


Abstract
Genetic evidence suggests that the Bacillus subtilis lrpC gene product participates in cell growth and sporulation. The purified LrpC protein, which has a predicted molecular mass of 16.4 kDa, is a tetramer in solution. LrpC binds with higher affinity (Kapp ~ 80 nM) to intrinsically curved DNA than to non-curved DNA (Kapp ~ 700 nM). DNase I footprinting and the supercoiling of relaxed circular plasmid DNA in the presence of topoisomerase I revealed that LrpC induces DNA bending and constrains DNA supercoils in vitro. The LrpC protein cooperatively increases DNA binding of the bona fide DNA-binding and DNA-bending protein Hbsu. LrpC forms inter- and intramolecular bridges on linear and supercoiled DNA molecules, resulting in a large network and DNA compactation. Collectively, these findings suggest that LrpC is an architectural protein and that its activities could provide a means to modulate DNA transactions.
INTRODUCTION
For the catalysis and regulation of a variety of DNA transactions the active components are often insufficient to generate the proper structure, and accessory factors are needed. The bacterial chromatin-associated proteins HU, IHF, Fis, H-NS, StpA and Hha and the eukaryotic HMG1-like proteins are some of such accessory factors (1–5). The best understood systems where these factors are required are some of the prokaryotic site-specific recombination and DNA transposition reactions (5).
Nucleotide sequence analysis of a SPβ-free Bacillus subtilis genome revealed the presence of only one gene coding for a sequence-independent chromatin-associated Hbsu protein, whereas genes coding for sequence-specific architectural proteins such as IHF, Fis, H-NS, StpA, CRP and Hha cannot be predicted (6). This could suggest that the B.subtilis Hbsu protein is involved in a larger number of DNA transactions than its Escherichia coli homologue HU, and that Hbsu could functionally replace the sequence-specific chromatin-associated proteins. This is consistent with the fact that Hbsu is an essential product (7). Alternatively, another protein(s) not defined in E.coli as having such a role could perform a role analogous to E.coli sequence-specific chromatin-associated proteins in B.subtilis. Candidates to begin our studies are the different B.subtilis Lrp-type proteins [LrpA and LrpB (8), LrpC (9), AzlB (10) and YezC, YwrC and YugG (6)]. Very little is known about the hypothetical Lrp-type products YezC, YwrC and YugG. Unless otherwise stated, the indicated genes and products are of B.subtilis origin. The LrpA and LrpB proteins are implicated in KinB-dependent sporulation, probably through regulation of glyA transcription (8). Recently, it has been postulated that AzlB negatively regulates the azlBCDEF operon (10). A possible role for LrpC in entry into the sporulation and growth phases was proposed (9). Since a lrpC null strain has a pleiotropic phenotype (9) we began with the biochemical characterisation of LrpC protein. The results presented provide the first biochemical evidence that the B.subtilis LprC protein is a sequence-independent DNA-binding and DNA-bending protein that promotes DNA bridging. The architectural role of LrpC could provide a means to regulate DNA transactions.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli strain BL21 (DE3) was used for lrpC overexpression (11) and JM103 for amplification of the plasmid DNA (12).
The plasmids pUC18 (12), pLysS (11), pT712lrp (9), pCB50 (13) and pCB8 (14) have been described previously. Plasmid pET3blrpC was a gift from F. LeHégarat and C. Beloin (Université d’Orsay, Paris, France).
Enzymes, proteins and and reagents
Dithiothreitol (DTT), isopropyl-β-d-thiogalactopyranoside, lysozyme, restriction and modification enzymes and protease inhibitors (PMSF, Bestatin, Leupeptin and Pepstatin) were from Merck, Calbiochem, Boehringer and Promega, respectively. The Hbsu and β proteins were purified as described previously (15). Phosphocellulose was from Whatman and Superose 12 from Pharmacia. Alumina was from Sigma.
[α-32P]dATP and [γ-32P]ATP were from Amersham Corp (Germany). The concentration of DNA was determined using a molar extinction coefficient of 6500 M–1 cm–1 at 260 nm. The amount of DNA is expressed as mol DNA molecules.
Purification of the LrpC protein
BL21(DE3)[pLysS pET3blrpC] cells were grown and induced as described previously (16). The cell paste (10 g) was lysed by grinding with alumina. The crude extract was resuspended in 50 ml of buffer A (50 mM Tris–HCl pH 7.5, 1 mM DTT, 1 mM EDTA, 1 mM PMSF, 1 mM Bestatin, 10 µM Leupeptin, 1 µM Pepstatin, 5% glycerol) containing 200 mM NaCl. The alumina and the cell debris were removed by centrifugation at 18 000 r.p.m. (SS 34 Sorvall rotor) for 30 min. The 16 kDa LrpC protein was present in the supernatant (fraction I).
Polyethylenimine (10%, pH 7.5) was slowly added to fraction I with constant stirring to a final concentration of 0.25% (A260 ~ 120). The solution was centrifuged at 15 000 r.p.m. (SS 34 Sorvall rotor) for 10 min, and the pellet discarded. The LrpC protein, which migrates in SDS–PAGE as a 16 kDa product, remains in the supernatant (fraction II). The proteins of fraction II were precipitated by addition of solid ammonium sulphate to a final saturation of 60%. The pellet was resuspended in a 70% ammonium sulphate solution and pelleted again (fraction III).
The protein pellet was resuspended in buffer B (50 mM Tris–HCl pH 7.5, 1 mM DTT, 1 mM EDTA, 5% glycerol) and loaded onto a 5 ml phosphocellulose column equilibrated with buffer B containing 100 mM NaCl. The column was extensively washed with buffer B containing 300 mM NaCl. Under these conditions the EcoLrp protein is eluted from the column. The LrpC protein was eluted using a step gradient from 350 to 450 mM NaCl in buffer B (fraction IV) (see Fig. 1A, lane 4). Fraction IV was concentrated by ammonium sulphate precipitation (70% saturation) and loaded onto a Superose 12 column equilibrated with buffer B containing 1 M NaCl. The fractions containing the LrpC protein, which elutes with a Mr of 60 000–70 000, were pooled and concentrated on a phosphocellulose column (fraction V), glycerol to a final concentration of 50% was added and the samples stored at –20°C.
Figure 1.

Purification and molecular mass determination of the LrpC protein. (A) SDS–PAGE (15%) stained with Coomassie Blue. (B) Western blot of the gel presented in (A) highlighted with polyclonal antibodies raised against EcoLrp. Lane 1, cell lysate (induced cells); lane 2, polyethylenimine supernatant; lane 3, ammonium sulphate (60% saturation) pellet; lane 4, eluate from the phosphocellulose column (400 mM NaCl); lane 5, eluate from the FPLC Superose 12 column (1 M NaCl). The molecular mass standards, in kDa, are indicated. (C) Calibration graph of molecular mass against Kav of several protein standards (filled circle) for a Superose 12 column using the same conditions as described for LrpC (open circle).
The N-terminal amino acid sequence of the purified protein was determined by automated Edman degradation in a pulsed liquid phase sequencer (model 477A; Applied Biosystems). The LrpC protein concentration was determined using a molar extinction coefficient of 7860 M–1 cm–1 at 280 nm and is expressed as mol protein tetramers.
Molecular mass determination
The native molecular mass of the LrpC protein was determined by gel filtration FPLC using a Superose 12 column (Pharmacia, Sweden). Chromatography was carried out in buffer B containing 100 mM or 1 M NaCl at 4°C with a flow rate of 0.4 ml/min, and the A280 was measured. An aliquot of 80 µg of LrpC protein was applied to the column. A standard curve of Kav versus log10 relative mobility was determined as recommended by Pharmacia. Protein standards were obtained from Pharmacia (RNase, 13 kDa; chymotrypsinogen A, 25 kDa; ovalbumin, 44 kDa; bovin serum albumin, 66 kDa; aldolase, 158 kDa).
Measurement of LrpC–DNA complexes
The formation of LrpC–DNA complexes was measured either by filter binding assay, using alkali-treated filters (type HAWP 0.45 µm; Millipore) or by electrophoretic mobility shift assay (EMSA). The intrinsically curved 331 bp DNA fragment from the 5′ intergenic region of the lrpC gene (coordinates 153–484; GenBank accession no. Z99106) was obtained by PCR from pT712lrp. The curved 597 bp DNA fragment from the 5′ intergenic region of the hbs gene (coordinates 31–628; GenBank accession no. X52418) was obtained from pCB50. The non-curved 213 bp DNA segment from the coding region of the lrpC gene (coordinates 490–705; GenBank accession no. Z99106) was obtained by PCR from pT712lrp.
For filter binding assays, the standard reaction (20 µl) contained the 331 bp curved [γ-32P]DNA (1 nM) (in the presence or absence of curved or non-curved cold DNA) and the indicated amount of the LrpC protein in buffer C (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 30 µg/ml BSA, 5% glycerol) containing 75 mM NaCl. The reaction was incubated for 15 min at 30°C and then diluted in ice-cold buffer C containing 75 mM NaCl (1 ml) and filtered through KOH-treated filters. Filters were dried and the amount of radioactivity retained on the filter was determined by scintillation counting. The DNA retained on the filter was corrected for the retention of radiolabelled DNA in the absence of LrpC protein (~2–3%). The specific activity of the labelled DNA was measured as TCA-precipitable material. All reactions were performed in duplicate.
For EMSA, the 597 bp intrinsically curved [α-32P]DNA fragment (1 nM) was incubated with increasing concentrations of LrpC or Hbsu protein or with a fixed amount of one of the proteins and increasing concentrations of the second one, in buffer C containing 75 mM NaCl for 15 min at 30°C. The protein–DNA complexes were separated by 4% non-denaturing PAGE in 0.5× TBE.
For DNase I footprinting, the 331 bp [γ-32P]DNA fragment (1 nM) was used. Labelling of only one strand was achieved using a [γ-32P]ATP-labelled primer and the DNA segment amplified by PCR. DNase I footprinting was performed essentially as described (17). Reaction mixtures (20 µl) were equilibrated at 37°C for 20 min with different concentrations of LrpC (3.5, 10, 30, 90, 250 and 850 nM). The DNA was precipitated with ethanol and analysed by 6% urea denaturing PAGE. Autoradiographs of the dried gels were subsequently taken.
Aggregation of DNA by LrpC was performed as described by Lee and Graigie (18). The standard reaction (20 µl) contained a mixture of either 70 nM 182 bp [γ-32P]DNA plus cold 182 bp curved DNA (coordinates 282–464; GenBank accession no. Z99106) obtained by PCR amplification from pT712lrp or 5 nM cold pUC18 supercoiled DNA and the indicated amount of LrpC protein in buffer C containing 100 mM NaCl. The samples were incubated for 10 min on ice and then centrifuged at 12 000 g for 20 min. After addition of 0.5% SDS, the DNA remaining in the supernatant was analysed by electrophoresis in 0.8% agarose gels run in TAE buffer and stained with ethidium bromide (pUC18 DNA) or by 5% non-denaturing PAGE in 0.5× TBE (182 bp segment), and autoradiographs of the dried gels were subsequently taken.
Circularisation assay
A 20 µl reaction mix containing the 182-bp [γ-32P]DNA fragment (0.5 nM) and various concentrations of LrpC protein (30–500 nM) was incubated in buffer C containing 50 mM NaCl and 1 mM ATP for 15 min at 37°C, and then 1 U of T4 DNA ligase (New England Biolabs) was added. The DNA ligase reaction was terminated after 30 min at 16°C. The sample was incubated with proteinase K (0.5 µg/µl) and then 0.5% SDS was added. The deproteinised ligation products were analysed by 4% non-denaturing PAGE in 0.5× TBE, and autoradiographs of the dried gels were subsequently taken.
Supercoiling assay
Relaxed plasmid DNA was produced by treating supercoiled pUC18 (10 nM) DNA with 2 U of calf thymus DNA topoisomerase I (Topo I) for 30 min at 37°C in buffer C containing 50 mM NaCl and aliquots of the mixture were added to various amounts of LrpC protein (50–1600 nM) in buffer C. After 30 min, the samples were deproteinised by incubation with proteinase K (0.5 µg/µl) and then 0.5% SDS. The DNA was analysed by gel electrophoresis (0.8% agarose gels run in TAE buffer) and stained with ethidium bromide.
In vitro assay for β-recombination
Plasmid pCB8, which contains two directly oriented target sites for β-recombinase (six sites) separated by ~2.2 kb, was used as the substrate for site-specific recombination (15,19). Reaction mixtures (25 µl), containing pCB8 DNA (10 nM) and 50 nM β-recombinase, in the presence or absence of Hbsu (50 nM) and increasing concentrations of LrpC (7–2000 nM) or a fixed amount of LrpC (400 nM) and increasing concentrations of Hbsu (50–1000 nM), in buffer D (50 mM Tris–HCl pH 7.5, 50 mM NaCl, 10 mM MgCl2), were incubated for 30 min at 30°C. The reaction was stopped by heating (70°C for 10 min). The DNA was digested with SalI and PstI and the recombinant DNA fragments generated were analysed by 0.8% agarose gel electrophoresis and stained with ethidium bromide.
RESULTS AND DISCUSSION
Purification and native state of LrpC
The LrpC protein was overexpressed in E.coli cells and purified as described in Materials and Methods. The LrpC polypeptide, under the expression conditions described, accounts for ~3% of total protein mass. Previously, it has been shown that LrpC protein cross-reacts with polyclonal antibodies raised against EcoLrp (9). As revealed in Figure 1A and B, the 16 kDa LrpC polypeptide is free of 18.8 kDa EcoLrp protein, which runs in SDS–PAGE as a protein with a Mr of 21 000 (see Fig. 1B, lanes 3 and 4). After the last step of purification the LrpC protein was >99% pure, as judged by SDS–PAGE (Fig. 1A, lane 5) and quantitative analysis of the N-terminal amino acid composition.
Identification of the final product as LrpC protein was confirmed by sequencing the N-terminus of the purified protein. The sequence of the first 10 N-terminal residues of the purified protein is in perfect agreement with the prediction from the nucleotide sequence of the lrpC gene, except for the Met residue of the initiation codon, which is absent in the purified protein.
The LrpC polypeptide consists of 144 amino acid residues with a predicted molecular mass of 16 449 Da. The molecular mass of native LrpC protein was estimated by gel filtration using a Superose 12 column. Under the experimental conditions described, and at lower salt conditions (100 mM NaCl), ~6% of the sample eluted as an aggregate. From the elution profile of LrpC protein and of a number of protein standards we estimate that the majority of LrpC eluted at a Mr of ~65 000 at a concentration of 6 µM. If we assume that LrpC has a spherical shape, it is likely that the native mass of the protein is four times that of a LrpC protomer (Fig. 1C). In contrast, EcoLrp protein has been shown to be a dimer at 10 µM concentration (20) and Pseudomonas putida Lrp-type BkdR protein is a tetramer in solution that binds as a dodecamer to its operator site (21).
LrpC binds preferentially to curved DNA
A computer-based analysis (Bend, DNASTAR v.4.02) of the nucleotide sequence of the 331 bp lrpC 5′ intergenic region (promoter region) revealed the presence of a discrete intrinsically curved DNA segment, whereas such intrinsic curvature cannot be predicted when the 213 bp lprC coding segment was analysed (data not shown).
The 331 bp [γ-32P]DNA fragment (1 nM) was incubated with increasing concentrations of LrpC (2–1200 nM) and the protein–DNA complex analysed by filter binding assay. As revealed in Figure 2B, at LrpC concentrations <20 nM, no binding was detected, whereas at higher protein concentrations the amount of LrpC–DNA complex formed increased exponentially until a plateau was observed. The exponential increase in complex formation suggests that LrpC protein binds to its target sequences in a cooperative manner. The apparent equilibrium constant (Kapp) of the LrpC–DNA complex was estimated to be 80 nM at pH 7.5 and 30°C (Fig. 2B). The presence of l-leucine (30 mM) does not modify LrpC DNA-binding activity (data not shown). Three other members of the Lrp-type family of transcriptional regulators have been biochemically characterised (EcoLrpC, P.putida BkdR and Agrobacterium tumefaciens PutR) (reviewed in 22–25). The four Lrp-type proteins bind DNA cooperatively. If we assume that the experimental designs are comparable, it can be deduced that EcoLrp binds to DNA with the highest affinity (Kapp 5 nM), then PutR (20 nM), then LrpC (80 nM) and then BkdR with the lowest affinity (300 nM). Unlike LrpC, the Kapp of these Lrp-type proteins was affected by the presence of amino acids (24–26; this work).
Figure 2.

LrpC binds preferentially to curved DNA. (A) Schematic representation of the DNA fragments used in this work. The lrpC gene is indicated with an arrow. Putative promoters P1 and P2 (pale grey box), the computer predicted curved region (dark grey box) and the ribosomal binding site (RBS) are indicated. A nucleotide sequence resembling the consensus EcoLrp binding site, within promoter P2, is indicated. The DNA fragments used in this work are shown. The relevant features of the nucleotide sequence that shows DNase I hypersensitive sites are indicated. The arrows denote DNase I hypersensitive sites in the upper and lower strand, respectively. (B). The 331 bp curved [γ-32P]DNA fragment (1 nM) was incubated with increasing LrpC concentrations (2–1200 nM) in buffer B containing 75 mM NaCl for 15 min at 30°C. The DNA retained on the filter was corrected for the retention of [32P]DNA in the absence of LrpC protein (2–3% of total input). (C) Competition assays of LrpC protein DNA binding activity. The 331 bp curved [γ-32P]DNA fragment (1 nM) was incubated with LrpC (100 nM) and the indicated fold excess of cold 331 bp curved DNA fragment (square) or cold 213 bp non-curved DNA fragment (diamond). Binding conditions and determination of retained LrpC–DNA were as in (B). Under these conditions, in the absence of competitor DNA, 60% of the labelled DNA was retained, and this value is given as 100%.
To learn whether LrpC distinguishes between curved and non-curved DNA we measured the ability of those DNAs to act as competitors for binding of the curved 331 bp DNA fragment using a filter binding assay. LrpC (100 nM) was incubated with the 331 bp curved [γ-32P]DNA (1 nM) and increasing concentrations of cold curved or non-curved DNA and the protein–DNA complex was trapped by filter binding. As revealed in Figure 2C, in the absence of cold DNA ~60% of labelled DNA is bound to LrpC protein (100% in Fig. 2C) The presence of an ~1.2-fold excess of cold curved DNA (331 bp DNA) was required to compete out 50% of the radiolabelled DNA substrate, whereas a 10-fold excess of non-curved DNA (213 bp DNA) was needed for such competition. Hence, LrpC seems to bind to curved DNA with an ~8.5-fold greater affinity than to non-curved DNA (Kapp ~ 700 nM). However, when the amount of non-curved DNA required for 50% competition is expressed as mol nucleotides LrpC binds to curved DNA with a 7-fold greater affinity than to non-curved DNA. Unlike EcoLrp, which binds to a 15 bp core sequence (reviewed in 22–25; see below), LrpC binds preferentially to curved DNA (Kapp ~ 80 nM) and with approximately nine times lower affinity to non-curved DNA (Kapp ~ 700 nM).
LrpC promotes DNA bending
To examine the type of complexes that LrpC makes with the 331 bp curved DNA (1 nM) a DNase I footprinting assay was used. As revealed in Figure 3, phosphodiester bonds hypersensitive to DNase I cleavage were observed at positions 17, 47 and 54 (coordinates 467, 437 and 430 of the upper strand of the lrpC promoter region; 9) in the presence of LrpC protein (4–850 nM). Three minor DNase I hypersensitive sites were observed at positions 25, 88 and 91 (coordinates 459, 396 and 393; 9). Previously, a 15 bp sequence (coordinates 297–311 of the lrpC promoter region; 9; Fig. 2A) that matches the DNA-binding consensus sequence of EcoLrp protein at 12 of 15 positions was predicted (9). Neither protected nor hypersensitive sites were observed in this predicted Lrp-like core sequence. When labelling the bottom strand, some minor hypersensitive sites were observed in the DNA region corresponding to the predicted curved DNA region (see Fig. 2A).
Figure 3.

LrpC promotes DNA bending. DNase I footprinting analysis of the 331 bp [γ-32P]DNA (1 nM, labelled on the top strand) incubated with increasing concentrations of LrpC (lanes 2–7, 4, 10, 30, 90, 250 and 850 nM) and partially digested with DNase I. The partially digested complexes were precipitated and separated by 6% denaturing PAGE, and the cleavage pattern visualised by autoradiography. The hypersensitive sites are indicated with arrowheads; the numbers correspond to the base pairs of the DNA fragment from the labelled site.
The addition of 30 mM l-leucine does not modify the LrpC footprint (data not shown). It is likely, therefore, that LrpC, which upon binding increases the angle of curvature of the analysed DNA segment, is not responsive to leucine (see above).
The Lrp-type proteins EcoLrp, BkdR and PutR cause an extended pattern of protected DNase I cleavage (>100 bp upstream of the transcription start sites) and hypersensitive sites having a periodicity of ~10 nt (24–26). Unlike these Lrp-type proteins from Gram-negative bacteria (24–26), tetrameric LrpC fails to protect either of the strands of the analysed DNA from DNase I attack and no periodicity of hypersensitive sites was obvious, suggesting that LrpC protein does not loop DNA and that the role of LrpC is mostly transient. This could explain the apparent discrepancy between the Kapp (at least 4 nM) using DNase I footprinting and the Kapp using filter binding (80 nM) or EMSA (~75 nM). Filter binding and EMSA assays directly measure those complexes stable enough to be trapped on a nitrocellulose filter or upon migration on a gel, however, the footprinting assay effectively detects even weakly bound protein–DNA complexes which are too unstable to be retained on a nitrocellulose filter or in a gel.
The ability of LrpC to promote DNA bending was confirmed by a T4 DNA ligase-mediated circularisation assay with a short DNA fragment (from the 331 bp internal segment, 182 bp in length) (Fig. 4). The identity of the circularised 182 bp DNA fragment (0.5 nM) was evident from its resistance to exonuclease III treatment (data not shown). No circular products could be detected in the absence or presence of LrpC up to 60 nM. LrpC promotes circularisation of the DNA fragment at concentrations of 120–250 nM, whereas a LrpC concentration of 500 nM or higher seems to preclude such contacts of the DNA ends (Fig. 4 and data not shown).
Figure 4.
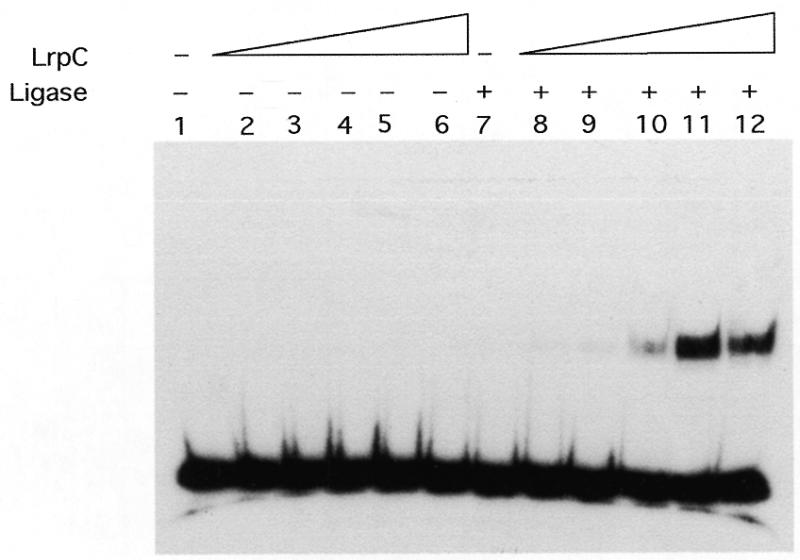
DNA circularisation by LrpC. The 182 bp [γ-32P]DNA fragment (0.5 nM) was incubated with increasing concentrations of the LrpC protein (30–500 nM, lanes 2–6 and 8–12) in buffer C containing 50 mM NaCl and 1 mM ATP for 15 min at 37°C. In samples 1–6 the DNA was further incubated with ligase buffer and in lanes 7–12 with 1 U of T4 DNA ligase for 30 min at 16°C. The samples were deproteinised and analysed by 4% non-denaturing PAGE in 0.5× TBE, and autoradiographs of the dried gels were subsequently taken.
Mutually cooperative binding of LrpC and Hbsu
Bacillus subtilis Hbsu and the highly homologous Bacillus stearothermophilus Hbst protein are bona fide sequence-independent DNA-binding and DNA-bending proteins (15,19,27,28). To examine the influence of Hbsu on binding of LrpC to curved DNA (a 597 bp hbs 5′ intergenic region), we incubated the curved [α-32P]DNA (1 nM) with Hbsu (40–200 nM), LrpC (6–30 nM), a fixed amount of Hbsu (40 nM) and varying amounts of LrpC (1.5–24 nM) or a fixed amount of LrpC (6 nM) and various concentrations of Hbsu (20–80 nM) for 15 min at 30°C. The reaction mixture was then subjected to non-denaturing PAGE. No Hbsu interaction with curved [α-32P]DNA was observed in the presence of up to 70 nM protein, whereas in the presence of concentrations >100 nM one discrete protein–DNA complex (CI) was detected (Fig. 5, lanes 2–5, and data not shown). The Kapp of the Hbsu–curved DNA complex was ~120 nM at pH 7.5 and 30°C (data not shown). In the presence of 6–24 nM LrpC, no or a small amount of protein–DNA complex was observed, whereas at protein concentrations >30 nM, a discrete complex (C1) and a high order complex that did not enter the gel were observed (Fig. 5, lanes 6 and 9). In quantitative terms, the Kapp of LrpC–curved DNA complexes was ~75 nM at pH 7.5 and 30°C (data not shown). Since no sequence similarity longer than six identical nucleotides was detected between the curved DNA substrates used [the 331 (Fig. 2) and 597 bp (Fig. 5) fragments] and the Kapp values of the LrpC–DNA complexes were similar, we could assume that such DNA binding might be sequence-independent.
Figure 5.
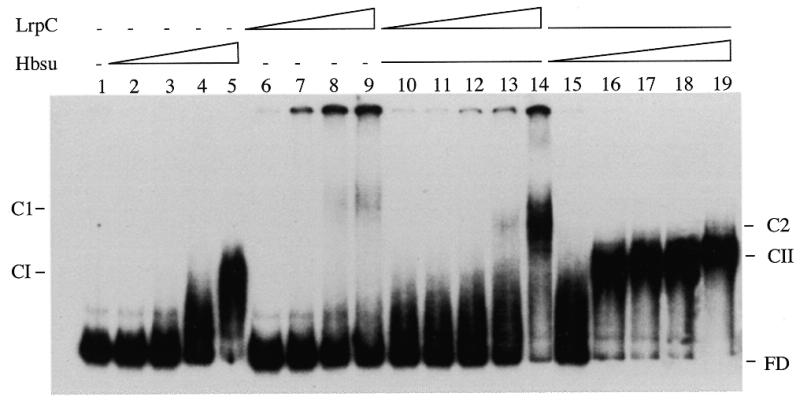
Mutually cooperative binding of LrpC and Hbsu. EMSA assay of the 597 bp curved DNA in the presence of the indicated proteins. The 597 bp curved [α-32P]DNA fragment (1 nM, lane 1) was incubated with increasing concentrations of Hbsu protein (lanes 2–5, 40, 80, 100 and 200 nM) or LrpC (lanes 6–9, 6, 12, 24 and 30 nM), with a constant amount of Hbsu (40 nM) and increasing concentrations of LrpC (lanes 10–14, 1.5, 3, 6, 12 and 24 nM) or with constant LrpC (6 nM) and increasing concentrations of Hbsu (lanes 15–19, 20, 40, 50, 60 and 80 nM). The Hbsu complex (CI), LrpC complex (C1), the complexes formed in the presence of both proteins (Hbsu + LrpC, complex CII; LrpC + Hbsu, complex C2) and the free DNA (FD) are denoted.
To examine the influence of Hbsu on the affinity of LrpC for curved DNA, we incubated the curved [α-32P]DNA (1 nM) with a sub-optimal concentration of Hbsu protein (40 nM) and varying concentrations of LrpC (1.5–24 nM). As shown in Figure 5, lanes 10–14, a diffuse complex migrating faster than LrpC–DNA was observed. This ternary complex, termed C2, contains both LrpC and Hbsu (data not shown). The Kapp of LrpC–DNA binding (C2 complex) was reduced at least 3-fold, from 75 to ~24 nM, when Hbsu (40 nM) was present in the reaction mixture. Since mutual cooperative binding of LrpC and mammalian HMG1 was observed when the latter was used instead of Hbsu (data not shown), we ruled out a direct protein–protein interaction (19). To measure the influence of LrpC on the affinity of Hbsu for curved DNA, we incubated the curved [α-32P]DNA (1 nM) with a sub-optimal concentration of LrpC protein (6 nM) and varying concentrations of Hbsu (20–80 nM). Figure 5, lanes 15–19, shows that the Kapp of Hbsu–DNA binding (ternary complex CII) was reduced at least 4-fold, from ~120 to ~30 nM, when LrpC (6 nM) was bound to the curved DNA.
Identical results to those presented in Figure 5 were observed when Hbsu or LrpC protein was bound to the DNA and the pre-formed complex Hbsu–DNA or LrpC–DNA was then incubated with LrpC or Hbsu, respectively (data not shown). It is likely, therefore, that the curvature promoted by one of the proteins stabilises a DNA structure that could be recognised with higher efficiency by the second protein. This is consistent with the fact that both LrpC and Hbsu make transient contacts with DNA (Fig. 3; 15,19) and that both proteins show greater affinity for curved DNA (Fig. 2; 13,19) and promote DNA bending (Figs 3 and 4; 13,15,19).
LrpC bridges DNA
Above we showed that LrpC forms higher order complexes that do not migrate into the gel (Fig. 5). To explain these observation we could propose that LrpC forms intermolecular bridges between different DNA segments that result in a large network. Using the assay described by Lee and Graigie (18), the 182 bp [γ-32P]DNA (70 nM; Fig. 6) or supercoiled circular pUC18 DNA (5 nM; data not shown) was incubated with increasing concentrations of LrpC protein (50–1600 nM) for 10 min on ice and the mixture centrifuged at 12 000 g for 20 min. After deproteinisation, the DNA remaining in the supernatant was analysed by agarose gel electrophoresis (pUC18 DNA) or non-denaturing PAGE (182 bp DNA). As revealed in Figure 6, incubation of linearised 182 bp DNA with LrpC (200 nM) at a stoichiometry of one LrpC tetramer per 64 bp resulted in ~65% of the DNA being pelleted by low speed centrifugation, while in the presence of one LrpC tetramer per 32 bp ~90% of the DNA substrate was precipitated. Similar results were obtained when supercoiled pUC18 DNA was used as substrate (data not shown). Hence, LrpC protein promotes intramolecular (pUC18 as substrate) as well as intermolecular bridging (182 bp fragment as substrate) with subsequent compactation of the DNA.
Figure 6.
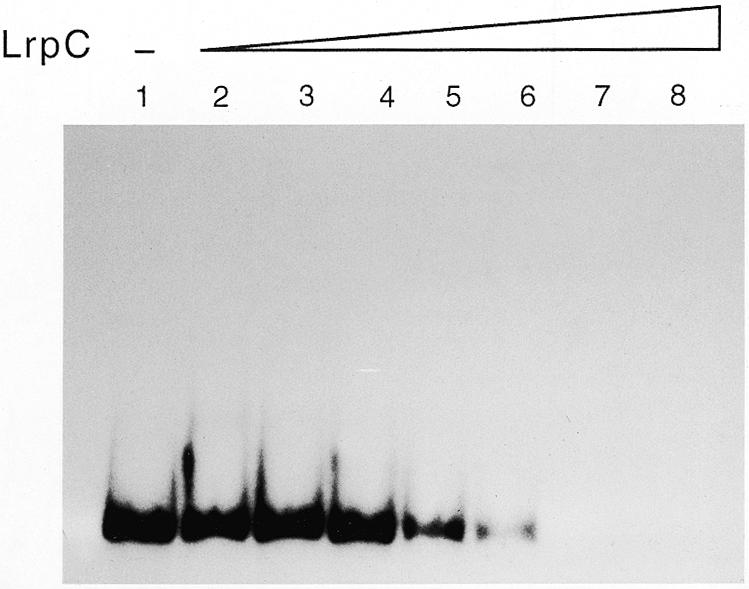
LrpC promotes DNA aggregation. The 182 bp γ-32P-labelled DNA (70 nM) was incubated with increasing amounts of LrpC (lanes 2–8, 25, 50, 100, 200, 400, 800 and 1600 nM) in buffer B containing 100 mM NaCl for 10 min on ice, and then centrifuged at 12 000 g for 20 min. After addition of 0.5% SDS, the DNA remaining in the supernatant was analysed by 4% non-denaturing PAGE in 0.5× TBE, and autoradiographs of the dried gels were subsequently taken.
Western blots analysis, with polyclonal antibodies against LrpC, of samples obtained from the supernatant and the pellet in the absence and presence of DNA showed that ~6% of the total protein was present in the pellet in the absence of DNA, whereas >95% of the protein was in the pellet in the presence of DNA (data not shown). Furthermore, similar results to those described in Figure 6 were obtained when LrpC was centrifuged prior to incubation with the DNA and the supernatant used for further analysis.
Recently, Lee and Craigie (18) showed that a host factor involved in protecting retroviral DNA from autointegration, termed barrier-autointegration factor (BAF), promotes intermolecular bridging between DNA segments in an open mesh in which the DNA molecules are bridged by BAF. Both BAF (one dimer per 75 bp; 18) and LrpC (one tetramer per 64 bp) promote intermolecular aggregation of DNA resulting in formation of a network. In contrast to LrpC, the other biochemically characterised Lrp-type proteins do not form DNA networks (22,23,29).
LrpC constrains DNA supercoils
To address whether the LrpC protein bound to supercoiled DNA (10 nM) could restrain the negative torsional tension, supercoiled plasmid DNA was incubated with Topo I (2 U) and increasing amounts of LrpC (50–1600 nM). In the absence of LrpC the DNA was partially relaxed by Topo I (Fig. 7, lane 2). As revealed in Figure 7, lanes 4–6, LrpC protein (100–400 nM) hinders DNA relaxation, implying that LrpC constrains supercoils and suggesting a role of this protein in bacterial chromatin. The maximum number of supercoils was constrained at a ratio of one LrpC tetramer per 67 bp. Such an affect, however, is not observed when higher concentrations of LrpC were added (800–1600 nM) (Fig. 7, lanes 7 and 8). These data can be interpreted as showing that LrpC constrains supercoils and that it does not inhibit the action of Topo I, because at high LrpC concentrations Topo I still has access to the DNA. At the highest LrpC concentration, at which supercoils were no longer constrained (1.6 µM LrpC, one LrpC tetramer per 16 bp), the DNA was fully compacted by LrpC (Fig. 6).
Figure 7.
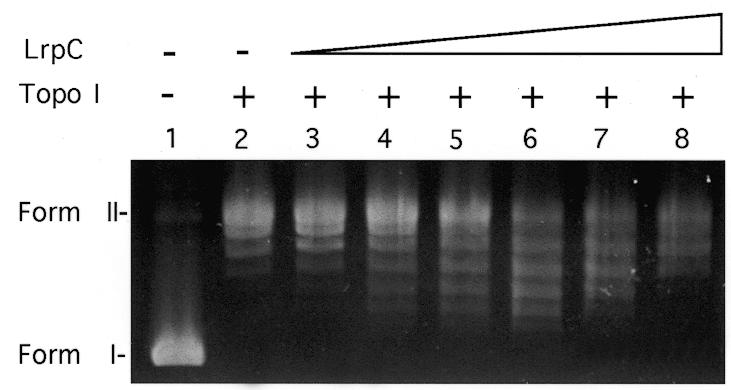
LrpC alters the topology of DNA. A supercoiling assay with pUC18 plasmid DNA (10 nM) and increasing concentrations of LrpC (lanes 3–8, 50, 100, 200, 400, 800 and 1600 nM) in the presence of Topo I (2 U). Lane 1, naked DNA; lane 2, in the presence of 2 U Topo I. The deproteinised DNA was analysed by agarose gel electrophoresis and ethidium bromide staining. The migration positions of relaxed (form II) and supercoiled (form I) plasmid DNA are indicated.
LrpC cannot replace Hbsu in β-recombinase-mediated site-specific recombination
The site-specific β-recombinase is unable to catalyse recombination between two directly oriented (deletions) or inversely oriented (inversions) six sites unless an assisting sequence-independent DNA-binding and DNA-bending protein (e.g. Hbsu, HU or HMG1) is present (15,19,30). Recently, it has been shown that the DNA-bending proteins, such as E.coli HU, Hbsu or mammalian or plant HMG1-like protein, facilitate the joining of already bent distant recombination sites to form a stable β-recombinase-mediated synaptic complex (15,19,30,31).
To examine whether LrpC protein, which has been shown to be a DNA-bending protein (this work) plays a role in assembly of the β-recombinase–six nucleoprotein complex we used the site-specific DNA recombination reaction (15). β-Recombinase (50 nM) catalyses DNA deletions between two directly oriented six sites located on the substrate pCB8 (10 nM) in the presence of 50–1000 nM Hbsu protein (15). However, when increasing amounts of LrpC protein (7–2000 nM) were used instead of Hbsu, β-recombinase failed to catalyse DNA deletions (data not shown). It is likely, therefore, that LrpC cannot replace Hbsu or eukaryotic HMG1 protein in β-recombinase-mediated site-specific recombination and that only a limited repertoire of DNA structures can be active for β-recombinase-mediated DNA resolution. Hence, the repertoire of DNA structures generated by LrpC are different from those generated by Hbsu protein. The presence of a constant amount of Hbsu and increasing concentrations of LrpC does not affect the yield of recombination products more than 2-fold, however, when we analysed the role of LrpC in the presence of increasing concentrations of Hbsu, we observed that in the presence of an excess of Hbsu, LrpC inhibited the β-recombinase-mediated recombination reaction ~4-fold (data not shown). Above we showed that LrpC forms a nucleoprotein complex that is recognised by Hbsu protein, affecting its DNA binding affinity. Since we have observed that, under these experimental conditions, LrpC does not prevent binding of the β-recombinase or Hbsu proteins to the DNA substrate (data not shown) but affects the supercoiling state of the DNA (Fig. 7) by compacting it (Fig. 6), and since β-recombinase-mediated deletion is strictly dependent on a narrow window of DNA superhelicity (31), we could assume that, in the presence of an excess of Hbsu, LrpC modifies the DNA topology making the substrate refractory to β-recombinase.
Like the chromatin-associated proteins HU, Hbsu, HMG1 and H-NS (reviewed in 1–5), we have shown that LrpC binds to a DNA structure specifically (Fig. 2), is capable of modulating DNA structures (Figs 3–5) and constrains DNA supercoils in vitro (Fig. 7). A parallel can be drawn between LrpC and the sequence-independent chromatin-associated proteins of both prokaryotic (dimeric EcoHU and Hbsu) and eukaryotic (monomeric HMG1) origin (1–5). It is also conceivable that LrpC is an architectural element that facilitates the formation of nucleoprotein structures. LrpC, which shares a significant degree of identity (30–35%) with Lrp-type proteins, seems to be a distant member of the family. We can hypothesise that LrpC is an architectural protein that did not acquire the more specialised functions that other members of the Lrp-type family have acquired (32) and can be considered as a chromatin-associated protein.
Acknowledgments
ACKNOWLEDGEMENTS
We thank F. Le Hégarat and C. Beloin for the gift of anti-EcoLrp polyclonal antibodies and plasmids. We are grateful to J. C. Alonso for continuous support and to F. Rojo, J. Barbé and L. Hirschbein for their interest in this project. This work was partially supported by grant PB 96-0817 from DGICYT and 06G-004/96 from the Consejería de Educación y Cultura de la Comunidad de Madrid to J. C. Alonso. S.A. was supported by a Comunidad de Madrid Training Grant; G.L. is holder of a fellowship from DGICYT (PB 96-0817).
REFERENCES
- 1.Bianchi M.E. (1994) Mol. Microbiol., 14, 1–5. [DOI] [PubMed] [Google Scholar]
- 2.Grosschedl R. (1995) Curr. Opin. Cell Biol., 7, 362–370. [DOI] [PubMed] [Google Scholar]
- 3.Pérez-Martín J., Rojo,F. and deLorenzo,V. (1994) Microbiol. Rev., 58, 268–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberto J., Drlica,K. and Rouvière-Yaniv,J. (1994) Biochemie, 76, 901–908. [DOI] [PubMed] [Google Scholar]
- 5.Nash H.A. (1996) In Lin,E.C.C. and Lynch,A.S. (eds), Regulation of Gene Expression in Escherichia coli. R.G.Landes Co., Austin, TX, pp. 149–179.
- 6.Kunst F., Ogasawara,N., Moszer,I., Albertini,A.M., Alloni,G.A., Azevedo,V., Bertero,M.G., Bessières,P., Bolotin,A., Borchert,S. et al. (1997) Nature, 390, 249–256. [DOI] [PubMed] [Google Scholar]
- 7.Micka B. and Marahiel,M.A. (1992) Biochemie, 74, 641–650. [DOI] [PubMed] [Google Scholar]
- 8.Dartois V., Liu,J. and Hoch,J.A. (1997) Mol. Microbiol. 25, 39–51. [DOI] [PubMed] [Google Scholar]
- 9.Beloin C., Ayora,S., Exley,R., Hirschbein,L., Ogasawara,N., Kasahara,Y., Alonso,J.C. and Le Hégarat,F. (1997) Mol. Gen. Genet., 256, 63–71. [DOI] [PubMed] [Google Scholar]
- 10.Belitsky B.R., Gustafsson,M.C., Sonenshein,A.L. and von Wachenfeldt,C. (1997) J. Bacteriol., 179, 5448–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Studier W.F., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 12.Yanisch-Perron C., Viera,J. and Messing,J. (1985) Gene, 33, 103–109. [DOI] [PubMed] [Google Scholar]
- 13.Fernández S. and Alonso,J.C (1999) Gene, 231, 187–193. [DOI] [PubMed] [Google Scholar]
- 14.Rojo F. and Alonso,J.C. (1994) J. Mol. Biol., 238, 159–172. [DOI] [PubMed] [Google Scholar]
- 15.Alonso J.C., Weise,F.. and Rojo,F. (1995) J. Biol. Chem., 270, 2938–2945. [DOI] [PubMed] [Google Scholar]
- 16.Ayora S., Rojo,F., Ogasawara,N., Nakai,S. and Alonso,J.C. (1996) J. Mol. Biol., 256, 301–318. [DOI] [PubMed] [Google Scholar]
- 17.Galas D.J. and Schmitz,A. (1978) Nucleic Acids Res., 5, 3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee M.S. and Craigie,R. (1998) Proc. Natl Acad. Sci. USA, 95, 1528–1533. [Google Scholar]
- 19.Alonso J.C., Gutierrez,C. and Rojo,F. (1995) Mol. Microbiol., 18, 471–478. [DOI] [PubMed] [Google Scholar]
- 20.Willis D.A., Ryan,C.W., Platko,J.V. and Calvo,J.M. (1991). J. Biol. Chem., 266, 10768–10774. [PubMed] [Google Scholar]
- 21.Huang N., Madhusudhan,K.T. and Sokatch,J.R. (1996) Biochem. Biophys. Res. Commun., 223, 315–319. [DOI] [PubMed] [Google Scholar]
- 22.Newman E.B. and Lin,R. T (1995) Annu. Rev. Microbiol., 49, 747–775. [DOI] [PubMed] [Google Scholar]
- 23.Newman E.B., Lin,R.T. and d’Ari,R. (1996). In Neidhardt,F.C., Ingraham,J.L., Low,K.B., Magasanik,B., Schaechter,M. and Umbarger,M.E. (eds), Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. American Society for Microbiology, Washington, DC, pp. 1513–1525.
- 24.Madhusudhan K.T., Hester,K.L., Friend,V. and Sokatch,J.R. (1997) J. Bacteriol., 179, 1992–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jafri S., Evoy,S., Cho,K., Graighead,H.G. and Winans,S.C. (1999) J. Mol. Biol., 288, 811–824. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q. and Calvo,J.M. (1993) EMBO J., 12, 2495–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka I., Appelt,K., Dijk,J., White,S.W. and Wilson,K.S. (1984) Nature, 310, 376–381. [DOI] [PubMed] [Google Scholar]
- 28.Vis H., Mariani,M., Vorgias,C.E., Wilson,K.S., Kaptein,R. and Boelens,R. (1995) J. Mol. Biol., 254, 692–703. [DOI] [PubMed] [Google Scholar]
- 29.Calvo J.M. and Matthews,R.G. (1994) Microbiol.. Rev., 58, 466–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ritt C., Grimm,R., Fernández,S., Alonso,J.C. and Grasser,K.D. (1998) Plant J., 14, 623–631. [DOI] [PubMed] [Google Scholar]
- 31.Canosa I., Lurz,R., Rojo,F. and Alonso,J.C. (1998) J. Biol. Chem., 273, 13886–13891. [DOI] [PubMed] [Google Scholar]
- 32.d’Ari R., Lin,R.T. and Newman,E.B. (1993) Trends Biochem. Sci., 18, 260–263. [DOI] [PubMed] [Google Scholar]