Scholar]
- 3.Kim T, Chelluboina B, Chokkalla AK, Vemuganti R. Age and sex differences in the pathophysiology of acute cns injury. Neurochem Int. 2019;127:22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu K, Mo Y, Li D, Yu Q, Wang L, Lin F, Kong C, Balelang MF, Zhang A, Chen S, et al. N(6)-methyladenosine demethylases alkbh5/fto regulate cerebral ischemia-reperfusion injury. Ther Adv Chronic Dis. 2020;11:2040622320916024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, et al. The obesity-associated fto gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fredriksson R, Hägglund M, Olszewski PK, Stephansson O, Jacobsson JA, Olszewska AM, Levine AS, Lindblom J, Schiöth HB. The obesity gene, fto, is of ancient origin, up-regulated during food deprivation and expressed in neurons of feeding-related nuclei of the brain. Endocrinology. 2008;149:2062–2071 [DOI] [PubMed] [Google Scholar]
- 7.Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Brönneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, et al. The fat mass and obesity associated gene (fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–1048 [DOI] [PubMed] [Google Scholar]
- 8.Chokkalla AK, Mehta SL, Vemuganti R. Epitranscriptomic regulation by m(6)a rna methylation in brain development and diseases. J Cereb Blood Flow Metab. 2020;40:2331–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merkurjev D, Hong WT, Iida K, Oomoto I, Goldie BJ, Yamaguti H, Ohara T, Kawaguchi SY, Hirano T, Martin KC, et al. Synaptic n(6)-methyladenosine (m(6)a) epitranscriptome reveals functional partitioning of localized transcripts. Nat Neurosci. 2018;21:1004–1014 [DOI] [PubMed] [Google Scholar]
- 10.Yu J, Chen M, Huang H, Zhu J, Song H, Zhu J, Park J, Ji SJ. Dynamic m6a modification regulates local translation of mrna in axons. Nucleic Acids Res. 2018;46:1412–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao W, Han X, Ge M, Chen C, Xiao X, Li H, Hei Z. N(6)-methyladenosine (m(6)a) methylation in ischemia-reperfusion injury. Cell Death Dis. 2020;11:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen W, Li H, Su H, Chen K, Yan J. Fto overexpression inhibits apoptosis of hypoxia/reoxygenation-treated myocardial cells by regulating m6a modification of mhrt. Mol Cell Biochem. 2021;476:2171–2179 [DOI] [PubMed] [Google Scholar]
- 13.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The arrive guidelines 2.0: Updated guidelines for reporting animal research. J Cereb Blood Flow Metab. 2020;40:1769–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez MS, Vemuganti R. Modeling transient focal ischemic stroke in rodents by intraluminal filament method of middle cerebral artery occlusion. Methods Mol Biol. 2018;1717:101–113 [DOI] [PubMed] [Google Scholar]
- 15.Yi D, Wang Q, Zhao Y, Song Y, You H, Wang J, Liu R, Shi Z, Chen X, Luo Q. Alteration of n (6) -methyladenosine mrna methylation in a rat model of cerebral ischemia-reperfusion injury. Front Neurosci. 2021;15:605654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear rna is a major substrate of the obesity-associated fto. Nat Chem Biol. 2011;7:885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen Z, Liu P, Sun Q, Li Y, Acharya R, Li X, Sun C. Fto inhibits upr(mt)-induced apoptosis by activating jak2/stat3 pathway and reducing m6a level in adipocytes. Apoptosis. 2021;26:474–487 [DOI] [PubMed] [Google Scholar]
- 18.Song H, Wang Y, Wang R, Zhang X, Liu Y, Jia G, Chen PR. Sfpq is an fto-binding protein that facilitates the demethylation substrate preference. Cell Chem Biol. 2020;27:283–291.e286 [DOI] [PubMed] [Google Scholar]
- 19.Engel M, Eggert C, Kaplick PM, Eder M, Röh S, Tietze L, Namendorf C, Arloth J, Weber P, Rex-Haffner M, et al. The role of m(6)a/m-rna methylation in stress response regulation. Neuron. 2018;99:389–403.e389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Xiu J, Zhu C, Meng K, Li C, Han R, Du T, Li L, Xu L, Liu R, et al. Fat mass and obesity-associated protein regulates rna methylation associated with depression-like behavior in mice. Nat Commun. 2021;12:6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu J, Zhang Y, Ma H, Zeng R, Liu R, Wang P, Jin X, Zhao Y. Epitranscriptomic profiling of n6-methyladenosine-related rna methylation in rat cerebral cortex following traumatic brain injury. Mol Brain. 2020;13:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen J, Yang L, Wei W. Role of fto on camkii/creb signaling pathway of hippocampus in depressive-like behaviors induced by chronic restraint stress mice. Behav Brain Res. 2021;406:113227 [DOI] [PubMed] [Google Scholar]
- 23.Li L, Zang L, Zhang F, Chen J, Shen H, Shu L, Liang F, Feng C, Chen D, Tao H, et al. Fat mass and obesity-associated (fto) protein regulates adult neurogenesis. Hum Mol Genet. 2017;26:2398–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spychala A, Rüther U. Fto affects hippocampal function by regulation of bdnf processing. PLoS One. 2019;14:e0211937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu PF, Han QQ, Chen FF, Shen TT, Li YH, Cao Y, Chen JG, Wang F. Erasing m(6)a-dependent transcription signature of stress-sensitive genes triggers antidepressant actions. Neurobiol Stress. 2021;15:100390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sohrabji F, Park MJ, Mahnke AH. Sex differences in stroke therapies. J Neurosci Res. 2017;95:681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee A, McCullough LD. Sex-specific immune responses in stroke. Stroke. 2022;53:1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demarest TG, Schuh RA, Waite EL, Waddell J, McKenna MC, Fiskum G. Sex dependent alterations in mitochondrial electron transport chain proteins following neonatal rat cerebral hypoxic-ischemia. J Bioenerg Biomembr. 2016;48:591–598 [DOI] [PubMed] [Google Scholar]
- 29.Salisbury DA, Casero D, Zhang Z, Wang D, Kim J, Wu X, Vergnes L, Mirza AH, Leon-Mimila P, Williams KJ, et al. Transcriptional regulation of n(6)-methyladenosine orchestrates sex-dimorphic metabolic traits. Nat Metab. 2021;3:940–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inano S, Takao H, Hayashi N, Abe O, Ohtomo K. Effects of age and gender on white matter integrity. AJNR Am J Neuroradiol. 2011;32:2103–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Adamson C, Yuan W, Altaye M, Rajagopal A, Byars AW, Holland SK. Sex differences in white matter development during adolescence: A dti study. Brain Res. 2012;1478:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sollmann N, Echlin PS, Schultz V, Viher PV, Lyall AE, Tripodis Y, Kaufmann D, Hartl E, Kinzel P, Forwell LA, et al. Sex differences in white matter alterations following repetitive subconcussive head impacts in collegiate ice hockey players. Neuroimage Clin. 2018;17:642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fakhran S, Yaeger K, Collins M, Alhilali L. Sex differences in white matter abnormalities after mild traumatic brain injury: Localization and correlation with outcome. Radiology. 2014;272:815–823 [DOI] [PubMed] [Google Scholar]
- 34.Raja R, Rosenberg G, Caprihan A. Review of diffusion mri studies in chronic white matter diseases. Neurosci Lett. 2019;694:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chamorro Á, Lo EH, Renú A, van Leyen K, Lyden PD. The future of neuroprotection in stroke. J Neurol Neurosurg Psychiatry. 2021;92:129–135 [DOI] [PubMed] [Google Scholar]
- 36.Lyden PD. Cerebroprotection for acute ischemic stroke: Looking ahead. Stroke. 2021;52:3033–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available in the article and online-only Supplementary material.