

Abstract
With rapid advances in computer algorithms and hardware, fast and accurate virtual screening has led to a drastic acceleration in selecting potent small molecules as drug candidates. Computational modeling of RNA-small molecule interactions has become an indispensable tool for RNA-targeted drug discovery. The current models for RNA-ligand binding have mainly focused on the docking-and-scoring method. Accurate docking and scoring should tackle four crucial problems: (1) conformational flexibility of ligand, (2) conformational flexibility of RNA, (3) efficient sampling of binding sites and binding poses, and (4) accurate scoring of different binding modes. Moreover, compared with the problem of protein-ligand docking, predicting ligand binding to RNA, a negatively charged polymer, is further complicated by additional effects such as metal ion effects. Thermodynamic models based on physics-based and knowledge-based scoring functions have shown highly encouraging success in predicting ligand binding poses and binding affinities. Recently, kinetic models for ligand binding have further suggested that including dissociation kinetics (residence time) in ligand docking would result in improved performance in estimating in vivo drug efficacy. More recently, the rise of deep-learning approaches has led to new tools for predicting RNA-small molecule binding. In this review, we present an overview of the recently developed computational methods for RNA-ligand docking and their advantages and disadvantages.
Graphical Abstract
RNA-targeted drug discovery requires the synergy of enhanced sampling and accurate scoring with fast computational speed. The distinct aspects of RNA-ligand docking compared to protein-ligand docking pose unique challenges, which demand a new generation of molecular docking models. This review presents an overview of recently developed RNA-ligand molecular docking methods for RNA-targeted drug discovery.
1. Introduction: targeting RNA with small molecules is a highly promising strategy for drug discovery
Ribonucleic acid (RNA) molecules are transcribed from DNA in the cell nucleus and play a variety of critical roles in gene expression and regulation at the level of transcription and translation. According to their cellular functions, RNA molecules can be categorized into two types: messenger (coding) RNAs (mRNAs) that encode the amino acid sequences and are translated into proteins, and noncoding RNAs (ncRNAs), which, instead of encoding amino acid sequence, serve as enzymatic, structural, and regulatory elements for gene expression. With the coding RNAs occupying only less than 3% of the human genome (1-3), the vast majority of the human genome sequences are transcribed to ncRNAs, such as ribosomal RNAs (rRNAs), microRNAs (miRNAs), small interfering RNAs (siRNAs), small nuclear RNAs (snRNAs), and various riboswitches (4; 5). With the ever increasing discoveries of new RNA structures and cellular functions and the continuous developments of powerful RNA structure determination methods, RNA-based therapeutics are becoming new promising methods to treat human disease. In general, RNA-based therapeutics can be classified into two types. In the first type, therapeutic RNAs—including RNA aptamers, antisense oligonucleotides (ASO), small interfering RNAs (siRNA), and guide RNAs (gRNA)—bind to the target (e.g., RNA transcripts, DNA targets, and protein targets) to inhibit or induce targeted biochemical reactions. This approach has attracted tremendous interest in the field of gene therapy and has been under very active development (6-9). In the second type of RNA-based therapeutics, an RNA molecule serves as the target for drug (small molecule) binding (2; 9-15). This second approach is analogous to protein-targeted drug discovery. However, only ~1.5% of the human genome encodes protein (2; 3; 13; 16-18), and of these protein-encoding genes, only 10-15% are disease-related (2; 3; 13; 19-21). The availability of druggable protein targets is further restricted by the structural and energetic fitness required for high-affinity drug binding. In contrast, genes that are undruggable or difficult to drug by targeting their associated proteins may be inhibited by drugs targeting the corresponding mRNA sequence. Therefore, compared to proteins, RNAs show much broader druggability. Additionally, non-coding RNAs play important roles in most human diseases from cancer to viral infection such as COVID-19. Targeting the large number of non-coding RNAs would open up remarkable new opportunities for drug discovery. For example, antibiotics targeting ribosomal RNA (rRNA), which forms an active site of a bacterial ribosome, effectively inhibit bacterial protein synthesis (22-25). Specific small molecules (ligands) bound to common riboswitches in bacterial cells regulate gene expression through ligand-induced conformational changes of the RNA (26-40). To inhibit viral replication, a potentially effective strategy is to use small molecule as a drug to target viral RNA motifs which are often highly structured (9; 11; 41), such as the HIV transactivation response (TAR) element in the 5’ untranslated region (42; 43), the internal ribosome entry site (IRES) element located in the hepatitis C virus (HCV) genome (44-48), and the influenza A virus RNA promoter (49; 50). Screening small molecule compounds selected from the compound library against an atypical three-stemmed RNA pseudoknot that stimulates −1 programmed ribosomal frameshifting (51; 52) in SARS-Cov RNA genome shows inhibition of the −1 ribosomal frameshifting of SARS-CoV with an IC50 at 210 μM (53; 54). In addition to the above examples, many precursor messenger RNAs and microRNAs have also shown great promise as therapeutic targets (9; 14; 15).
Compared with predicting protein-ligand interactions, which remains a challenging problem, modeling binding interactions between RNA and small ligand molecules presents three unique challenges. First, unlike a protein, RNA is highly charged, with each phosphate group carrying one electronic charge. Thus, RNA folding and ligand binding require the participation of metal ions such as Mg2+ and water molecules to stabilize the binding pocket structure of the RNA and to mediate ligand-RNA interactions (55-58). Second, RNA molecules are often quite flexible, capable of folding into multiple stable conformations, and ligand binding often induces structural switches between different conformers or change the structure of an RNA receptor. Compared with protein-ligand binding, ligand binding sites on RNA can be less deep and more polar, solvated, and conformationally flexible (3; 11; 58), which adds further complexity to predicting RNA-small molecule interactions. Third, the fact, that we have a limited number of experimentally determined structures for RNA molecules and RNA-ligand complexes makes pure knowledge-based approaches less effective for RNA-ligand predictions. In this regard, a physics-based approach or a hybrid knowledge-based and physics-based approach can yield unique advantages (57; 59-71).
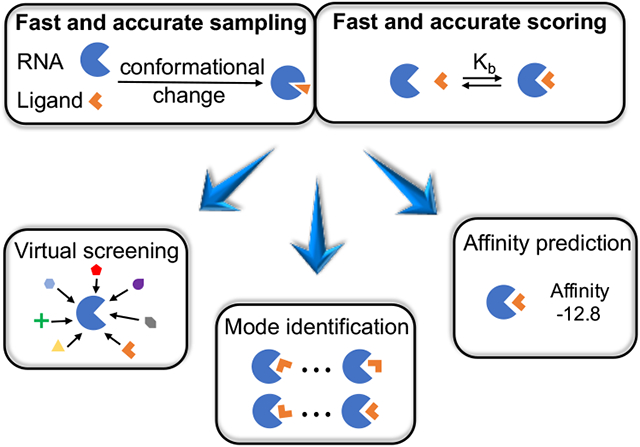
Although successful computational tools have been developed for protein-ligand binding (72-75), the difference in chemical structure and energetics between RNA and proteins demands new methods for RNA-ligand interactions. These new methods for small molecule selection, shown in Figure 1; are necessary for virtual screening, binding mode identification, and binding affinity prediction of specific RNA targets. A successful computational drug discovery requires the integration of three key components: (i) a method to identify the druggable RNA target, (ii) a computationally efficient, sampling algorithm for RNA conformations, ligand conformers, and ligand binding poses, (iii) accurate scoring functions to assess the RNA-ligand complex structures and evaluate the binding affinity. In this review, we focus on computational challenges in predicting RNA-ligand interactions, with specific emphasis on recent advances.
Figure 1:

Three major applications of an RNA-ligand interaction model. Virtual screening involves docking against, small molecules in a large library and scoring every docked pose. Top-scored selections are treated as the most, promising candidates for putative binders. For a given RNA-ligand pair, computational models for ligand binding pose identification and RNA-ligand binding affinity prediction rely on scoring the possible RNA-ligand complex structures. An ideal scoring function for ligand binding pose identification should have the ability to distinguish the native pose from a large pool of docked decoy poses, while achieving the maximum correlation between the predicted scores and the experimental affinities for different. RNA-ligand pairs.
2. Methods for identifying druggable RNA targets and binding sites
2.1. Identifying druggable target RNAs
The druggability of a particular RNA target depends on the answers to three questions. First, does the inhibition/enhancement of the target RNA function lead to effective control of the disease? Second, is the RNA target accessible for the small molecule binding in the cellular environment? Multiple factors can affect the accessibility of the target RNA, such as the abundance and lifetime of the target RNA in disease-related cellular processes (2; 9; 13). Third, does the target RNA adopt a binding site that enables small molecule binding with high affinity and high specificity? Small molecule targeting the particular RNA with high specificity can reduce the off-target side effects. An effective way to achieve high specificity is to target RNA that is unique in the diseased cells, pathogenic viruses or bacteria, such as riboswitches which are common in bacteria but rare in eukaryotes. Another way is to computationally identify RNA motifs that is able to form unique and high-affinity pockets capable of small molecule binding (2; 13; 76).
Inforna (77; 78) is a template-based method capable of selecting RNA targets according to RNA secondary structure motifs such as hairpins, symmetric and asymmetric internal loops, and bulges. The current Inforna 2.0 template database (77) contains 1936 pairs of known RNA secondary structure motif-ligand bound complexes (78). For a given RNA target, Inforna 2.0 (78) identifies RNA secondary structure motifs and from the template database, for a given motif, finds the corresponding ligand partners with fitness scores (79; 80). The fitness score (79; 80) provides a measure of RNA-ligand binding affinity as well as the selectivity of the RNA motif against many other small molecules (78). RNAs of high selectivity and affinity fitness scores are more likely to be druggable. With the top scored small molecules as lead compounds, chemical similarity screening of compound library gives potential potent binders. Inforna (77; 78) has been proved to be successful in various studies (13), such as identifying small molecules that target oncogenic miRNA precursors (81; 82) and an A bulge in the (iron responsive element) IRE (83) of the SNCA mRNA related to Parkinson’s disease (84).
On the basis of RNA secondary structures, Warner et al. (2) showed that information content (85; 86) can be used to identify druggable RNA motifs for potentially high-specificity and high-potency binding (87). Information content measures the amount of information (in bits) required to specify the sequence and structure complexity of an RNA motif, where motifs with high bits (~30 bits) are more complex and more likely to be unique in the transcriptome (2). In experiments, RNA structural information content is attainable through chemical probing techniques such as selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) (88-91). Focusing on RNA motifs with sufficient complexity (high information content) can lead us to RNAs with high binding specificity and affinity thus higher druggability. As an example, the binding affinities of GTP (86; 92) and targaprimir-96 (93) both show a strong correlation with the information content of the RNA motifs, where a 10-bit increase in information content results in a 10-fold increase in binding affinity (2).
2.2. Identifying binding sites for a given target RNA
An overall assessment of binding pockets.
A recent statistical analysis demonstrates that many RNAs indeed fold into structures that form pockets amenable to selective small molecule binding (76). To identify potential RNA suitable for ligand binding, Hewitt et al. (76) have evaluated RNA binding pockets using PocketFinder (87) for 1552 structured RNAs and all the proteins in the Protein Data Bank (PDB) (94), where a binding pocket is described by the volume and the solvent exposure of the pocket (buriedness) and the fraction of the pocket considered to be hydrophobic (hydrophobicity). The results suggest that although ligand-bound pockets on RNAs and proteins show overall similar physical properties, RNA pockets are on average less hydrophobic than their protein counterparts (76). Moreover, compared to the unbound pockets of RNA, the ligand-bound pockets are generally larger in volume, more buried, and more hydrophobic (76).
Geometry-based methods.
In search for binding sites based on RNA-ligand shape complementarity, DOCK 6 (65) selects the binding pockets from a negative image of the receptor surface, where each cavity is characterized by a set of overlapping spheres (95). Similarly, rDock (67) applies a two-sphere mapping algorithm to identify the binding sites. Within the defined docking space, large spherical probes are placed on each grid point to rule out superficial and shallow sites. Then, small spherical probes are placed on the remaining unallocated grid points to map the cavities that serve as the possible binding sites (67). Wide and shallow minor grooves of RNA, which can geometrically accommodate a wide range of ligand shapes and serve as non-specific binding sites, are often identified as putative binding pockets and cause false-positive predictions.
Energy-based methods.
Other programs find binding sites by estimating the overall probe-pocket interaction energies, where the probes are virtual atoms and traverse the surface of the receptor. PocketFinder (87) and AutoLigand (96) are such cases and are equipped in ICM (97) and AutoDock (98), respectively. PocketFinder uses a Lennard-Jones (LJ) potential to describe the interactions between probe atoms and receptor atoms, and grid maps generated from the calculated interactions are used to identify the binding sites. AutoLigand uses a similar approach but involves an extra iterative step to identify the optimal binding sites from the grid maps, and it accounts for connection between the neighboring possible pockets.
Network and machine-learning approaches.
By treating RNA-ligand interaction as a network of contacting atoms, network-based approaches have shown great promise in the prediction of the functional sites in RNA-ligand interactions. For example, using inter-nucleotide Euclidean (hamming) distance network for a 3D or 2D structure Rsite (99) and Rsite2 (100) predict the functional sites for RNA-ligand binding from the maximally closely clustered nucleotides. However, since the inter-nucleotide networks in Rsite and Rsite2 do not distinguish the different connection types between the nucleotides, these models often lead to false positive predictions. To distinguish the different connection types, RBind (101; 102) transforms an RNA structure into a graph, where a node and a edge denote a nucleotide and a non-covalent contact between the nucleotides, respectively, and predict the functional sites as regions formed by nucleotides of the maximum closeness. On a test set with 19 RNA-ligand complexes, RBind (average positive predictive value PPV = 0.67) outperforms Rsite (average PPV = 0.42) and Rsite2 (average PPV = 0.40) (101). The result suggests that the different types of inter-nucleotide interactions encoded in the RNA structure provide important information for the prediction of the functional sites. RNAsite (103), a random forest-based model, uses sequence-based and/or structure-based descriptors to predict whether a given nucleotide belongs to the functional sites. In the model, a nucleotide is defined as part of a functional site if it contains an atom within 4 Å to the target ligand. In RNAsite, four different sets of features for each nucleotide are extracted: geometrical features of local surface convexity/concavity (Laplacian norm), topological features of the RNA nucleotide interaction network similar to the one used in RBind (101; 102), nucleotide-specific accessible surface areas, and position-specific evolutionary conservation of the nucleotide calculated from multiple sequence alignment. The model is trained on 60 RNAs with five-fold cross validation and tested on two separate sets with 19 (RB19) and 18 (TE18) RNAs, respectively. By using Mathews correlation coefficient (MCC) and area under the receiver operating characteristic curve (AUC), RNAsite (103) shows better performance compared to Rsite (99), Rsite2 (100) and RBind (101; 102), with 0.253 and 0.567 for MCC, and 0.776 and 0.877 for AUC on TE18 and RB19 sets, respectively. The promising results indicate the necessity to include more independent features as descriptors. Although these models have been trained specifically for RNA-small molecule complexes, further improvements are possible, for example, through a combination with machine learning methods (104-107).
3. Methods for efficient sampling of ligand binding modes with flexible conformations — a major challenge in RNA-ligand docking
Exhaustive sampling of possible RNA-ligand complex structures is challenging due to the flexible nature of RNA and small molecules. Additionally, following the induced-fit effect or the conformational selection mechanism, RNA targets often undergo conformational changes in response to ligand binding (40; 108-110) (see Figure 2a). This leads to the coupling between RNA folding, including cotranscriptional folding, and ligand binding when virtual screening is performed (40; 108-111). A widely used approach to tackle this problem is ensemble docking (42; 43; 55; 112), where a ligand docks to an ensemble of RNA structures. An alternative approach is to sample the conformational changes on the fly in the docking process. Various methods (60; 64-67; 70) have been developed to treat flexible docking. However, in part due to the required computational time for large-scale virtual screening, predicting large conformational changes remains a challenge.
Figure 2:

RNA conformational changes and binding interactions mediated by water molecules and ions. (a) The local structure difference of preQ1 riboswitches between apo (ligand-free) and holo (ligand-bound) states. The structure in orange denotes the apo state (PDB code: 6VUH (113)) and the structure in blue denotes the holo state (PDB code: 3Q50 (114)) with its bound small molecule (PRF) colored in magenta. Upon binding, the small molecule displaces residue A14 (colored in green for both apo and holo states) and causes the local structural transition. (b) Water molecules mediated RNA-ligand interactions. Water molecules form a bridge between small molecule Neomycin B (NEM, magenta) and 16S-rRNA A-site (PDB code: 2ET4 (115)). The isolated red dots denote the oxygen atoms in water molecules. The black dashed lines show the water-mediated hydrogen bonding contacts that promote NEM binding to the RNA receptor. (c) Metal ions in RNA-small molecule interactions. The ligand benfotiamine (BTP, magenta) interacts with residues G60, C77, and G78 of the Thi-box riboswitch through two magnesium ions (green) and the G42-A43 base stack (PDB code: 2HOO (116)). The black solid lines represent the inner sphere metal ion coordination. The polyanionic RNA recognizes the positively charged metal ion complex made up of the monophosphorylated compound and cations.
For ligand docking to RNA targets, such as ribosomal RNAs and riboswitches with reliable binding site information (117; 118), local sampling with a rigorous energy scoring function can often provide accurate predictions for the ligand binding pose. However, for a broad range of therapeutic RNAs including viral genomic RNAs (119; 120), the lack of the binding site information poses a great challenge to drug screening. For RNAs, unlike proteins, we have limited examples of RNA-ligand bound structures and scarce knowledge about the binding sites. This fact highlights the importance of blind docking (vs. local docking), where a small molecule is docked to the entire surface of the receptor without any prior knowledge of the binding site (see Figure 3). Although computational models—including AutoDock Vina (66), GOLD (59), and Glide (61)—or models originally developed for protein-ligand docking, but optimized for RNA targets—such as AutoDock (98), ICM (97), DOCK 6 (65), and FITTED (62)—can be adopted for RNA-ligand docking, methods developed specifically for RNA targets, such as RiboDock (60), rDock (67), MORDOR (64), and RLDOCK (70; 71), have demonstrated advantages for RNA systems. See Table 1 for a list of docking software.
Figure 3:

The difference between local and blind docking. A complex of an aminoglycoside antibiotic, gentamicin (green) and the 16S-rRNA A site of bacterial ribosome is used for illustration (PDB code: 2ET3 (115)). In this example, both docking (local & blind) processes are carried out using the RLDOCK model (70; 71). In local docking, the binding pocket is predefined and the sampling is contained within the red dashed box. The small magenta spheres denote candidate binding sites predicted by RLDOCK. In blind docking, the binding site detection is performed across the whole surface of the RNA. The small yellow and magenta spheres denote the predicted high- and low-probability binding sites, respectively. Two cavities identified by RLDOCK (anchored by yellow spheres) are zoomed out separately.
Table 1:
Docking programs available for RNA. Docking programs without dedicated binding site detection module are shown with a dash.
| Docking program | Target | Conformational search algorithm |
Binding site prediction |
|---|---|---|---|
| AutoDock Vina (66) | protein | Monte Carlo & quasi-Newton | - |
| GOLD (59) | protein | genetic algorithm | - |
| Glide (61) | protein | Monte Carlo | SiteMap (121) |
| AutoDock (98) | protein/RNA | genetic algorithm | AutoLigand (96) |
| ICM (97) | protein/RNA | Monte Carlo | PocketFinder (87) |
| DOCK 6 (65) | protein/RNA | incremental construction | sphgen module (95; 122) |
| FITTED (62) | protein/RNA | genetic algorithm | - |
| RiboDock (60) | RNA | genetic algorithm | two-sphere filter |
| rDock (67) | protein/RNA | genetic algorithm Monte Carlo simplex minimization | two-sphere filter |
| MORDOR (64) | RNA | molecular dynamics | grid-based systematic search |
| RLDOCK (70; 71) | RNA | multi-conformer docking | grid-based systematic search |
3.1. Sampling of ligand conformations
There are three general ways to treat flexible ligand conformations in docking (123-125): multi-conformer docking, incremental construction, and stochastic optimization. These three strategies differ in their computational speed and the conformational adaptability of the docked ligand to the geometric features of the binding-site.
Multi-conformer docking.
The multi-conformer docking algorithm prepares a conformational ensemble for a ligand (small molecule) and performs rigid docking for each the ligand conformers against the same target (126). For a given binding pocket, this method can be computationally fast if a limited number of conformers are docked. A key determinant for the success of this approach is that the near native conformations of the ligand must be included in the ligand conformer ensemble. Currently, there exist a number of ligand conformer generators, such as OMEGA (127; 128), RDKit (129), and Open Babel (130). These models have been shown in benchmark studies to reproduce reliable conformational ensembles of small molecules within seconds (130; 131). Combining a new molecular dynamics approach and a quantum mechanically-refined ligand-RNA interaction force field, a recently released conformer generator has led to improved accuracy for in silico drug design (132; 133). Its web-based server and the database of bioactive conformational ensembles not only speed up the process of finding experimentally favorable ligand conformations through massive docking but also provide proper initial structures for further optimization (134). In addition, in the docking process, a ligand conformer ensemble is constructed prior to conformational sampling, thus, the conformer ensemble can be appropriately built for the small molecules in question. For example, a ligand conformer ensemble can be generated with bias toward the low-energy states (70; 71) or with maximum diversity in the conformational space.
RNA-Ligand DOCKing (RLDOCK) (70; 71) is a recently developed docking model for flexible ligands using a multi-conformer approach. In RLDOCK, the ligand-binding mode is described by four variables (R, L, A, O), where L denotes the ligand conformer, A denotes the ligand atom placed at (anchor site) R, and O is the 3D rotation angle of L about A (at position R). For each RNA-ligand pair, the RLDOCK algorithm generates an ensemble of flexible ligand conformers and binding poses through the following steps.
The algorithm generates an ensemble of viable anchor sites R based on the following two criteria: (a) there should be no steric clash between ligand and RNA atoms and (b) the RNA structural environment around R should form a pocket geometry.
Based on the viable anchor sites R generated above, by exhaustively enumerating all the different combinations of (R, L, A, O), RLDOCK samples all the possible ligand binding sites and binding poses. The results are stored for subsequent refinement. Before applying the scoring function to rank order all the binding modes, to accelerate computational speed, RLDOCK first sieves the exhaustive ensemble by removing those with high LJ potential between RNA and ligand.
All R sites with low LJ potentials ULJ(R) (below the threshold) are selected as preferred R sites. Here ULJ(R) is the minimum LJ potential over all possible (L, A, O) values for a given R.
For each preferred R, preferred ligand conformers L are selected from low LJ potentials ULJ(R, L), the minimum LJ potential over all possible (A, O) values for a given set (R, L).
Similarly, for each preferred R and L, preferred ligand atoms A are selected from the low LJ potentials ULJ(R, L, A), the minimum LJ potential over all possible O values for a given set (R, L, A).
After the above procedure, a preferred (R, L, A, O) ensemble with all the possible orientations (O) of the ligand is generated and subsequently ranked by the scoring function. To speed up the LJ potential calculation, RLDOCK employs a grid-based energy calculation, where each grid stores the LJ energy between RNA and a probe atom on the grid for fast computation of LJ energy for a given binding mode. Through the above procedure, RLDOCK generates millions of possible ligand configurations through exhaustive rotation and translation transformations at each putative binding site for each pre-configured conformer (see Figure 4). Compared with other models, RLDOCK has the unique merit of using complete sampling for ligand conformers and binding modes.
Figure 4:

Illustration of conformational sampling methods used in RLDOCK, using the docking of 2’-deoxyguanosine to 2’-deoxyguanosine riboswitch (PDB code: 3SKL(135)) as an example. An ensemble of different conformers of the 2’-deoxyguanosine (dG) is constructed for flexible docking. The sampling and scoring procedures are shown in order and labeled through A to E. (A) First, the regions of possible anchor sites within the riboswitch, colored in magenta, are determined by the geometric features of the target RNA. (B) Second, with exhaustive sampling of these prepared conformers through translation and rotation around the anchor sites, (C) binding sites (yellow dots) are selected according to Lennard-Jones potential between RNA and ligand atoms. (D) Finally, the sampled ligand conformations associated with the selected binding sites are ranked (E) by a physics-based scoring function.
Incremental construction.
By anchoring rigid fragments through geometric matching and then incrementally building the ligand structure, on-the-fly ligand conformer sampling allows the local environment of the binding pocket to guide the growth of the small molecule. An inherent drawback of this approach is that small errors in the early steps can be amplified throughout the process, especially for large ligands. DOCK 6 (65) adopts this incremental construction strategy for ligand conformational sampling and search (53). Unlike the original greedy algorithm (cluster-based pruning) to sieve the sampled ligand structures followed by clustering and ranking at each step, an improved algorithm, which skips the conformational clustering step in order to retain the original, diverse conformations of the flexible bonds, has led to an increase in the success rate by 10% for the prediction of binding poses (65). The results have demonstrated the importance of maintaining the diversity of ligand conformers.
Stochastic optimization.
The stochastic optimization method searches for binding modes on-the-fly by optimizing flexible torsional angles, orientation, and position of the small molecule. The Monte Carlo (MC) (61; 66) and Genetic algorithms (GA) are the most widely used stochastic optimization algorithms. A combination of different stochastic methods often lead to improved sampling and optimization results. For example, AutoDock Vina (66) adopts a hybrid approach with MC for global optimization and Broyden-Fletcher-Goldfarb-Shanno (BFGS) for local optimization (66). Other examples include ICM (97) and RiboDock (60), which employ MC coupled with simulated annealing. Several protein docking programs, such as GOLD (59), AutoDock (98), and FITTED (62), use GA to sample and search for ligand conformations. Modifications to some of these methods for RNA targets have led to highly promising results. For example, through parameterizing the scoring function (136) or adding a new solvation term to the original scoring function (137), AutoDock can treat RNA-ligand interactions. Similarly, through proper optimization (58), FITTED can be used to predict RNA-ligand docking. The accuracy may be further improved once all possible RNA hydrogen bond donors and acceptors are considered, and after metal ions such as Mg2+ and Mn2+ are included as part of the receptor (58). Like other stochastic optimization algorithms, a shortcoming of MC and GA is that the optimization process may become trapped in the local minima. This may pose a severe challenge due to the rugged energy landscape of RNA-ligand complexes. The problem can be alleviated through repetitive docking with random placement of the small molecule (ligand) and implementation of algorithms such as tabu search (138; 139) and stochastic tunneling (140) to accelerate the de-trapping from the local minima. By minimizing the likelihood of poses being trapped in a local minimum in the early stages of the conformational search, rDock (67), a model for both nucleic acid and protein docking, employs a GA/MC hybrid method to enhance efficient sampling of ligand binding poses. The GA/MC hybrid method involves three rounds of GA, low-temperature MC, and Simplex minimization, each of which adopts an independent scoring function. An optimized set of scoring functions has been shown to significantly enhance the efficiency of sampling even with an unfavorable initial pose by minimizing the possibility of being trapped in a local minimum during the conformational search.
In summary, while multi-conformer docking provides a fast way to consider ligand flexibility prior to the docking calculation, its performance depends on the quality of the generated conformer ensemble. In contrast, stochastic and incremental sampling approaches can treat ligand flexibility during the docking process. However, such on-the-fly sampling approaches suffer from the problem that a small error in the early steps can be amplified in the later steps, and stochastic approaches suffer from the problem that poses can be potentially trapped in a local minima while docking. In addition, both approaches require additional energy terms to account for intra-ligand interactions. Although the conformational sampling modules in RNA-ligand docking software (57; 67; 70; 71; 141-143) have shown promising results for recovering native or near-native ligand poses, for a flexible RNA that undergoes conformational changes upon ligand binding, a search for a fast and accurate sampling method by combining folding and binding algorithms for both ligand and RNA continues.
3.2. Incorporation of RNA flexibility
It has been shown that for protein-ligand docking, ignoring the protein (receptor) flexibility can cause incorrectly predicted binding modes (144). The problem can be more severe for ligand binding to an RNA, whose structure can be more flexible than a protein. To address this important issue in RNA-ligand docking, several successful approaches have been developed to incorporate RNA conformational changes in the docking algorithm. These approaches can be classified into three types: soft docking, ensemble docking, and fully flexible docking (See Figure 5).
Figure 5:

Different approaches to modeling RNA flexibility in RNA-ligand interactions illustrated using HIV-TAR RNA (PDB: 1ANR (145)) as an example. The orange and blue regions correspond to rigid and flexible portions of RNA, respectively. From left to right, a) bases from the active site are allowed to partially overlap with atoms from ligand through soft potential, b) an ensemble of various RNA conformations is used to perform docking, and c) RNA with full flexibility. Computational efficiency decreases from left to the right.
Soft docking.
A mathematically convenient way for soft-docking is to decrease the energy penalties for steric clashes, thus tolerating some degrees of overlap between RNA and ligand (146). Glide (61) and GOLD (59) offer such options for users. In earlier work, Moitessier et al. (55) have employed this strategy to dock aminoglycosides in ribosomal A-site RNA, where RNA flexibility was considered using a set of soft van der Waals potentials, and the approach led to increased average accuracy. Soft docking is attractive for its convenience of implementation. However, the limited sampling space without the adjustment of backbone prevents its application to large conformational changes.
Ensemble docking.
In ensemble docking, a given ligand docks into an ensemble of RNA conformations or an ensemble-averaged RNA conformation. Ensemble docking is found to be useful in several RNA-targeted studies (42; 43; 55), and has also been proved successful in protein-small molecule docking (147; 148). In an attempt to reproduce the experimentally determined RNA-aminoglycoside complexes, soft docking to an ensemble-averaged RNA structure gives the best performance with an average RMSD of 2.49Å between the predicted and the experimentally measured binding mode (55). Ensemble docking-based virtual screening with the ICM docking model (42) for HIV-TAR (42; 43) has predicted a TAR-targeting compound with high specificity. Furthermore, virtual screening for an experimentally derived TAR conformational ensemble against a ligand library composed of ~100,000 drug-like organic compounds (43) provided an enriched family of TAR-targeting binders. From a practical perspective, the number of receptor conformations used in the ensemble docking is usually limited due to computational feasibility, thus receptor conformation selection can influence the accuracy of the prediction. Using only the conformational ensemble in the lowest-energy basin may not be the optimal strategy as ligand binding can stabilize and selectively enrich the population of conformations in other basins on the energy landscape (149-151). Therefore, ensemble docking should not ignore low-populated ligand-free RNA conformations.
Molecular dynamics.
Molecular dynamics (MD) simulation (152-159) can not only refine conformations of RNA-ligand complexes and generate ligand and RNA structures, but also shed light on the trajectory and the folding/unfolding of possible metastable states for both RNA and ligand in the docking process (43; 46; 53). However, in practice, ligand binding events can occur in the timescale up to seconds (108; 110; 160), and an all-atom MD simulation for the process goes beyond the capacity of available computing power, especially when virtual screening for drug molecules is considered. Powered by advanced sampling techniques, several computational methods have enabled the characterization of RNA conformational changes upon ligand binding (161-165). A non-equilibrium MD simulation (166) and an umbrella-sampling-based MD simulation (167) both have revealed the competitive relationship between the formation of the kissing-loop and the binding of the small molecule. A recent explicit-solvent MD simulation has shown a small molecule-induced stabilization effect in an adenine riboswitch and the ability of the riboswitch in the near-native states to attract small molecules through hydrogen bonding and base-stacking interaction (168). These results demonstrated the unique advantage of MD simulations for the investigation of physical mechanisms in RNA-ligand binding.
Fully flexible RNA.
Molecular dynamics simulation with proper force field can provide reliable sampling of RNA-ligand complex conformations. For example, by applying an RMSD penalty term to the conventional potential energy, MORDOR (64) (MOlecular Recognition with a Driven dynamics OptimizeR), by simulating ligand docking trajectories, can give conformational sampling and show ligand-induced conformational changes for RNA (64). As an application, a MORDOR-based virtual screening has found a small family of binders targeting human telomerase RNAs (hTR) (169). However, further applications of MORDOR are limited by the high computational cost, which can take up to hours for a docking run. To accelerate simulation, Supervised Molecular Dynamics (SuMD) has been proposed to sample the conformations of RNA-drug complexes (170). SuMD accelerates the simulation by applying a tabu-like algorithm to guide the docking when the ligand is far away from the binding site and a conventional MD simulation when the ligand is close to the binding site. The hybrid method enables efficient simulation of the binding process within an affordable timescale. Although the simulated trajectory does not necessarily represent the physical binding process, SuMD may capture possible conformational changes. The reliability of SuMD for RNA-ligand docking is supported by success in predicting binding modes for several pharmaceutically important RNAs (170), where SuMD predicts RNA-ligand docking mode with a minimum RMSD of 0.34Å for the best case.
Similarly, another method based on elastic potential grids was initially proposed for modeling protein flexibility during docking (171) and later extended to RNA targets (112). In this type of method, a 3D grid of the potential field of the initial RNA conformation is calculated in advance using DrugScoreRNA (172). After determining the potential grids, AutoDock (98) is used as a docking engine with precalculated elastic potential grids for docking. Due to its ability to account for RNA flexibility, docking to the deformable potential grids generated from unbound RNA has a much better performance than simply docking to unbound RNA alone. However, one of the limitations of the approach is that it requires a priori knowledge of the available end states of deformation. Moreover, the model cannot treat conformational changes caused by rotational flip motion and 2D structural rearrangements.
In summary, Molecular dynamics-based methods are time-consuming and thus not suitable for large-scale virtual screening. Rigid docking is fast but lacks accuracy. Soft docking and ensemble docking are in the middle ground between the two extremes as they sacrifice the ability of a more complete sampling of conformations in order to reduce the computational time. At the current stage, a versatile approach to accurately treat receptor flexibility awaits to be developed.
4. Accurate scoring functions for RNA-ligand docking: challenges and promises
Selecting a native ligand binding pose from an ensemble of candidates requires a reliable scoring function. There are three different approaches to the development of a scoring function: physics-based approach, knowledge-based approach, and machine-learning approach; see Table 2 and Table 3 for a list of reviewed scoring functions and the summary of the benchmark results, respectively.
Table 2:
Summary of the reviewed scoring functions used in different models for predicting small molecule binding. a Some scoring functions optimized for protein may also be used for RNA. b Some models contain more than one scoring function, only the default one is listed. c The year that the original model was first published.
| Category | Model | Targeta | Score typeb | Yearc |
|---|---|---|---|---|
| Physics-based | MORDOR (64) | RNA | force fields | 2008 |
| DOCK 6 (65) | RNA | force fields | 2009 | |
| GOLD (59) | protein | empirical terms | 1997 | |
| Glide (61) | protein | empirical terms | 2004 | |
| RiboDock (60) | RNA | empirical terms | 2004 | |
| AutoDock 4 (63) | protein | empirical terms | 2007 | |
| AutoDock Vina (66) | protein | empirical terms | 2010 | |
| iMDLScore1 (57) iMDLScore2 (57) |
RNA | empirical terms | 2012 | |
| rDock (67) | protein nucleic acid | empirical terms | 2014 | |
| RLDOCK (70; 71) | RNA | empirical terms | 2020 | |
| Knowledge-based | DrugScoreRNA (112; 173) | RNA | statistical potentials | 2000 |
| KScore (174) | protein nucleic acid | statistical potentials | 2008 | |
| LigandRNA (175) | RNA | statistical potentials | 2013 | |
| SPA-LN (176) | nucleic acid | iterative statistical potentials | 2017 | |
| ITScore-NL (142) | nucleic acid | iterative statistical potentials | 2020 | |
| Machine-learning | T-Bind (177) | protein | gradient boosting trees | 2018 |
| RNAPosers (141) | RNA | random forest | 2020 | |
| RNAmigos (178) | RNA | graph neural network | 2020 |
Table 3:
Summary of the benchmark results reported in the literature. Results of the different methods tested on the same test set are grouped together for comparison. The first column “Test set” shows the number of test cases and the original references (shown in parentheses) reporting the test results. a Performance of affinity prediction is reported in terms of Pearson correlation coefficient, R2. Correlation coefficient is calculated between experimental binding affinities and predicted binding affinities. Benchmarks without affinity prediction are shown as dashes. b Performance of pose identification is reported as success rate. The criteria for a correct prediction is shown in the (rank, RMSD) format. For example, (1, 2.5Å) means the top-1 prediction that has RMSD less than 2.5 Å to the native pose. Benchmarks without pose identification are shown as dashes. c RLDOCK, rDock, rDock_solv, AutoDock Vina use 38 instead of 42 complexes. MORDOR uses 32 instead of 42 complexes. d Only several top performing models evaluated in literature (57) are listed for this benchmark dataset. e Three outliers 3GX3, 2ESI and 1F1T are excluded in the binding affinity calculation. f Near native poses are sampled through rDock reference ligand method (67) and g Average and standard deviation from 100 sets of 100 random docking poses out of a pool of 1000 decoy conformations. h Native pose is included in pose identification. are included in pose identification. i RNA-adapted AutoDock scoring function (137) is used. j Scoring function is used to guide the docking instead of using default Vina scoring function.
| Test set | Scoring function |
Docking engine |
Affinitya prediction(R2) |
Poseb identification(%) |
|
|---|---|---|---|---|---|
| 42 (70; 71; 142; 175) complexes | Correlation | (1, 2.0Å) | (3, 2.0Å) | ||
| RLDOCK | RLDOCK | - | 55.3c | 60.5c | |
| ITScore-NL | DOCK6 | - | 50.0 | 54.7 | |
| LigandRNA+DOCK6 | DOCK6 | - | 47.6 | 54.8 | |
| rDock_solv | rDock 2014 | - | 39.5c | 55.3c | |
| DOCK6 | DOCK6 | - | 35.7 | 45.2 | |
| LigandRNA | DOCK6 | - | 35.7 | 42.9 | |
| AutoDock Vina | AutoDock Vina | - | 31.6c | 44.7c | |
| DrugScoreRNA | DOCK6 | - | 31.0 | 42.9 | |
| rDock | rDock 2014 | - | 28.9c | 47.4c | |
| MORDOR | MORDOR | - | - | 62.5c | |
| 34 (57; 142; 176) complexesd | Correlatione | (3, 1.5Å) | (5, 3.0Å | ||
| SPA-LN | rDock 2014 | 0.36 | 50.6 | 76.6 | |
| Gold Fitness | GOLD5.0.1 | 0.25 | 42.9 | 73.2 | |
| ASP | GOLD5.0.1 | 0.29 | 42.9 | 66.1 | |
| rDock_solv | rDock 2006.2 | 0.18 | 41.1 | 73.2 | |
| rDock | rDock 2006.2 | 0.15 | 33.9 | 60.7 | |
| ITScore-NL | - | 0.41 | - | - | |
| 56 (67; 141; 176) complexes | Correlation | - | (1, 2.5Å) | ||
| RNAPosers | rDock 2014 | - | - | 62.5f | |
| rDock_solv | rDock 2014 | - | - | 54±3g | |
| SPA-LN | rDock 2014 | - | - | 54±3g,h | |
| AutoDock Vina | AutoDock Vina | - | - | 29±2g | |
| GlideScore | Glide (v.57111) | - | - | 17.8 | |
| 31 (137; 141; 173) | Correlation | (1, 2.0Å) | (1, 2.5Å) | ||
| RNAPosers | rDock 2014 | - | 57.1f | 61.9f | |
| DrugScoreRNA | AutoDock 3.0.5j | - | 41.9 | 45.2 | |
| AutoDocki | AutoDock 3.0.5j | - | 25.8 | 35.5 | |
| 77 (142; 176; 235) complexes | Correlation | (3, 1.5Å) | (5, 3.0Å) | ||
| ITScore-NL | AutoDock 4.2 | 0.41 | 71.4h | 90.9h | |
| SPA-LN | rDock 2014 | 0.33 | 50.6h | 76.6h | |
4.1. Physics-based methods: physical principles of RNA-ligand binding lead to accurate scoring functions
Force-field approach.
Atom-based physical force fields, originally derived from thermodynamic data and ab initio calculations, have enabled molecular dynamics simulations for nucleic acids-targeted drug discovery (179-183). One of the key issues in physical force field-based computations for molecular docking is the solvent effect. Since the virtual screening of ligands against an RNA target demands high computational efficiency for the docking calculation, implicit solvent models, such as Poisson-Boltzmann surface area (PB/SA) model (184-190) and the Generalized-Born surface area (GB/SA) model (191-199), would be highly promising due to the optimal balance between speed and accuracy. A hybrid force field that combines an implicit solvent model and an all-atom force field can often lead to accurate and efficient simulation of an RNA-ligand binding process. As shown by the success of DOCK 6 (65), generalized Born and Poisson Boltzmann implicit solvent models combined with the AMBER force fields can provide an effective energy model for an RNA-ligand docking system (65). MORDOR (64), which combines an implicit solvent model GBSW (Generalized Born with Simple sWitching) (200) with the CHARMM-27 (201) force fields for the receptor and AMBER force fields (202) for ligand molecules, demonstrated how the hybrid energy function can lead to successful modeling of receptor-ligand binding. By using root-mean-square-distance constraints in energy minimization, the model allows local flexibility of the receptor to accommodate possible conformational changes induced by ligand binding and in the meantime, to guide the ligand to probe the surface of the target RNA.
In summary, physical force field-based scoring functions have the advantage of providing insights into the underlying physical mechanism of RNA-small molecule interaction, however, computational costs and the need for expert knowledge in simulating a specific system hinders the application of these models in large-scale virtual screening for drug discovery.
Empirical energy approach.
Physically, different interactions in an RNA-ligand complex are correlated thus nonadditive. A simplified approach is to evaluate the total energy as a weighted sum of the component interactions such as van der Waals, electrostatic, desolvation and hydrogen-bond interactions:
| (1) |
where the weight coefficients wi can be fitted by optimizing the success rate of the computational prediction for the training set. The above empirical scoring function has the advantage of high computational efficiency and adaptivity, which makes accurate prediction possible for specific types of RNA targets and ligands. Compared to the more rigorous force-field approach, empirical scoring functions, which often use “softer” energy forms, are more tolerant for minor clashes and suboptimal interactions during docking, thus partially alleviate the problem of incomplete sampling for receptors and ligand conformations. It is important to note that due to the different physical interactions and correlations between the different interactions in protein-ligand and RNA-ligand systems, parameters fitted from protein-ligand docking may not be transferable to RNA-ligand docking.
The semi-empirical free energy function in AutoDock 4 (63), the fully empirical scoring function of AutoDock Vina (66), and other models such as GoldScore in GOLD (59) and GlideScore in Glide (61) have demonstrated success in predicting protein-ligand docking. These docking software packages, not specifically designed for nucleic acids, may not give optimal results for RNA-ligand docking. RNA-specific scoring functions such as iMDLScore1 and iMDLScore2 (57) have optimized the weight coefficients of the scoring terms (63) using multilinear regression (MLR) methods. In a comprehensive evaluation and comparison for eleven other scoring functions, iMDLScore1 and iMDLScore2 (57) have shown better performance in both binding mode and affinity predictions.
Several RNA-ligand docking software packages have incorporated their respective built-in empirical scoring functions (60; 67; 70; 71) specifically for RNA/DNA-small molecule interactions. The scoring function of rDock (67), the successor of the original model RiboDock (60), contains a weighted sum of various intermolecular and intramolecular interaction energies, including the van der Waals potential (vdW), an empirical energy term for attractive and repulsive polar interactions and the desolvation energy. Because virtual drug screening can benefit from our knowledge, such as pharmacophoric points and shape similarity, derived from known RNA-ligand complexes, rDock has added pseudo-energy restraint terms as an empirical bias, such as pharmacophoric restraints. Pharmacophoric restraints used in rDock ensures the generated ligand poses to satisfy the pharmacophores derived from the known RNA-ligand complexes or the hot-spot mapping methods. Applications to the virtual screening against Hsp90, both rDock and Glide show significant improvements with the inclusion of the bias (67). Since rDock has been optimized for RNA docking, it outperforms Vina and Glide for RNA-ligand docking: For a set of 56 RNA-ligand complexes, the top-ranked poses predicted by rDock show a 54 ± 3 % success rate with a 2.5Å RMSD cut-off compared to 29± 2% for Vina and around 17.8% for Glide (67).
RLDOCK (70; 71) trained the weight coefficients for the different interaction terms such as van der Waals (vdW), electrostatic, polar and nonpolar hydration energies, and hydrogen-bond interactions for a set of 30 RNA-small molecule complexes. To enhance computational efficiency. RLDOCK adopts a two-step screening algorithm: In the first step, using a computationally efficient, crude estimation for the Born radii in the electrostatic energy calculation and the solvent-accessible surface area in the hydration energy, the model selects an initial pool of potential binding poses; in the second step, a rigorous scoring function is used to re-rank the binding poses to identify the top-ranked poses. Test on a separate set of 200 RNA-small molecule complexes indicates that the success rate of identifying the native/near-native binding modes increases significantly from the crude scoring function to the more refined scoring function, with 8.3%, 22.2%, and 29.6% for the crude scoring function and 17%, 40.4%, and 49.1% for the more rigorous scoring function within RMSD thresholds 1Å, 2Å, and 3Å, respectively. Considering the fluctuations in the ligand pose, RLDOCK groups similar ligand poses (according to the mutual RMSD) into clusters and rank the clustered poses. With the RLDOCK-ranked ligand poses (clusters), 44.3%, 74.3%, and 82.2% of the top-10 are within 1, 2, and 3Å (RMSD), respectively, to the native pose (70; 71), Furthermore, tested on a previously proposed set of 38 RNA-small molecule complexes (175), RLDOCK has demonstrated a higher success rate compared to other models for recovering the native ligand binding poses within RMSD of 2Å to the native pose. Specifically, RLDOCK shows a success rate of 55.3% (60.5%) for the top-1 (top-3) predicted poses as compared to 28.9% (39.5%), 36.8% (44.7%), 39.5% (47.4%), and 50.0% (57.9%) for DrugScoreRNA(112; 172), DOCK 6(65), LigandRNA(175; 203), and a combined LigandRNA(175; 203) and DOCK 6(65) approach, respectively. Since RLDOCK distinguishes itself from other models by a global, complete sampling of all the possible binding sites and poses, the results above demonstrate the importance of high-quality sampling for ligand poses.
In summary, compared to atomistic force field-based approaches, the empirical energy function methods manage to reduce the computational burden using simple functional forms for RNA-small molecule interactions. However, this approach is subject to two main limitations: (a) neglecting the correlation between different interactions and (b) transferability of the weight coefficients between different RNA-ligand systems. The success of the model depends on the quality of the curated training set, thus the accuracy of the predictions is limited by the lack of available high-quality data for RNA-ligand complexes.
4.2. Knowledge-based scoring functions: statistical potential provides efficient scoring of binding modes
Statistical potential approach based on reference states.
A statistical potential approach uses the inverse Boltzmann law to extract energy-like potential for user-defined interacting pairs from the experimental data:
| (2) |
where and ρij is the relative frequency of the user-defined interacting pair (i, j) between the receptor R and ligand L. Before being applied to RNA-ligand docking models (112; 142; 172; 174-176), the statistical potential approach has been demonstrated to be effective for predicting protein-ligand docking (72; 73; 75; 173; 204-216). Different variants of the statistical potential approach have been proposed for RNA-ligand systems since the development of DrugScoreRNA (112; 172). These statistical potential approaches mainly differ in two aspects: the choice of reference state and functional forms of potential energy terms. As early attempts, DrugScoreRNA (112; 172), Kscore (174), and LigandRNA (175) have constructed the reference state by treating all the relevant atoms in the RNA-ligand complex as non-interacting particles, with different atom types differentiated or undifferentiated. In addition to the distance-dependent pairwise potential used in Kscore and DrugScoreRNA, LigandRNA, by taking into consideration the relative orientations between different atom pairs, has added a three-body anisotropic potential. Combined with DOCK 6 (65), this orientation-dependent potential shows a higher success rate than DrugScoreRNA in predicting the native binding modes. Specifically, for a test set consisting of 42 RNA-small molecule complexes, with the 2Å RMSD criteria for a correctly predicted ligand pose, DrugScoreRNA, DOCK 6, and LigandRNA show a success rate of 31.0%, 35.7% and 35.7%, respectively. A DOCK6 and LigandRNA hybrid score scheme further gives the success rate of 47.6%. The results show that the knowledge-based approach can benefit from a more accurate potential that accounts for more detailed information such as distance and angular correlations between the different interactions.
Iterative statistical potential approach.
A major limitation of the above traditional approach is that the reference state ignores the many-body correlations between the different interactions. One way to circumvent this problem is to iteratively refine the energy function until the simulated probability distribution of the different atom pairs agrees with that observed from the experimental data (142; 176; 217-222). Because the simulated distribution is based on sampling over the full energy landscape, an iterative approach can account for both native and nonnative interactions.
SPA-LN (176) is an iterative statistical potential model for predicting nucleic acid-small molecule interactions. Using intrinsic specificity ratio (ISR), a measure of the native vs. nonnative binding modes discriminative power, the energy-like scoring function can account for both affinity and specificity. For binding affinity prediction, using Pearson correlation coefficient between the predicted and the experimentally measured affinity as a measure, for a set of 77 complexes from version 2014 of PDBbind database (223) and a separate set of 34 nucleic acid-small molecule complexes, SPA-LN gives Pearson correlation coefficients 0.58 and 0.60, respectively. For the binding pose prediction, for a test set of 56 nucleic acid-small molecule complexes (67), for the top-scored poses with 2.5Å RMSD cutoff for a pose considered to be native or near-native, SPA-LN (176), rDock (67), AutoDock Vina (66) and Glide (61) give a success rate of 54(±3)%, 54(±3)%, 29(±2)%, and 17.8%, respectively. The performance of SPA-LN suggests the importance of considering not only affinity but also the specificity of RNA-ligand binding.
To capture the stacking and electrostatic interactions in nucleic acids, ITScore-NL (142), an iterative statistical potential approach (219), adds an extra distance-dependent stacking potential term and an electrostatic potential energy term to the scoring function. Stacking potentials were calculated for all carbon-carbon atom pairs involved in stacking interactions between nucleobases and planar aromatic groups of a small molecule. Electrostatic potential was calculated for the polar atom pairs using the Debye-Hückel approximation. The model was compared with other methods in two datasets to validate the performance on native pose recovery and binding affinity prediction. With the same set of 77 nucleic acid-small molecule complexes used in the test of SPA-LN, ITScore-NL achieved a higher Pearson correlation coefficient (R = 0.64) than that shown in SPA-LN (R = 0.58). As for the success rate of native pose recovery, ITScore-NL (142) was able to correctly identify native binding mode of 71.43% (50.64% for SPA-LN (176)) complexes with RMSD cutoff 1.5Å if the top-3 predictions are selected and 90.90% (76.62% for SPA-LN (176)) complexes with RMSD cutoff 3.0Å if the top-5 predictions are selected. Compared to LigandRNA (175) on a 42 RNA-small molecule complexes with only top-scored poses being selected and poses with RMSD cutoff 2.0Å being used, the success rates of LigandRNA (175) and ITScore-NL (142) are 35.7% and 50.0%, respectively. The results indicate the importance of including stacking and electrostatic interactions in RNA-ligand docking.
In summary, compared to atomistic force field methods, the statistical potential approach is associated with a much higher computational efficiency. However, the choice of reference state places obstacles for accurate modeling of RNA-small molecule interactions. Even though iterative approaches have been developed to circumvent the reference state problem, constructing diverse and complete decoy sets for training remains a challenge for RNA-ligand complexes. Furthermore, because data-driven approaches rely on experimentally determined structural data, the success of the models suffers from limited structure data for RNAs and RNA-ligand complexes.
4.3. Machine-learning based scoring method for RNA-ligand docking: an emerging scoring approach with high promise
Machine-learning approach.
With the success of machine-learning methods in various fields, a variety of machine-learning models such as support vector machine (SVM), random forest (RF), neural network (NN), and convolutional neural network (CNN) have been proposed and shown success to predict protein-small molecule interactions (224-229). Machine learning approaches not only have the advantage of utilizing the experimental data and making fast predictions but also with a large number of trainable parameters, can leverage experimental data better than traditional knowledge-based methods. Furthermore, the relation between input features and output results is learned through the training process. Therefore, a machine-learning method can be readily adopted across different types of tasks by simply changing the input features and output format, which can be engineered for the corresponding task. Figure 6 shows a typical workflow of training, validating, and testing a machine-learning model.
Figure 6:

The typical workflow of a machine-learning approach. Training and validation cycle usually needs to be performed many times before the performance on the validation set reaches an acceptable level. After the training-validation cycle, the trained model is used to make predictions on the test set.
Importance of feature engineering.
Although the quality and amount of the training data is vital to the performance of machine-learning methods, input feature engineering, which is often overlooked, is also critical to the success of the model. Generally, input features extracted from structure or geometry-based models for RNA-ligand binding often contain a large amount of detailed structural information, resulting in noise and excessively high dimensions in the parameter space. For example, there are many CNN-based approaches (230-234) that simply extend the 2D image in the original CNN model by treating the binding site as a 3D image. However, this type of 3D image is not rotational invariant hence requires rotational augmentation when used in training and prediction. The extra dimensions added would significantly increase computer time for making a single prediction and for performing large-scale virtual screening for drug discovery. Additionally, many atoms which are shown as pixels in the image may not even contribute to the binding, thus further complicate the learning process. An optimal engineered feature should maximally simplify the input information while capturing the key features that determine RNA-ligand docking results.
T-Bind (177) is a method for protein-small molecule binding affinity prediction. What makes it interesting for its possible application to RNA-ligand binding is not only the machine-learning model but also, more importantly, its feature extraction method. In T-Bind (177), Cang et al. (177) introduces a novel mathematical concept, element specific persistent homology (ESPH) or multicomponent persistent homology, to capture the crucial topological information around the binding site. This feature extraction method offers a new way to embed geometric information into topological invariants and simplify the input features while still capturing the key information. Benchmark tests using PDBbind database (223; 235-239), a comprehensive collection of protein-small molecule binding affinity data together with 3D structures, have yielded Pearson correlation coefficients 0.818 and 0.767 for PDBbind v2007 core set (237) and PDBbind v2013 core set (238), respectively, and the T-Bind outperforms other scoring functions (177). The result shows the merit of this feature extraction method and the promise of applying the same approach to the prediction of RNA-small molecule interactions.
RNAmigos (178) is a machine-learning model designed for the prediction of RNA-small molecule interactions. In RNAmigos (178), the base-pairing network around the binding site is simplified as a connected 2D graph with vertices and edges, where a nucleotide is represented by a vertex and backbone connectivity and base-pairing are represented by the different types of edges. The base-pairing interactions encoded in the 2D graph provide a signature for predicting the fingerprint of the small molecule that wifi most likely bind to the site. Furthermore, RNAmigos (178) shows the versatility of the machine-learning method. The model combines RNA base-pairing information at the binding site (in a 2D graph format) and graph neural network (240) (designed for data with 2D graph structure) to directly predict the fingerprint for the small molecule. RNAmigos encodes the predicted fingerprint as a 166 bit MDL Molecular Access keys (MACCS) (241). The small molecule can be found in a compound library simply by a similarity search against the predicted fingerprint. This procedure circumvents the traditional virtual screening route (docking then scoring) and substantially reduces the time for the search of a putative drug. RNAmigos (178) was trained with a set of 773 RNA-small molecule binding sites associated to 270 unique small molecules. Test results on an enrichment dataset with 176 unique RNA chains against 82 unique ligands have shown that RNAmigos outperforms a template-based method (Inforna 2.0 (77; 78)). Although RNAmigos shows promising results, it has two major limitations. First, RNAmigos requires prior knowledge of the binding site in order to generate a base-pairing network, and a misplaced binding site could led to a degradation in the predictive power. However, accurate determination of the binding site itself can be a challenging task. Second, RNAmigos considers different RNA-small molecule complexes to occur equally likely. Such treatment can cause an effective bias in the training process because the binding affinities of the different RNA-small molecule complexes can span across a large range of values.
As another machine-learning model for RNA-small molecule binding, RNAPosers (141) contains a set of trained pose classifiers that can estimate the “nativeness” of a ligand for a given structure of the RNA and the ligand. The classifiers are based on the random forest method (242) with an ensemble of 1000 decision trees. For a given ligand, RNAPosers takes a pose fingerprint as its input, where the pose fingerprint is calculated as the sum of a Gaussian function multiplied by a cosine damping factor over the RNA-ligand interacting atom pairs. The comparison between RNAPosers and two knowledge-based methods, DrugScoreRNA (112; 173) and SPA-LN (176), shows that RNAPosers (141) is able to yield higher success rates for the prediction of the native binding pose. Specifically, for a set of 31 RNA-small molecule complexes used in DrugScoreRNA (112; 173), RNAPosers gives a success rate of 61.9% as compared to 57.1% for DrugScoreRNA. For another set of 56 RNA-small molecule complexes used as a validation set in SPA-LN (176), RNAPosers gives a success rate of 62.5% compared to 54.0% for SPA-LN. These results show the advantage of the machine learning method over traditional knowledge-based approaches.
Coarse-grained conformational representation, traditionally implemented in RNA folding models, can lead to a unique method for feature engineering. Recently Stefaniak and Bujnicki developed a new machine-learning model, AnnapuRNA (143), specifically for RNA-ligand interactions. The feature engineering algorithm in AnnapuRNA employs a coarse-grained representation of both RNA and ligand to derive RNA-ligand contact statistics. Specifically, RNA structure is coarse-grained with each nucleotide replaced with five beads (243), and a ligand is represented with the concept of pharmacophores (244). Contact statistics collected from the coarse-grained representation with the assumption that the coarse-grained contact statistics can represent the core RNA-ligand interaction data. Five different machine-learning algorithms (Random Forest-RF, k Nearest Neighbors-kNN, Gaussian Naïve Bayes-GNB, Support Vector Machines with RBF kernel-SVM, and Deep-Learning-multi-layer feedforward artificial neural network-DL) have been trained on the coarse-grained statistics and each RNA-ligand complex is evaluated using a scoring function for the nativeness probability of the contacts and the ligand internal energy (202). Benchmark test with a set of 33 RNA-ligand complexes has shown that kNN and DL algorithms give the best results and more extensive tests with 4 docking methods and 9 scoring functions have demonstrated that AnnapuRNA outperformed other programs tested. The results has indicated that the coarse-grained representation combined with the concept of pharmacophores can indeed provide an effective, simplified way of feature engineering for RNA-ligand binding.
In summary, although machine-learning models for protein folding (245-250) and protein-small molecule interactions (72-75) have shown significant success, modeling RNA-small molecule interactions using machine learning is a relatively new adventure. The machine learning approaches have several intrinsic limitations. Because the model involves a large number of trainable parameters, the training process is prone to overfitting, especially for cases with only limited training data. The problem is more important for RNA-small molecule binding due to the lack of a comprehensive and high-quality curated database. Although protein-focused libraries, such as PDB (94), PDBbind (223; 235-239), can contain data for RNA-small molecule complexes, a more comprehensive, dedicated NDB-like database (251; 252) for RNA-ligand complexes is needed.
4.4. Accounting for solvent-mediated interactions
The sugar-phosphate backbone is negatively charged and polar, resulting in an accumulation of water molecules, cations, and water/ion-mediated RNA-ligand interactions. However, most molecular docking models do not explicitly consider the bridging effects of water molecules (Figure 2b) and metal ions (Figure 2c). The neglect of such solvent effects is a notable drawback that can cause inaccurate predictions for RNA-ligand interactions. A viable approach is to use simulations with explicit waters and/or ions to refine RNA structures (53; 55). Then, the positions of important water molecules can be retained for further docking against ligands. In this approach, the results are sensitive to the selection of the important water molecules. Because ligand-RNA interactions are sensitive to the positions and orientations of the water molecules and ions within the cavity space, achieving robust accuracy can be challenging with this approach.
An alternative approach is to predict the binding of water molecules and bound metal ions to the RNA prior to docking (253-260) then treat the predicted bound water molecules and/or ions as part of the receptor for RNA-small molecule docking. The Tightly Binding Ion (TBI) (256; 261; 262) model and the Monte Carlo TBI (MCTBI) model (260; 263) predict the ion distribution around an RNA structure. Through explicit sampling of the discrete ion distributions, TBI and MCTBI go beyond the mean-field Poisson-Boltzmann theory by accounting for the correlation between the different ions. 3D-RISM is another promising model for predicting the distribution of both solvent and ions around a given macromolecule (258). In combination with a force field calibrated by prototype ionophores, 3D-RISM is able to recapitulate the water distribution around guanine quadruplexes by considering the correlation between solvent particles and treating the system as macromolecules in equilibrium with a bulk solvent at constant composition (and chemical potential). For the Oxytricha nova telomeric G-quadruplex structure as a test case, 35% (80%) of the 3D-RISM-predicted water binding modes are within 1Å (2 Å) from the crystallographic modes, indicating that the model may be reliable for treating solvent effects (264). Splash’Em (Solvation Potential Laid around Statistical Hydration on Entire Macromolecules) is a model for predicting bridging water molecules in nucleic acid-ligand complexes. Using the statistical information of water molecules around nucleotides in the PDB (94) and a scoring function containing a hydrogen-bonding potential with both directionality and polarization, Splash’Em has identified 62% of water molecules in nucleic acid-ligand complexes within 1Å (265).
5. Current achievements of RNA-ligand docking models: a performance comparison
Table 3 summarizes the benchmark test results of various computational methods for RNA-ligand docking. The data is adopted from the original publications of the models and the results are grouped according to the test sets. Cautions should be taken when comparing the performance of the different models. First of all, RNAs and ligands in different test systems can have different structural and physical features, thus a direct performance comparison of the different models based on the results from different test systems may not be appropriate. Second, even for methods evaluated based on the same test dataset, the interpretation of the performance comparison can be complicated. For example, for the same benchmark dataset, the decoy poses generated for pose identification can be quite different for the different tests. A robust and objective comparison between the different scoring functions demands a consistent and systematic benchmark test protocol for the generation of the decoy binding poses (57).
Nevertheless, the benchmark test results in Table 3 provides useful insights for the selection of computational methods. First, for pose identification, we find a clear trend that RNA-specific methods consistently outperform those developed for proteins or generic macromolecules. The result shows the importance of considering RNA-specific interactions and structural features for RNA-ligand docking. Second, recent advances in RNA-ligand scoring function have been mainly focused on knowledge-based/machine learning-based approaches (141-143; 176). The knowledge-based/machine learning-based approaches provide equal or better performance (especially for affinity prediction) than the traditional physics-based/empirical approaches (59; 63; 65-67), except for MORDOR (64) and RLDOCK (70; 71). Third, even with the knowledge-based/machine learning-based approaches, the current success rate for affinity prediction is quite low. To improve the prediction accuracy, with the currently limited available data for RNA-ligand binding affinities and complex structures, new physics-based models that can accurately capture RNA-ligand interactions and conformational ensembles would be highly needed.
6. Non-docking based methods for modeling receptor-ligand binding
In addition to the scoring functions discussed above, other physics-based methods can also achieve high accuracy for determining the binding modes and affinities. Some of these methods, however, are not suitable for docking software due to either the expensive computational cost or the technical difficulty of incorporation of the method into a software. Because extensive reviews have been reported for free-energy methods (266), such as Molecular Mechanics/Poisson Boltzmann Surface Area (MM/PBSA) and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) (267-274), Linear Interaction Energy method (LIE) (275-277) Free-Energy Perturbation (FEP) (277; 278) and Thermodynamic Integration (TI) (272; 279), we here focus on several more recently developed physics-based methods for modeling small molecule binding problems, with the purpose of applying the methods to predict RNA-ligand binding.
Quantum mechanical methods.
Quantum mechanical approaches, which can treat polarization, charge transfer, and many-body effects, are considered to be more accurate than force-fields in molecular mechanics. Methods to model small molecule binding range from less accurate semi-empirical methods such as density functional theory (DFT) to more sophisticated methods, such as second-order Møller-Plesset perturbation theory (MP2), configuration interaction (CT) and strict coupled-cluster calculations (CC) (280). Improvements in computer hardware have substantially reduced the computational time for quantum mechanical calculations. However, the cost is still relatively high, and systems under investigation usually need to be significantly simplified or divided into smaller fragments. Although fragmentation calculation enables the use of quantum mechanics-based methods for computing the energies of large biomolecules, such as proteins (281-283), the method has not been extensively applied to RNA-small molecule interacting systems. The difficulties may stem from long-range electrostatic interactions and many-body quantum mechanical effects in RNAs (283; 284).
Chen et al. (285) have extended molecular fractionation with conjugate caps (MFCC) scheme using quantum mechanical calculations to study DNA/RNA-small molecule interactions. Through the division of the system at each phosphate group, three oligo-nucleic acid interaction systems are decomposed into smaller subsystems, and the calculated interaction energy is found to be in excellent agreement with the results obtained from ab initio calculation for the original full system. In another study, Mlýnský et al. (286) have compared the abilities of various hybrid QM/MM methods, including ab initio, DFT and semi-empirical approaches, to investigate the possible catalytic mechanism for the hairpin ribozyme. By using various hybrid QM/MM methods, Mlýnský et al. have computationally reconstructed potential and free energy surfaces for the catalysis reaction system. Among the tested methods, the activation barriers calculated from spin-component scaled Møller-Plesset (SCS-MP2) method and hybrid MPW1K functional show the best agreement with those derived from the experimental rate constant data. Recently, Bezerra et al. (68) have applied MFCC fragmentation scheme (287-289) within a density functional theory framework to calculate the interaction energy between ribosomal RNA and aminoglycoside hygromycin B. The calculation has revealed the regions where the drug molecule interacts strongly with the ribosome, and the result provides guidance for the improvement of drug-receptor affinity.
Compared to MM/MD based approaches, the quantum-mechanics approaches above are able to carry out more accurate calculations at the expense of expensive computational cost and hence the methods may not be suitable for large-scale virtual screening in drug discovery.
3D-RISM theory.
By treating the distribution of bound ligands around the receptor as a receptor-ligand two-body correlation problem, three-dimensional reference interaction site model (3D-RISM) with Kovalenko-Hirata (KH) closure relation predicts the spatial distribution function of the ligand in the field created by the receptor (290-292). The probable binding sites are identified from the peaks of the ligand spatial distribution function and the binding mode is determined based on the superposition approximation (69). The 3D-RISM/KH approach has three unique advantages. First, it identifies the ligand-binding site from the distribution function for the mixture of solvent and ligand. Because the calculation circumvents the sampling and scoring in the traditional docking process, the 3D-RISM/KH approach avoids the limitations of sampling methods. Second, the theory explicitly accounts for the solvent effects. Therefore, the theory can treat ion and water-mediated interactions in receptor-ligand binding. Third, unlike the traditional continuum models, 3D-RISM/KH calculation can take the hydrogen bond between the solute and solvents into consideration. Several studies (293-298) have employed 3D-RISM/KH theory to predict the binding sites and binding modes of small molecules in proteins. In a recent study, Sugita and coworkers (69) have tested their 3D-RISM/KH approach on 18 different types of proteins and successfully predicted the native binding modes for half of the systems.
However, the 3D-RISM/KH theory is not without limitations. First, there is an upper limit of the query small molecules for a computationally feasible calculation. In practice, a large ligand needs to be divided into fragments which are later re-connected based on the calculated distributions. Second, 3D-RISM/KH takes longer than traditional docking calculation to evaluate tens of thousands of ligands as drug candidates for a given target. Third, the theory is based on correlation functions and may not be able to account for certain discrete configurations and interactions that are important for an accurate prediction.
Kinetic effects.
For many drug molecules, binding equilibrium might not even be reached or maintained in the in vivo system. As a result, the thermodynamic equilibrium binding affinity may not be a proper indicator for the in vivo efficacy of the drug. In contrast, residence time, or the lifetime of the binary drug-target, complex, measured by the inverse of the unbinding rate constant koff, may be a better indicator for drug efficacy in vivo (299-303). Indeed, a growing amount of evidence points to the direct correlation between the residence time of a drug molecule and its in vivo efficacy (299-309); See Figure 7 for a schematic visualization of the binding process for systems with simple and complex transition and intermediate states.
Figure 7:

A simplified representation of the binding kinetics between the unbound receptor (R), unbound ligand (L) and the bound receptor-ligand complex (RL). (a) The binding kinetics of a system with only one transient state (TS) along the binding reaction coordinate. The figure shows a binding scenario where both receptor and ligand undergo conformational changes in the binding process. The kinetic residence time (i.e., the inverse of the RNA-ligand dissociation constant koff) depends on the free energy difference (ΔGoff) between the bound state and transient state, while the thermodynamic binding energy (ΔGbind) is determined by the free energy difference between the unbound state (R+L) and bound state (RL). (b) In practice, often the binding kinetic profile of a system contains multiple transient states (TS) and intermediate states (IS) with a much more complicated kinetic mechanism.
Currently, kinetic models such as unbiased MD, Markov state model, and weighted ensemble and metadynamics (MTD) have demonstrated the success of kinetic modeling in protein-targeted drug discovery (183; 302; 303; 310). Similar kinetic studies for RNA-ligand binding is expected to provide a novel strategy for RNA-targeted drug design (311).
7. Conclusions and future perspectives
The rapidly growing therapeutic interest in RNA-targeted drug discovery causes an increasing demand for computational tools for predicting RNA-ligand interactions. Virtual screening remains an important first step in novel drug design when only the targeted RNA information is available. Various docking (sampling) methods and scoring functions have been developed to accelerate this process and in the meantime, have deepened our understanding of RNA-ligand binding mechanisms. Physics-based and knowledge-based approaches have shown promising success in predicting ligand binding poses and binding affinities. However, powered by advanced algorithms, machine-learning methods, although still in their infancy for RNA-ligand docking, have begun to show highly encouraging improved performance compared to traditional approaches.
Computer-aided drug discovery has come a long way. Past efforts have been mainly protein-centered. New RNA-based therapeutic design and computational methods have emerged. With the growth of the database of known structures and kinetic and thermodynamic measurements (e.g., binding affinity), data-driven methods, especially machine-learning methods, are expected to play a more and more important role. Furthermore, with the appreciation of RNA-ligand binding kinetic effects on in vivo efficacy of the drug, kinetics-based models, although currently have not been fully explored for RNA-drug binding, would be developed at an accelerated pace. With the development of various computational tools developed for RNA-targeted drug discovery, a CASP- and D3R-like (312; 313) community-wide events with blind tests and well-curated benchmark datasets, similar to the benchmarks widely used in the protein-ligand modeling community (216; 314-317), would be much needed.
Acknowledgments
This work was supported by the National Institutes of Health under Grants R35-GM134919 and R01-GM117059 to S-J. C. The authors thank Travis Hurst for critical reading of the manuscript.
References
- [1].Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012. Sep;489(7414):101–8. Available from: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Warner KD, Hajdin CE, Weeks KM. Principles for targeting RNA with drug-like small molecules. Nature Reviews Drug Discovery. 2018. Aug;17(8):547–58. Available from: 10.1038/nrd.2018.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tessaro F, Scapozza L. How ‘Protein-Docking’ Translates into the New Emerging Field of Docking Small Molecules to Nucleic Acids? Molecules. 2020;25(12). Available from: https://www.mdpi.com/1420-3049/25/12/2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sharp PA. The Centrality of RNA. Cell. 2009;136(4):577–80. Available from: https://www.sciencedirect.com/science/article/pii/S0092867409001433. [DOI] [PubMed] [Google Scholar]
- [5].Chappell J, Takahashi MK, Meyer S, Loughrey D, Watters KE, Lucks J. The centrality of RNA for engineering gene expression. Biotechnology Journal. 2013;8(12):1379–95. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/biot.201300018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-Targeted Therapeutics. Cell Metabolism. 2018;27(4):714 739. Available from: http://www.sciencedirect.com/science/article/pii/S1550413118301827. [DOI] [PubMed] [Google Scholar]
- [7].Yin W, Rogge M. Targeting RNA: A Transformative Therapeutic Strategy. Clinical and Translational Science. 2019;12(2):98–112. Available from: https://ascpt.onlinelibrary.wiley.com/doi/abs/10.1111/cts.12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yu AM, Jian C, Yu AH, Tu MJ. RNA therapy: Are we using the right molecules? Pharmacology & Therapeutics. 2019;196:91 104. Available from: http://www.sciencedirect.com/science/article/pii/S016372581830216X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu AM, Choi YH, Tu MJ. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacological Reviews. 2020;72(4):862–98. Available from: https://pharmrev.aspetjournals.org/content/72/4/862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Connelly C, Moon M, Schneekloth J. The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chemical Biology. 2016;23(9):1077 1090. Available from: http://www.sciencedirect.com/science/article/pii/S2451945616302525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hermann T. Small molecules targeting viral RNA. WIREs RNA. 2016;7(6):726–43. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wrna.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Donlic A, Hargrove AE. Targeting RNA in mammalian systems with small molecules. WIREs RNA. 2018;9(4):e1477. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wrna.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Costales MG, Childs-Disney JL, Haniff HS, Disney MD. How We Think about Targeting RNA with Small Molecules. Journal of Medicinal Chemistry. 2020;63(17):8880–900. Available from: 10.1021/acs.jmedchem.9b01927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Meyer SM, Williams CC, Akahori Y, Tanaka T, Aikawa H, Tong Y, et al. Small molecule recognition of disease-relevant RNA structures. Chem Soc Rev. 2020;49:7167–99. Available from: 10.1039/D0CS00560F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shao Y, Zhang Q. Targeting RNA structures in diseases with small molecules. Essays in Biochemistry. 2020. Dec;64(6):955–66. Available from: 10.1042/EBC20200011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Clamp M, Fry B, Kamal M, Xie X, Cuff J, Lin MF, et al. Distinguishing protein-coding and noncoding genes in the human genome. Proceedings of the National Academy of Sciences. 2007;104(49):19428–33. Available from: https://www.pnas.org/content/104/49/19428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ezkurdia I, Juan D, Rodriguez JM, Frankish A, Diekhans M, Harrow J, et al. Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Human Molecular Genetics. 2014. June;23(22):5866–78. Available from: 10.1093/hmg/ddu309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Somody JC, MacKinnon SS, Windemuth A. Structural coverage of the proteome for pharmaceutical applications. Drug Discovery Today. 2017;22(12):1792–9. Available from: https://www.sciencedirect.com/science/article/pii/S1359644617301642. [DOI] [PubMed] [Google Scholar]
- [19].Hopkins AL, Groom CR. The druggable genome. Nature Reviews Drug Discovery. 2002. Sep;1(9):727–30. Available from: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- [20].Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature Reviews Drug Discovery. 2006. Dec;5(12):993–6. Available from: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- [21].Dixon SJ, Stockwell BR. Identifying druggable disease-modifying gene products. Current Opinion in Chemical Biology. 2009;13(5):549 555. Omics/Biopolymers/Model Systems. Available from: http://www.sciencedirect.com/science/article/pii/S1367593109001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Poehlsgaard J, Douthwaite S. The bacterial ribosome as a target for antibiotics. Nature Reviews Microbiology. 2005. Nov;3(11):870–81. Available from: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- [23].Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proceedings of the National Academy of Sciences. 2010;107(40):17158–63. Available from: https://www.pnas.org/content/107/40/17158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Deak D, Outterson K, Powers JH, Kesselheim AS. Progress in the Fight Against Multidrug-Resistant Bacteria? A Review of U.S. Food and Drug Administration–Approved Antibiotics, 2010–2015. Annals of Internal Medicine. 2016. Sep;165(5):363–72. Available from: https://www.acpjournals.org/doi/abs/10.7326/M16-0291. [DOI] [PubMed] [Google Scholar]
- [25].Lin J, Zhou D, Steitz TA, Polikanov YS, Gagnon MG. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Annual Review of Biochemistry. 2018;87(1):451–78. Available from: 10.1146/annurev-biochem-062917-011942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mandal M, Breaker RR. Gene regulation by riboswitches. Nature Reviews Molecular Cell Biology. 2004. Jun;5(6):451–63. Available from: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- [27].Tucker BJ, Breaker RR. Riboswitches as versatile gene control elements. Current Opinion in Structural Biology. 2005;15(3):342 348. Sequences and topology/Nucleic acids. Available from: http://www.sciencedirect.com/science/article/pii/S0959440X05000874. [DOI] [PubMed] [Google Scholar]
- [28].Blount KF, Breaker RR. Riboswitches as antibacterial drug targets. Nature Biotechnology. 2006. Dec;24(12):1558–64. Available from: 10.1038/nbt1268. [DOI] [PubMed] [Google Scholar]
- [29].Montange RK, Batey RT. Riboswitches: Emerging Themes in RNA Structure and Function. Annual Review of Biophysics. 2008;37(1):117–33. Available from: 10.1146/annurev.biophys.37.032807.130000. [DOI] [PubMed] [Google Scholar]
- [30].Roth A, Breaker RR. The Structural and Functional Diversity of Metabolite-Binding Riboswitches. Annual Review of Biochemistry. 2009;78(1):305–34. Available from: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Garst AD, Edwards AL, Batey RT. Riboswitches: Structures and Mechanisms. Cold Spring Harbor Perspectives in Biology. 2011;3(6). Available from: http://cshperspectives.cshlp.org/content/3/6/a003533.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Breaker RR. Riboswitches and the RNA World. Cold Spring Harbor Perspectives in Biology. 2012;4(2). Available from: http://cshperspectives.cshlp.org/content/4/2/a003566.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Serganov A, Nudler E. A Decade of Riboswitches. Cell. 2013;152(1):17 24. Available from: http://www.sciencedirect.com/science/article/pii/S0092867412015462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Peselis A, Serganov A. Themes and variations in riboswitch structure and function. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2014;1839(10):908 918. Riboswitches. Available from: http://www.sciencedirect.com/science/article/pii/S1874939914000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Berens C, Suess B. Riboswitch engineering — making the all-important second and third steps. Current Opinion in Biotechnology. 2015;31:10 15. Analytical Biotechnology. Available from: http://www.sciencedirect.com/science/article/pii/S0958166914001426. [DOI] [PubMed] [Google Scholar]
- [36].McCown PJ, Corbino KA, Stav S, Sherlock ME, Breaker RR. Riboswitch diversity and distribution. RNA. 2017;23(7):995–1011. Available from: http://rnajournal.cshlp.org/content/23/7/995.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hallberg ZF, Su Y, Kitto RZ, Hammond MC. Engineering and In Vivo Applications of Riboswitches. Annual Review of Biochemistry. 2017;86(1):515–39. Available from: 10.1146/annurev-biochem-060815-014628. [DOI] [PubMed] [Google Scholar]
- [38].Breaker RR. Riboswitches and Translation Control. Cold Spring Harbor Perspectives in Biology. 2018;10(11). Available from: http://cshperspectives.cshlp.org/content/10/11/a032797.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pavlova N, Kaloudas D, Penchovsky R. Riboswitch distribution, structure, and function in bacteria. Gene. 2019;708:38–48. Available from: https://www.sciencedirect.com/science/article/pii/S0378111919304998. [DOI] [PubMed] [Google Scholar]
- [40].Bédard ASV, Hien EDM, Lafontaine DA. Riboswitch regulation mechanisms: RNA, metabolites and regulatory proteins. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2020;1863(3):194501. Available from: https://www.sciencedirect.com/science/article/pii/S1874939919302834. [DOI] [PubMed] [Google Scholar]
- [41].Di Giorgio A, Duca M. Synthetic small-molecule RNA ligands: future prospects as therapeutic agents. Med Chem Commun. 2019;10:1242–55. Available from: 10.1039/C9MD00195F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nature Chemical Biology. 2011. Aug;7(8):553–9. Available from: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ganser LR, Lee J, Rangadurai A, Merriman DK, Kelly ML, Kansal AD, et al. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nature Structural & Molecular Biology. 2018. May;25(5):425–34. Available from: 10.1038/s41594-018-0062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Seth PP, Miyaji A, Jefferson EA, Sannes-Lowery KA, Osgood SA, Propp SS, et al. SAR by MS: Discovery of a New Class of RNA-Binding Small Molecules for the Hepatitis C Virus: Internal Ribosome Entry Site IIA Subdomain. Journal of Medicinal Chemistry. 2005;48(23):7099–102. Available from: 10.1021/jm050815o. [DOI] [PubMed] [Google Scholar]
- [45].Carnevali M, Parsons J, Wyles DL, Hermann T. A Modular Approach to Synthetic RNA Binders of the Hepatitis C Virus Internal Ribosome Entry Site. ChemBioChem. 2010;11(10):1364–7. Available from: https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/cbic.201000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Paulsen RB, Seth PP, Swayze EE, Griffey RH, Skalicky JJ, Cheatham TE, et al. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proceedings of the National Academy of Sciences. 2010;107(16):7263–8. Available from: https://www.pnas.org/content/107/16/7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dibrov SM, Ding K, Brunn ND, Parker MA, Bergdahl BM, Wyles DL, et al. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proceedings of the National Academy of Sciences. 2012;109(14):5223–8. Available from: https://www.pnas.org/content/109/14/5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dibrov SM, Parsons J, Carnevali M, Zhou S, Rynearson KD, Ding K, et al. Hepatitis C Virus Translation Inhibitors Targeting the Internal Ribosomal Entry Site. Journal of Medicinal Chemistry. 2014;57(5):1694–707. Available from: 10.1021/jm401312n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lee MK, Bottini A, Kim M, Bardaro MF, Zhang Z, Pellecchia M, et al. A novel small-molecule binds to the influenza A virus RNA promoter and inhibits viral replication. Chem Commun. 2014;50:368–70. Available from: 10.1039/C3CC46973E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bottini A, De SK, Wu B, Tang C, Varani G, Pellecchia M. Targeting Influenza A Virus RNA Promoter. Chemical Biology & Drug Design. 2015;86(4):663–73. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/cbdd.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Plant EP, Pérez-Alvarado GC, Jacobs JL, Mukhopadhyay B, Hennig M, Dinman JD. A Three-Stemmed mRNA Pseudoknot in the SARS Coronavirus Frameshift Signal. PLOS Biology. 2005. May;3(6). Available from: 10.1371/journal.pbio.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Su MC, Chang CT, Chu CH, Tsai CH, Chang KY. An atypical RNA pseudoknot stimulator and an upstream attenuation signal for −1 ribosomal frameshifting of SARS coronavirus. Nucleic Acids Research. 2005. January;33(13):4265–75. Available from: 10.1093/nar/gki731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Park SJ, Kim YG, Park HJ. Identification of RNA Pseudoknot-Binding Ligand That Inhibits the −1 Ribosomal Frameshifting of SARS-Coronavirus by Structure-Based Virtual Screening. Journal of the American Chemical Society. 2011;133(26):10094–100. Available from: 10.1021/ja1098325. [DOI] [PubMed] [Google Scholar]
- [54].Ritchie DB, Soong J, Sikkema WKA, Woodside MT. Anti-frameshifting Ligand Reduces the Conformational Plasticity of the SARS Virus Pseudoknot. Journal of the American Chemical Society. 2014;136(6):2196–9. Available from: 10.1021/ja410344b. [DOI] [PubMed] [Google Scholar]
- [55].Moitessier N, Westhof E, Hanessian S. Docking of Aminoglycosides to Hydrated and Flexible RNA. Journal of Medicinal Chemistry. 2006;49(3):1023–33. Available from: 10.1021/jm0508437. [DOI] [PubMed] [Google Scholar]
- [56].Li Y, Shen J, Sun X, Li W, Liu G, Tang Y. Accuracy Assessment of Protein-Based Docking Programs against RNA Targets. Journal of Chemical Information and Modeling. 2010;50(6):1134–46. Available from: 10.1021/ci9004157. [DOI] [PubMed] [Google Scholar]
- [57].Chen L, Calin GA, Zhang S. Novel Insights of Structure-Based Modeling for RNA-Targeted Drug Discovery. Journal of Chemical Information and Modeling. 2012;52(10):2741–53. Available from: 10.1021/ci300320t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Luo J, Wei W, Waldispühl J, Moitessier N. Challenges and current status of computational methods for docking small molecules to nucleic acids. European Journal of Medicinal Chemistry. 2019;168:414 425. Available from: http://www.sciencedirect.com/science/article/pii/S0223523419301606. [DOI] [PubMed] [Google Scholar]
- [59].Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. Journal of Molecular Biology. 1997;267(3):727 748. Available from: http://www.sciencedirect.com/science/article/pii/S0022283696908979. [DOI] [PubMed] [Google Scholar]
- [60].Morley SD, Afshar M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock®. Journal of Computer-Aided Molecular Design. 2004. Mar;18(3):189–208. Available from: 10.1023/B:JCAM.0000035199.48747.1e. [DOI] [PubMed] [Google Scholar]
- [61].Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. Journal of Medicinal Chemistry. 2004;47(7):1739–49. Available from: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- [62].Corbeil CR, Englebienne P, Moitessier N. Docking Ligands into Flexible and Solvated Macromolecules. 1. Development and Validation of FITTED 1.0. Journal of Chemical Information and Modeling. 2007;47(2):435–49. Available from: 10.1021/ci6002637. [DOI] [PubMed] [Google Scholar]
- [63].Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. Journal of Computational Chemistry. 2007;28(6):1145–52. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- [64].Guilbert C, James TL. Docking to RNA via Root-Mean-Square-Deviation-Driven Energy Minimization with Flexible Ligands and Flexible Targets. Journal of Chemical Information and Modeling. 2008;48(6):1257–68. Available from: 10.1021/ci8000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, et al. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA. 2009;15(6):1219–30. Available from: http://rnajournal.cshlp.org/content/15/6/1219.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010;31(2):455–61. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, et al. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLOS Computational Biology. 2014. April;10(4):1–7. Available from: 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bezerra KS, Fulco UL, Esmaile SC, Lima Neto JX, Machado LD, Freire VN, et al. Ribosomal RNA–Aminoglycoside Hygromycin B Interaction Energy Calculation within a Density Functional Theory Framework. The Journal of Physical Chemistry B. 2019;123(30):6421–9. Available from: 10.1021/acs.jpcb.9b04468. [DOI] [PubMed] [Google Scholar]
- [69].Sugita M, Hamano M, Kasahara K, Kikuchi T, Hirata F. New Protocol for Predicting the Ligand-Binding Site and Mode Based on the 3D-RISM/KH Theory. Journal of Chemical Theory and Computation. 2020;16(4):2864–76. Available from: 10.1021/acs.jctc.9b01069. [DOI] [PubMed] [Google Scholar]
- [70].Sun LZ, Jiang Y, Zhou Y, Chen SJ. RLDOCK: A New Method for Predicting RNA–Ligand Interactions. Journal of Chemical Theory and Computation. 2020;16(11):7173–83. Available from: 10.1021/acs.jctc.0c00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jiang Y, Chen SJ. RLDOCK method for predicting RNA-small molecule binding modes. Methods. 2021. Available from: https://www.sciencedirect.com/science/article/pii/S1046202321000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sousa SF, Fernandes PA, Ramos MJ. Protein–ligand docking: Current status and future challenges. Proteins: Structure, Function, and Bioinformatics. 2006;65(1):15–26. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- [73].Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein–ligand docking: recent advances and future directions. Phys Chem Chem Phys. 2010;12:12899–908. Available from: 10.1039/C0CP00151A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Colwell LJ. Statistical and machine learning approaches to predicting protein–ligand interactions. Current Opinion in Structural Biology. 2018;49:123 128. Theory and simulation Macromolecular assemblies. Available from: http://www.sciencedirect.com/science/article/pii/S0959440X17301525. [DOI] [PubMed] [Google Scholar]
- [75].Li J, Fu A, Zhang L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdisciplinary Sciences: Computational Life Sciences. 2019. Jun;11(2):320–8. Available from: 10.1007/s12539-019-00327-w. [DOI] [PubMed] [Google Scholar]
- [76].Hewitt WM, Calabrese DR, Schneekloth JS. Evidence for ligandable sites in structured RNA throughout the Protein Data Bank. Bioorganic & Medicinal Chemistry. 2019;27(11):2253–60. Available from: https://www.sciencedirect.com/science/article/pii/S0968089618321473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Velagapudi SP, Gallo SM, Disney MD. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nature Chemical Biology. 2014. Apr;10(4):291–7. Available from: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M, Childs-Disney JL. Inforna 2.0: A Platform for the Sequence-Based Design of Small Molecules Targeting Structured RNAs. ACS Chemical Biology. 2016;11(6):1720–8. Available from: 10.1021/acschembio.6b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Velagapudi S, Seedhouse S, Disney M. Structure-Activity Relationships through Sequencing (StARTS) Defines Optimal and Suboptimal RNA Motif Targets for Small Molecules. Angewandte Chemie International Edition. 2010;49(22):3816–8. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.200907257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Velagapudi SP, Seedhouse SJ, French J, Disney MD. Defining the RNA Internal Loops Preferred by Benzimidazole Derivatives via 2D Combinatorial Screening and Computational Analysis. Journal of the American Chemical Society. 2011;133(26):10111–8. Available from: 10.1021/ja200212b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Velagapudi SP, Luo Y, Tran T, Haniff HS, Nakai Y, Fallahi M, et al. Defining RNA-Small Molecule Affinity Landscapes Enables Design of a Small Molecule Inhibitor of an Oncogenic Noncoding RNA. ACS Central Science. 2017;3(3):205–16. Available from: 10.1021/acscentsci.7b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Disney MD, Velagapudi SP, Li Y, Costales MG, Childs-Disney JL. Chapter Three - Identifying and validating small molecules interacting with RNA (SMIRNAs). In: Hargrove AE, editor. RNA Recognition. vol. 623 of Methods in Enzymology. Academic Press; 2019. p. 45–66. Available from: https://www.sciencedirect.com/science/article/pii/S0076687919301429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhang P, Park HJ, Zhang J, Junn E, Andrews RJ, Velagapudi SP, et al. Translation of the intrinsically disordered protein α-synuclein is inhibited by a small molecule targeting its structured mRNA. Proceedings of the National Academy of Sciences. 2020;117(3):1457–67. Available from: https://www.pnas.org/content/117/3/1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kim WS, Kågedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Research & Therapy. 2014. Oct;6(5):73. Available from: 10.1186/s13195-014-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. Journal of Molecular Biology. 1986;188(3):415–31. Available from: https://www.sciencedirect.com/science/article/pii/0022283686901658. [DOI] [PubMed] [Google Scholar]
- [86].Carothers JM, Oestreich SC, Davis JH, Szostak JW. Informational Complexity and Functional Activity of RNA Structures. Journal of the American Chemical Society. 2004;126(16):5130–7. Available from: 10.1021/ja031504a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].An J, Totrov M, Abagyan R. Pocketome via Comprehensive Identification and Classification of Ligand Binding Envelopes*. Molecular & Cellular Proteomics. 2005;4(6):752–61. Available from: https://www.sciencedirect.com/science/article/pii/S1535947620314742. [DOI] [PubMed] [Google Scholar]
- [88].Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA Structure Analysis at Single Nucleotide Resolution by Selective 2’-Hydroxyl Acylation and Primer Extension (SHAPE). Journal of the American Chemical Society. 2005;127(12):4223–31. Available from: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- [89].Mortimer SA, Trapnell C, Aviran S, Pachter L, Lucks JB. SHAPE-Seq: High-Throughput RNA Structure Analysis. Current Protocols in Chemical Biology. 2012;4(4):275–97. Available from: https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/9780470559277.ch120019. [DOI] [PubMed] [Google Scholar]
- [90].Loughrey D, Watters KE, Settle AH, Lucks JB. SHAPE-Seq 2.0: systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing. Nucleic Acids Research. 2014. October;42(21):e165–5. Available from: 10.1093/nar/gku909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nature Methods. 2014. Sep;11(9):959–65. Available from: 10.1038/nmeth.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Carothers JM, Davis JH, Chou JJ, Szostak JW. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA. 2006;12(4):567–79. Available from: http://rnajournal.cshlp.org/content/12/4/567.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Velagapudi SP, Cameron MD, Haga CL, Rosenberg LH, Lafitte M, Duckett DR, et al. Design of a small molecule against an oncogenic noncoding RNA. Proceedings of the National Academy of Sciences. 2016;113(21):5898–903. Available from: https://www.pnas.org/content/113/21/5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Research. 2000. January;28(1):235–42. Available from: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. Journal of Molecular Biology. 1982;161(2):269–88. Available from: https://www.sciencedirect.com/science/article/pii/002228368290153X. [DOI] [PubMed] [Google Scholar]
- [96].Harris R, Olson AJ, Goodsell DS. Automated prediction of ligand-binding sites in proteins. Proteins: Structure, Function, and Bioinformatics. 2008;70(4):1506–17. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.21645. [DOI] [PubMed] [Google Scholar]
- [97].Abagyan R, Totrov M, Kuznetsov D. ICM-A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. Journal of Computational Chemistry. 1994;15(5):488–506. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.540150503. [Google Scholar]
- [98].Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19(14):1639–62. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/%28SICI%291096-987X%2819981115%2919%3A14%3C1639%3A%3AAID-JCC10%3E3.0.CO%3B2-B. [Google Scholar]
- [99].Zeng P, Li J, Ma W, Cui Q. Rsite: a computational method to identify the functional sites of noncoding RNAs. Scientific Reports. 2015. Mar;5(1):9179. Available from: 10.1038/srep09179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zeng P, Cui Q. Rsite2: an efficient computational method to predict the functional sites of noncoding RNAs. Scientific Reports. 2016. Jan;6(1):19016. Available from: 10.1038/srep19016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang K, Jian Y, Wang H, Zeng C, Zhao Y. RBind: computational network method to predict RNA binding sites. Bioinformatics. 2018. April;34(18):3131–6. Available from: 10.1093/bioinformatics/bty345. [DOI] [PubMed] [Google Scholar]
- [102].Wang H, Zhao Y. RBinds: A user-friendly server for RNA binding site prediction. Computational and Structural Biotechnology Journal. 2020;18:3762 3765. Available from: http://www.sciencedirect.com/science/article/pii/S2001037020304657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Su H, Peng Z, Yang J. Recognition of small molecule–RNA binding sites using RNA sequence and structure. Bioinformatics. 2021. January. Btaa1092. Available from: 10.1093/bioinformatics/btaa1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Jiménez J, Doerr S, Martínez-Rosell G, Rose AS, De Fabritiis G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics. 2017. May;33(19):3036–42. Available from: 10.1093/bioinformatics/btx350. [DOI] [PubMed] [Google Scholar]
- [105].Krivák R, Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics. 2018. Aug;10(1):39. Available from: 10.1186/s13321-018-0285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kozlovskii I, Popov P. Spatiotemporal identification of druggable binding sites using deep learning. Communications Biology. 2020. Oct;3(1):618. Available from: 10.1038/s42003-020-01350-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zhao J, Cao Y, Zhang L. Exploring the computational methods for protein-ligand binding site prediction. Computational and Structural Biotechnology Journal. 2020;18:417–26. Available from: https://www.sciencedirect.com/science/article/pii/S2001037019304465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Al-Hashimi HM, Walter NG. RNA dynamics: it is about time. Current Opinion in Structural Biology. 2008;18(3):321–9. Nucleic acids / Sequences and topology. Available from: https://www.sciencedirect.com/science/article/pii/S0959440X08000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ganser LR, Kelly ML, Herschlag D, Al-Hashimi HM. The roles of structural dynamics in the cellular functions of RNAs. Nature Reviews Molecular Cell Biology. 2019. Aug;20(8):474–89. Available from: 10.1038/s41580-019-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Scull CE, Dandpat SS, Romero RA, Walter NG. Transcriptional Riboswitches Integrate Timescales for Bacterial Gene Expression Control. Frontiers in Molecular Biosciences. 2021;7:480. Available from: https://www.frontiersin.org/article/10.3389/fmolb.2020.607158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zhao P, Zhang W, Chen SJ. Cotranscriptional folding kinetics of ribonucleic acid secondary structures. The Journal of Chemical Physics. 2011;135(24):245101. Available from: 10.1063/1.3671644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Krüger DM, Bergs J, Kazemi S, Gohlke H. Target Flexibility in RNA-Ligand Docking Modeled by Elastic Potential Grids. ACS Medicinal Chemistry Letters. 2011;2(7):489–93. Available from: 10.1021/ml100217h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Schroeder GM, Dutta D, Cavender CE, Jenkins J, Pritchett EM, Baker CD, et al. Analysis of a preQ1-I riboswitch in effector-free and bound states reveals a metabolite-programmed nucleobase-stacking spine that controls gene regulation. Nucleic Acids Research. 2020. June;48(14):8146–64. Available from: 10.1093/nar/gkaa546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jenkins JL, Krucinska J, McCarty RM, Bandarian V, Wedekind JE. Comparison of a PreQ1 Riboswitch Aptamer in Metabolite-bound and Free States with Implications for Gene Regulation*. Journal of Biological Chemistry. 2011;286(28):24626–37. Available from: https://www.sciencedirect.com/science/article/pii/S0021925819486262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].François B, Russell RJM, Murray JB, Aboul-ela F, Masquida B, Vicens Q, et al. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Research. 2005. January;33(17):5677–90. Available from: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Edwards TE, Ferré-D’Amaré AR. Crystal Structures of the Thi-Box Riboswitch Bound to Thiamine Pyrophosphate Analogs Reveal Adaptive RNA-Small Molecule Recognition. Structure. 2006;14(9):1459–68. Available from: https://www.sciencedirect.com/science/article/pii/S0969212606003303. [DOI] [PubMed] [Google Scholar]
- [117].Tok JBH, Bi L. Aminoglycoside and its Derivatives as Ligands to Target the Ribosome. Current Topics in Medicinal Chemistry. 2003;3(9):1001–19. Available from: https://sci-hub.se/http://www.eurekaselect.com/node/81184/article. [DOI] [PubMed] [Google Scholar]
- [118].Daldrop P, Reyes F, Robinson D, Hammond C, Lilley D, Batey R, et al. Novel Ligands for a Purine Riboswitch Discovered by RNA-Ligand Docking. Chemistry & Biology. 2011;18(3):324–35. Available from: https://www.sciencedirect.com/science/article/pii/S1074552111000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Göertz GP, Abbo SR, Fros JJ, Pijlman GP. Functional RNA during Zika virus infection. Virus Research. 2018;254:41–53. Advances in ZIKA Research. Available from: https://www.sciencedirect.com/science/article/pii/S016817021730521X. [DOI] [PubMed] [Google Scholar]
- [120].Rangan R, Zheludev IN, Hagey RJ, Pham EA, Wayment-Steele HK, Glenn JS, et al. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: a first look. RNA. 2020;26(8):937–59. Available from: http://rnajournal.cshlp.org/content/26/8/937.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Halgren T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chemical Biology & Drug Design. 2007;69(2):146–8. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- [122].DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. Journal of Medicinal Chemistry. 1988;31(4):722–9. Available from: 10.1021/jm00399a006. [DOI] [PubMed] [Google Scholar]
- [123].Rognan D. 6. In: Docking Methods for Virtual Screening: Principles and Recent Advances. John Wiley & Sons, Ltd; 2011. p. 153–76. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/9783527633326.ch6. [Google Scholar]
- [124].Waszkowycz B, Clark DE, Gancia E. Outstanding challenges in protein–ligand docking and structure-based virtual screening. WIREs Computational Molecular Science. 2011;1(2):229–59. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.18. [Google Scholar]
- [125].Wehler T, Brenk R. In: Garner AL, editor. Structure-Based Discovery of Small Molecules Binding to RNA. Cham: Springer International Publishing; 2018. p. 47–77. Available from: 10.1007/7355_2016_29. [DOI] [Google Scholar]
- [126].Kearsley SK, Underwood DJ, Sheridan RP, Miller MD. Flexibases: A way to enhance the use of molecular docking methods. Journal of Computer-Aided Molecular Design. 1994. Oct;8(5):565–82. Available from: 10.1007/BF00123666. [DOI] [PubMed] [Google Scholar]
- [127].OMEGA 4.1.0.0: OpenEye Scientific Software. Santa Fe, NM;. Available from: http://www.eyesopen.com. [Google Scholar]
- [128].Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. Journal of Chemical Information and Modeling. 2010;50(4):572–84. Available from: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].RDKit: Open-Source Cheminformatics Software;. Available from: http://www.rdkit.org.
- [130].Yoshikawa N, Hutchison GR. Fast, efficient fragment-based coordinate generation for Open Babel. Journal of Cheminformatics. 2019. Aug;11(1):49. Available from: 10.1186/s13321-019-0372-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Hawkins PCD. Conformation Generation: The State of the Art. Journal of Chemical Information and Modeling. 2017;57(8):1747–56. Available from: 10.1021/acs.jcim.7b00221. [DOI] [PubMed] [Google Scholar]
- [132].Moreno D, Zivanovic S, Colizzi F, Hospital A, Aranda J, Soliva R, et al. DFFR: A New Method for High-Throughput Recalibration of Automatic Force-Fields for Drugs. Journal of Chemical Theory and Computation. 2020;16(10):6598–608. Available from: 10.1021/acs.jctc.0c00306. [DOI] [PubMed] [Google Scholar]
- [133].Zivanovic S, Colizzi F, Moreno D, Hospital A, Soliva R, Orozco M. Exploring the Conformational Landscape of Bioactive Small Molecules. Journal of Chemical Theory and Computation. 2020;16(10):6575–85. Available from: 10.1021/acs.jctc.0c00304. [DOI] [PubMed] [Google Scholar]
- [134].Zivanovic S, Bayarri G, Colizzi F, Moreno D, Gelpi JL, Soliva R, et al. Bioactive Conformational Ensemble Server and Database. A Public Framework to Speed Up In Silico Drug Discovery. Journal of Chemical Theory and Computation. 2020;16(10):6586–97. Available from: 10.1021/acs.jctc.0c00305. [DOI] [PubMed] [Google Scholar]
- [135].Pikovskaya O, Polonskaia A, Patel DJ, Serganov A. Structural principles of nucleoside selectivity in a 2’-deoxyguanosine riboswitch. Nature Chemical Biology. 2011. Oct;7(10):748–55. Available from: 10.1038/nchembio.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Barbault F, Zhang L, Zhang L, Fan BT. Parametrization of a specific free energy function for automated docking against RNA targets using neural networks. Chemometrics and Intelligent Laboratory Systems. 2006;82(1):269–75. Selected Papers from the International Conference on Chemometrics and Bioinformatics in Asia. Available from: https://www.sciencedirect.com/science/article/pii/S0169743905001280. [Google Scholar]
- [137].Detering C, Varani G. Validation of Automated Docking Programs for Docking and Database Screening against RNA Drug Targets. Journal of Medicinal Chemistry. 2004;47(17):4188–201. Available from: 10.1021/jm030650o. [DOI] [PubMed] [Google Scholar]
- [138].Tongcheng C, Tonghua L. A combination of numeric genetic algorithm and tabu search can be applied to molecular docking. Computational Biology and Chemistry. 2004;28(4):303–12. Available from: https://www.sciencedirect.com/science/article/pii/S1476927104000623. [Google Scholar]
- [139].Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery. 2004. Nov;3(11):935–49. Available from: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- [140].Flick J, Tristram F, Wenzel W. Modeling loop backbone flexibility in receptor-ligand docking simulations. Journal of Computational Chemistry. 2012;33(31):2504–15. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.23087. [DOI] [PubMed] [Google Scholar]
- [141].Chhabra S, Xie J, Frank AT. RNAPosers: Machine Learning Classifiers for Ribonucleic Acid–Ligand Poses. The Journal of Physical Chemistry B. 2020;124(22):4436–45. Available from: 10.1021/acs.jpcb.0c02322. [DOI] [PubMed] [Google Scholar]
- [142].Feng Y, Huang SY. ITScore-NL: An Iterative Knowledge-Based Scoring Function for Nucleic Acid–Ligand Interactions. Journal of Chemical Information and Modeling. 2020;60(12):6698–708. Available from: 10.1021/acs.jcim.0c00974. [DOI] [PubMed] [Google Scholar]
- [143].Stefaniak F, Bujnicki JM. AnnapuRNA: A scoring function for predicting RNA-small molecule binding poses. PLOS Computational Biology. 2021. February;17(2):1–31. Available from: 10.1371/journal.pcbi.1008309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Current Opinion in Structural Biology. 2008;18(2):178–84. Theory and simulation / Macromolecular assemblages. Available from: https://www.sciencedirect.com/science/article/pii/S0959440X08000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Aboul-ela F, Karn J, Varani G. Structure of HIV-1 TAR RNA in the Absence of Ligands Reveals a Novel Conformation of the Trinucleotide Bulge. Nucleic Acids Research. 1996. October;24(20):3974–81. Available from: 10.1093/nar/24.20.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Ferrari AM, Wei BQ, Costantino L, Shoichet BK. Soft Docking and Multiple Receptor Conformations in Virtual Screening. Journal of Medicinal Chemistry. 2004;47(21):5076–84. Available from: 10.1021/jm049756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Huang SY, Zou X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins: Structure, Function, and Bioinformatics. 2007;66(2):399–421. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- [148].Amaro RE, Baudry J, Chodera J, Özlem Demir, McCammon JA, Miao Y, et al. Ensemble Docking in Drug Discovery. Biophysical Journal. 2018;114(10):2271–8. Available from: https://www.sciencedirect.com/science/article/pii/S0006349518303242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Santner T, Rieder U, Kreutz C, Micura R. Pseudoknot Preorganization of the PreQ1 Class I Riboswitch. Journal of the American Chemical Society. 2012;134(29):11928–31. Available from: 10.1021/ja3049964. [DOI] [PubMed] [Google Scholar]
- [150].Reining A, Nozinovic S, Schlepckow K, Buhr F, Fürtig B, Schwalbe H. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature. 2013. Jul;499(7458):355–9. Available from: 10.1038/nature12378. [DOI] [PubMed] [Google Scholar]
- [151].Zhao B, Hansen AL, Zhang Q. Characterizing Slow Chemical Exchange in Nucleic Acids by Carbon CEST and Low Spin-Lock Field R1ρ NMR Spectroscopy. Journal of the American Chemical Society. 2014;136(1):20–3. Available from: 10.1021/ja409835y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, et al. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society. 1995;117(19):5179–97. Available from: 10.1021/ja00124a002. [DOI] [Google Scholar]
- [153].Case DA, Cheatham TE III, Darden T, Gohlke H, Luo R, Merz KM Jr, et al. The Amber biomolecular simulation programs. Journal of Computational Chemistry. 2005;26(16):1668–88. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: The biomolecular simulation program. Journal of Computational Chemistry. 2009;30(10):1545–614. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Denning EJ, Priyakumar UD, Nilsson L, Mackerell AD Jr. Impact of 2’-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. Journal of Computational Chemistry. 2011;32(9):1929–43. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, et al. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. European Biophysics Journal. 2011. Apr;40(7):843. Available from: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- [157].Šponer J, Bussi G, Krepl M, Banáš P, Bottaro S, Cunha RA, et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chemical Reviews. 2018;118(8):4177–338. Available from: 10.1021/acs.chemrev.7b00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Banáš P, Sklenovský P, Wedekind JE, Šponer J, Otyepka M. Molecular Mechanism of preQ1 Riboswitch Action: A Molecular Dynamics Study. The Journal of Physical Chemistry B. 2012;116(42):12721–34. Available from: 10.1021/jp309230v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Kührová P, Mlýnský V, Zgarbová M, Krepl M, Bussi G, Best RB, et al. Improving the Performance of the Amber RNA Force Field by Tuning the Hydrogen-Bonding Interactions. Journal of Chemical Theory and Computation. 2019;15(5):3288–305. Available from: 10.1021/acs.jctc.8b00955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Haller A, Soulière MF, Micura R. The Dynamic Nature of RNA as Key to Understanding Riboswitch Mechanisms. Accounts of Chemical Research. 2011;44(12):1339–48. Available from: 10.1021/ar200035g. [DOI] [PubMed] [Google Scholar]
- [161].Sharma M, Bulusu G, Mitra A. MD simulations of ligand-bound and ligand-free aptamer: Molecular level insights into the binding and switching mechanism of the add A-riboswitch. RNA. 2009;15(9):1673–92. Available from: http://rnajournal.cshlp.org/content/15/9/1673.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Villa A, Wöhnert J, Stock G. Molecular dynamics simulation study of the binding of purine bases to the aptamer domain of the guanine sensing riboswitch. Nucleic Acids Research. 2009. June;37(14):4774–86. Available from: 10.1093/nar/gkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Priyakumar UD, MacKerell AD. Role of the Adenine Ligand on the Stabilization of the Secondary and Tertiary Interactions in the Adenine Riboswitch. Journal of Molecular Biology. 2010;396(5):1422–38. Available from: https://www.sciencedirect.com/science/article/pii/S0022283609015290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Gong Z, Zhao Y, Chen C, Xiao Y. Role of Ligand Binding in Structural Organization of Add A-riboswitch Aptamer: A Molecular Dynamics Simulation. Journal of Biomolecular Structure and Dynamics. 2011;29(2):403–16. Available from: 10.1080/07391102.2011.10507394. [DOI] [PubMed] [Google Scholar]
- [165].Di Palma F, Colizzi F, Bussi G. Ligand-induced stabilization of the aptamer terminal helix in the add adenine riboswitch. RNA. 2013;19(11):1517–24. Available from: http://rnajournal.cshlp.org/content/19/11/1517.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Nguyen PH, Derreumaux P, Stock G. Energy Flow and Long-Range Correlations in Guanine-Binding Riboswitch: A Nonequilibrium Molecular Dynamics Study. The Journal of Physical Chemistry B. 2009;113(27):9340–7. Available from: 10.1021/jp902013s. [DOI] [PubMed] [Google Scholar]
- [167].Allnér O, Nilsson L, Villa A. Loop-loop interaction in an adenine-sensing riboswitch: A molecular dynamics study. RNA. 2013;19(7):916–26. Available from: http://rnajournal.cshlp.org/content/19/7/916.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Bao L, Wang J, Xiao Y. Molecular dynamics simulation of the binding process of ligands to the add adenine riboswitch aptamer. Phys Rev E. 2019. Aug;100:022412. Available from: https://link.aps.org/doi/10.1103/PhysRevE.100.022412. [DOI] [PubMed] [Google Scholar]
- [169].Gómez Pinto I, Guilbert C, Ulyanov NB, Stearns J, James TL. Discovery of Ligands for a Novel Target, the Human Telomerase RNA, Based on Flexible-Target Virtual Screening and NMR. Journal of Medicinal Chemistry. 2008;51(22):7205–15. Available from: 10.1021/jm800825n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Bissaro M, Sturlese M, Moro S. Exploring the RNA-Recognition Mechanism Using Supervised Molecular Dynamics (SuMD) Simulations: Toward a Rational Design for Ribonucleic-Targeting Molecules? Frontiers in Chemistry. 2020;8:107. Available from: https://www.frontiersin.org/article/10.3389/fchem.2020.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kazemi S, Krüger D, Sirockin F, Gohlke H. Elastic Potential Grids: Accurate and Efficient Representation of Intermolecular Interactions for Fully Flexible Docking. ChemMedChem. 2009;4(8):1264–8. Available from: https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/cmdc.200900146. [DOI] [PubMed] [Google Scholar]
- [172].Pfeffer P, Gohlke H. DrugScoreRNAKnowledge-Based Scoring Function To Predict RNA-Ligand Interactions. Journal of Chemical Information and Modeling. 2007;47(5):1868–76. Available from: 10.1021/ci700134p. [DOI] [PubMed] [Google Scholar]
- [173].Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. Journal of Molecular Biology. 2000;295(2):337 356. Available from: http://www.sciencedirect.com/science/article/pii/S0022283699933715. [DOI] [PubMed] [Google Scholar]
- [174].Zhao X, Liu X, Wang Y, Chen Z, Kang L, Zhang H, et al. An Improved PMF Scoring Function for Universally Predicting the Interactions of a Ligand with Protein, DNA, and RNA. Journal of Chemical Information and Modeling. 2008;48(7):1438–47. Available from: 10.1021/ci7004719. [DOI] [PubMed] [Google Scholar]
- [175].Philips A, Milanowska K, Lach G, Bujnicki JM. LigandRNA: computational predictor of RNA–ligand interactions. RNA. 2013;19(12):1605–16. Available from: http://rnajournal.cshlp.org/content/19/12/1605.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Yan Z, Wang J. SPA-LN: a scoring function of ligand-nucleic acid interactions via optimizing both specificity and affinity. Nucleic Acids Research. 2017. April;45(12):e110–0. Available from: 10.1093/nar/gkx255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Cang Z, Wei GW. Integration of element specific persistent homology and machine learning for protein-ligand binding affinity prediction. International Journal for Numerical Methods in Biomedical Engineering. 2018;34(2):e2914. E2914 cnm.2914. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/cnm.2914. [DOI] [PubMed] [Google Scholar]
- [178].Oliver C, Mallet V, Gendron RS, Reinharz V, Hamilton W, Moitessier N, et al. Augmented base pairing networks encode RNA-small molecule binding preferences. Nucleic Acids Research. 2020. July;48(14):7690–9. Available from: 10.1093/nar/gkaa583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].De Vivo M, Cavalli A. Recent advances in dynamic docking for drug discovery. WIREs Computational Molecular Science. 2017;7(6):e1320. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1320. [Google Scholar]
- [180].Ganesan A, Coote ML, Barakat K. Molecular dynamics-driven drug discovery: leaping forward with confidence. Drug Discovery Today. 2017;22(2):249 269. Available from: http://www.sciencedirect.com/science/article/pii/S1359644616304147. [DOI] [PubMed] [Google Scholar]
- [181].Katiyar RS, Jha PK. Molecular simulations in drug delivery: Opportunities and challenges. WIREs Computational Molecular Science. 2018;8(4):e1358. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1358. [Google Scholar]
- [182].Liu X, Shi D, Zhou S, Liu H, Liu H, Yao X. Molecular dynamics simulations and novel drug discovery. Expert Opinion on Drug Discovery. 2018;13(1):23–37. Available from: 10.1080/17460441.2018.1403419. [DOI] [PubMed] [Google Scholar]
- [183].Decherchi S, Cavalli A. Thermodynamics and Kinetics of Drug-Target Binding by Molecular Simulation. Chemical Reviews. 2020;120(23):12788–833. Available from: 10.1021/acs.chemrev.0c00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Warwicker J, Watson HC. Calculation of the electric potential in the active site cleft due to α-helix dipoles. Journal of Molecular Biology. 1982;157(4):671–9. Available from: https://www.sciencedirect.com/science/article/pii/0022283682905058. [DOI] [PubMed] [Google Scholar]
- [185].Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proceedings of the National Academy of Sciences. 2001;98(18):10037–41. Available from: https://www.pnas.org/content/98/18/10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Grant JA, Pickup BT, Nicholls A. A smooth permittivity function for Poisson–Boltzmann solvation methods. Journal of Computational Chemistry. 2001;22(6):608–40. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.1032. [Google Scholar]
- [187].Rocchia W, Sridharan S, Nicholls A, Alexov E, Chiabrera A, Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: Applications to the molecular systems and geometric objects. Journal of Computational Chemistry. 2002;23(1):128–37. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- [188].Morreale A, Gil-Redondo R, Ortiz Ár. A new implicit solvent model for protein–ligand docking. Proteins: Structure, Function, and Bioinformatics. 2007;67(3):606–16. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.21269. [DOI] [PubMed] [Google Scholar]
- [189].Ren P, Chun J, Thomas DG, Schnieders MJ, Marucho M, Zhang J, et al. Biomolecular electrostatics and solvation: a computational perspective. Quarterly Reviews of Biophysics. 2012;45(4):427–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Wang C, Greene D, Xiao L, Qi R, Luo R. Recent Developments and Applications of the MMPBSA Method. Frontiers in Molecular Biosciences. 2018;4:87. Available from: https://www.frontiersin.org/article/10.3389/fmolb.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. Journal of the American Chemical Society. 1990;112(16):6127–9. [Google Scholar]
- [192].Hawkins GD, Cramer CJ, Truhlar DG. Pairwise solute descreening of solute charges from a dielectric medium. Chemical Physics Letters. 1995;246(1):122 129. Available from: http://www.sciencedirect.com/science/article/pii/000926149501082K. [Google Scholar]
- [193].Hawkins GD, Cramer CJ, Truhlar DG. Parametrized Models of Aqueous Free Energies of Solvation Based on Pairwise Descreening of Solute Atomic Charges from a Dielectric Medium. The Journal of Physical Chemistry. 1996;100(51):19824–39. Available from: 10.1021/jp961710n. [DOI] [Google Scholar]
- [194].Qiu D, Shenkin PS, Hollinger FP, Still WC. The GB/SA Continuum Model for Solvation. A Fast Analytical Method for the Calculation of Approximate Born Radii. The Journal of Physical Chemistry A. 1997;101(16):3005–14. Available from: 10.1021/jp961992r. [DOI] [Google Scholar]
- [195].Onufriev A, Bashford D, Case DA. Modification of the Generalized Born Model Suitable for Macromolecules. The Journal of Physical Chemistry B. 2000;104(15):3712–20. Available from: 10.1021/jp994072s. [DOI] [Google Scholar]
- [196].Tsui V, Case DA. Molecular Dynamics Simulations of Nucleic Acids with a Generalized Born Solvation Model. Journal of the American Chemical Society. 2000;122(11):2489–98. Available from: 10.1021/ja9939385. [DOI] [Google Scholar]
- [197].Feig M, Im W, Brooks CL. Implicit solvation based on generalized Born theory in different dielectric environments. The Journal of Chemical Physics. 2004;120(2):903–11. Available from: 10.1063/1.1631258. [DOI] [PubMed] [Google Scholar]
- [198].Liu HY, Kuntz ID, Zou X. Pairwise GB/SA Scoring Function for Structure-based Drug Design. The Journal of Physical Chemistry B. 2004;108(17):5453–62. Available from: 10.1021/jp0312518. [DOI] [Google Scholar]
- [199].Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins: Structure, Function, and Bioinformatics. 2004;55(2):383–94. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- [200].Im W, Lee MS, Brooks CL III. Generalized born model with a simple smoothing function. Journal of Computational Chemistry. 2003;24(14):1691–702. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- [201].MacKerell AD Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56(4):257–65. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/1097-0282%282000%2956%3A4%3C257%3A%3AAID-BIP10029%3E3.0.CO%3B2-W. [DOI] [PubMed] [Google Scholar]
- [202].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. Journal of Computational Chemistry. 2004;25(9):1157–74. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- [203].Philips A, Lach G, Bujnicki JM. Chapter Eleven - Computational Methods for Prediction of RNA Interactions with Metal Ions and Small Organic Ligands. In: Chen SJ, Burke-Aguero DH, editors. Computational Methods for Understanding Riboswitches. vol. 553 of Methods in Enzymology. Academic Press; 2015. p. 261–85. Available from: https://www.sciencedirect.com/science/article/pii/S0076687914000585. [DOI] [PubMed] [Google Scholar]
- [204].Muegge I, Martin YC. A General and Fast Scoring Function for Protein-Ligand Interactions: A Simplified Potential Approach. Journal of Medicinal Chemistry. 1999;42(5):791–804. Available from: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- [205].Mooij WTM, Verdonk ML. General and targeted statistical potentials for protein–ligand interactions. Proteins: Structure, Function, and Bioinformatics. 2005;61(2):272–87. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.20588. [DOI] [PubMed] [Google Scholar]
- [206].Velec HFG, Gohlke H, Klebe G. DrugScoreCSD Knowledge-Based Scoring Function Derived from Small Molecule Crystal Data with Superior Recognition Rate of Near-Native Ligand Poses and Better Affinity Prediction. Journal of Medicinal Chemistry. 2005;48(20):6296–303. Available from: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- [207].Zhang C, Liu S, Zhu Q, Zhou Y. A Knowledge-Based Energy Function for Protein-Ligand, Protein-Protein, and Protein-DNA Complexes. Journal of Medicinal Chemistry. 2005;48(7):2325–35. Available from: 10.1021/jm049314d. [DOI] [PubMed] [Google Scholar]
- [208].Huang SY, Zou X. An iterative knowledge-based scoring function to predict protein–ligand interactions: I. Derivation of interaction potentials. Journal of Computational Chemistry. 2006;27(15):1866–75. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.20504. [DOI] [PubMed] [Google Scholar]
- [209].Huang SY, Zou X. An iterative knowledge-based scoring function to predict protein–ligand interactions: II. Validation of the scoring function. Journal of Computational Chemistry. 2006;27(15):1876–82. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.20505. [DOI] [PubMed] [Google Scholar]
- [210].Yang CY, Wang R, Wang S. M-Score: A Knowledge-Based Potential Scoring Function Accounting for Protein Atom Mobility. Journal of Medicinal Chemistry. 2006;49(20):5903–11. Available from: 10.1021/jm050043w. [DOI] [PubMed] [Google Scholar]
- [211].Huang SY, Zou X. Inclusion of Solvation and Entropy in the Knowledge-Based Scoring Function for Protein-Ligand Interactions. Journal of Chemical Information and Modeling. 2010;50(2):262–73. Available from: 10.1021/ci9002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Neudert G, Klebe G. DSX: A Knowledge-Based Scoring Function for the Assessment of Protein–Ligand Complexes. Journal of Chemical Information and Modeling. 2011;51(10):2731–45. Available from: 10.1021/ci200274q. [DOI] [PubMed] [Google Scholar]
- [213].Zheng Z, Merz KM. Development of the Knowledge-Based and Empirical Combined Scoring Algorithm (KECSA) To Score Protein–Ligand Interactions. Journal of Chemical Information and Modeling. 2013;53(5):1073–83. Available from: 10.1021/ci300619x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Huang SY, Zou X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Research. 2014. January;42(7):e55–5. Available from: 10.1093/nar/gku077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Chen P, Ke Y, Lu Y, Du Y, Li J, Yan H, et al. DLIGAND2: an improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. Journal of Cheminformatics. 2019. Aug;11(1):52. Available from: 10.1186/s13321-019-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Su M, Yang Q, Du Y, Feng G, Liu Z, Li Y, et al. Comparative Assessment of Scoring Functions: The CASF-2016 Update. Journal of Chemical Information and Modeling. 2019;59(2):895–913. Available from: 10.1021/acs.jcim.8b00545. [DOI] [PubMed] [Google Scholar]
- [217].Thomas PD, Dill KA. Statistical Potentials Extracted From Protein Structures: How Accurate Are They? Journal of Molecular Biology. 1996;257(2):457 469. Available from: http://www.sciencedirect.com/science/article/pii/S0022283696901758. [DOI] [PubMed] [Google Scholar]
- [218].Thomas PD, Dill KA. An iterative method for extracting energy-like quantities from protein structures. Proceedings of the National Academy of Sciences. 1996;93(21):11628–33. Available from: https://www.pnas.org/content/93/21/11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Huang SY, Zou X. An iterative knowledge-based scoring function for protein–protein recognition. Proteins: Structure, Function, and Bioinformatics. 2008;72(2):557–79. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.21949. [DOI] [PubMed] [Google Scholar]
- [220].Zhang D, Chen SJ. IsRNA: An Iterative Simulated Reference State Approach to Modeling Correlated Interactions in RNA Folding. Journal of Chemical Theory and Computation. 2018;14(4):2230–9. Available from: 10.1021/acs.jctc.7b01228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Hurst T, Zhang D, Zhou Y, Chen SJ. A Bayes-inspired theory for optimally building an efficient coarse-grained folding force field. Communications in Information and Systems. 2021;21(1):65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Zhang D, Li J, Chen SJ. IsRNA1: De Novo Prediction and Blind Screening of RNA 3D Structures. Journal of Chemical Theory and Computation. 2021;17(3):1842–57. Available from: 10.1021/acs.jctc.0c01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Liu Z, Li Y, Han L, Li J, Liu J, Zhao Z, et al. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics. 2014. October;31(3):405–12. Available from: 10.1093/bioinformatics/btu626. [DOI] [PubMed] [Google Scholar]
- [224].Wojcikowski M, Ballester PJ, Siedlecki P. Performance of machine-learning scoring functions in structure-based virtual screening. Scientific Reports. 2017. Apr;7(1):46710. Available from: 10.1038/srep46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Yang X, Wang Y, Byrne R, Schneider G, Yang S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chemical Reviews. 2019;119(18):10520–94. Available from: 10.1021/acs.chemrev.8b00728. [DOI] [PubMed] [Google Scholar]
- [226].Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, et al. Applications of machine learning in drug discovery and development. Nature Reviews Drug Discovery. 2019. Jun;18(6):463–77. Available from: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Nguyen DD, Cang Z, Wu K, Wang M, Cao Y, Wei GW. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges. Journal of Computer-Aided Molecular Design. 2019. Jan;33(1):71–82. Available from: 10.1007/s10822-018-0146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Li H, Sze KH, Lu G, Ballester PJ. Machine-learning scoring functions for structure-based virtual screening. WIREs Computational Molecular Science. 2020;11(1):e1478. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1478. [Google Scholar]
- [229].Li H, Sze KH, Lu G, Ballester PJ. Machine-learning scoring functions for structure-based drug lead optimization. WIREs Computational Molecular Science. 2020;10(5):e1465. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1465. [Google Scholar]
- [230].Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein-Ligand Scoring with Convolutional Neural Networks. Journal of Chemical Information and Modeling. 2017;57(4):942–57. Available from: 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Jiménez J, Škalič M, Martínez-Rosell G, De Fabritiis G. KDEEP: Protein–Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. Journal of Chemical Information and Modeling. 2018;58(2):287–96. Available from: 10.1021/acs.jcim.7b00650. [DOI] [PubMed] [Google Scholar]
- [232].Imrie F, Bradley AR, van der Schaar M, Deane CM. Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. Journal of Chemical Information and Modeling. 2018;58(11):2319–30. Available from: 10.1021/acs.jcim.8b00350. [DOI] [PubMed] [Google Scholar]
- [233].Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics. 2018. May;34(21):3666–74. Available from: 10.1093/bioinformatics/bty374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Sunseri J, King JE, Francoeur PG, Koes DR. Convolutional neural network scoring and minimization in the D3R 2017 community challenge. Journal of Computer-Aided Molecular Design. 2019. Jan;33(1):19–34. Available from: 10.1007/s10822-018-0133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Wang R, Fang X, Lu Y, Wang S. The PDBbind Database: Collection of Binding Affinities for Protein-Ligand Complexes with Known Three-Dimensional Structures. Journal of Medicinal Chemistry. 2004;47(12):2977–80. Available from: 10.1021/jm030580l. [DOI] [PubMed] [Google Scholar]
- [236].Wang R, Fang X, Lu Y, Yang CY, Wang S. The PDBbind Database: Methodologies and Updates. Journal of Medicinal Chemistry. 2005;48(12):4111–9. Available from: 10.1021/jm048957q. [DOI] [PubMed] [Google Scholar]
- [237].Cheng T, Li X, Li Y, Liu Z, Wang R. Comparative Assessment of Scoring Functions on a Diverse Test Set. Journal of Chemical Information and Modeling. 2009;49(4):1079–93. Available from: 10.1021/ci9000053. [DOI] [PubMed] [Google Scholar]
- [238].Li Y, Liu Z, Li J, Han L, Liu J, Zhao Z, et al. Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. Journal of Chemical Information and Modeling. 2014;54(6):1700–16. Available from: 10.1021/ci500080q. [DOI] [PubMed] [Google Scholar]
- [239].Liu Z, Su M, Han L, Liu J, Yang Q, Li Y, et al. Forging the Basis for Developing Protein–Ligand Interaction Scoring Functions. Accounts of Chemical Research. 2017;50(2):302–9. Available from: 10.1021/acs.accounts.6b00491. [DOI] [PubMed] [Google Scholar]
- [240].Schlichtkrull M, Kipf TN, Bloem P, van den Berg R, Titov I, Welling M. Modeling Relational Data with Graph Convolutional Networks. In: Gangemi A, Navigli R, Vidal ME, Hitzler P, Troncy R, Hollink L, et al. , editors. The Semantic Web. Cham: Springer International Publishing; 2018. p. 593–607. [Google Scholar]
- [241].Durant JL, Leland BA, Henry DR, Nourse JG. Reoptimization of MDL Keys for Use in Drug Discovery. Journal of Chemical Information and Computer Sciences. 2002;42(6):1273–80. Available from: 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- [242].Breiman L. Random forests. Machine learning. 2001;45(1):5–32. [Google Scholar]
- [243].Boniecki MJ, Lach G, Dawson WK, Tomala K, Lukasz P, Soltysinski T, et al. SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Research. 2015. December;44(7):e63–3. Available from: 10.1093/nar/gkv1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Taminau J, Thijs G, De Winter H. Pharao: Pharmacophore alignment and optimization. Journal of Molecular Graphics and Modelling. 2008;27(2):161–9. Available from: https://www.sciencedirect.com/science/article/pii/S109332630800048X. [DOI] [PubMed] [Google Scholar]
- [245].Wei L, Zou Q. Recent Progress in Machine Learning-Based Methods for Protein Fold Recognition. International Journal of Molecular Sciences. 2016;17(12). Available from: https://www.mdpi.com/1422-0067/17/12/2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins: Structure, Function, and Bioinformatics. 2019;87(12):1141–8. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.25834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Kandathil SM, Greener JG, Jones DT. Recent developments in deep learning applied to protein structure prediction. Proteins: Structure, Function, and Bioinformatics. 2019;87(12):1179–89. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.25824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Kuhlman B, Bradley P. Advances in protein structure prediction and design. Nature Reviews Molecular Cell Biology. 2019. Nov;20(11):681–97. Available from: 10.1038/s41580-019-0163-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Gao W, Mahajan SP, Sulam J, Gray JJ. Deep Learning in Protein Structural Modeling and Design. Patterns. 2020;1(9):100142. Available from: http://www.sciencedirect.com/science/article/pii/S2666389920301902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Noé F, De Fabritiis G, Clementi C. Machine learning for protein folding and dynamics. Current Opinion in Structural Biology. 2020;60:77 84. Folding and Binding - Proteins. Available from: http://www.sciencedirect.com/science/article/pii/S0959440X19301447. [DOI] [PubMed] [Google Scholar]
- [251].Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, et al. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophysical journal. 1992. Sep;63(3):751–9. Available from: https://pubmed.ncbi.nlm.nih.gov/1384741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Coimbatore Narayanan B, Westbrook J, Ghosh S, Petrov AI, Sweeney B, Zirbel CL, et al. The Nucleic Acid Database: new features and capabilities. Nucleic Acids Research. 2013. October;42(D1):D114–22. Available from: 10.1093/nar/gkt980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Gebala M, Bonilla S, Bisaria N, Herschlag D. Does Cation Size Affect Occupancy and Electrostatic Screening of the Nucleic Acid Ion Atmosphere? Journal of the American Chemical Society. 2016;138(34):10925–34. Available from: 10.1021/jacs.6b04289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Zhou H. Macromolecular electrostatic energy within the nonlinear Poisson-Boltzmann equation. The Journal of Chemical Physics. 1994;100(4):3152–62. Available from: 10.1063/1.466406. [DOI] [Google Scholar]
- [255].Misra VK, Draper DE. The interpretation of Mg2+ binding isotherms for nucleic acids using Poisson-Boltzmann theory. Journal of Molecular Biology. 1999;294(5):1135–47. Available from: https://www.sciencedirect.com/science/article/pii/S002228369993334X. [DOI] [PubMed] [Google Scholar]
- [256].Tan ZJ, Chen SJ. Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. The Journal of Chemical Physics. 2005;122(4):044903. Available from: 10.1063/1.1842059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Mak CH, Henke PS. Ions and RNAs: Free Energies of Counterion-Mediated RNA Fold Stabilities. Journal of Chemical Theory and Computation. 2013;9(1):621–39. Available from: 10.1021/ct300760y. [DOI] [PubMed] [Google Scholar]
- [258].Giambaşu G, Luchko T, Herschlag D, York D, Case D. Ion Counting from Explicit-Solvent Simulations and 3D-RISM. Biophysical Journal. 2014;106(4):883–94. Available from: https://www.sciencedirect.com/science/article/pii/S0006349514000915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Hayes RL, Noel JK, Mandic A, Whitford PC, Sanbonmatsu KY, Mohanty U, et al. Generalized Manning Condensation Model Captures the RNA Ion Atmosphere. Phys Rev Lett. 2015. Jun;114:258105. Available from: https://link.aps.org/doi/10.1103/PhysRevLett.114.258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Sun LZ, Chen SJ. Monte Carlo Tightly Bound Ion Model: Predicting Ion-Binding Properties of RNA with Ion Correlations and Fluctuations. Journal of Chemical Theory and Computation. 2016;12(7):3370–81. Available from: 10.1021/acs.jctc.6b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Tan ZJ, Chen SJ. Predicting Electrostatic Forces in RNA Folding. In: Biophysical, Chemical, and Functional Probes of RNA Structure, Interactions and Folding: Part B. vol. 469 of Methods in Enzymology. Academic Press; 2009. p. 465–87. Available from: https://www.sciencedirect.com/science/article/pii/S0076687909690224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Zhu Y, He Z, Chen SJ. TBI Server: A Web Server for Predicting Ion Effects in RNA Folding. PLOS ONE. 2015. March;10(3):1–13. Available from: 10.1371/journal.pone.0119705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Sun LZ, Zhang JX, Chen SJ. MCTBI: a web server for predicting metal ion effects in RNA structures. RNA. 2017;23(8):1155–65. Available from: http://rnajournal.cshlp.org/content/23/8/1155.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Giambaşu GM, Case DA, York DM. Predicting Site-Binding Modes of Ions and Water to Nucleic Acids Using Molecular Solvation Theory. Journal of the American Chemical Society. 2019;141(6):2435–45. Available from: 10.1021/jacs.8b11474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Wei W, Luo J, Waldispühl J, Moitessier N. Predicting Positions of Bridging Water Molecules in Nucleic Acid–Ligand Complexes. Journal of Chemical Information and Modeling. 2019;59(6):2941–51. Available from: 10.1021/acs.jcim.9b00163. [DOI] [PubMed] [Google Scholar]
- [266].Limongelli V. Ligand binding free energy and kinetics calculation in 2020. WIREs Computational Molecular Science. 2020;10(4):e1455. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1455. [Google Scholar]
- [267].Kuhn B, Kollman PA. Binding of a Diverse Set of Ligands to Avidin and Streptavidin: An Accurate Quantitative Prediction of Their Relative Affinities by a Combination of Molecular Mechanics and Continuum Solvent Models. Journal of Medicinal Chemistry. 2000;43(20):3786–91. Available from: 10.1021/jm000241h. [DOI] [PubMed] [Google Scholar]
- [268].Gouda H, Kuntz ID, Case DA, Kollman PA. Free energy calculations for theophylline binding to an RNA aptamer: Comparison of MM-PBSA and thermodynamic integration methods. Biopolymers. 2003;68(1):16–34. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/bip.10270. [DOI] [PubMed] [Google Scholar]
- [269].Murray JB, Meroueh SO, Russell RJM, Lentzen G, Haddad J, Mobashery S. Interactions of Designer Antibiotics and the Bacterial Ribosomal Aminoacyl-tRNA Site. Chemistry & Biology. 2006;13(2):129–38. Available from: https://www.sciencedirect.com/science/article/pii/S1074552105003753. [DOI] [PubMed] [Google Scholar]
- [270].Meroueh SO, Mobashery S. Conformational Transition in the Aminoacyl t-RNA Site of the Bacterial Ribosome both in the Presence and Absence of an Aminoglycoside Antibiotic. Chemical Biology & Drug Design. 2007;69(5):291–7. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1747-0285.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- [271].Freedman H, Huynh LP, Le L, Cheatham TE, Tuszynski JA, Truong TN. Explicitly Solvated Ligand Contribution to Continuum Solvation Models for Binding Free Energies: Selectivity of Theophylline Binding to an RNA Aptamer. The Journal of Physical Chemistry B. 2010;114(6):2227–37. Available from: 10.1021/jp9059664. [DOI] [PubMed] [Google Scholar]
- [272].Hu G, Ma A, Wang J. Ligand Selectivity Mechanism and Conformational Changes in Guanine Riboswitch by Molecular Dynamics Simulations and Free Energy Calculations. Journal of Chemical Information and Modeling. 2017;57(4):918–28. Available from: 10.1021/acs.jcim.7b00139. [DOI] [PubMed] [Google Scholar]
- [273].Peddi SR, Sivan SK, Manga V. Molecular dynamics and MM/GBSA-integrated protocol probing the correlation between biological activities and binding free energies of HIV-1 TAR RNA inhibitors. Journal of Biomolecular Structure and Dynamics. 2018;36(2):486–503. Available from: 10.1080/07391102.2017.1281762. [DOI] [PubMed] [Google Scholar]
- [274].Chen F, Sun H, Wang J, Zhu F, Liu H, Wang Z, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 8. Predicting binding free energies and poses of protein–RNA complexes. RNA. 2018;24(9):1183–94. Available from: http://rnajournal.cshlp.org/content/24/9/1183.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Engineering, Design and Selection. 1994. March;7(3):385–91. Available from: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- [276].Åqvist J, Luzhkov VB, Brandsdal BO. Ligand Binding Affinities from MD Simulations. Accounts of Chemical Research. 2002;35(6):358–65. Available from: 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- [277].Sund J, Lind C, Åqvist J. Binding Site Preorganization and Ligand Discrimination in the Purine Riboswitch. The Journal of Physical Chemistry B. 2015;119(3):773–82. Available from: 10.1021/jp5052358. [DOI] [PubMed] [Google Scholar]
- [278].Zwanzig RW. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. The Journal of Chemical Physics. 1954;22(8):1420–6. Available from: 10.1063/1.1740409. [DOI] [Google Scholar]
- [279].Kirkwood JG. Statistical Mechanics of Fluid Mixtures. The Journal of Chemical Physics. 1935;3(5):300–13. Available from: 10.1063/1.1749657. [DOI] [Google Scholar]
- [280].Ryde U, Söderhjelm P. Ligand-Binding Affinity Estimates Supported by Quantum-Mechanical Methods. Chemical Reviews. 2016;116(9):5520–66. Available from: 10.1021/acs.chemrev.5b00630. [DOI] [PubMed] [Google Scholar]
- [281].Collins MA, Bettens RPA. Energy-Based Molecular Fragmentation Methods. Chemical Reviews. 2015;115(12):5607–42. Available from: 10.1021/cr500455b. [DOI] [PubMed] [Google Scholar]
- [282].Raghavachari K, Saha A. Accurate Composite and Fragment-Based Quantum Chemical Models for Large Molecules. Chemical Reviews. 2015;115(12):5643–77. Available from: 10.1021/cr500606e. [DOI] [PubMed] [Google Scholar]
- [283].Jin X, Zhang JZH, He X. Full QM Calculation of RNA Energy Using Electrostatically Embedded Generalized Molecular Fractionation with Conjugate Caps Method. The Journal of Physical Chemistry A. 2017;121(12):2503–14. Available from: 10.1021/acs.jpca.7b00859. [DOI] [PubMed] [Google Scholar]
- [284].Albuquerque EL, Fulco UL, Freire VN, Caetano EWS, Lyra ML, de Moura FABF. DNA-based nanobiostructured devices: The role of quasiperiodicity and correlation effects. Physics Reports. 2014;535(4):139 209. DNA-based nanobiostructured devices: The role of quasiperiodicity and correlation effects. Available from: http://www.sciencedirect.com/science/article/pii/S0370157313003797. [Google Scholar]
- [285].Chen XH, Zhang JZH. Theoretical method for full ab initio calculation of DNA/RNA–ligand interaction energy. The Journal of Chemical Physics. 2004;120(24):11386–91. Available from: 10.1063/1.1737295. [DOI] [PubMed] [Google Scholar]
- [286].Mlýnský V, Banáš P, Šponer J, van der Kamp MW, Mulholland AJ, Otyepka M. Comparison of ab Initio, DFT, and Semiempirical QM/MM Approaches for Description of Catalytic Mechanism of Hairpin Ribozyme. Journal of Chemical Theory and Computation. 2014;10(4):1608–22. Available from: 10.1021/ct401015e. [DOI] [PubMed] [Google Scholar]
- [287].da Costa RF, Freire VN, Bezerra EM, Cavada BS, Caetano EWS, de Lima Filho JL, et al. Explaining statin inhibition effectiveness of HMG-CoA reductase by quantum biochemistry computations. Phys Chem Chem Phys. 2012;14:1389–98. Available from: 10.1039/C1CP22824B. [DOI] [PubMed] [Google Scholar]
- [288].Mota KB, Lima Neto JX, Lima Costa AH, Oliveira JIN, Bezerra KS, Albuquerque EL, et al. A quantum biochemistry model of the interaction between the estrogen receptor and the two antagonists used in breast cancer treatment. Computational and Theoretical Chemistry. 2016;1089:21 27. Available from: http://www.sciencedirect.com/science/article/pii/S2210271X16301773. [Google Scholar]
- [289].de Sousa BG, Oliveira JIN, Albuquerque EL, Fulco UL, Amaro VE, Blaha CAG. Molecular modelling and quantum biochemistry computations of a naturally occurring bioremediation enzyme: Alkane hydroxylase from Pseudomonas putida P1. Journal of Molecular Graphics and Modelling. 2017;77:232 239. Available from: http://www.sciencedirect.com/science/article/pii/S1093326317304588. [DOI] [PubMed] [Google Scholar]
- [290].Kovalenko A, Hirata F. Three-dimensional density profiles of water in contact with a solute of arbitrary shape: a RISM approach. Chemical Physics Letters. 1998;290(1):237 244. Available from: http://www.sciencedirect.com/science/article/pii/S0009261498004710. [Google Scholar]
- [291].Kovalenko A, Hirata F. Self-consistent description of a metal–water interface by the Kohn–Sham density functional theory and the three-dimensional reference interaction site model. The Journal of Chemical Physics. 1999;110(20):10095–112. Available from: 10.1063/1.478883. [DOI] [Google Scholar]
- [292].Kovalenko A, Hirata F. Hydration free energy of hydrophobic solutes studied by a reference interaction site model with a repulsive bridge correction and a thermodynamic perturbation method. The Journal of Chemical Physics. 2000;113(7):2793–805. Available from: 10.1063/1.1305885. [DOI] [Google Scholar]
- [293].Yoshida N, Phongphanphanee S, Maruyama Y, Imai T, Hirata F. Selective Ion-Binding by Protein Probed with the 3D-RISM Theory. Journal of the American Chemical Society. 2006;128(37):12042–3. Available from: 10.1021/ja0633262. [DOI] [PubMed] [Google Scholar]
- [294].Imai T, Hiraoka R, Seto T, Kovalenko A, Hirata F. Three-Dimensional Distribution Function Theory for the Prediction of Protein-Ligand Binding Sites and Affinities: Application to the Binding of Noble Gases to Hen Egg-White Lysozyme in Aqueous Solution. The Journal of Physical Chemistry B. 2007;111(39):11585–91. Available from: 10.1021/jp074865b. [DOI] [PubMed] [Google Scholar]
- [295].Imai T, Oda K, Kovalenko A, Hirata F, Kidera A. Ligand Mapping on Protein Surfaces by the 3D-RISM Theory: Toward Computational Fragment-Based Drug Design. Journal of the American Chemical Society. 2009;131(34):12430–40. Available from: 10.1021/ja905029t. [DOI] [PubMed] [Google Scholar]
- [296].Imai T, Miyashita N, Sugita Y, Kovalenko A, Hirata F, Kidera A. Functionality Mapping on Internal Surfaces of Multidrug Transporter AcrB Based on Molecular Theory of Solvation: Implications for Drug Efflux Pathway. The Journal of Physical Chemistry B. 2011;115(25):8288–95. Available from: 10.1021/jp2015758. [DOI] [PubMed] [Google Scholar]
- [297].Kiyota Y, Yoshida N, Hirata F. A New Approach for Investigating the Molecular Recognition of Protein: Toward Structure-Based Drug Design Based on the 3D-RISM Theory. Journal of Chemical Theory and Computation. 2011;7(11):3803–15. Available from: 10.1021/ct200358h. [DOI] [PubMed] [Google Scholar]
- [298].Nikolić D, Blinov N, Wishart D, Kovalenko A. 3D-RISM-Dock: A New Fragment-Based Drug Design Protocol. Journal of Chemical Theory and Computation. 2012;8(9):3356–72. Available from: 10.1021/ct300257v. [DOI] [PubMed] [Google Scholar]
- [299].Tummino PJ, Copeland RA. Residence Time of Receptor-Ligand Complexes and Its Effect on Biological Function. Biochemistry. 2008;47(20):5481–92. Available from: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- [300].Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Current Opinion in Chemical Biology. 2010;14(4):467 474. Next Generation Therapeutics. Available from: http://www.sciencedirect.com/science/article/pii/S1367593110000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Zhang R, Monsma F. The importance of drug-target residence time. Current opinion in drug discovery & development. 2009;12(4):488. [PubMed] [Google Scholar]
- [302].Copeland RA. The drug-target residence time model: a 10-year retrospective. Nature Reviews Drug Discovery. 2016. Feb;15(2):87–95. Available from: 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- [303].Schuetz DA, de Witte WEA, Wong YC, Knasmueller B, Richter L, Kokh DB, et al. Kinetics for Drug Discovery: an industry-driven effort to target drug residence time. Drug Discovery Today. 2017;22(6):896 911. Available from: http://www.sciencedirect.com/science/article/pii/S1359644617300958. [DOI] [PubMed] [Google Scholar]
- [304].Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nature Reviews Drug Discovery. 2006. Sep;5(9):730–9. Available from: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- [305].Swinney DC. Applications of Binding Kinetics to Drug Discovery. Pharmaceutical Medicine. 2008. Jan;22(1):23–34. Available from: 10.1007/BF03256679. [DOI] [Google Scholar]
- [306].Swinney DC. The role of binding kinetics in therapeutically useful drug action. Current opinion in drug discovery & development. 2009;12(1):31. [PubMed] [Google Scholar]
- [307].Pan AC, Borhani DW, Dror RO, Shaw DE. Molecular determinants of drug–receptor binding kinetics. Drug Discovery Today. 2013;18(13):667 673. Available from: http://www.sciencedirect.com/science/article/pii/S1359644613000627. [DOI] [PubMed] [Google Scholar]
- [308].Yin N, Pei J, Lai L. A comprehensive analysis of the influence of drug binding kinetics on drug action at molecular and systems levels. Mol BioSyst. 2013;9:1381–9. Available from: 10.1039/C3MB25471B. [DOI] [PubMed] [Google Scholar]
- [309].Bernetti M, Cavalli A, Mollica L. Protein-ligand (un)binding kinetics as a new paradigm for drug discovery at the crossroad between experiments and modelling. Med Chem Commun. 2017;8:534–50. Available from: 10.1039/C6MD00581K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Bernetti M, Masetti M, Rocchia W, Cavalli A. Kinetics of Drug Binding and Residence Time. Annual Review of Physical Chemistry. 2019;70(1):143–71. Available from: 10.1146/annurev-physchem-042018-052340. [DOI] [PubMed] [Google Scholar]
- [311].Umuhire Juru A, Patwardhan NN, Hargrove AE. Understanding the Contributions of Conformational Changes, Thermodynamics, and Kinetics of RNA-Small Molecule Interactions. ACS Chemical Biology. 2019;14(5):824–38. Available from: 10.1021/acschembio.8b00945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins: Structure, Function, and Bioinformatics. 1995;23(3):ii–iv. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- [313].Gathiaka S, Liu S, Chiu M, Yang H, Stuckey JA, Kang YN, et al. D3R grand challenge 2015: Evaluation of protein–ligand pose and affinity predictions. Journal of Computer-Aided Molecular Design. 2016. Sep;30(9):651–68. Available from: 10.1007/s10822-016-9946-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Li Y, Han L, Liu Z, Wang R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. Journal of Chemical Information and Modeling. 2014;54(6):1717–36. Available from: 10.1021/ci500081m. [DOI] [PubMed] [Google Scholar]
- [315].Li Y, Su M, Liu Z, Li J, Liu J, Han L, et al. Assessing protein–ligand interaction scoring functions with the CASF-2013 benchmark. Nature Protocols. 2018. Apr;13(4):666–80. Available from: 10.1038/nprot.2017.114. [DOI] [PubMed] [Google Scholar]
- [316].Huang N, Shoichet BK, Irwin JJ. Benchmarking Sets for Molecular Docking. Journal of Medicinal Chemistry. 2006;49(23):6789–801. Available from: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. Journal of Medicinal Chemistry. 2012;55(14):6582–94. Available from: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]