

Abstract
Zoonomia is the largest comparative genomics resource for mammals produced to date. By aligning genomes for 240 species, we identify bases that, when mutated, are likely to affect fitness and alter disease risk. At least 332 million bases (~10.7%) in the human genome are unusually conserved across species (evolutionarily constrained) relative to neutrally evolving repeats, and 4552 ultraconserved elements are nearly perfectly conserved. Of 101 million significantly constrained single bases, 80% are outside protein-coding exons and half have no functional annotations in the Encyclopedia of DNA Elements (ENCODE) resource. Changes in genes and regulatory elements are associated with exceptional mammalian traits, such as hibernation, that could inform therapeutic development. Earth’s vast and imperiled biodiversity offers distinctive power for identifying genetic variants that affect genome function and organismal phenotypes.
One-Sentence Summary:
We compare genomes from hundreds of mammals to explore features conserved by evolution and the origins of exceptional traits.
Placental mammals, the evolutionary lineage that includes humans, are exceptionally diverse, with more than 6100 extant species (1), from the 2-g bumblebee bat to the 150,000-kg blue whale (2, 3). Over the past 100 million years, mammals have adapted to almost every habitat on Earth (Fig. 1A) (4). Zoonomia is the largest comparative genomics resource for mammals produced to date, with whole genomes aligned for 240 diverse species [2.3-fold more families and 3.9fold more species than the mammals included in the earlier 100 Vertebrates alignment (5)] and protein-coding sequences aligned for 427 species (6). Using this resource, we can find elements that are conserved in the genomes of all placental mammals, elements that are changing unusually quickly in particular lineages, and elements that are associated with particular traits. All three approaches address a primary challenge in genomics: identifying genomic elements that affect genome function and organismal phenotypes (7).
Fig. 1. New placental mammal phylogeny supports the long-fuse model of diversification.
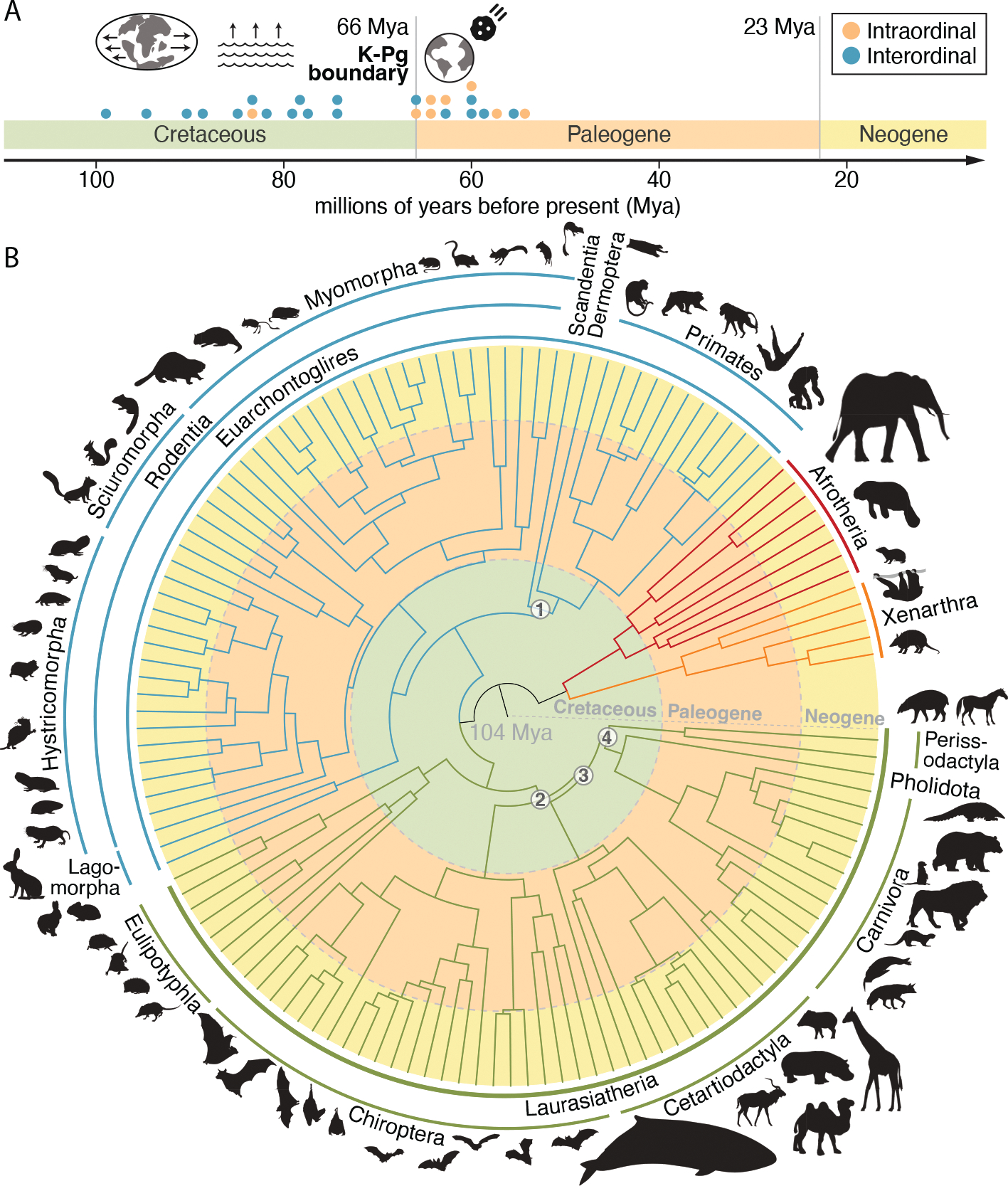
(A) Most interordinal diversification occurred in the Cretaceous, coincident with continental fragmentation and sea level changes. A pulse of intraordinal diversification occurred after the mass extinction event at the Cretaceous-Paleogene (K-Pg) boundary. Green, orange, and yellow shading bounded by gray lines demarcates different time periods. (B) A phylogeny based on divergence times estimated using ~470 kb of near-neutrally evolving sequence for 240 species resolves recalcitrant relationships in the placental mammal phylogeny (black numbers in white circles), including (1) Euarchonta (primates, colugos, and treeshrews), (2) Scrotifera [Perissodactyla (odd-toed ungulates), Cetartiodactyla (terrestrial even-toed ungulates and cetaceans), carnivorans, and bats], (3) Fereuungulata (perissodactyls, cetartiodactyls, carnivorans, pangolins), and (4) Zoomata [perissodactyls and Ferae (carnivorans and pangolins)]. [Species silhouettes are from PhyloPic]
Species evolve through selection on both small, sequence-level mutations and larger structural changes to the genome (e.g., translocation of transposable elements, inversions, deletions, and duplications), as well as through hybridization with other species (8–10). Mutations are assumed to arise by random chance and then rise and fall in frequency within a population as a consequence of both neutral drift and selection. Mutations that disrupt characteristics that are essential for survival tend to be lost, whereas those conferring an advantage are more likely to be retained, eventually resulting in genetic differences that differentiate species. By aligning the genomes of many different species, we can measure whether mutations at a given position in the genome are retained more or less often than expected under neutral drift (11–13). Fewer differences between species than expected suggests evolutionary constraint (dearth of variation due to purifying selection; also referred to as conservation), whereas more differences than expected in some lineages suggests acceleration (rapid evolution that may be clade-specific) (12, 13). Both metrics indicate that the given position has a role in molecular function. Measures of constraint and acceleration do not vary with cell type or developmental time point sampled, which simplifies sample collection and data generation. They are complementary to methods for annotating the functional genome (14, 15).
Previous studies have used comparative genomics analyses to associate protein-coding changes with specific adaptations (16), such as diet type (17), echolocation (18), and subterranean habitation (19). However, these studies included few species relative to Zoonomia. As a result, they lacked the power and resolution required to investigate changes in genes and noncoding regulatory elements on a genome-wide level. Studying the evolution of regulatory elements, which make up much of the functional sequence in the genome, is particularly challenging because they tend to evolve more quickly and be less strongly conserved than coding elements (15, 20, 21). By substantially increasing the number and diversity of species in our comparative genomic analyses, we increase the sensitivity and specificity of methods used for detecting evolutionary signals and associating these signals with species level phenotypes (22, 23).
Evolutionary constraint is a powerful tool for determining which genomic variants are causally implicated in human diseases. We explore this in detail in our companion paper (24), where we show that constrained positions are enriched for variants that explain common disease heritability more than any other functional annotation and that using the Zoonomia constraint scores improves polygenic risk scoring and fine-mapping of candidate disease loci.
Here, we use the new comparative genomics resources produced by Zoonomia to explore placental mammal evolution, including the origins of exceptional traits. We also synthesize the discoveries described by the compendium of papers in the Zoonomia package.
Evolutionary constraint and acceleration in mammals
We selected species for inclusion in Zoonomia to maximize the evolutionary branch length represented and thereby increase the power to detect constraint (4). The updated 241-way reference-free Cactus alignment with 240 species (domestic dog has two representatives) overcomes limitations of reference-based alignments (table S1) (4, 11). It includes genomic elements lost in humans, allows detection of multiple-orthology relationships, and captures complex rearrangements and copy-number variation. We observed 3.6 million perfectly conserved sites, which is 19,000-fold more than expected by chance, assuming a uniform substitution rate (4), and is consistent with purifying selection on functional positions in the genome. We measured constraint across the human, chimpanzee, mouse, dog, and little brown bat reference genomes by projecting the Cactus alignment onto each species and then measuring sequence constraint with phyloP (Fig. 2, A and B, and table S2) (11, 12). The chimpanzee referenced alignment supports the investigation of bases deleted in only humans. Mouse, dog, and little brown bat have well-annotated reference genomes and represent diverse branches of the mammalian lineage, supporting comparative research in a wide range of organisms. We measured sequence constraint in the primate subset of the Cactus alignment (43 species) using PhastCons, which offers more power with fewer species by scoring multibase elements rather than single bases (24, 25).
Fig 2. Comparing 240 species resolves mammalian constraint to single bases and identifies elements under selection.
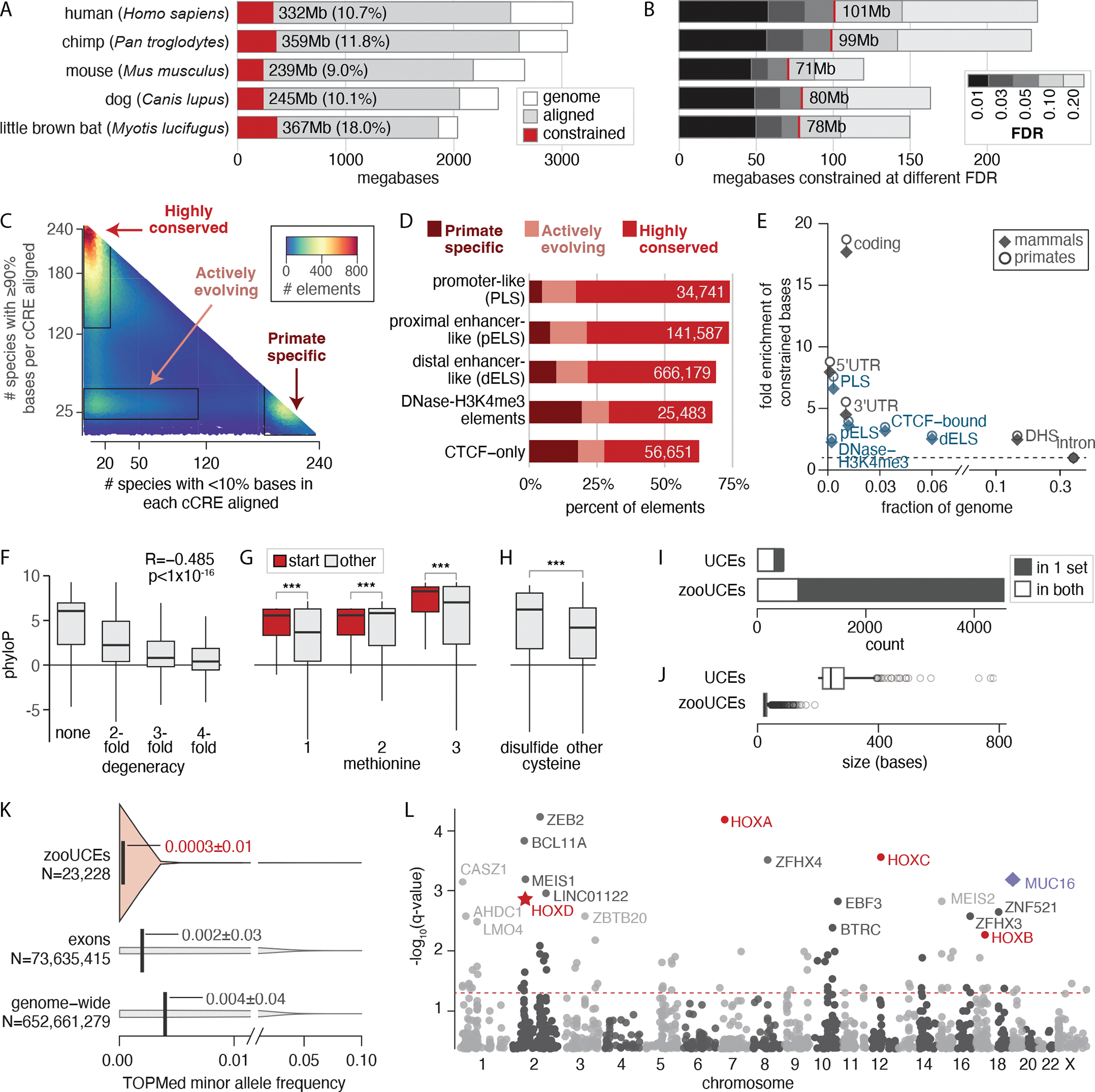
(A and B) We estimated a lower-bound on the total amount of the genome under constraint (A) and the number of single bases constrained at different FDR thresholds (B). The red lines in (B) indicate the 5% FDR threshold, with the amount of sequence below this threshold given. (C and D) Comparing the number of species with poor alignments (x axis) with those with good alignments (y axis) at 924,641 human candidate cis-regulatory elements (14) (C) reveals three clusters that are nonrandomly distributed across element types (all chi-square test p < 2.2 × 10−308) (D). (E) Functional elements are enriched for constraint, with candidate cis-regulatory elements in blue and other element types in black. The dashed line indicates no enrichment. DHS, DNase hypersensitivity site; 3′UTR, 3′ untranslated region; 5′UTR, 5′ untranslated region. (F) Constraint is negatively correlated with degeneracy across 59,504,353 protein-coding positions. (G) Methionine codons functioning as start sites in protein-coding sequence are more constrained at each of the three codon positions. (H) Cysteines in disulfide bridges are more constrained than other cysteines. In (F) to (H), the box boundaries represent 25 and 75% quartiles, with a horizontal line at the median and the vertical line demarcating an additional 1.5 times interquartile range (IQR) above and below the box boundaries. *** pWilcoxon< 1 × 10−16. (I) Most zooUCEs are new and do not overlap ultraconserved elements in the original set (73). (J) All zooUCEs are shorter than the original ultraconserved elements. Box and whisker parameters are the same as in (F), with outlier zooUCEs (>1.5 times IQR below or above the box boundaries) plotted as open circles. (K) Human variants in zooUCEs (light orange) have lower minor allele frequencies than they do in exons or genome-wide (gray). The vertical lines are at the means. The filled area is the distribution of allele frequencies. (L) Constraint measured in 100-kb bins genome-wide. The most constrained 100-kb bins include the HOX clusters (red). HOXD (red star) overlaps the longest synteny block shared across mammals (174). Rearrangements in this locus can lead to limb malformations and other damaging outcomes. One bin containing MUC16 (purple diamond) significantly lacks constraint. MUC16 provides a mucosal barrier that protects epithelial cells from pathogens. The red dashed line indicates q = 0.05. Labeled bins have q < 0.006.
We inferred a new phylogeny of placental mammals that we used for subsequent analyses that require a tree (26) (Fig. 1B). This phylogeny used only bases from the alignment that scored as near-neutrally evolving with phyloP (N = 466,232). It places interordinal diversification before the major extinction event marking the end of the Cretaceous period, addressing a long-standing debate in the field (27–30). A divergence time analysis of the phylogeny supports the “long-fuse”‘ model of mammalian diversification, with interordinal diversification in the Cretaceous and most intraordinal diversification after the CretaceousPaleogene mass extinction event (31–33), and not the fossil record-derived “explosive” model, which places all interand intraordinal diversification after the Cretaceous-Paleogene event, or other scenarios (34–36).
At any given site in the genome, the number of species aligned can vary from just one to all 240. The variation in alignment depth distinguishes regulatory regions with differing evolutionary histories (37). In the human-referenced alignment, 91% of the human genome aligns to at least five species, but only 11% aligns to ≥95% (≥228) of species (fig. S1). Candidate cis-regulatory elements are 926,535 putative regulatory elements in the human genome defined by the Encyclopedia of DNA Elements (ENCODE) resource (14) using DNA accessibility and chromatin modification data. In the alignment at candidate cis-regulatory elements, we discern three common patterns (Fig. 2C). In highly conserved elements, most bases align in most species, including distantly related species. In actively evolving elements, most species have a partial alignment to humans. Primate-specific elements align exceptionally well in only a small number of species. Promoter-like and enhancer-like elements tend to be highly conserved. Elements that specifically bind the transcription factor CTCF or are marked by H3K4me3 (trimethylated histone H3 lysine 4) are more likely to be evolving actively, and about 20% are primate-specific (Fig. 2D).
Estimate of genome-wide constraint
We estimate that a minimum of 332 Mb (10.7%) of the human genome is under constraint through purifying selection (Fig. 2A) (12). We computed this lower-bound of the percentage under constraint by comparing the observed genome-wide phyloP score distribution to that expected in the absence of selection (modeled using ancestral repeats) (fig. S2A). Using bootstrapping, we show that the sample of ancestral repeats used had little effect on the lower-bound constraint estimate that was achieved; a 95% confidence interval spans only 1.9 mega–base pairs (Mbp). Ancestral repeats are a reasonable proxy for neutrally evolving sequence and can help account for local factors such as GC-content and mutation rate variation that might affect the phyloP score distribution (12, 38, 39). Our estimate of 10.7% falls at the upper end of previous estimates, which ranged from 3 to 12% (40). It is substantially higher than estimates of at least 5% that were calculated using similar methods but much smaller mammalian datasets (12, 13). With more species, we have more power to detect both weaker constraint across mammals and lineage-specific constraint, although these scenarios are not readily distinguished by the phyloP scores (fig. S2, B and C).
The lower-bound estimates for constraint in chimp-, mouse-, dog-, and bat-referenced projections of the alignment range from 239 Mb in the mouse (9.0%) to 359 Mb in the chimp (11.8%) (Fig. 2A and table S2). We are unable to determine whether the total amount of constraint truly varies between species. Both the species composition of the dataset and technical confounders, including differences in assembly contiguity and quality, could explain the differences observed. The amount of sequence detected as significantly constrained [false discovery rate (FDR) < 0.05] correlates with the average branch length to the nine closest species [Spearman’s correlation coefficient (r) = −0.975; p = 0.0048], with more constraint detected in species with more closely related species in the alignment (table S3). This suggests that the amount of the genome under detectable constraint in mouse, dog, and bat will increase as additional species are added to the alignment.
Genes enriched for constraint and acceleration
Genes with highly constrained protein-coding sequences are enriched in biological processes that function similarly across species, whereas those that are changing more quickly are enriched in processes that vary between species, consistent with previous studies (41–45). We tested the top 5% most accelerated and most conserved genes as measured by mean phyloP score of coding sequence (data S1) against a nonredundant representative set of Gene Ontology (GO) biological processes using WebGestalt and identified overrepresented gene sets (46–48). The most constrained genes are involved in posttranscriptional regulation of gene expression (“mRNA processing”; GO:0006397; 81 of 487 genes; pFDR < 0.0002) and embryonic development (“cell-cell signaling by wnt”; GO:0198738, 79 of 460 genes, pFDR < 0.0002) (fig. S3A and table S4). RNA processing is essential for regulating cellular responses to environmental change (49), and defects can cause debilitating diseases (50). “Pattern specification process” ranks third and includes all four HOX gene clusters (GO:0007389, 76 of 433; pFDR < 0.0002). The most accelerated genes shape an animal’s interaction with its environment, including innate and adaptive immune responses, skin development, smell, and taste (fig. S3B).
We leveraged the large number of species in the Zoonomia alignments to show that a well described gene inactivation, originally speculated to be human-specific (51), is found in 10 different lineages of mammals. The gene CMAH is inactivated in humans by a 92-bp frameshifting exon deletion but is intact in other great apes (52). CMAH encodes an enzyme that converts the sialic acid Neu5Ac to Neu5Gc, and its loss restricts infection by pathogens dependent on Neu5Gc [e.g., malaria parasite Plasmodium reichenowi (53)] but increases susceptibility to viruses that bind Neu5Ac [e.g., severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (54)]. When first observed, the loss of CMAH in humans was speculated to explain human-specific brain expansion (55, 56), but other mammals were subsequently shown to lack CMAH function (57–59). We combined the Cactus whole-genome alignment with analyses of read coverage and coding sequence alignment and found that CMAH has been inactivated in 40 of 239 species analyzed, representing 10 lineages (five newly discovered), including three rodent lineages and three bat lineages (fig. S4) (58). We confirm that CMAH loss occurred in the ancestor of all mustelids and pinnipeds using 11 species (compared with three originally) and that, among the primates, only humans and platyrrhine (New World) monkeys have lost CMAH (57). The role of CMAH in pathogen response suggests that its loss could shape the zoonotic potential of Neu5Gc-dependent pathogens, but further investigation is needed (60). Correlating CMAH inactivation with susceptibility to infection by SARS-CoV-2 or other viruses will require measuring infection susceptibility for a larger and more diverse set of mammals than has been studied to date.
Single-base resolution of constraint
Coding regions are the most strongly enriched for evolutionarily constrained positions, but most (80%) constrained positions are noncoding (Fig. 2E). We defined a “constrained base” as a position that has a positive phyloP score with FDR < 5%. Constrained bases comprise 3.26% (101 Mb) of the human genome (Fig. 2B and table S2) and tend to cluster together, as previously described (13, 61). Most (80%) are within 5 bp of another constrained base, and 30%are in blocks ≥5 bp.The conservative FDR < 5% threshold limits the number of false positives but may miss weakly constrained bases or bases constrained in just a subset of mammals. Using a threshold of FDR < 20% increases the estimated percentage of bases constrained from 3.26 to 7.56% (Fig. 2B and table S2).
The phyloP scores have three-base periodicity in coding sequence, consistent with the genetic code (62, 63). The Zoonomia phyloP scores are strongly correlated with the codon degeneracy at individual positions. Nondegenerate sites are farmore likely to be constrained bases than fourfold degenerate sites (74.1 versus 18.5%). The median phyloP score exomewide is 4.9 [interquartile range (IQR) = 5.8] in the first position (nondegenerate for 17 of 20 amino acids), 6.0 (IQR = 4.0) in the second (nondegenerate in 19 of 20), and 0.68 (IQR = 2.7) in the third (nondegenerate for 2 of 20) (fig. S5). The more functionally equivalent nucleotide options a coding base has in the genetic code, the weaker its phyloP score (Spearman’s r = −0.51, p < 2.2 × 10−16) (Fig. 2F). Our ability to demonstrate expected patterns of constraint in coding sequence suggests that we have achieved sufficient power to resolve constraint to single bases in the human genome. This is unprecedented. The 29 Mammals project alignment resolved constraint to ~12 bases (13), and studies with more species examined only a subset of the genome (12). Comparing exomes for 141,456 humans achieved only gene or exon-level resolution (64).
We discern stronger constraint at critical positions in peptides than at other protein-coding positions, supporting the utility of the Zoonomia phyloP scores for predicting functional importance. Whereas previous work had shown broadly that splice sites are often located in constrained regions (61), we discern enrichment of constraint at start codons, stop codons, and splice sites specifically (24 times, 19 times, and 25 times greater than genomewide; chi-square test, p < 2.2 × 10−16). Methionine codons that function as start codons are more conserved than methionines elsewhere in the peptide (Fig. 2G). Cysteines in intrapeptide disulfide bridges, which can cause misfolding when mutated (65), are more conserved than other cysteines (Fig. 2H).
Bases constrained in mammals are less likely to be variable in humans, consistent with purifying selection (64, 66–68). Previous work showed that variants in functional positions have lower minor allele frequencies among humans in the Trans-Omics for Precision Medicine dataset (TOPMed) (69). Positions designated as evolutionarily constrained in Zoonomia similarly have lower minor allele frequencies in TOPMed, consistentwith functional importance [constrained: frequency = 0.0026 ± 0.02 (±SD) and N = 20,718,868; unconstrained: 0.0040 ± 0.04 and N = 601,458,551; pWilcoxon= 9.5× 10−13] (69). The less variable the position is in humans, the stronger its constraint across mammals (Spearman’s r = 0.78, p = 0.00014; N = 622,177,419; fig. S6A).
Incorporating mammalian constraint into functional predictions will likely be particularly informative for poorly annotated positions. The correlation between the percentage of variants that are very rare in humans (minor allele frequency <0.005 variants) and phyloP scores is strongest for positions that are scored as having unknown functional impact by SnpEff (70) (Spearman’s r = 0.98, p = 5.45 × 10−7; N = 608,227,093; fig. S6B). SnpEff already considers 100-way vertebrate constraint scores in scoring variants, suggesting that constraint within mammals provides functional information that is not available through other sources.
Using versions of the reference-free Cactus alignment projected onto species other than human, we can assess constraint at positions that are deleted in the human genome and thus missing from previous resources (5, 13). We identified 10,032 human-specific deletions that overlap conserved elements and functionally assessed their regulatory effects using massively parallel reporter assays (71). Subsetting on just human-specific deletions constrained in chimp (phyloP score > 1) substantially increased concordance between measured regulatory change and predicted transcription factor binding differences [Pearson’s correlation coefficient (r) increases from 0.25 (p = 0.0037) to 0.37 (p = 0.00019); Spearman’s r increases from 0.24 (p = 0.00614) to 0.32 (p = 0.00158)].
New catalogs of conserved elements
We expanded and refined the catalog of ultraconserved elements in the human genome by 13-fold using the Cactus alignment, providing a rich new resource for exploring essential mammalian traits (72). The original set of 481 mammal ultraconserved elements consists of elements >200 bp long with identical sequence between human, mouse, and rat (73). Most are noncoding, and many function as enhancers during embryonic development (74–76). We defined Zoonomia ultraconserved elements (zooUCEs) as regions 20 bp or longer where every position is identical in at least 235 of 240 (98%) species in the alignment. Of the 4552 zooUCEs [average size 28.9 ± 13.0 bp (±SD)], 753 overlap 318 of the original ultraconserved elements, whereas 3799 are new (Fig. 2, I and J). Twenty-seven zooUCEs are longer than 100 bp (fig. S7A). Most of the zooUCEs are noncoding (69% are outside of proteincoding exons). Like the original ultraconserved elements, they are enriched near genes whose products are involved in transcription-related and developmental biological processes (table S5 and data S1) (73). The longest two zooUCEs (190 and 161 bp) are separated by a single base and are in an intron of POLA1, which encodes the catalytic subunit of DNA polymerase α.
HumanTOPMed variants are rare in zooUCEs compared with the rest of the genome, suggesting purifying selection within humans similar to the original UCEs (25, 72, 77, 78). ZooUCEs have fewer positions that are variable in humans (17.6%) than the coding sequences of genes (22.7%), which are known to be exceptionally constrained (69). When variants do occur in zooUCEs, their allele frequencies tend to be extremely low compared with those of variants that occur elsewhere in the genome. Average minor allele frequencies were 12.97 and 7.72 times lower in zooUCEs [N = 23,228; mean = 0.0003 ± 0.01 (±SD)] compared with genome-wide (N = 652,661,279; mean = 0.004 ± 0.04) and within exons (N = 73,635,415; mean = 0.002 ± 0.03), respectively (Fig. 2K).
We also cataloged constrained regions in the human genome using a phyloP score–based metric that allowed for more variability in constraint across mammals than the zooUCE criteria. Regions of contiguous constraint are regions of at least 20 bases where every individual base has a phyloP score above the FDR < 5% threshold (fig. S7B). Of the 595,536 such regions that we identified, most are short (median size = 32, IQR = 27), but 273 are longer than 500 bp and six are longer than 1 kb. The longest (1.36 kb) is in an intron of the gene METAP1D (chr2:172071926-172073285) and encompasses four distal enhancer-like candidate cis-regulatory elements. METAP1D encodes an essential mitochondrial protein that is conserved at least back to the common ancestor of human and zebrafish (79). This locus physically interacts with at least one transcription start site for each of METAP1D (FastHiC q = 2.23 × 10−2), TLK1 (FastHiC q = 7.62 × 10−3), and HAT1 (FastHiC q = 3.92 × 10−2) in human adult cortex Hi-C data (80–82). The synteny between these three genes is preserved in the Xenopus frog (83, 84). TLK1 regulates chromatin structure (85), HAT1 coordinates histone production and acetylation (86), and both are expressed in the cerebral cortex of 19 (TLK1) or 21 (HAT1) out of 19 or 21 mammals analyzed in a previous study, respectively (87).
We identified broad regions of unusually high constraint by scoring 100-kb nonoverlapping bins (N = 28,218) across the genome based on the fraction of bases that were constrained (data S2). We identified 53 bins with significantly elevated constraint (q < 0.05; average 17.8% constrained bases versus 3.5% for the genome; table S6). These bins are enriched for transcription-related biological processes and overlap the four HOX gene clusters (Fig. 2L). Five are in gene deserts, and two neighbor highly constrained developmental transcription factors (LMO4 and BCL11A) (88, 89).
Constraint suggests regulatory function
Zoonomia’s metrics of constraint can help detect positions likely to have regulatory function both within and outside of coding regions. In coding sequence, fourfold degenerate sites that overlap ENCODE3 transcription factor binding sites (N = 2,647,541) (90) show moderately higher constraint than other fourfold degenerate sites (N = 2,420,610; chi-square test, p < 2.2 × 10−16; fig. S8). Noncoding constrained bases are enriched in regulatory elements across mammals and within primates, including at promoter-like signatures, enhancer-like signatures, sites bound by CTCF, and sites marked by H3K4me3 (Fig. 2E) (20, 91). The proportion of bases under constraint is higher in the subset of gene deserts (the longest 5%of intergenic regions) that neighbor developmental transcription factors (224 of 873 regions; pWilcoxon=2.15× 10−15) (92, 93) than in other gene deserts and is particularly high in candidate cis-regulatory elements within such gene deserts (N = 38,065; pWilcoxon= 6.95 × 10−280 compared with elements in other gene deserts; table S7).
Zoonomia constraint scores can distinguish which regulatory elements are likely to be functionally conserved across species. We identified transcription factor binding sites genome-wide for 367 transcription factors using convolutional neural networks and publicly available data for more than 600 ENCODE3 (14) transcription factor binding experiments spanning hundreds of cell and tissue types (37). This is a more comprehensive assessment of the regulatory landscape in mammals than was performed in previous work, which focused on two or three different transcription factors in five or six species (94, 95). We used a two-component Gaussian mixture model to classify sites as constrained or unconstrained. Of 15.6 million unique binding sites, covering 5.7% of the human genome, 1.9 million (0.8% of the genome) are constrained (table S8). Minor allele frequencies at sites variable in humans are significantly lower in constrained (mean = 0.0022, SD = 0.032) than in unconstrained (mean = 0.0036, SD = 0.041) binding sites (one-sided pWilcoxon < 2.2 × 10−16), consistent with strong purifying selection on these sites. The fraction of binding sites constrained varies by transcription factor and ranges from 1.5% (ZNF250) to 59.8%(YY2) (fig. S10A). The orthologs of the constrained binding sites are enriched for active histone marks [H3K4me3 and H3K27ac (acetylated histone H3 lysine 27)] in macaque, dog, mouse, and rat compared with unconstrained binding sites, suggesting that constrained sites are more likely to be functional in other species (fig. S9).
The correlation of constraint with both motif information content and functional state is evident in transcription factor binding sites for CTCF. CTCF is a highly conserved and ubiquitously expressed transcription factor that mediates genome three-dimensional (3D) structure (96–98). Overall, 14.8% of CTCF’s binding sites are constrained (Fig. 3A). Motif information content for individual bases is significantly more correlated with base-level constraint in constrained sites than in unconstrained sites, showing that Zoonomia achieved single-base resolution constraint in noncoding regulatory elements that were missing from earlier analyses (95, 99) (Fig. 3B and fig. S10). This pattern persists across constrained binding sites for all evaluated transcription factors (Fig. 3C and fig. S10, B and C), advancing earlier work that lacked single base–level resolution (37, 95, 99). The motif logos calculated from constrained CTCF binding sites are nearly identical across species, unlike unconstrained sites (Fig. 3D), suggesting that constrained binding sites are more likely to be functional in other mammals (Fig. 3, E and F).
Fig. 3. Conserved function of constrained transcription factor binding sites.
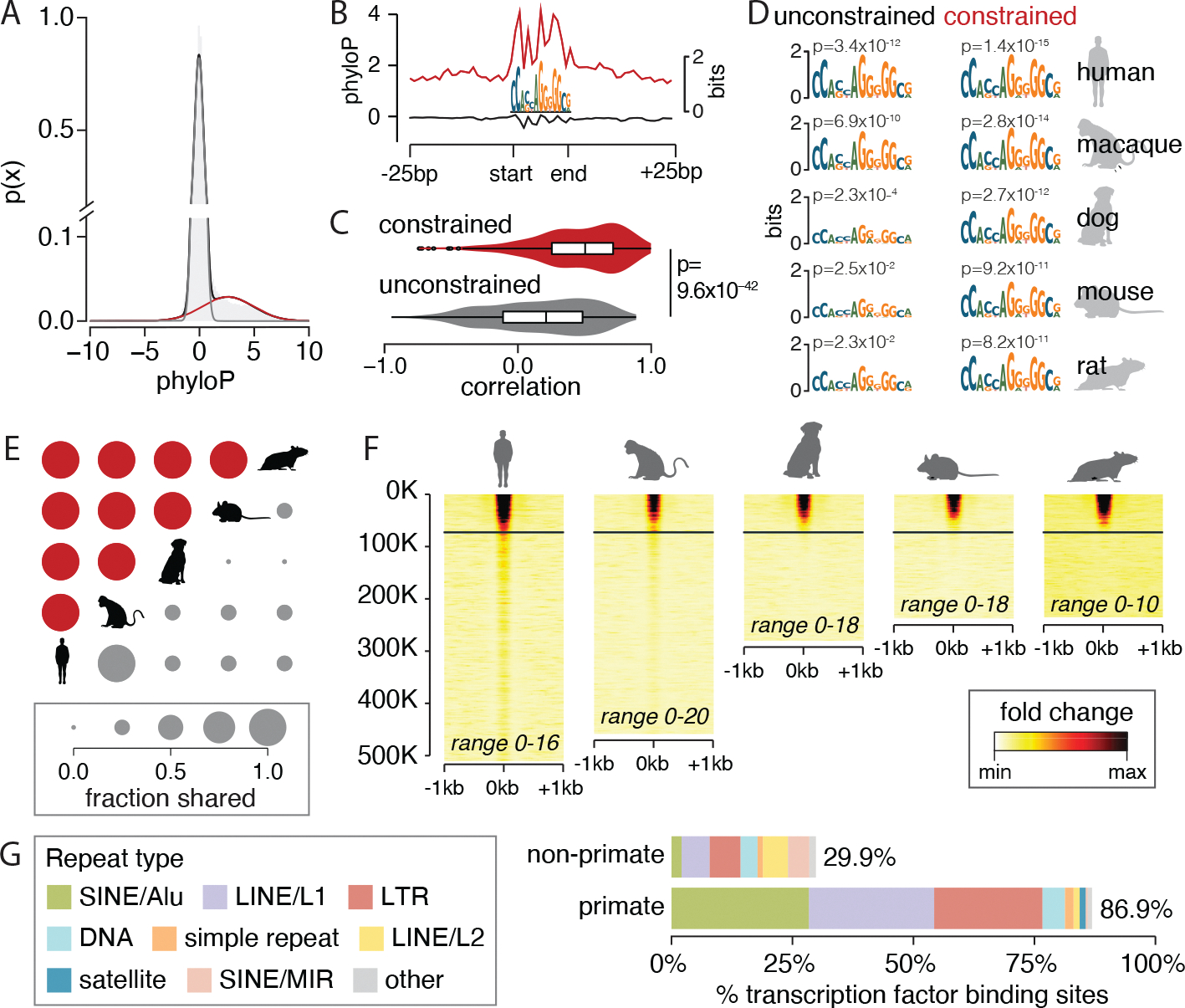
(A) A two-component Gaussian mixture model fit over average phyloP scores across binding sites for CTCF distinguishes the distribution for evolutionarily constrained sites (red) from others (gray). (B) At CTCF binding sites, aggregate phyloP scores are high for constrained binding sites (red, 61,832 sites) but not for unconstrained binding sites (gray, 424,177 sites). The same pattern is observed for other transcription factors (fig. S10). (C) Across all transcription factors, aggregate phyloP scores are more strongly correlated (Pearson’s correlation) with binding site information content for constrained sites than for unconstrained sites. Boxes and whiskers represent 25% quartile, 75% quartile, minimum, and maximum, with a horizontal line at the median. The shading indicates the density of the data. (D) CTCF logos of constrained and unconstrained sets for four species made by lifting over human transcription factor binding sites. (E) Fraction of constrained (red) and unconstrained (gray) CTCF binding sites that are shared between pairs of species. (F) CTCF transcription factor chromatin immunoprecipitation sequencing (ChiP-seq) signal over binding sites in mammalian livers sorted by average phyloP scores. Each row is a binding site; in nonhuman species, only aligned sites are shown. The horizontal lines indicate significant constraint. Ranges give the minimum and maximum ChIP-seq fold change over input for each species. (G) Percentage of primate-specific and non–primate-specific transcription factor binding sites that are derived from individual transposable element classes. LINE, long interspersed nuclear element; LTR, long terminal repeat; MIR, mammalian-wide interspersed repeat; SINE, short interspersed nuclear element. [Species silhouettes are from PhyloPic]
Unannotated constraint
Almost half of all constrained bases (48.5%) are in regions with no annotations in the thousands of cell types, tissues, or conditions assayed by ENCODE3 (table S9) (14). We grouped constrained bases (phyloP FDR < 5%) fewer than 5 bp apart in unannotated intergenic regions (excluding repeats, centromeres, and telomeres) to define 423,586 elements, which we term unannotated intergenic constrained regions (UNICORNs) (median size = 20 bp; IQR = 23; 95th percentile = 131 bp; 0.5% of genome; Fig. 4A and fig. S7C). Most (77.0%) of these unannotated elements are within 500 kb of the transcription start site for a protein-coding gene. They tend to contain fewer variants (pWilcoxon < 2.2 × 10−16) with lower minor allele frequencies (pWilcoxon< 2.2 × 10−16) than other intergenic regions (Fig. 4B).
Fig. 4. Constraint highlights unannotated regions that are likely functional.
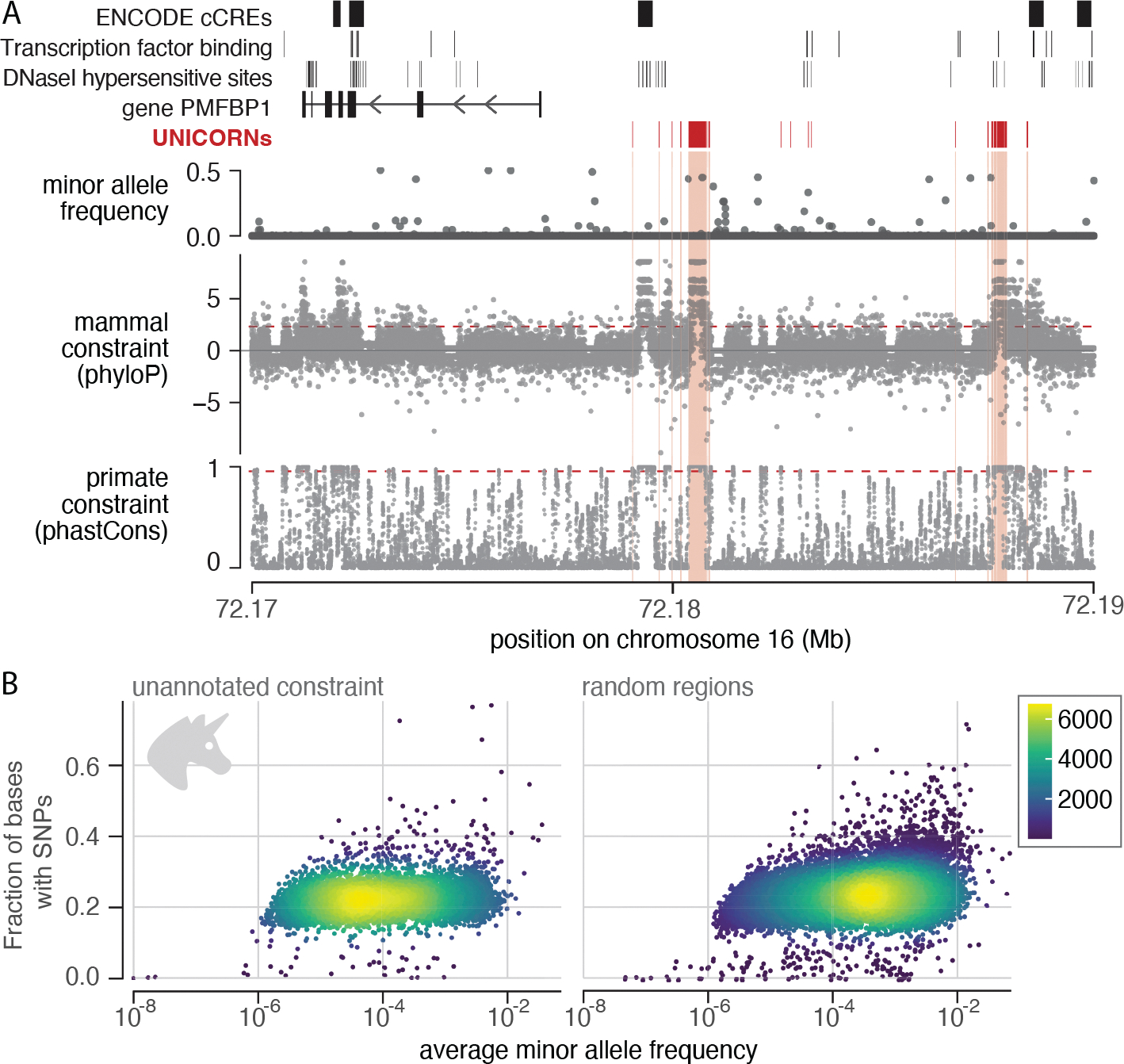
(A) Example UNICORNs on human chromosome 16. The largest is 418 bp and located 3.5 kb upstream of the transcription start site of the gene PMFBP1; the second largest is 174 bp. Gray dots represent single bases. Red dashed lines represent the FDR < 5% threshold for phyloP and the threshold for phastCons that captures equivalent genome proportion (phastCons base score ≥ 0.961). UNICORNs lack coding or regulatory annotations in ENCODE (top track), and most have low diversity in human populations (second track). (B) UNICORNs contain fewer variants, and those present have lower allele frequencies than those in the random set (Wilcoxon rank sum test, p < 2.2 × 10−16). The fraction of bases with single-nucleotide polymorphisms (SNPs) versus mean minor allele frequency for human SNPs within UNICORNs (left) or within a random set of unannotated sequences (right) is shown. Allele frequencies were log10 transformed. Human variants and allele frequencies were obtained from TOPMed data freeze 8 (69).
Many unannotated regions are likely to be functional under conditions that were not assayed in human ENCODE3 (table S9) (14). For example, open chromatin regions (a proxy for candidate enhancers) in developing brain tissues (100), adult motor cortical neuron cell types (101), and narrowly defined regions of young adult brain (102) overlap 8.8, 7.1, and 8.6% of UNICORNs respectively (17% collectively; 5.4, 2.7, and 4.2% are active in only developing brain, adult motor cortical neurons, and young adult brain regions, respectively). As resources like ENCODE expand to include more difficult-to-access time points, cell types, and tissues, we anticipate that the function of many UNICORNs will be elucidated.
Regions of accelerated evolution
Recent evolution in the human lineage may have occurred in part by modifying the 3D structure of the genome, which can alter gene regulation (103). We developed an automated pipeline for identifying “accelerated” regions that are highly constrained across mammals but exceptionally variable in particular lineages (104). We found 312 regions accelerated in humans and 141 in chimpanzees, most of which are noncoding. Human (82%) and chimpanzee (86%) accelerated regions tend to have signatures of positive selection (after accounting for other factors such as GC-biased gene conversion); these accelerated regions also tend to reside near developmental and neurological genes, consistent with previous work (105–108). In domains that contain human accelerated regions, we show that the 3D genome structure is altered by human-specific structural variants, suggesting a role for enhancer hijacking in the species-specific evolution of these loci (109).
Evolution through transposable elements
We cataloged transposable elements in the genomes of 248 species (fig. S11) (110). Transposable elements are mobile DNA sequences 100 to 10,000 bp long that can accumulate to >1 million copies per genome. Despite their potential to influence genome structure and function (111, 112), they are difficult to analyze, and most studies have focused on human and mouse (113). We analyzed transposable element class, number, and distribution in 248 species (table S1). There is little variation between mammals in the fraction of the genome in transposable elements [N= 248; 49.0 ± 7.5% (±SD)], consistent with counterbalancing accumulation with DNA loss (114). Recent accumulation, especially retrotransposon accumulation, is positively correlated with genome size [hierarchical Bayesian model, coefficient of determination (R2) = 0.54 (95% high probability density 0.42, 0.64)], suggesting insufficient time to purge insertions after a surge of activity, and negatively correlated with transposable element diversity, suggesting that genomic control mechanisms may limit the repertoire of active elements (110, 115). Younger transposable element families are more likely to include insertions that are polymorphic in the species and thus may be subsequently lost. However, any family with multiple members is likely a permanent feature of the species because there is no known mechanism to target an entire family for elimination. Bats are a hotspot for horizontal transfer of DNA transposons, with more than 200 such events, compared with just 11 transferred into other lineages (table S10) (116).
Overall, about 11% of constrained human bases are in transposable elements, with constraint enriched in simple repeats and DNA transposons and depleted in short interspersed nuclear elements, long terminal repeats, and satellite repeats (fig. S12A). This likely reflects the absence of function within more recently inserted transposable elements. DNA transposons are an ancient class of repeats known to acquire functional roles, such as the transcription factor ZBED5 (70% constrained) (117). By contrast, the repeat classes depleted in constraint have been activemore recently during primate evolution and are therefore less likely to be functional (118). In simple repeats, constraint is negatively correlated with distance to the nearest gene. Simple repeats near genes, where they are more likely to influence gene expression (119), are more constrained (Spearman’s r = −0.13, p < 2.2 × 10−16; fig. S12B).
Most (87%) primate-specific transcription factor binding sites overlap transposable elements, unlike most non–primate-specific sites (30%) (Fig. 3G). Sites in transposable elements, and especially those in younger elements, tend to be less conserved and change more quickly (fig. S13). Our results suggest that transposable elements may be a driver of recent regulatory innovations in primates (120–122), with the caveat that the binding sites have not been confirmed to have regulatory function (123). Transposable element–derived CTCF binding sites found only in primates are enriched near genes involved in vision, reproduction, immunity, lower extremity development, and social behavior [enrichment analysis of cis-regulatory regions with Genomic Regions Enrichment of Annotations Tool (GREAT) (108); table S11].
Connecting genotype to phenotype
The Zoonomia resource offers an unprecedented opportunity to explore the evolution of exceptional mammalian traits by associating genomic variation with species-level phenotypes in hundreds of diverse species. For many traits, phenotype annotations are sparse, limiting the application of these methods. Here, we illustrate the potential of this approach using traits that vary within multiple clades of mammals and for which we have species-level phenotypes for a large number of Zoonomia species. We apply tests for different modes of evolution, including changes in gene number, gene sequence, and gene regulation.
Olfactory ability
Mammals have widely varying olfactory abilities, reflecting adaptation to different ecological niches (124–128). Olfactory receptor gene repertoire is a proxy for olfactory ability in mammals (128). We investigated olfactory evolution by first identifying olfactory receptor genes in genome assemblies of 249 mammalian species through genome annotation by means of a set of mammalian receptor profile hidden Markov models (table S12) (127). This increases by 10-fold the number of species with olfactory gene annotations. Our annotated gene counts do not vary with genome quality, as measured by contig N50 (Spearman’s r = 0.065, p = 0.31, N = 249), scaffold N50 (Spearman’s r =0.0091, p = 0.89,N=249), or genome completeness (129) (Spearman’s r = 0.10, p = 0.11, N = 249), and capture the wide variation across species [mean count = 1218 ± 683 (±SD), N = 249] (Fig. 5A and fig. S14).
Fig. 5. Associating coding and regulatory change with species phenotypes.
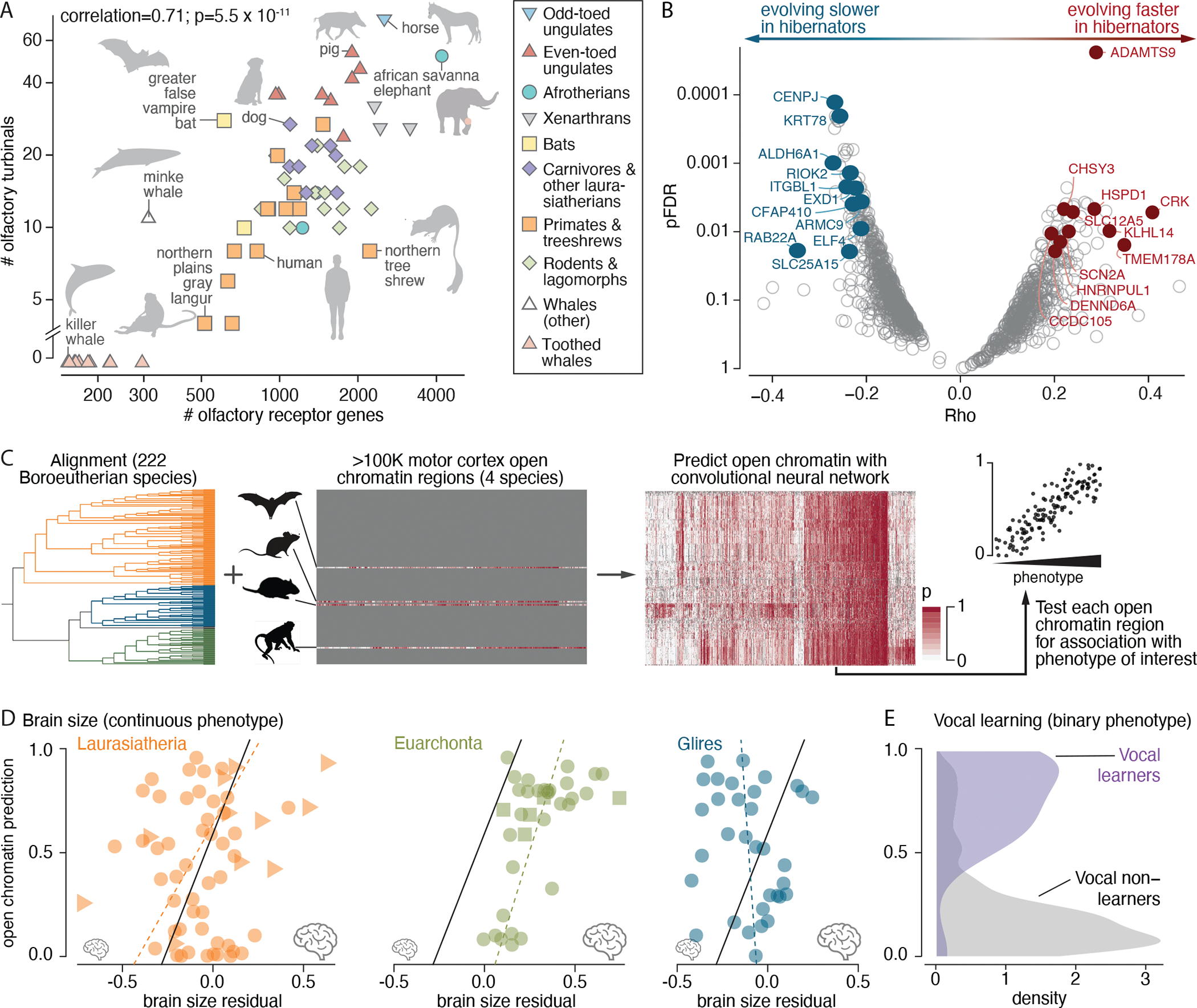
(A) Olfactory receptor gene count (x axis) is associated with the number of olfactory turbinals (y axis) in 64 species. Labels and silhouettes mark outliers and species of interest. (B) Testing the coding sequence of 16,209 genes identified 341 genes that are evolving faster or slower in hibernators (pFDR < 0.05; gray open circles), and 22 are significant after phylogeny-aware permutation testing (permutation pFDR < 0.05; labeled), including 11 evolving faster (red filled circles) and 11 evolving slower (blue filled circles). (C) TACIT first trains a predictive classifier on sequences that underlie open chromatin regions from tissues or cell types in a few species and then predicts open chromatin in many others and tests for phenotype associations. (D) TACIT associated a motor cortex open chromatin region with brain size (a continuous value trait), driven by associations within Laurasiatheria (59 species) and Euarchonta (36 species) but not within Glires (33 species). Results are for a rhesus macaque open chromatin region (chr10:48660711-48661679) near MACROD2. The phylolm line of best fit is shown for all species [solid line; phylolm coefficient (slope) = 0.45, permutation pFDR = 0.11] and, as a visual aid, for each clade (dashed line). Triangles represent cetaceans (highest variation in brain size residual), squares represent great apes (highest variation in brain size residual within Euarchonta), and circles represent other species. (E) TACIT associated a motor cortex open chromatin region with vocal learning (a binary trait) in the GALC locus (phylolm coefficient = 6.51, permutation pFDR = 0.045) (137). Results are for an Egyptian fruit bat open chromatin region (PVIL01002568.1:139004-139596). [Species silhouettes are from PhyloPic]
By improving representation within lineages, most notably rodents (N=55), cetaceans (N = 17), and xenarthrans (N = 8), we discern variation in olfaction that was missed in earlier studies (fig. S15). Rodents have more olfactory receptor genes on average than other mammals [55 rodents versus 194 others, mean = 1434 ± 466 (±SD) versus 1156 ± 721, t = 3.4, pt-test = 0.0008]. The top rodent is the Central American agouti (3233 genes), which has more genes than all but three other species (Hoffmann’s two-toed sloth, the nine-banded armadillo, and the African savanna elephant). Cetaceans have the narrowest variation of any order. All cetaceans (17 species) have exceptionally small olfactory receptor gene repertoires relative to other mammals (225 ± 75 genes compared with 1290 ± 650 genes, t = −22.9, pt-test = 5.8 × 10−60). Baleen whales retain olfactory structures that were lost in toothed whales (130, 131), and, consistent with this anatomic evidence for olfactory ability, the four baleen whale species in Zoonomia have more olfactory receptor genes than the 13 toothed whales (339 ± 36 versus 190 ± 40, t = −6.96, pt-test = 0.00064) (fig. S14).
The association of olfactory turbinal number with olfactory receptor gene repertoire across placental mammals suggests that both evolve in response to selection on olfactory capacity. Olfactory turbinals are an anatomic feature of the nasal cavity that is known to affect olfactory capacity (132–134). In 64 species that were phenotyped for both traits, the number of olfactory turbinals correlates with the number of olfactory receptor genes (Spearman’s r = 0.71, p = 5.50 × 10−11) (Fig. 5A). This relationship remains significant after accounting for species relationships by applying a phylogenetic generalized least squares method (phylolm coefficient = 0.014, p = 4.31 × 10−10) and a permutation approach that preserves the tree topology (permutation p = 0.0013) (fig. S16) (135–137). We also confirm earlier observations that the number of genes is negatively associated with group living (phylolm coefficient = −0.0013, phylogeny-aware permutation p = 0.022) (127, 138), possibly because social animals are less dependent on smell. The association between the number of genes and solitary living fails to reach significance (phylolm coefficient = 0.00086, phylogeny-aware permutation p = 0.099).
Hibernation
Zoonomia includes the largest mammal protein-coding alignment completed to date, with 17,795 human genes aligned in up to 488 assemblies of 427 distinct species (6). This alignment complements the Cactus whole-genome alignment (4, 11). It integrates gene annotation, ortholog detection, and classification of genes as intact or inactivated and can join orthologous fragments of genes split in fragmented assemblies.
Our protein-coding alignment includes 22 deep hibernators (species capable of core temperature depression below 18°C for >24 hours) and 154 strict homeotherms (species that maintain constant body temperature), offering an opportunity to explore the genomic origins of hibernation. Forms of torpor are found in every deep mammalian lineage, suggesting that metabolic depression through heterothermy existed in some form in the ancestor of all mammals (139, 140). Modifications, including the capacity for seasonal hibernation, may be derived. Understanding the genomics of hibernation, including cellular recovery from repeated cooling and rewarming without apparent long-term harm, could inform therapeutics, critical care, and long-distance spaceflight (141, 142).
Comparing hibernators and strict homeotherms to the reconstructed ancestral mammal protein-coding sequence using generalized least squares forward genomics (23) identified 28 100-bp regions (pFDR < 0.05) in 20 genes where hibernators are less diverged from the placental mammalian ancestor (table S13). Two of these genes, MFN2 and PINK1, overlap four GO Biological Process gene sets related to depolarization and degradation of damaged mitochondria, an organelle essential for metabolic depression (table S14) (143), although the process’s enrichment is only nominally significant (top geneset p = 7.5 × 10−5; pFDR = 0.39). A third, TXNIP, also regulates mitophagy (144) and shows torpor-responsive gene expression in rodents (145–147) and bats (148).
Testing with RERconverge identified an additional 22 genes as evolving unusually fast or slow in hibernators compared with homeotherms (Fig. 5B and data S3) (149–151). RERconverge tests for associations between relative evolutionary (substitution) rates of genes and the evolution of traits. We controlled for the high proportion of hibernators in the bat lineage, a potential confounder, through a Bayes factor analysis that quantified the amount of signal arising from hibernators and from bats and excluded genes with a hibernator signal less than fivefold larger than the bat signal (fig. S17). The top-scoring genes (pFDR < 0.05 and phylogeny-aware permutation pFDR < 0.05) included 11 that are evolving faster and 11 that are evolving slower in hibernating species (fig. S18). Faster-evolving genes are nominally enriched in gene sets related to temperature response and immunity (fig. S18A and table S15). Among the genes that are evolving faster in hibernators are HSPD1 [involved in stress adaptation underlying mammalian torpor (152)], the mTor pathway inhibitor ADAMST9 [also implicated in longevity based on sequence convergence in microbats and naked mole rats (153)], and two genes connected to neurodevelopmental disorders [the voltage-gated sodium channel gene SCN2A (154) and the membrane K-Cl cotransporter gene SLC12A5 (155)].
There is no overlap between the two methods inthe genes that score as significant (phylogenyaware permutation pFDR ≤ 0.05), suggesting that their distinct methodologies are sensitive to different types of sequence change. One gene (the neurodevelopmental gene NCDN) is nominally significant in both sets (p < 0.05 and permutation p < 0.05 in both analyses).
Neurological traits
We developed a toolkit for associating differences in cis-regulatory elements, an important driver of phenotype divergence (156–158), with differences in phenotypes that include brain size and vocal learning (159, 160). This TissueAware Conservation Inference Toolkit (TACIT) does not require tissue-specific cis-regulatory element data from every species, which is costly and logistically challenging to obtain. Instead, it uses cis-regulatory sequence features in a tissue or cell type of interest from a few species to train machine-learning models that can be used to predict activity in that tissue or cell type at cis-regulatory element orthologs in many species (Fig. 5C) (15). Models trained in one species can identify species and tissue-specific cis-regulatory element activity in others, including for elements not used in training, demonstrating the feasibility of this approach (15). We then associated the predictions with phenotypes. We ran TACIT on traits that are phenotyped in more than 80 Zoonomia species and are proposed to involve neural cell types for which we have cis-regulatory element data from multiple species (motor cortex and parvalbumin neurons) (101, 161–163).
Brain size, measured relative to body size, is associated with predicted activity at cis-regulatory elements that are active in the motor cortex (49 out of 98,912 elements tested, four species with training data, 158 species tested) and parvalbumin neurons (15 out of 35,034 elements tested, two species with training data, 72 species tested) (phylogeny-aware permutation pFDR < 0.15) (159, 164–166). This includes a region near the gene MACROD2, a nervous system development gene implicated in microcephaly and intellectual disability in humans (Fig. 5D) (167, 168). Motor cortex cis-regulatory elements near genes previously implicated in microcephaly or macrocephaly tend to have more significant associations with brain size across mammals (one-sided pWilcoxon= 0.013).
In an analysis of 175 phenotyped species, both protein-coding changes and cis-regulatory changes were associated with capacity for vocal learning (160). Vocal learning is the ability to mimic noninnate sounds and likely evolved convergently in humans, bats, cetaceans, and pinnipeds (169). Our analysis of candidate cis-regulatory elements active in motor cortex (N = 94,444) and parvalbumin neurons (N = 35,557) identified motor cortex elements near GALC (Fig. 5E) (170), TSHZ3 (171), and other speech disorder-related genes.
Applying genomics to biodiversity conservation
In addition to illuminating mammalian evolutionary history, Zoonomia’s alignment and measures of constraint can help efforts to protect biodiversity for the future. Evolutionary constraint scores enable empirical estimation of deleterious genetic load and its demographic drivers across diverse species. We find that Zoonomia species with smaller historical effective population sizes carry higher fixed genetic load, with proportionally more missense substitutions (phylolm p = 7.76 × 10−5) and substitutions at constrained sites (phylolm p = 9.63 × 10−3). Species with a smaller historical effective population size are also more likely to be classified as threatened by the International Union for Conservation of Nature (IUCN) (phylolm p < 3.3 × 10−5), suggesting that historical processes are predictive of species’ contemporary extinction risk status. Our analysis showed that threatened species have fewer substitutions at extremely constrained sites (phylolm p = 0.001), particularly in primates, whereas the opposite is true of missense substitutions, possibly because severely deleterious alleles have been purged or lost to drift (172) (Fig. 6). As the number of species with reference genomes grows, so will the power to leverage genomic data for identifying those most susceptible to the impacts of rapid environmental changes that characterize the Anthropocene.
Fig. 6. Genomic metrics distinguish at-risk primate species.
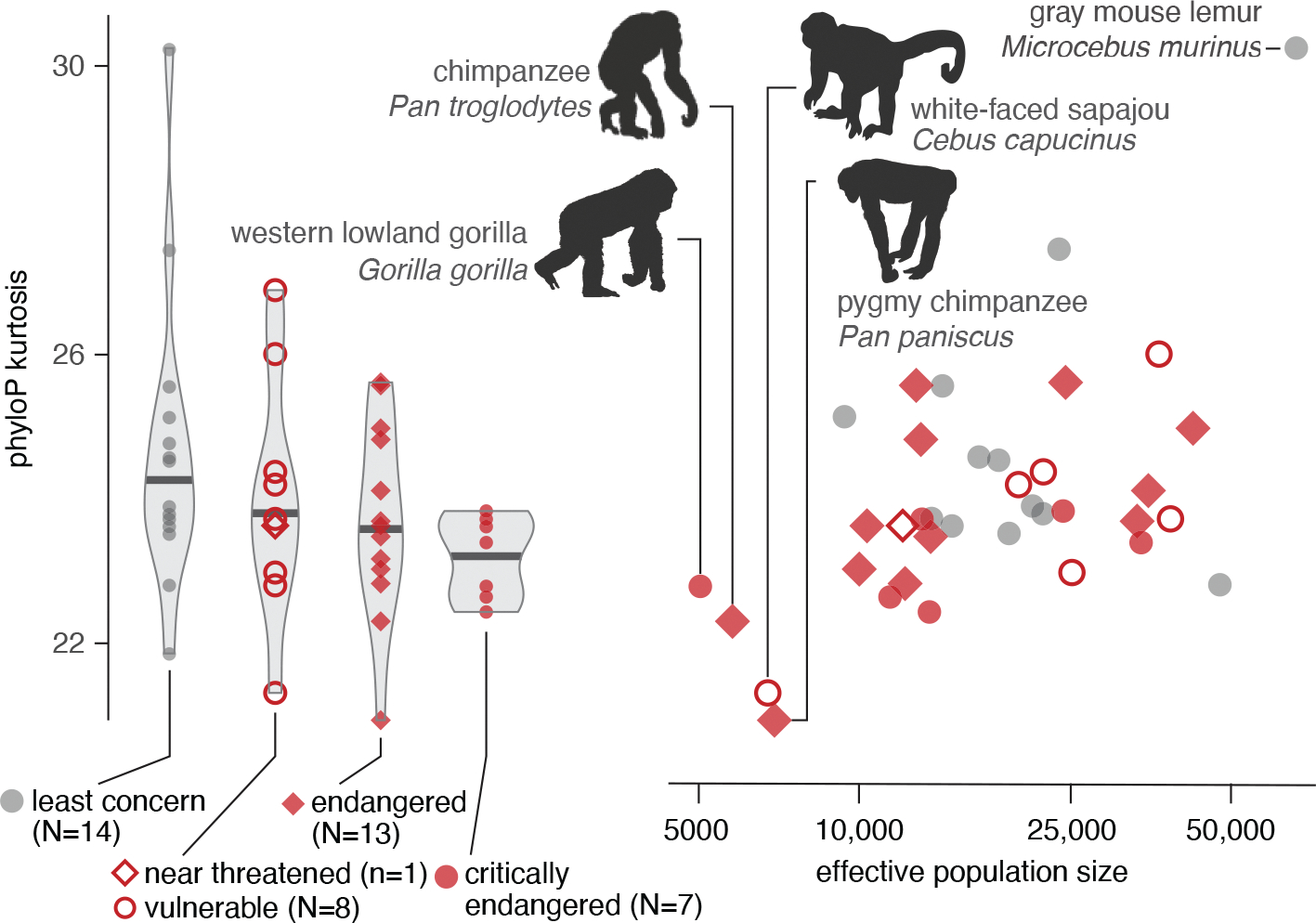
Primates that are categorized at increasing levels of extinction risk and with smaller effective population sizes have fewer substitutions at extremely constrained sites,measured as kurtosis (which describes the tail of the distribution) of phyloP scores (phylolm p =7.9 × 10−4 and p = 0.024, respectively). Four at-risk species with the smallest effective population size (labeled with silhouettes) have low kurtosis (i.e., fewer phyloP outliers), and a species categorized as “least concern” with the largest effective population size has high kurtosis (gray mouse lemur; labeled). [Species silhouettes are from PhyloPic]
Discussion
By aligning hundreds of mammalian genomes, Zoonomia realizes the vision of the landmark 29 Mammals paper (13) to achieve single-base resolution of constraint across the human genome. This resource, which includes even deeper coverage of protein-coding regions (6), addresses a central goal of medical genomics: to identify genetic variants that influence disease risk and understand their biological mechanisms (7, 24, 37, 71, 173). It also opens new opportunities for exploring the evolution of mammalian genomes as species diverged and adapted to a wide range of ecological niches (15, 26, 110, 116, 160, 174) and for discovering what is distinctively human (104).
Zoonomia illustrates how new sequencing technology and analysis methods are transforming comparative genomics while underscoring the critical need for high-quality phenotype annotations. Studies into the genomic origins of exceptional mammalian traits have the potential to inform human therapeutic development (141) but are limited by sparse and inconsistent phenotype data. Here, we focus on a handful of traits for which we could define phenotypes consistently in large numbers of species, including hibernation (174 species), brain size (158 species), and vocal learning (175 species). Achieving the richer datasets that are needed to study other traits, evaluate pattern robustness, and address broader prospects requires collaborations between genomics researchers and scientists with expertise in morphology, physiology, and behavior to develop standardized phenotype definitions that apply across species (175). It also requires proper collection, annotation, and data-handling practices that facilitate discovery, evaluation, and reuse of data (176).
Comparative genomics projects are classically motivated by the potential to advance human biomedicine, but they rely on biodiversity imperiled by human activity (177). Our analysis suggests that even a single reference genome per species may help conservation scientists identify potentially threatened populations earlier when management efforts can be more efficient and effective, butmore work is needed to develop these methods (172). Through close and enduring partnerships with researchers working in biodiversity conservation, resources from Zoonomia and other comparative genomics projects can address questions in human health and basic biology while simultaneously guiding efforts to protect the biodiversity that is essential to these discoveries (178).
Methods summary
Alignment and annotation
We finalized the Zoonomia Cactus alignment by updating the initial Progressive Cactus alignment used in (11) to remove a mislabeled genome. We identified genes in Zoonomia genomes using halLiftover in conjunction with the Zoonomia Cactus alignment, identifying sequences orthologous to the protein-coding sequence of human exons from ENSEMBL across each of the 241 assemblies. We also developed an alternative reference-based approach described in our companion paper (6), which we applied to 427 species. We used a combination of two approaches using short sequencing reads and genome assemblies to determine whether the CMAH gene had been lost in mammalian genomes. We considered putative CMAH gene loss events to be cases where both these approaches indicated loss of the same part of the gene.
Constraint scoring
We used the Zoonomia alignment and a randomly selected set of ancestral repeat positions (100 kb total) to generate three different neutral models: one for autosomes and one each for the two sex chromosomes. We used PhyloFit from Phast v1.5 to estimate branch lengths. We used this same method to estimate primate-neutral models, but with the ancestral branch reconstruction based on the 43 primates from the alignment. We used phyloP (part of the PHAST v1.5 package) to calculate per-base constraint and acceleration p values. We calculated phyloP scores on the human-, chimpanzee-, mouse-, dog-, and batreferenced 241-way alignments, as well as for a human-referenced, primates-only alignment (43-way). We computed a mammalian phyloP threshold by converting the p values corresponding to the phyloP scores into q values using a FDR correction. We considered any column with a resulting q ≤ 0.05 to be significantly evolutionarily constrained or accelerated, as determined by the sign of the score.
Analyzing constraint
Proportion of genome under constraint
We estimated lower bounds for the fraction of sites under purifying selection across the human, chimpanzee, dog, house mouse, and little brown bat genomes by comparing the empirical cumulative distribution functions of phyloP scores across each genome to the those of ancestral repeats, following the same method detailed in (12).
Constraint in functional elements
We extracted phyloP scores for all positions in protein-coding genes (GENCODE v.36) including 5′ and 3′ untranslated regions, and compared constraint between different positions within coding sequences. We summarized mean and standard deviation phyloP scores for positions within codons, degenerate and nondegenerate positions, methionines that act as and do not act as start codons, and cysteines that form and do not form intrapeptide disulfide bridges. We calculated constraint enrichment for several genome features (coding sequences, 5′ untranslated regions, 3′untranslated regions, introns, DNase hypersensitivity sites, and the five types of cCREs [ENCODE candidate cis-regulatory regions (14)], where we calculated constraint enrichment as the constrained fraction of the feature divided by the constrained fraction of the genome.
Highly constrained regions
We identified all positions where the number of species aligned was ≥235 and the base was the same among all species aligned at that position. We then merged neighboring positions, creating zooUCEs ranging in size from 20 to 190 bp. We assessed overlap between our zooUCEs and previously defined UCEs. We also defined regions of contiguous constraint as regions of at least 20 contiguous base pairs with phyloP scores above the FDR > 0.05 threshold and identified 100-kb bins with significantly high or low constraint.
Constraint in unannotated regions
We subsetted the human genome, removing all regions with the following annotations: GENCODE v37 exons (untranslated regions and exons for all protein-coding genes), promoters (transcription start site ±1 kb), introns, ENCODE3 cCREs, DNase hypersensitivity sites (including transcription factor binding sites), chromatin interaction analysis with paired-end tag sequencing (ChIA-PET) anchors, three promoter annotation sets, and six enhancer annotation sets (table S9). Within the remaining unannotated sequence, we identified closely located constraint positions to define a set of 423,586 UNICORNs.
Olfaction
We explored the olfactory receptor gene family across the Zoonomia species set, independently of alignment-based annotation. We mined all genomes for olfactory receptor gene sequences using the olfactory receptor assigner (179). We classified sequences as “pseudogenes” if they contained in-frame stop codons or were shorter than 650 bp and therefore not long enough to form the seven-transmembrane domain. We curated species-specific numbers of olfactory turbinals from both sides of the nasal cavity (table S12), obtaining turbinal numbers for 64 species in our sample. We tested for an association between the total number of olfactory receptor genes with the number of olfactory turbinals using phylolm (136), solitary living status, and group living status while accounting for the Zoonomia phylogenetic tree (26, 138).
Hibernation
We investigated genomic differences between mammals that we defined as hibernators and as strict homeotherms (table S1), with 22 species defined as deep hibernators and 154 species defined as strict homeotherms. We used generalized least squares forward genomics to identify genes that are more similar to the mammalian ancestor than they are to non-hibernators as well as to identify regions conserved in hibernators relative to the placental ancestor. We also used RERconverge (149) to identify genes with significant evolutionary rate shifts in hibernating mammals versus nonhibernating mammals. Such genes are putative hibernation-related genes.
Supplementary Material
ACKNOWLEDGMENTS
We thank everyone who collected the samples and phenotype data essential for this project. We thank the San Diego Zoo Wildlife Alliance staff, E. Baitchman, R. Johnston, Zoo New England, Broad Institute Genomics Platform, and the SNP&SEQ Technology Platform (National Genomics Infrastructure Sweden and Science for Life Laboratory). We thank K. Hinde, N. Upham, and the March Mammal Madness team for useful discussions. Compute resources were provided by the Broad Institute, Swedish National Infrastructure for Computing (SNIC) at UPPMAX, the Texas A&M High Performance Research Computing Center, the Extreme Science and Engineering Discovery Environment (XSEDE) through the Pittsburgh Supercomputing Center Bridges and Bridges-2 Compute Clusters, and Lehigh University’s Research Computing infrastructure. Funding: This work was funded by National Institutes of Health (NIH) grant R37CA218570; NIH grant R01HG008742; NIH grant R01HG010485; NIH grant RO1HG002939; NIH grant U01HG010961; NIH grant U24-HG010136; NIH grant U24HG009446; NIH grant U41HG002371; NIH grant U41HG007234; NIH grant U19AG057377; NIH grant DP1DA046585; NIH grant F30DA053020; NIH grant R24OD018250; National Science Foundation (NSF) grant DEB-1753760; NSF grant DEB2150664; NSF grant DBI-2046550; NSF grant DEB 1838283; NSF grant DEB-1457735; NSF grant DGE-1252522; NSF grant DGE1745016; NSF grant IOS-2032006; NSF grant IOS-1929592; NSF grant IOS-2022007; NSF grant IOS-2029774; NSF grant TGBIO200055; NSF grant ACI-1548562; NSF Postdoctoral Fellowship in Biology 2011038 (C.F.); European Research Council under the European Union’s Horizon 2020 research and innovation program grant 864203 (T.M.-B.); MINECO/FEDER, UE grant PID2021126004NB-100 (T.M.-B.); FEDER/UE grant AEI-PGC2018-101927BI00 704 (A.N.C.), AEI grant CEX2018-000792-M (A.N.C. and T.M.-B.), Science Foundation Ireland 19/FFP/6790 (E.C.T.); Irish Research Council Laureate grant (E.C.T.); Distinguished professorship from the Swedish Research Council (K.L.-T.); Swedish Research Council Vetenskapsrådet grant D0886501 (P.F.S.); Carnegie Mellon University Computational Biology Department Lane Postdoctoral Fellowship (I.M.K.); Carnegie Mellon University SURF grant (D.E.S.); Gift from Ed and Pam Taft, Roddenberry Foundation, Gladstone Institutes (K.S.P.); LOEWECentre for Translational Biodiversity Genomics (M.H.); Robert and Rosabel Osborne Endowment, UC Davis (H.A.L.); SFI Centre for Research Training in Genomics Data Science grant 18/CRT/6214 (L.R.); Sloan Foundation grant (A.R.P.); UMaine Institute of Medicine Seed Grant (D.L.L.); University College Dublin Ad Astra Fellowship (G.M.H.); Knut and Alice Wallenberg Foundation (K.L.-T.); NSF grant 2019035 (Lehigh University Research Computing Infrastructure); NSF grant TG-BIO200055 [The Extreme Science and Engineering Discovery Environment (XSEDE)]; and Swedish Research Council grant 2018-05973 [Swedish National Infrastructure for Computing (SNIC) at UPPMAX]. Author contributions: Conceptualization: A.N., A.R.P., B.P., D.A.R., E.K.K., H.A.L., K.L.-T., K.S.P., M.H., O.A.R., W.J.M.; Data curation: A.G.H., A.M.B., A.R.B., B.K., C.F., C.L., D.L.L., D.P.G., F.W., G.M.H., I.M.K., I.R., J.C.A., L.R., M.D., M.J.C., M.X.D., P.F.S., R.S., W.K.M.; Investigation or formal analysis: A.G.H., A.K., A.J.L., A.M.B., A.L.L., A.O., A.P.W., A.V., B.N.P., B.K., C.F., C.L., D.L.L., D.B.G., E.K.K., F.W., G.A., G.M.H., I.M.K., I.R., J.C.A., J.Ga., J.Gr., J.R.S.M., J.R.X., K.C.K., L.G., L.R.M., L.R., M.A.S., M.B., M.D., M.H., M.J.C., M.S., M.X.D., N.M.F., N.S.P., O.W., P.F.S., R.W.R., R.S., S.G., S.K.R., S.V.K., V.D.M., X.L.; Methodology: A.F.A.S., A.G.H., A.J.L., G.M.H., J.C.A., K.C.K., L.G., M.J.C., M.X.D., P.F.S., R.M.H.; Project administration: D.P.G., E.K.K., J.J., J.R.S.M., K.L.-T.; Resources: A.F.A.S., B.K., C.S., G.M.H., J.C.A., J.J., K.C.K., K.-P.K., M.D., M.N., M.X.D., N.M.F., R.S.; Supervision: A.F.A.S., A.G.H., A.K., A.N., A.R.P., B.P., B.S., B.W.B., D.A.R., E.K.K., E.C.T., G.M.H., H.A.L., I.M.K., J.R.S.M., K.L.-T., K.-P.K., K.S.P., M.H., M.S., O.A.R., P.F.S., R.M.H., T.M.-B., W.J.M., Z.W.; Visualization: A.K., E.K.K., G.A., K.M.M., I.M.K., K.F., L.R.M., M.J.C., N.M.F., X.L., Z.W.; Writing – original draft: A.G.H., A.J.L., A.N., A.P.W., A.R.P., D.A.R., D.P.G., E.K.K., G.M.H., H.A.L., I.M.K., I.R., J.D., K.C.K., K.L.-T., K.S.P., M.A.S., M.E.W., M.H., M.J.C., N.M.F., P.F.S., W.J.M., X.L.; Writing – review and editing: A.J.L., A.K., A.O., B.N.P., B.P., B.S., C.F., D.E.S., E.C.T., F.W., G.A., P.C.S., J.R.X., K.-P.K., L.R.M., M.D., M.E.W., M.S., M.X.D., N.S.P., O.A.R., O.W., R.S., S.G., S.K.R., W.J.M., X.L. Diversity and inclusion: One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. Competing interests: P.F.S. is a consultant and shareholder for Neumora. L.G. and K.C.K. are employees of Fauna Bio, Inc. E.K.K. and A.G.H. are advisors of Fauna Bio, Inc. P.C.S. is a cofounder, consultant, and shareholder of Sherlock Biosciences and DelveDx and a board member and shareholder of Danaher Corporation. P.C.S. holds several patents related to genomic sequencing and characterization. Data and materials availability: Scripts are archived at Zenodo (180). The Cactus alignment and constraint scores are available at https://cglgenomics.ucsc.edu/data/cactus/ and at https://genome.ucsc.edu/cgi-bin/hgTrackUi?db=hg38&g=cons241way. The protein-coding sequence alignment is at http://genome.senckenberg.de/download/TOGA/. Information regarding genome assemblies and specimen biosamples is provided in (4) and at https://zoonomiaproject.org/. License information: Copyright © 2023 the authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original US government works. https://www.science.org/about/science-licenses-journal-article-reuse
Zoonomia Consortium
Gregory Andrews1, Joel C. Armstrong2, Matteo Bianchi3, Bruce W. Birren4, Kevin R. Bredemeyer5, Ana M. Breit6, Matthew J. Christmas3, Hiram Clawson2, Joana Damas7, Federica Di Palma8,9, Mark Diekhans2, Michael X. Dong3, Eduardo Eizirik10, Kaili Fan1, Cornelia Fanter11, Nicole M. Foley5, Karin Forsberg-Nilsson12,13, Carlos J. Garcia14, John Gatesy15, Steven Gazal16, Diane P. Genereux4, Linda Goodman17, Jenna Grimshaw14, Michaela K. Halsey14, Andrew J. Harris5, Glenn Hickey18, Michael Hiller19,20,21, Allyson G. Hindle11, Robert M. Hubley22, Graham M. Hughes23, Jeremy Johnson4, David Juan24, Irene M. Kaplow25,26, Elinor K. Karlsson1,4,27, Kathleen C. Keough17,28,29, Bogdan Kirilenko19,20,21, Klaus-Peter Koepfli30,31,32, Jennifer M. Korstian14, Amanda Kowalczyk25,26, Sergey V. Kozyrev3, Alyssa J. Lawler4,26,33, Colleen Lawless23, Thomas Lehmann34, Danielle L. Levesque6, Harris A. Lewin7,35,36, Xue Li1,4,37, Abigail Lind28,29, Kerstin Lindblad-Toh3,4, Ava Mackay-Smith38, Voichita D. Marinescu3, Tomas Marques-Bonet39,40,41,42, Victor C. Mason43, Jennifer R. S. Meadows3, Wynn K. Meyer44, Jill E. Moore1, Lucas R. Moreira1,4, Diana D. Moreno-Santillan14, Kathleen M. Morrill1,4,37, Gerard Muntané24, William J. Murphy5, Arcadi Navarro39,41,45,46, Martin Nweeia47,48,49,50, Sylvia Ortmann51, Austin Osmanski14, Benedict Paten2, Nicole S. Paulat14, Andreas R. Pfenning25,26, BaDoi N. Phan25,26,52, Katherine S. Pollard28,29,53, Henry E. Pratt1, David A. Ray14, Steven K. Reilly38, Jeb R. Rosen22, Irina Ruf54, Louise Ryan23, Oliver A. Ryder55,56, Pardis C. Sabeti4,57,58, Daniel E. Schäffer25, Aitor Serres24, Beth Shapiro59,60, Arian F. A. Smit22, Mark Springer61, Chaitanya Srinivasan25, Cynthia Steiner55, Jessica M. Storer22, Kevin A. M. Sullivan14, Patrick F. Sullivan62,63, Elisabeth Sundström3, Megan A. Supple59, Ross Swofford4, Joy-El Talbot64, Emma Teeling23, Jason Turner-Maier4, Alejandro Valenzuela24, Franziska Wagner65, Ola Wallerman3, Chao Wang3, Juehan Wang16, Zhiping Weng1, Aryn P. Wilder55, Morgan E. Wirthlin25,26,66, James R. Xue4,57, Xiaomeng Zhang4,25,26
1Program in Bioinformatics and Integrative Biology, UMass Chan Medical School, Worcester, MA 01605, USA. 2Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 3Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Uppsala 751 32, Sweden. 4Broad Institute of MIT and Harvard, Cambridge, MA 02139, USA. 5Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 6School of Biology and Ecology, University of Maine, Orono, ME 04469, USA. 7The Genome Center, University of California Davis, Davis, CA 95616, USA. 8Genome British Columbia, Vancouver, BC, Canada. 9School of Biological Sciences, University of East Anglia, Norwich, UK. 10School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619-900, Brazil. 11School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. 12Biodiscovery Institute, University of Nottingham, Nottingham, UK. 13Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala 751 85, Sweden. 14Department of Biological Sciences, Texas Tech University, Lubbock, TX 79409, USA. 15Division of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA. 16Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 17Fauna Bio Incorporated, Emeryville, CA 94608, USA. 18Baskin School of Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 19Faculty of Biosciences, GoetheUniversity, 60438 Frankfurt, Germany. 20LOEWE Centre for Translational Biodiversity Genomics, 60325 Frankfurt, Germany. 21Senckenberg Research Institute, 60325 Frankfurt, Germany. 22Institute for Systems Biology, Seattle, WA 98109, USA. 23School of Biology and Environmental Science, University College Dublin, Belfield, Dublin 4, Ireland. 24Department of Experimental and Health Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 25Department of Computational Biology, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 26Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 27Program in Molecular Medicine, UMass Chan Medical School, Worcester, MA 01605, USA. 28Department of Epidemiology & Biostatistics, University of California San Francisco, San Francisco, CA 94158, USA. 29Gladstone Institutes, San Francisco, CA 94158, USA. 30Center for Species Survival, Smithsonian’s National Zoo and Conservation Biology Institute, Washington, DC 20008, USA. 31Computer Technologies Laboratory, ITMO University, St. Petersburg 197101, Russia. 32Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA 22630, USA. 33Department of Biological Sciences, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 34Senckenberg Research Institute and Natural History Museum Frankfurt, 60325, Frankfurt am Main, Germany. 35Department of Evolution and Ecology, University of California Davis, Davis, CA 95616, USA. 36John Muir Institute for the Environment, University of California Davis, Davis, CA 95616, USA. 37Morningside Graduate School of Biomedical Sciences, UMass Chan Medical School, Worcester, MA 01605, USA. 38Department of Genetics, Yale School of Medicine, New Haven, CT 06510, USA. 39Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain. 40CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08036, Spain. 41Department of Medicine and Life Sciences, Institute of Evolutionary Biology (UPFCSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain.42Institut Català de Paleontologia Miquel Crusafont, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. 43Institute of Cell Biology, University of Bern, 3012 Bern, Switzerland. 44Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA. 45BarcelonaBeta Brain Research Center, Pasqual Maragall Foundation, Barcelona 08005, Spain. 46CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08003, Spain. 47Department of Comprehensive Care, School of Dental Medicine, Case Western Reserve University, Cleveland, OH 44106, USA. 48Department of Vertebrate Zoology, Canadian Museum of Nature, Ottawa, ON K2P 2R1, Canada. 49Department of Vertebrate Zoology, Smithsonian Institution, Washington, DC 20002, USA. 50Narwhal Genome Initiative, Department of Restorative Dentistry and Biomaterials Sciences, Harvard School of Dental Medicine, Boston, MA 02115, USA. 51Department of Evolutionary Ecology, Leibniz Institute for Zoo and Wildlife Research, 10315 Berlin, Germany. 52Medical Scientist Training Program, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA. 53Chan Zuckerberg Biohub, San Francisco, CA 94158, USA. 54Division of Messel Research and Mammalogy, Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 55Conservation Genetics, San Diego Zoo Wildlife Alliance, Escondido, CA 92027, USA. 56Department of Evolution, Behavior and Ecology, School of Biological Sciences, University of California San Diego, La Jolla, CA 92039, USA. 57Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA. 58Howard Hughes Medical Institute, Harvard University, Cambridge, MA 02138, USA. 59Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 60Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 61Department of Evolution, Ecology and Organismal Biology, University of California Riverside, Riverside, CA 92521, USA. 62Department of Genetics, University of North Carolina Medical School, Chapel Hill, NC 27599, USA. 63Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden. 64Iris Data Solutions, LLC, Orono, ME 04473, USA. 65Museum of Zoology, Senckenberg Natural History Collections Dresden, 01109 Dresden, Germany. 66Allen Institute for Brain Science, Seattle, WA 98109, USA.
References and Notes
- 1.Burgin CJ, Colella JP, Kahn PL, Upham NS, How many species of mammals are there? J. Mammal. 99, 1–14 (2018). doi: 10.1093/jmammal/gyx147 [DOI] [Google Scholar]
- 2.Jones KE, Safi K, Ecology and evolution of mammalian biodiversity. Philos. Trans. R. Soc. London Ser. B 366, 2451–2461 (2011). doi: 10.1098/rstb.2011.0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones KE et al. , PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals: Ecological Archives E090-184. Ecology 90, 2648–2648 (2009). doi: 10.1890/08-1494.1 [DOI] [Google Scholar]
- 4.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245 (2020). doi: 10.1038/s41586-020-2876-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.University of California Santa Cruz Genomics Institute, Conservation track settings; http://genome.ucsc.edu/cgibin/hgTrackUi?g=cons100way.
- 6.Kirilenko BM et al. , Integrating gene annotation with orthology inference at scale. Science 380, eabn3107 (2023). doi: 10.1126/science.abn3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lappalainen T, MacArthur DG, From variant to function in human disease genetics. Science 373, 1464–1468 (2021). doi: 10.1126/science.abi8207 [DOI] [PubMed] [Google Scholar]
- 8.Taylor SA, Larson EL, Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3, 170–177 (2019). doi: 10.1038/s41559-018-0777-y [DOI] [PubMed] [Google Scholar]
- 9.Kazazian HH Jr., Mobile elements: Drivers of genome evolution. Science 303, 1626–1632 (2004). doi: 10.1126/science.1089670 [DOI] [PubMed] [Google Scholar]
- 10.Feulner PGD, De-Kayne R, Genome evolution, structural rearrangements and speciation. J. Evol. Biol. 30, 1488–1490 (2017). doi: 10.1111/jeb.13101 [DOI] [PubMed] [Google Scholar]
- 11.Armstrong J et al. , Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature 587, 246–251 (2020). doi: 10.1038/s41586-020-2871-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A, Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010). doi: 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindblad-Toh K et al. , A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). doi: 10.1038/nature10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore JE et al. , Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). doi: 10.1038/s41586-020-2493-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplow IM et al. , Inferring mammalian tissue-specific regulatory conservation by predicting tissue-specific differences in open chromatin. BMC Genomics 23, 291 (2022). doi: 10.1016/j.celrep.2012.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiller M et al. , A “forward genomics” approach links genotype to phenotype using independent phenotypic losses among related species. Cell Rep. 2, 817–823 (2012). doi: 10.1016/j.celrep.2012.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner F et al. , Reconstruction of evolutionary changes in fat and toxin consumption reveals associations with gene losses in mammals: A case study for the lipase inhibitor PNLIPRP1 and the xenobiotic receptor NR1I3. J. Evol. Biol. 35, 225–239 (2022). doi: 10.1111/jeb.13970 [DOI] [PubMed] [Google Scholar]
- 18.Marcovitz A et al. , A functional enrichment test for molecular convergent evolution finds a clear protein-coding signal in echolocating bats and whales. Proc. Natl. Acad. Sci. U.S.A. 116, 21094–21103 (2019). doi: 10.1073/pnas.1818532116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Partha R et al. , Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling. eLife 6, e25884 (2017). doi: 10.7554/eLife.25884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villar D et al. , Enhancer evolution across 20 mammalian species. Cell 160, 554–566 (2015). doi: 10.1016/j.cell.2015.01.006; doi: 10.1186/s12864-022-08450-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong ES et al. , Deep conservation of the enhancer regulatory code in animals. Science 370, eaax8137 (2020). doi: 10.1126/science.aax8137 [DOI] [PubMed] [Google Scholar]
- 22.Meadows JRS, Lindblad-Toh K, Dissecting evolution and disease using comparative vertebrate genomics. Nat. Rev. Genet. 18, 624–636 (2017). doi: 10.1038/nrg.2017.51 [DOI] [PubMed] [Google Scholar]
- 23.Prudent X, Parra G, Schwede P, Roscito JG, Hiller M, Controlling for phylogenetic relatedness and evolutionary rates improves the discovery of associations between species’ phenotypic and genomic differences. Mol. Biol. Evol. 33, 2135–2150 (2016). doi: 10.1093/molbev/msw098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan PF et al. , Leveraging base pair mammalian constraint to understand genetic variation and human disease. Science 380, eabn2937 (2023). doi: 10.1123/science.abn2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siepel A et al. , Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). doi: 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foley NM et al. , A genomic timescale for placental mammal evolution. Science 380, eabl8189 (2023). doi: 10.1017/pab.2017.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies TW, Bell MA, Goswami A, Halliday TJD, Completeness of the eutherian mammal fossil record and implications for reconstructing mammal evolution through the Cretaceous/Paleogene mass extinction. Paleobiology 43, 521–536 (2017). doi: 10.1017/pab.2017.20 [DOI] [Google Scholar]
- 28.Springer MS, Foley NM, Brady PL, Gatesy J, Murphy WJ, Evolutionary models for the diversification of placental mammals across the KPg boundary. Front. Genet. 10, 1241 (2019). doi: 10.3389/fgene.2019.01241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foley NM, Springer MS, Teeling EC, Mammal madness: Is the mammal tree of life not yet resolved? Philos. Trans. R. Soc. London Ser. B 371, 20150140 (2016). doi: 10.1098/rstb.2015.0140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.dos Reis M et al. , Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. Biol. Sci. 279, 3491–3500 (2012). doi: 10.1098/rspb.2012.0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meredith RW et al. , Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334, 521–524 (2011). doi: 10.1126/science.1211028 [DOI] [PubMed] [Google Scholar]
- 32.Hedges SB, Parker PH, Sibley CG, Kumar S, Continental breakup and the ordinal diversification of birds and mammals. Nature 381, 226–229 (1996). doi: 10.1038/381226a0 [DOI] [PubMed] [Google Scholar]
- 33.Eizirik E, Murphy WJ, O’Brien SJ, Molecular dating and biogeography of the early placental mammal radiation. J. Hered. 92, 212–219 (2001). doi: 10.1093/jhered/92.2.212 [DOI] [PubMed] [Google Scholar]
- 34.O’Leary MA et al. , The placental mammal ancestor and the post-K-Pg radiation of placentals. Science 339, 662–667 (2013). doi: 10.1126/science.1229237 [DOI] [PubMed] [Google Scholar]
- 35.Esselstyn JA, Oliveros CH, Swanson MT, Faircloth BC, Investigating difficult nodes in the placental mammal tree with expanded taxon sampling and thousands of ultraconserved elements. Genome Biol. Evol. 9, 2308–2321 (2017). doi: 10.1093/gbe/evx168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Archibald JD, Deutschman DH, Quantitative analysis of the timing of the origin and diversification of extant placental orders. J. Mamm. Evol. 8, 107–124 (2001). doi: 10.1023/A:1011317930838 [DOI] [Google Scholar]
- 37.Andrews G et al. , Mammalian evolution of human cis-regulatory elements and transcription factor binding sites. Science 380, eabn7930 (2023). doi: 10.1126/science.abn7930 [DOI] [PubMed] [Google Scholar]
- 38.Lunter G, Ponting CP, Hein J, Genome-wide identification of human functional DNA using a neutral indel model. PLOS Comput. Biol. 2, e5 (2006). doi: 10.1371/journal.pcbi.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eory L, Halligan DL, Keightley PD, Distributions of selectively constrained sites and deleterious mutation rates in the hominid and murid genomes. Mol. Biol. Evol. 27, 177–192 (2010). doi: 10.1093/molbev/msp219 [DOI] [PubMed] [Google Scholar]
- 40.Ponting CP, Hardison RC, What fraction of the human genome is functional? Genome Res. 21, 1769–1776 (2011). doi: 10.1101/gr.116814.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchmann K, Evolution of innate immunity: Clues from invertebrates via fish to mammals. Front. Immunol. 5, 459 (2014). doi: 10.3389/fimmu.2014.00459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espregueira Themudo G et al. , Losing genes: The evolutionary remodeling of cetacea skin. Front. Mar. Sci. 7, 592375 (2020). doi: 10.3389/fmars.2020.592375 [DOI] [Google Scholar]
- 43.Antinucci M, Risso D, A matter of taste: Lineage-specific loss of function of taste receptor genes in vertebrates. Front. Mol. Biosci. 4, 81 (2017). doi: 10.3389/fmolb.2017.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conboy CM et al. , Cell cycle genes are the evolutionarily conserved targets of the E2F4 transcription factor. PLOS ONE 2, e1061 (2007). doi: 10.1371/journal.pone.0001061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alberts B, Johnson A, Lewis J,Raff M, Roberts K, Walter P, Eds., “Universal mechanisms of animal development” in Molecular Biology of the Cell (Garland Science, ed. April, 2002), chap. 21. [Google Scholar]
- 46.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B, WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019). doi: 10.1093/nar/gkz401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carbon S et al. , The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49, D325–D334 (2021). doi: 10.1093/nar/gkaa1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashburner M et al. , Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). doi: 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pai AA, Luca F, Environmental influences on RNA processing: Biochemical, molecular and genetic regulators of cellular response. Wiley Interdiscip. Rev. RNA 10, e1503 (2019). doi: 10.1002/wrna.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scotti MM, Swanson MS, RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32 (2016). doi: 10.1038/nrg.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chou HH et al. , A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl. Acad. Sci. U.S.A. 95, 11751–11756 (1998). doi: 10.1073/pnas.95.20.11751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Irie A, Koyama S, Kozutsumi Y, Kawasaki T, Suzuki A, The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 273, 15866–15871 (1998). doi: 10.1074/jbc.273.25.15866 [DOI] [PubMed] [Google Scholar]
- 53.Dankwa S et al. , Ancient human sialic acid variant restricts an emerging zoonotic malaria parasite. Nat. Commun. 7, 11187 (2016). doi: 10.1038/ncomms11187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Unione L et al. , The SARS-CoV-2 spike glycoprotein directly binds exogeneous sialic acids: A NMR view. Angew. Chem. Int. Ed. 61, e202201432 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chou H-H et al. , Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 11736–11741 (2002). doi: 10.1073/pnas.182257399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayakawa T, Aki I, Varki A, Satta Y, Takahata N, Fixation of the human-specific CMP-N-acetylneuraminic acid hydroxylase pseudogene and implications of haplotype diversity for human evolution. Genetics 172, 1139–1146 (2006). doi: 10.1534/genetics.105.046995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Springer SA, Diaz SL, Gagneux P, Parallel evolution of a self-signal: Humans and new world monkeys independently lost the cell surface sugar Neu5Gc. Immunogenetics 66, 671–674 (2014). doi: 10.1007/s00251-014-0795-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peri S, Kulkarni A, Feyertag F, Berninsone PM, Alvarez-Ponce D, Phylogenetic distribution of CMPNeu5Ac hydroxylase (CMAH), the enzyme synthetizing the proinflammatory human xenoantigen Neu5Gc. Genome Biol. Evol. 10, 207–219 (2018). doi: 10.1093/gbe/evx251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ng PSK et al. , Ferrets exclusively synthesize Neu5Ac and express naturally humanized influenza A virus receptors. Nat. Commun. 5, 5750 (2014). doi: 10.1038/ncomms6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carlson CJ et al. , The future of zoonotic risk prediction. Philos. Trans. R. Soc. London Ser. B 376, 20200358 (2021). doi: 10.1098/rstb.2020.0358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cooper GM et al. , Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913 (2005). doi: 10.1101/gr.3577405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohta T, Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 40, 56–63 (1995). doi: 10.1007/BF00166595 [DOI] [PubMed] [Google Scholar]
- 63.Crick FH, Barnett L, Brenner S, Watts-Tobin RJ, General nature of the genetic code for proteins. Nature 192, 1227–1232 (1961). doi: 10.1038/1921227a0 [DOI] [PubMed] [Google Scholar]
- 64.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiedemann C, Kumar A, Lang A, Ohlenschläger O, Cysteines and disulfide bonds as structure-forming units: Insights from different domains of life and the potential for characterization by NMR. Front Chem. 8, 280 (2020). doi: 10.3389/fchem.2020.00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tennessen JA et al. , Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012). doi: 10.1126/science.1219240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Backman JD et al. , Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021). doi: 10.1038/s41586-021-04103-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Castle JC, SNPs occur in regions with less genomic sequence conservation. PLOS ONE 6, e20660 (2011). doi: 10.1371/journal.pone.0020660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taliun D et al. , Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cingolani P et al. , A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). doi: 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xue JR et al. , The functional and evolutionary impacts of human-specific deletions in conserved elements. Science 380, eabn2253 (2023). doi: 10.1126/science.abn2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Snetkova V, Pennacchio LA, Visel A, Dickel DE, Perfect and imperfect views of ultraconserved sequences. Nat. Rev. Genet. 23, 182–194 (2022). doi: 10.1038/s41576-021-00424-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bejerano G et al. , Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004). doi: 10.1126/science.1098119 [DOI] [PubMed] [Google Scholar]
- 74.Pennacchio LA et al. , In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006). doi: 10.1038/nature05295 [DOI] [PubMed] [Google Scholar]
- 75.Visel A et al. , Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet. 40, 158–160 (2008). doi: 10.1038/ng.2007.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.de la Calle-Mustienes E et al. , A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 15, 1061–1072 (2005). doi: 10.1101/gr.4004805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drake JA et al. , Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat. Genet. 38, 223–227 (2006). doi: 10.1038/ng1710 [DOI] [PubMed] [Google Scholar]
- 78.Habic A et al. , Genetic variations of ultraconserved elements in the human genome. OMICS 23, 549–559 (2019). doi: 10.1089/omi.2019.0156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma ACH et al. , Methionine aminopeptidase 2 is required for HSC initiation and proliferation. Blood 118, 5448–5457 (2011). doi: 10.1182/blood-2011-04-350173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giusti-Rodríguez P et al. , Using three-dimensional regulatory chromatin interactions from adult and fetal cortex to interpret genetic results for psychiatric disorders and cognitive traits. bioRxiv 406330 [Preprint] (2019); 10.1101/406330. [DOI] [Google Scholar]
- 81.HUGIn2; http://hugin2.genetics.unc.edu/Project/hugin/.
- 82.Xu Z, Zhang G, Wu C, Li Y, Hu M, FastHiC: A fast and accurate algorithm to detect long-range chromosomal interactions from Hi-C data. Bioinformatics 32, 2692–2695 (2016). doi: 10.1093/bioinformatics/btw240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kent WJ et al. , The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). doi: 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee BT et al. , The UCSC Genome Browser database: 2022 update. Nucleic Acids Res. 50, D1115–D1122 (2022). doi: 10.1093/nar/gkab959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Segura-Bayona S, Stracker TH, The Tousled-like kinases regulate genome and epigenome stability: Implications in development and disease. Cell. Mol. Life Sci. 76, 3827–3841 (2019). doi: 10.1007/s00018-019-03208-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gruber JJ et al. , HAT1 coordinates histone production and acetylation via H4 promoter binding. Mol. Cell 75, 711–724.e5 (2019). doi: 10.1016/j.molcel.2019.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bastian FB et al. , The Bgee suite: Integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Res. 49, D831–D847 (2021). doi: 10.1093/nar/gkaa793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bauer DE, Orkin SH, Hemoglobin switching’s surprise: The versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Curr. Opin. Genet. Dev. 33, 62–70 (2015). doi: 10.1016/j.gde.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ochoa SD, Salvador S, LaBonne C, The LIM adaptor protein LMO4 is an essential regulator of neural crest development. Dev. Biol. 361, 313–325 (2012). doi: 10.1016/j.ydbio.2011.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang J et al. , Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22, 1798–1812 (2012). doi: 10.1101/gr.139105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davydov EV et al. , Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput. Biol. 6, e1001025 (2010). doi: 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ovcharenko I et al. , Evolution and functional classification of vertebrate gene deserts. Genome Res. 15, 137–145 (2005). doi: 10.1101/gr.3015505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Laat W, Duboule D, Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 502, 499–506 (2013). doi: 10.1038/nature12753 [DOI] [PubMed] [Google Scholar]
- 94.Schmidt D et al. , Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148, 335–348 (2012). doi: 10.1016/j.cell.2011.11.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmidt D et al. , Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040 (2010). doi: 10.1126/science.1186176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tang Z et al. , CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015). doi: 10.1016/j.cell.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vietri Rudan M et al. , Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 10, 1297–1309 (2015). doi: 10.1016/j.celrep.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zuin J et al. , Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. U.S.A. 111, 996–1001 (2014). doi: 10.1073/pnas.1317788111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boeva V, Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic cells. Front. Genet. 7, 24 (2016). doi: 10.3389/fgene.2016.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Markenscoff-Papadimitriou E et al. , A chromatin accessibility atlas of the developing human telencephalon. Cell 182, 754–769.e18 (2020). doi: 10.1016/j.cell.2020.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bakken TE et al. , Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 598, 111–119 (2021). doi: 10.1038/s41586-021-03465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fullard JF et al. , An atlas of chromatin accessibility in the adult human brain. Genome Res. 28, 1243–1252 (2018). doi: 10.1101/gr.232488.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tena JJ, Santos-Pereira JM, Topologically associating domains and regulatory landscapes in development, evolution and disease. Front. Cell Dev. Biol. 9, 702787 (2021). doi: 10.3389/fcell.2021.702787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Keough KC et al. , Three-dimensional genome rewiring in loci with human accelerated regions. Science 380, eabm1696 (2023). doi: 10.1126/science.abm1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hubisz MJ, Pollard KS, Exploring the genesis and functions of Human Accelerated Regions sheds light on their role in human evolution. Curr. Opin. Genet. Dev. 29, 15–21 (2014). doi: 10.1016/j.gde.2014.07.005 [DOI] [PubMed] [Google Scholar]
- 106.Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, Pollard KS, Many human accelerated regions are developmental enhancers. Philos. Trans. R. Soc. London Ser. B 368, 20130025 (2013). doi: 10.1098/rstb.2013.0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kostka D, Hubisz MJ, Siepel A, Pollard KS, The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol. 29, 1047–1057 (2012). doi: 10.1093/molbev/msr279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McLean CY et al. , GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). doi: 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kronenberg ZN et al. , High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018). doi: 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Osmanski AB et al. , Insights into mammalian TE diversity via the curation of 248 mammalian genome assemblies. Science 380, eabn1430 (2023). doi: 10.1126/science.abn1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bourque G et al. , Ten things you should know about transposable elements. Genome Biol. 19, 199 (2018). doi: 10.1186/s13059-018-1577-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Branco MR, Chuong EB, Crossroads between transposons and gene regulation. Philos. Trans. R. Soc. London Ser. B 375, 20190330 (2020). doi: 10.1098/rstb.2019.0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Senft AD, Macfarlan TS, Transposable elements shape the evolution of mammalian development. Nat. Rev. Genet. 22, 691–711 (2021). doi: 10.1038/s41576-021-00385-1 [DOI] [PubMed] [Google Scholar]
- 114.Kapusta A, Suh A, Feschotte C, Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. U.S.A. 114, E1460–E1469 (2017). doi: 10.1073/pnas.1616702114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang L, Scott L, Wichman HA, Tracing the history of LINE and SINE extinction in sigmodontine rodents. Mob. DNA 10, 22 (2019). doi: 10.1186/s13100-019-0164-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Paulat NS et al. , Chiropterans are a hotspot for horizontal transfer of DNA transposons in Mammalia. Mol. Biol. Evol. 10.1093/molbev/msad092 (2023).doi: 10.1093/molbev/msad092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hayward A, Ghazal A, Andersson G, Andersson L, Jern P, ZBED evolution: Repeated utilization of DNA transposons as regulators of diverse host functions. PLOS ONE 8, e59940 (2013). doi: 10.1371/journal.pone.0059940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cechova M et al. , High satellite repeat turnover in great apes studied with shortand long-read technologies. Mol. Biol. Evol. 36, 2415–2431 (2019). doi: 10.1093/molbev/msz156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fotsing SF et al. , The impact of short tandem repeat variation on gene expression. Nat. Genet. 51, 1652–1659 (2019). doi: 10.1038/s41588-019-0521-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trizzino M et al. , Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 27, 1623–1633 (2017). doi: 10.1101/gr.218149.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sundaram V, Wysocka J, Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. London Ser. B 375, 20190347 (2020). doi: 10.1098/rstb.2019.0347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sundaram V et al. , Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 24, 1963–1976 (2014). doi: 10.1101/gr.168872.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cusanovich DA, Pavlovic B, Pritchard JK, Gilad Y, The functional consequences of variation in transcription factor binding. PLOS Genet. 10, e1004226 (2014). doi: 10.1371/journal.pgen.1004226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu A et al. , Convergent degeneration of olfactory receptor gene repertoires in marine mammals. BMC Genomics 20, 977 (2019). doi: 10.1186/s12864-019-6290-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Niimura Y, Matsui A, Touhara K, Extreme expansion of the olfactory receptor gene repertoire in African elephants and evolutionary dynamics of orthologous gene groups in 13 placental mammals. Genome Res. 24, 1485–1496 (2014). doi: 10.1101/gr.169532.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kishida T, Kubota S, Shirayama Y, Fukami H, The olfactory receptor gene repertoires in secondary-adapted marine vertebrates: Evidence for reduction of the functional proportions in cetaceans. Biol. Lett. 3, 428–430 (2007). doi: 10.1098/rsbl.2007.0191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hughes GM et al. , The birth and death of olfactory receptor gene families in mammalian niche adaptation. Mol. Biol. Evol. 35, 1390–1406 (2018). doi: 10.1093/molbev/msy028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nei M, Niimura Y, Nozawa M, The evolution of animal chemosensory receptor gene repertoires: Roles of chance and necessity. Nat. Rev. Genet. 9, 951–963 (2008). doi: 10.1038/nrg2480 [DOI] [PubMed] [Google Scholar]
- 129.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM, BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). doi: 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- 130.Thewissen JGM, George J, Rosa C, Kishida T, Olfaction and brain size in the bowhead whale (Balaena mysticetus). Mar. Mamm. Sci. 27, 282–294 (2011). doi: 10.1111/j.17487692.2010.00406. [DOI] [Google Scholar]
- 131.Springer MS, Gatesy J, Inactivation of the olfactory marker protein (OMP) gene in river dolphins and other odontocete cetaceans. Mol. Phylogenet. Evol. 109, 375–387 (2017). doi: 10.1016/j.ympev.2017.01.020 [DOI] [PubMed] [Google Scholar]
- 132.Bird DJ, Amirkhanian A, Pang B, Van Valkenburgh B, Quantifying the cribriform plate: Influences of allometry, function, and phylogeny in Carnivora. Anat. Rec. 297, 2080–2092 (2014). doi: 10.1002/ar.23032 [DOI] [PubMed] [Google Scholar]
- 133.Martinez Q et al. , Convergent evolution of olfactory and thermoregulatory capacities in small amphibious mammals. Proc. Natl. Acad. Sci. U.S.A. 117, 8958–8965 (2020). doi: 10.1073/pnas.1917836117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Larochelle R, Baron G, Comparative morphology and morphometry of the nasal fossae of four species of North American shrews (Soricinae). Am. J. Anat. 186, 306–314 (1989). doi: 10.1002/aja.1001860307 [DOI] [PubMed] [Google Scholar]
- 135.Grafen A, The phylogenetic regression. Philos. Trans. R. Soc. London Ser. B 326, 119–157 (1989). doi: 10.1098/rstb.1989.0106 [DOI] [PubMed] [Google Scholar]
- 136.Tung Ho L, Ané C, A linear-time algorithm for Gaussian and non-Gaussian trait evolution models. Syst. Biol. 63, 397–408 (2014). doi: 10.1093/sysbio/syu005 [DOI] [PubMed] [Google Scholar]
- 137.Saputra E, Kowalczyk A, Cusick L, Clark N, Chikina M, Phylogenetic permulations: a statistically rigorous approach to measure confidence in associations in a phylogenetic context. Mol. Biol. Evol. 38, 3004–3021 (2021). doi: 10.1093/molbev/msab068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lukas D, Clutton-Brock TH, The evolution of social monogamy in mammals. Science 341, 526–530 (2013). doi: 10.1126/science.1238677 [DOI] [PubMed] [Google Scholar]
- 139.Lovegrove BG, The evolution of endothermy in Cenozoic mammals: A plesiomorphic-apomorphic continuum. Biol. Rev. Camb. Philos. Soc. 87, 128–162 (2012). doi: 10.1111/j.1469185X.2011.00188.x [DOI] [PubMed] [Google Scholar]
- 140.Lovegrove BG, Lobban KD, Levesque DL, Mammal survival at the Cretaceous-Palaeogene boundary: Metabolic homeostasis in prolonged tropical hibernation in tenrecs. Proc. Biol. Sci. 281, 20141304 (2014). doi: 10.1098/rspb.2014.1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Carey HV et al. , Elucidating nature’s solutions to heart, lung, and blood diseases and sleep disorders. Circ. Res. 110, 915–921 (2012). doi: 10.1161/CIRCRESAHA.111.255398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nordeen CA, Martin SL, Engineering human stasis for long-duration spaceflight. Physiology 34, 101–111 (2019). doi: 10.1152/physiol.00046.2018 [DOI] [PubMed] [Google Scholar]
- 143.Staples JF, Metabolic suppression in mammalian hibernation: The role of mitochondria. J. Exp. Biol. 217, 2032–2036 (2014). doi: 10.1242/jeb.092973 [DOI] [PubMed] [Google Scholar]
- 144.Huang C et al. , Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci. Rep. 6, 29196 (2016). doi: 10.1038/srep29196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hand LE et al. , Induction of the metabolic regulator Txnip in fasting-induced and natural torpor. Endocrinology 154, 2081–2091 (2013). doi: 10.1210/en.2012-2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fu R et al. , Dynamic RNA regulation in the brain underlies physiological plasticity in a hibernating mammal. Front. Physiol. 11, 624677 (2021). doi: 10.3389/fphys.2020.624677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schwartz C, Hampton M, Andrews MT, Seasonal and regional differences in gene expression in the brain of a hibernating mammal. PLOS ONE 8, e58427 (2013). doi: 10.1371/journal.pone.0058427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sun H, Wang J, Xing Y, Pan Y-H, Mao X, Gut transcriptomic changes during hibernation in the greater horseshoe bat (Rhinolophus ferrumequinum). Front. Zool. 17, 21 (2020). doi: 10.1186/s12983-020-00366-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kowalczyk A et al. , RERconverge: An R package for associating evolutionary rates with convergent traits. Bioinformatics 35, 4815–4817 (2019). doi: 10.1093/bioinformatics/btz468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Partha R, Kowalczyk A, Clark NL, Chikina M, Robust method for detecting convergent shifts in evolutionary rates. Mol. Biol. Evol. 36, 1817–1830 (2019). doi: 10.1093/molbev/msz107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chikina M, Robinson JD, Clark NL, Hundreds of genes experienced convergent shifts in selective pressure in marine mammals. Mol. Biol. Evol. 33, 2182–2192 (2016). doi: 10.1093/molbev/msw112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang Y, Storey KB, “Life in suspended animation: Role of chaperone proteins in vertebrate and invertebrate stress adaptation” in Regulation of Heat Shock Protein Responses, Asea AAA, Kaur P, Eds. (Springer, 2018), pp. 95–137. [Google Scholar]
- 153.Lambert MJ, Portfors CV, Adaptive sequence convergence of the tumor suppressor ADAMTS9 between small-bodied mammals displaying exceptional longevity. Aging (Albany NY) 9, 573–582 (2017). doi: 10.18632/aging.101180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sanders SJ et al. , Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 41, 442–456 (2018). doi: 10.1016/j.tins.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fukuda A, Watanabe M, Pathogenic potential of human SLC12A5 variants causing KCC2 dysfunction. Brain Res. 1710, 1–7 (2019). doi: 10.1016/j.brainres.2018.12.025 [DOI] [PubMed] [Google Scholar]
- 156.King MC, Wilson AC, Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (1975). doi: 10.1126/science.1090005 [DOI] [PubMed] [Google Scholar]
- 157.Pfenning AR et al. , Convergent transcriptional specializations in the brains of humans and song-learning birds. Science 346, 1256846 (2014). doi: 10.1126/science.1256846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wray GA et al. , The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20, 1377–1419 (2003). doi: 10.1093/molbev/msg140 [DOI] [PubMed] [Google Scholar]
- 159.Kaplow IM et al. , Relating enhancer genetic variation across mammals to complex phenotypes using machine learning. Science 380, eabm7993 (2023). doi: 10.1126/science.aay5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wirthlin ME et al. , Vocal learning-associated convergent evolution in mammalian proteins and regulatory elements. bioRxiv 2022.12.17.520895 [Preprint] (2022); 10.1101/2022.12.17.520895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Srinivasan C et al. , Addiction-associated genetic variants implicate brain cell typeand region-specific cis-regulatory elements in addiction neurobiology. J. Neurosci. 41, 9008–9030 (2021). doi: 10.1523/JNEUROSCI.2534-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wirthlin M et al. , The regulatory evolution of the primate finemotor system. bioRxiv 2020.10.27.356733 [Preprint] (2020); 10.1101/2020.10.27.356733. [DOI] [Google Scholar]
- 163.Li YE et al. , An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136 (2021). doi: 10.1038/s41586-021-03604-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hofman MA, Evolution of the human brain: When bigger is better. Front. Neuroanat. 8, 15 (2014). doi: 10.3389/fnana.2014.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bjerke IE et al. , Densities and numbers of calbindin and parvalbumin positive neurons across the rat and mouse brain. iScience 24, 101906 (2020). doi: 10.1016/j.isci.2020.101906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang X, Kaplow IM, Wirthlin M, Park TY, Pfenning AR, HALPER facilitates the identification of regulatory element orthologs across species. Bioinformatics 36, 4339–4340 (2020). doi: 10.1093/bioinformatics/btaa493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ito H et al. , Biochemical and morphological characterization of a neurodevelopmental disorder-related mono-ADPribosylhydrolase, MACRO domain containing 2. Dev. Neurosci. 40, 278–287 (2018). doi: 10.1159/000492271 [DOI] [PubMed] [Google Scholar]
- 168.Lombardo B et al. , Intragenic deletion in MACROD2: A family with complex phenotypes including microcephaly, intellectual disability, polydactyly, renal and pancreatic malformations. Cytogenet. Genome Res. 158, 25–31 (2019). doi: 10.1159/000499886 [DOI] [PubMed] [Google Scholar]
- 169.Janik VM, Slater PJB, Vocal learning in mammals. Adv. Stud. Behav. 26, 59–99 (1997). doi: 10.1016/S00653454(08)60377-0 [DOI] [Google Scholar]
- 170.Orsini JJ, Escolar ML, Wasserstein MP, Caggana M, “Krabbe disease” in GeneReviews, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, Eds. (Univ. Washington, Seattle, 2000). [PubMed] [Google Scholar]
- 171.Caubit X et al. , TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat. Genet. 48, 1359–1369 (2016). doi: 10.1038/ng.3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wilder AP et al. , The contribution of historical processes to contemporary extinction risk in placental mammals. Science 380, eabn5856 (2023). doi: 10.1126/science.aay5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Roy A, Sakthikumar S, Kozyrev SV, Nordin J, Pensch R, Pettersson M, Karlsson E, Lindblad-Toh K, Forsberg-Nilsson K; Zoonomia Consortium, Using evolutionary constraint to define novel candidate driver genes in medulloblastoma. bioRxiv 2022.11.02.514465 [Preprint] (2022); 10.1101/2022.11.02.514465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Damas J et al. , Evolution of the ancestral mammalian karyotype and syntenic regions. Proc. Natl. Acad. Sci. U.S.A. 119, e2209139119 (2022). doi: 10.1073/pnas.2209139119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stephan T et al. , Darwinian genomics and diversity in the tree of life. Proc. Natl. Acad. Sci. U.S.A. 119, e2115644119 (2022). doi: 10.1073/pnas.2115644119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wilkinson MD et al. , The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016). doi: 10.1038/sdata.2016.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ceballos G et al. , Accelerated modern human-induced species losses: Entering the sixth mass extinction. Sci. Adv. 1, e1400253 (2015). doi: 10.1126/sciadv.1400253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lewin HA et al. , The Earth BioGenome Project 2020: Starting the clock. Proc. Natl. Acad. Sci. U.S.A. 119, e2115635118 (2022). doi: 10.1073/pnas.2115635118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hayden S et al. , Ecological adaptation determines functional mammalian olfactory subgenomes. Genome Res. 20, 1–9 (2010). doi: 10.1101/gr.099416.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Christmas M, Kaplow I, Lind A, MattChristmas/Zoonomia: Zoonomia Flagship one v1.0. Zenodo; (2023); 10.5281/zenodo.7295354. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.