


Abstract
In enteric bacteria, several small RNAs (sRNAs) including MicC employ endoribonuclease RNase E to stimulate target RNA decay. A current model proposes that interaction of the sRNA 5′ monophosphate (5′P) with the N-terminal sensing pocket of RNase E allosterically activates cleavage of the base-paired target in the active site. In vivo evidence supporting this model is lacking. Here, we engineered a genetic tool allowing us to generate 5′ monophosphorylated sRNAs of choice in a controllable manner in the cell. Four sRNAs were tested and none performed better in target destabilization when 5′ monophosphorylated. MicC retains full activity even when RNase E is defective in 5′P sensing, whereas regulation is lost upon removal of its scaffolding domain. Interestingly, sRNAs MicC and RyhB that originate with a 5′ triphosphate group are dramatically destabilized when 5′ monophosphorylated, but stable when in 5′ triphosphorylated form. In contrast, the processing-derived sRNAs CpxQ and SroC, which carry 5′P groups naturally, are highly stable. Thus, the 5′ phosphorylation state determines stability of naturally triphosphorylated sRNAs, but plays no major role for target RNA destabilization in vivo. In contrast, the RNase E C-terminal half is crucial for MicC-mediated ompD decay, suggesting that interaction with Hfq is mandatory.
INTRODUCTION
In bacteria, small RNAs (sRNAs) provide an additional layer of gene regulation at the post-transcriptional level. A major group of sRNAs cooperates with the RNA chaperone Hfq to regulate translation and/or stability of target RNAs by base-pairing (1–3). Several sRNAs stimulate target RNA decay actively, independent of translation (4,5). To this end, Gram-negative sRNAs recruit endoribonuclease RNase E to promote cleavage of the base-paired target (6–8).
RNase E together with helicase RhlB, PNPase and enolase forms the degradosome complex, which is important for bulk RNA turnover (9–11). RNase E initiates RNA degradation through cleavage at single-stranded AU rich sites with strong preference for U at position +2 (12,13). Full-length RNase E assembles into a homotetramer, but the N-terminal half (NTH) can form a catalytically active dimer on its own (14). The C-terminal half of RNase E (CTH; residues 530–1061) scaffolds the degradosome components and provides RNA binding domains as well as a membrane attachment site (15,16). The NTH of RNase E contains the active site and a distinct 5′ monophosphate (5′P) sensing pocket, which binds the 5′P of RNA ends (17,18). The 5′P is recognized through hydrogen bonding with residues Arg169 and Thr170 in the 5′P sensing pocket of one protomer. This interaction favors the transition of RNase E to a closed conformation, which allows for substrate orientation and boosts its cleavage by the other protomer in the principal dimer (19–21). Therefore, RNase E strongly prefers 5′ monophosphorylated over 5′ triphosphorylated substrates (22). Cleavage generates 5′ monophosphorylated products that are vulnerable to further RNase E attack. Conversion of 5′ triphosphorylated to 5′ monophosphorylated transcripts is catalyzed by the pyrophosphohydrolase RppH (23,24). A second class of RNase E substrates is cleaved independent of the 5′ phosphorylation status (25). RNA secondary structures or adaptor proteins may guide RNase E in this case (26–29). The existence of these two classes of substrates was inferred from in vitro studies and from the properties of mutants affected globally in either of the degradation pathways (23,25,30). However, analysis of selected transcripts is difficult in vivo, as it is technically challenging to produce a particular RNA in a completely 5′ monophosphorylated state without affecting RNA decay in general.
Different mechanisms are discussed for how sRNAs guide RNase E to their targets. Initial work on the sRNA/mRNA pair SgrS/ptsG demonstrated that Hfq plays a critical role (7). Several studies reported interaction of Hfq with a region in the CTH of RNase E, either dependent or independent of RNA (16,31–33). Truncation of the RNase E-CTH often slows down sRNA-induced target RNA degradation (6,7,31,34,35), suggesting that the CTH is involved in recognition or presentation of the RNA duplex to the catalytic domain. Cleavage sites and Hfq binding sites in target RNAs are often close or overlap (36,37). Thus, following hybridization on Hfq, the RNA duplex may be released and rapidly transferred to RNase E, which binds and cleaves the target downstream of the duplex (18,37,38). Recent evidence supports a model wherein the AR2 RNA-binding domain of the RNase E-CTH interacts with Hfq-bound target mRNAs, thereby recruiting the cognate sRNA to RNase E in a second step (39).
Another model proposes that RNase E is guided by the 5′P group of the sRNA to cleave the target (40). Based on in vitro experiments using interaction of sRNA MicC with ompD as example, the 5′P of the sRNA was proposed to interact with the sensing pocket in RNase E, thereby boosting cleavage of the base-paired target in the active site (Figure 1, bottom right; (28)). In vitro, the catalytic domain of RNase E is sufficient for this stimulated cleavage (28,40). Recently, sRNAs were discovered that are generated through processing of parental mRNAs and therefore carry 5′P ends naturally. Theoretically, this feature could endow these sRNAs by default with the ability to destabilize their targets through interaction with the RNase E 5′ sensor pocket (13,41,42). Again, results obtained in vitro support this idea (43).
Figure 1.

Inducible two-plasmid system for generation of 5′ monophosphorylated sRNAs in vivo. Based on in vitro experiments, sRNAs such as MicC and CpxQ were proposed to activate cleavage of target RNAs through interaction of their 5′P groups with the 5′ sensory pocket of RNase E (box bottom right; (40)). To investigate whether this mechanism operates in vivo, a two-plasmid system was engineered allowing to release sRNAs of interest in a 5′P state through RNase E/RapZ-mediated cleavage of parental fusion RNAs. (A) The cleavage system consists of the sRNA of interest (in red) fused to the 3′ end of the first 146 nt of sRNA GlmZ comprising the first two stem loop structures (designated GlmZ’; in blue; construct II), which are recognized and bound by the RNase E adaptor protein RapZ. RapZ recruits RNase E by interaction resulting in cleavage 7 nt downstream of GlmZ’ (i.e. at position 153) regardless of sequence composition at the cleavage site (44). Consequently, the sRNAs are released with their authentic 5′ ends in a monophosphorylated state, when inserting a 7 nt spacer sequence between GlmZ’ and the sRNA start nucleotide. Plasmid pYG215 transcribing a glmZ’-gfp fusion RNA from the IPTG-inducible PLlacO-1 promoter (construct I), served as negative control as well as parental plasmid for construction of derivatives. An AgeI restriction site overlapping with the last three nucleotides of the GlmZ’ 3 end and an XbaI site in the 3′ UTR allows construction of glmZ’-sRNA fusions of choice (construct II). Substitution of the AatII-XbaI fragment results in an isogenic plasmid, which expresses the sRNA of interest as 5′-PPP transcript (construct III). (B) For inducible cleavage of the fusion RNAs, cells additionally carried the compatible plasmid pYG205 carrying rapZ under control of the arabinose-inducible PAra promoter. To avoid interference with endogenous genes, the constructs were tested in cells deleted for the chromosomal copies of glmZ, glmY, rapZ and the sRNA gene under investigation.
The model proposing a key role for the 5′P groups of sRNAs has received much attention as it intuitively explains how sRNAs may stimulate target decay. However, in vivo evidence is lacking as it is difficult to produce selected transcripts in a completely 5′ monophosphorylated state in a controllable manner in vivo. Here, we adapted a device based on the RNase E adaptor protein RapZ and the sRNA GlmZ aptamer from Escherichia coli (44) to release 5′ monophosphorylated variants of four sRNAs in the living cell. Upon comparison with their 5′ triphosphorylated counterparts, we could not detect a major stimulatory role of the sRNA 5′P group for target degradation. In contrast, the RNase E-CTH is essential for MicC-promoted ompD decay, supporting the idea that Hfq-RNase E interaction is decisive. Interestingly, we find that a 5′P group dramatically destabilizes sRNAs MicC and RyhB, which are naturally produced in 5′ triphosphorylated state. In contrast, the processing-derived sRNAs CpxQ and SroC remain stable, suggesting they possess features allowing them to escape the 5′P-dependent pathway of RNA degradation.
MATERIALS AND METHODS
Bacterial strains, plasmids and growth conditions
Bacteria were routinely cultivated at 37°C in LB medium containing the required antibiotics. Strains, plasmids and oligonucleotides used in this study are listed in Supplementary Tables I, II and III, respectively. Construction of plasmids is described under ‘Supplementary Material’. Established alleles tagged with an antibiotic resistance gene were moved between strains by T4GT7 phage transduction (45). Deletions of chromosomal sRNA genes were generated using the λ red recombination system (46). Briefly, DNA fragments comprising the kan resistance gene flanked by the required homologous extensions were amplified by PCR using plasmid pKD4 as template and the oligonucleotides itemized in Supplementary Table III. The PCR fragments were introduced by electroporation into target strains carrying plasmid pKD46 and recombinants were obtained upon kanamycin selection. If required, resistance genes flanked by FLP recombinase sites were removed by using the temperature sensitive plasmid pCP20 (46). FLAG-tagging of the chromosomal skp gene was performed using oligonucleotides BG1615/BG1616 and plasmid pSUB11 as described (47). The Ptac-ompD expression cassette was ectopically integrated into the λattB site on the E. coli chromosome following a described procedure (48). Diagnostic PCRs were used to verify chromosomal constructions.
In the experiments using the GlmZ aptamer for sRNA release, respective transformants were grown overnight in LB supplemented with 0.2% l-arabinose to express rapZ from pYG205, if applicable. On the next day, the overnight cultures were used to inoculate subcultures in the same medium to an OD600 = 0.1. For induction of the PLlacO-1 promoter, which initiates transcription of sRNA genes and GlmZ aptamer-sRNA fusions (49), IPTG was added at the desired concentration (10 μM, 50 μM, 100 μM or 1 mM) when the cultures reached an OD600 = 0.3. For determination of RNA steady state levels, cells were harvested for total RNA extraction when cultures attained an OD600 = 0.5–0.6. Harvested cells were immediately cooled on ice and centrifuged for 3 min, 12 851 × g at 4°C. After centrifugation supernatants were subsequently removed and pellets were frozen in liquid nitrogen followed by RNA extraction and Northern blotting.
RNA half-life determination
For RNA half-life determination, 500 μg/ml rifampicin was added to bacterial cultures when reaching an OD600 of 0.5–0.6. Samples harvested at the time of rifampicin addition corresponded to t = 0 min. Additional samples for RNA extraction were harvested at indicated time points after rifampicin addition. Bacteria were immediately cooled on ice after harvesting and were centrifuged for 3 min, 12 851 × g at 4°C. After centrifugation supernatants were subsequently removed and pellets were frozen in liquid nitrogen followed by RNA extraction and Northern blotting.
Extraction of total RNA and Northern blotting
Total RNA was extracted from pelleted cells using the ReliaPrep RNA Cell Miniprep System (Promega). The total RNA (usually 1.5–3 μg) was mixed with 2× RNA loading dye (95% Formamide, 0.5 mM EDTA, 0.025% SDS, 0.025% Bromophenol Blue, 0.025% Xylene Cyanol) and separated by denaturing gel electrophoresis (7 M urea, 5% or 8% acrylamide,1x TBE) for 1.5–2 h at 100V using 0.5× TBE as running buffer. Subsequently, the RNA was transferred to a Hybond N+ nylon membrane (GE Healthcare) via semi-dry electroblotting for 1h at 120 mA using 0.5× TBE as transfer buffer. For detection of the longer nhaB mRNA, total RNA (3–5 μg) was mixed with 2× RNA loading dye containing EtBr (0.2 mg/ml) and separated on an 1% agarose gel, containing 1× MOPS and 18% Formaldehyde, for 3–4 h at 100 V in 1× MOPS as running buffer. RNA transfer to the membrane was carried out using a vacuum blotter (Vacugene XL, GE Healthcare) for 4 h at 90 mbar. During blotting the gel was covered first for 20 min with denaturation solution (50 mM NaOH, 10 mM NaCl), then 20 min with neutralization solution (100 mM Tris/HCl pH 7.4) and for the remaining time with 20× SSC. After blotting, the RNA was crosslinked to the membrane by exposure to 254 nm UV radiation. RNA probes were generated and labeled with Digoxigenin (DIG) by in vitro transcription using T7 RNA polymerase (NEB) and DIG RNA labeling mix (Roche). The templates for probe synthesis were obtained by PCR using primers BG230/BG231 for GlmZ, BG287/BG288 for rrfD (5S rRNA), BG1701/BG1702 for MicC, BG1492/BG1490 for RyhB, BG1496/BG1494 for CpxQ, BG1500/BG1498 for SroC, BG1727/BG1728 for ompD, BG1703/BG1704 for sodB, BG1799/BG1800 for nhaB and BG1613/BG1614 for GcvB. Cross-linked membranes were incubated with the respective probes (50–100 ng/ml) overnight at 68°C in hybridization buffer (50 ml Formamide, 25 ml 20× SSC, 20 ml 10% Blocking Reagent (Roche), 1 ml 10% N-laurylsarcosine, 200 μl 10% SDS, ad 100 ml with ddH20). On the next day, membranes were washed 2 × 5 min at room temperature (RT) in 2× SSC/0.1% SDS and 2 × 15 min at 68°C in 0.1× SSC/0.1% SDS. Afterwards, membranes were incubated for at least 30 min at RT in 1× Buffer I (0.1 M Malic acid, 0.15 M NaCl, pH 7.5) containing 1% Blocking Reagent (Roche). Then, an anti-DIG AP-conjugated antibody (1:10 000 diluted) was added for at least 30 min. Prior to detection, the membranes were washed 2 × 15 min in 1x Buffer I and for 3–5 min in Buffer III (0.1 M Tris, 0.1 M NaCl, pH 9.5). Signal detection was achieved in a ChemiDoc (BioRad) by using CDP* (Roche) as substrate.
Quantification of Northern blots
Signals of Northern blots from steady state and half-life experiments were quantified using the GelQuant software from BiochemLabSolutions (http://biochemlabsolutions.com/GelQuantNET.html). For quantification of RNA half-life experiments, RNA signal intensities were normalized to the corresponding 5S rRNA signals and plotted semi-logarithmically in percent against time. The resulting graphs present the average values of usually 2–5 independent experiments, as indicated in the figure legends. Half-life values are presented as mean ± standard deviation (SD). For steady state experiments, target RNA signal intensities, in presence of the different sRNA variants, were compared to target RNA signal intensities obtained in presence of the glmZ’-gfp fusion RNA (pYG215) on the same blot and normalized to the corresponding 5S or 23S rRNA signals. For comparison with target RNA signals in presence of the glmZ’-gfp fusion RNA, the average of all target signals in presence of glmZ’-gfp on the same membrane was used for calculations. Therefore, it was first excluded that the addition of arabinose and/or IPTG has a systematic effect on target RNA levels in presence of the glmZ’-gfp fusion RNA (Supplementary Figure S1). sRNA signal intensities were compared to the strongest sRNA signal on the same membrane followed by 5S rRNA normalization. The resulting bar graphs present the average normalized RNA signal intensities and SD from usually 2–7 independent experiments, as indicated in the individual figures.
RESULTS
An engineered two-plasmid system for generation of 5′ monophosphorylated sRNAs on demand in vivo
To generate 5′ monophosphorylated sRNAs in vivo, we adapted a tool based on the RNase E adaptor protein RapZ and sRNA GlmZ in E. coli (44). GlmZ is a 207 nt long sRNA, which activates translation of the glmS mRNA by base-pairing (50,51). GlmZ consists of three stem loop structures and a single-stranded region (nt 147–172) between stem loops 2 and 3. When not required, GlmZ is cleaved by RNase E in the single-stranded region comprising the base-pairing site (44,52). As a peculiarity, cleavage strictly depends on protein RapZ, which binds GlmZ and recruits RNase E for cleavage (27,44,53,54). Cleavage is independent of the 5′ end phosphorylation state of GlmZ and requires only the NTH of RNase E. Assisted by RapZ, RNase E cleaves 6 or 7 nt downstream of stem loop 2 regardless of the sequence following this structure. Consequently, it can be replaced with any sequence without affecting cleavage position. Thereby, the GlmZ aptamer can be used to release an RNA of interest with its regular 5′ end in a monophosphorylated state (44).
To trigger conditional cleavage of GlmZ fusion RNAs by RNase E, plasmid pYG205 was utilized, which carries the rapZ gene under control of the arabinose-inducible PAra promoter (Figure 1B). The sRNA gene of interest was located on a compatible plasmid and transcribed either autonomously (i.e. as 5′ triphosphorylated (5′PPP) primary transcript) or as fusion with the glmZ aptamer from the IPTG-inducible PLlacO-1 promoter (Figure 1A, constructs III and II). In the latter case, the sRNA genes including 4 nt of their upstream sequences were fused to the 3′ end of the first two stem loops of GlmZ (GlmZ nt 1–146; designated GlmZ’). This places the sRNA 7 nt downstream of GlmZ’ leading to its release with its regular 5′ end in a 5′ monophosphorylated state (5′P). Plasmid pYG215 served as negative control and as parental plasmid for the derivatives transcribing the various sRNA constructs (Figure 1A, construct I). It carries the gfpmut3* gene (encoding a Gfp variant) including its 5′ UTR fused to the 3′ end of GlmZ’. We previously demonstrated that expression of gfpmut3* originating from this fusion RNA is strictly regulated by RapZ (44). Presence of appropriate restriction sites allows for convenient substitution of the glmZ’-gfpmut3* sequence or only the gfpmut3* sequence with the sRNA sequence of choice (Figure 1A).
Thus, test strains were co-transformed with the sRNA plasmid of choice and plasmid pYG205 to allow for regulated cleavage of fusion RNAs. To evaluate the regulatory potential of the sRNAs, double transformants carrying plasmids pYG205/pYG215 served as negative controls. To avoid interference with our analysis, the chromosomal copies of rapZ, glmY, glmZ and the sRNA under investigation were deleted.
5′P-MicC is unstable in vivo and unable to destabilize its target ompD more efficiently than 5′PPP-MicC
In Salmonella, the first 12 nt of sRNA MicC pair with codons 23–26 of the ompD mRNA to accelerate its degradation (Figure 2A; (6)). RNase E cleaves ompD 4 or 5 residues downstream of the MicC-ompD duplex (Figure 2A), followed by rapid degradation of the 5′ monophosphorylated cleavage product (6). Cleavage of ompD by RNase E was reconstituted in vitro and observed to be faster in presence of 5′P-MicC as compared to 5′PPP-MicC (40). MicC and its primary target ompC are conserved in E. coli, but the ompD gene is absent (6,55). To enable its analysis in E. coli, we placed ompD under control of an operator-less Ptac promoter on the E. coli chromosome. The resulting strain transcribes the ∼1245 nt long ompD mRNA constitutively, allowing for its detection by Northern blotting (Figure 2B, lane 1).
Figure 2.

Comparison of the regulatory strength of 5′PPP- and 5′P-MicC towards ompD mRNA in vivo. (A) RNA duplex formed by MicC with the coding region of ompD (6). Previously mapped RNase E cleavage sites (6,40) are indicated with arrows. Residues U11/U12 of MicC were exchanged with cytidines to prevent cleavage by RNase E at position +9 (Supplementary Figure S3). (B) Northern blot addressing ompD steady state levels in presence of the GlmZ’-gfp control fusion, 5′PPP-MicC and 5′P-MicC, respectively. Strain Z1129 (ΔmicC ΔglmY ΔglmZ ΔrapZ λattB::Ptac-ompD, lane 1) carried either plasmid pYG215 (glmZ’-gfp, lanes 2, 3), pYG314 (micC, lanes 4–8) or pYG313 (glmZ’-micC, lanes 9–13). IPTG (10 μM, 50 μM, 100 μM or 1 mM) was added to induce expression of corresponding RNA constructs. Expression of rapZ from plasmid pYG205 was induced using 0.2% arabinose where indicated (+). MicC and its decay products are indicated with arrows. Total RNAs were isolated and 3 μg of each sample was separated on two denaturing PAA gels containing 5% and 8% acrylamide, respectively. After blotting, the membranes were consecutively hybridized with probes for ompD (first panel from top) or for MicC and GlmZ (third and fourth panel from top). Both membranes were re-probed for 5S rRNA to obtain loading controls. (C) Quantification of Northern blots. Bar graphs presenting the average normalized signal intensities and SD of 2–6 independent experiments. ompD mRNA signal intensities, in presence of MicC (light grey) and GlmZ’-MicC (dark grey), were compared to ompD signal intensities obtained in presence of GlmZ’-gfp (lanes 2 and 3; =100%) and normalized to the corresponding 5S rRNA loading controls (top graph). MicC signal intensities were compared to the strongest signal obtained for MicC (lane 8; = 100%) and normalized to the 5S rRNA loading controls (bottom graph).
Next, we tested the transformants transcribing the various RNA constructs from the PLlacO-1 promoter. Total RNAs were extracted and analyzed for steady state levels of ompD, GlmZ’, MicC and 5S rRNA transcripts. A strong accumulation of GlmZ’ was observable in the strain transcribing the glmZ’-gfp control fusion, but exclusively in the presence of both inducers, demonstrating cleavage of the fusion RNA by RapZ/RNase E (Figure 2B, lanes 2 and 3; Supplementary Figure S2, lanes 2–4). The levels of ompD mRNA remained unaffected by the glmZ’-gfp mRNA, as expected. The plasmid transcribing micC triggered strong accumulation of MicC upon full induction of the PLlacO-1 promoter with 1 mM IPTG (Figure 2B, lanes 4–8). Concomitantly, ompD levels were strongly reduced reflecting repression by MicC.
Full induction of glmZ’-micC transcription using 1 mM IPTG also reduced ompD levels, but to a lesser extent (Figure 2B, cf. lanes 8 and 13; Figure 2C, top graph). The GlmZ’ signal strongly increased with incremental IPTG concentrations, indicating successful cleavage of the majority of glmZ’-micC fusion RNAs. A slower migrating band likely referring to the uncleaved GlmZ’-MicC fusion RNA became only visible at highest IPTG concentrations and was of much weaker intensity (Figure 2B, lanes 9–13 in GlmZ blot). Concomitantly, 5′P-MicC became detectable but at much lower amounts when compared to 5′PPP-MicC (Figure 2B, MicC blot). Induction of RapZ synthesis successfully released the GlmZ’ aptamer from the fusion RNA as indicated by strong accumulation of GlmZ’ upon addition of arabinose. Interestingly, 5′P-MicC amounts increased only slightly concomitantly, with no additional effect on ompD (Supplementary Figure S2, lanes 7 and 8). This observation implies that most of the 5′P-MicC molecules that are additionally produced upon full induction of rapZ, become rapidly degraded. For better comparison we analyzed ompD repression under comparable steady state levels of the two MicC variants. Induction of 5′PPP-MicC with 0.05–0.1 mM IPTG and of 5′P-MicC with 1 mM IPTG produced steady state levels of mature full-length MicC sRNA that were comparable to each other (Figure 2B, cf. lanes 6 and 7 with lane 13; Figure 2C bottom graph). Under those conditions, the two sRNA variants achieved a comparable repression of ompD (Figure 2C, top graph).
To determine whether differences in steady state levels reflect RNA stabilities, we studied RNA turnover. Following rifampicin addition, total RNAs were extracted at various times and analyzed by northern blot (Figure 3; Table 1). A weak signal for 5′P-MicC was only detectable within the first 2 min, indicating rapid decay (t1/2 = 2 ± 0.8 min). In contrast, the initial levels of 5′PPP-MicC were much higher and the sRNA was slowly degraded over time (t1/2 > 16 min). As 5′P-MicC becomes rapidly degraded, it can be concluded that the signal referring to 5′-PPP MicC indeed represents mostly tri- or diphosphorylated MicC species. The initial ompD level observed at the time of rifampicin addition (t = 0 min) was higher and decay rates were minimally lower when 5′P-MicC was produced (t1/2 = 6.2 ± 1.9 min versus t1/2 = 5.5 ± 2.3 min in case of 5′PPP-MicC). In both cases, ompD levels decreased mainly within the first two minutes followed by a slower degradation kinetics. Taken together, the data show that both MicC variants achieve a comparable repression of ompD when compared at similar steady state levels. However, most of the produced 5′P-MicC molecules are instantly degraded, while 5′PPP-MicC is fairly stable, meaning that on a one-to-one basis, more 5′P-MicC than 5′PPP-MicC molecules must be produced to compensate for lower stability of the former and to achieve a comparable repression of ompD.
Figure 3.

Impact of 5′PPP- and 5′P-MicC on ompD mRNA decay rates and comparison of their stabilities. (A) Northern blot assessing ompD decay rates in presence 5′PPP-MicC and 5′P-MicC and determination of MicC stability. Transcription was stopped by addition of rifampicin (t0) and samples were removed at indicated times for RNA extraction and Northern blotting. Strain Z1129 (ΔmicC ΔglmY ΔglmZ ΔrapZ λattB::Ptac-ompD), containing plasmid pYG205 (rapZ), carried additionally either plasmid pYG314 (micC) or pYG313 (glmZ’-micC). Transcription of micC and glmZ’-micC was induced with 1 mM IPTG and 0.2% arabinose was added to trigger RapZ-mediated cleavage of the fusion RNAs. Isolated total RNAs (3 μg each) were separated on two denaturing PAA gels, containing 5% or 8% acrylamide, respectively. After blotting, the membranes were probed for ompD (first panel from top) or MicC (third panel from top) and GlmZ (fourth panel from top). Both membranes were re-probed for 5S rRNA to obtain loading controls. MicC and its decay products are indicated with arrows. (B) Semi-logarithmic plots of ompD and MicC signal intensities for half-life determination. The data are presented as mean, n = 4–5. Half-lives values are presented as mean ± SD.
Table 1.
Overview of RNA half-life measurements performed in this study. Half-life experiments were performed and quantified as described in Materials and Methods and in the corresponding figure legends. For glmZ’-gfp RNA control experiments one representative blot from each strain background is shown in Supplementary Figure S15
| Figure/strain | sRNA | RNA half-life (min) | Target RNA | RNA half-life (min) |
|---|---|---|---|---|
| Figure 3 | ||||
| E. coli (Z1129) | 5′PPP MicC | >16 | ompD | 5.5 ± 2.3 |
| 5′P MicC | 2 ± 0.8 | ompD | 6.2 ± 1.9 | |
| GlmZ’-gfp | ompD | 7.4 ± 1.0 | ||
| Figure S12 | ||||
| E. coli Δ rppH (Z1153) | 5′PPP MicC | >16 | ompD | 3.3 |
| 5′P MicC | 1.3 | ompD | 4.6 | |
| GlmZ’-gfp | ompD | 5.7 | ||
| Fig. S13 | ||||
| S. typhimurium rneR169K (Z1267) | 5′PPP MicC | 6.2 ± 1.6 | ompD | 3.4 ± 1.1 |
| GlmZ’-gfp | ompD | 11.2 ± 6.6 | ||
| S. typhimurium rne WT (Z1266) | 5′PPP MicC | 3.4 ± 0.6 | ompD | 5.6 ± 0.2 |
| GlmZ’-gfp | ompD | 6.3 ± 1.1 | ||
| Fig. S14 | ||||
| E. coli rne598 (Z1245) | 5′PPP MicC | >16 | ompD | 13.9 ± 1.5 |
| 5′P MicC | 4.4 ± 0.6 | ompD | >16 | |
| GlmZ’-gfp | ompD | >16 | ||
| Figure S5 | ||||
| E. coli (Z1013) | 5′PPP RyhB | 15.2 ± 2.1 | sodB | 8.4 ± 2.6 |
| 5′P RyhB | - | sodB | 8.9 ± 3.4 | |
| GlmZ’-gfp | sodB | >16 | ||
| Figure 6 | ||||
| E. coli (Z1012) | 5′�PPP CpxQ | >16 | nhaB | 2.4 ± 0.6 |
| 5′P CpxQ | >16 | nhaB | 2.6 ± 1.3 | |
| GlmZ’-gfp | nhaB | 3.8 ± 1.7 | ||
| Figure 7 | ||||
| E. coli (Z1014) | 5′PPP SroC | >16 | GcvB | 3.8 ± 1.3 |
| 5′P SroC | >16 | GcvB | 3.8 ± 0.9 | |
| GlmZ’-gfp | GcvB | >16 | ||
| Fig. S11 | ||||
| E. coli Δ rppH (Z1077) | 5′PPP SroC | >16 | GcvB | 4.7 ± 0.3 |
| 5′P SroC | >16 | GcvB | 6.5 ± 1.9 | |
| GlmZ’-gfp | GcvB | >16 |
The weak regulatory strength of 5′P-MicC is not caused by imprecise cleavage of the glmZ’-micC fusion RNA
The base-pairing site of MicC is provided by its immediate 5′ end (Figure 2A). Albeit RNase E was observed to cleave with high precision (44), it cannot be excluded per se that GlmZ’-MicC was cleaved at least partially a few nucleotides further downstream. Such imprecise cleavage would result in MicC variants lacking residues from the base-pairing site. This may not only affect binding of 5′P-MicC to ompD, but could also destabilize the sRNA as base-pairing provides protection (40). To address this possibility, we constructed two additional glmZ’-micC fusion genes, in which one or two guanine nucleotides were inserted upstream of the micC sequence (designated MicCG and MicCGG), thereby increasing the distance between glmZ’ and the first micC nucleotide to 8 and 9 residues, respectively. Release of the 5′P-MicCG and 5′P-MicCGG variants also repressed ompD only weakly, showing virtually no difference to the original 5′P-MicC construct (Supplementary Figure S2, cf. lanes 6–14). Inspection of the blots probed for GlmZ and MicC confirmed release of the 5′P-MicC variants in presence of IPTG and arabinose. Thus, the weak regulatory impact of 5′P-MicC cannot be explained by imprecise cleavage of the parental fusion RNA.
Rapid elimination of 5′P-MicC through cleavage by RNase E explains its weak regulatory activity
The levels of the MicC variants generated through processing were much lower as compared to 5′PPP-MicC, indicating that they are unstable (Supplementary Figure S2). Northern blotting detected two shorter species in addition to full-length MicC. Whereas the full-length species is predominant in case of 5′PPP-MicC, the slightly shorter species is equally or more abundant in case of the 5′P-MicC variants (Figure 2B, cf. lane 8 with 13; Supplementary Figure S2, cf. lane 5 with 8, 11, 14). Previous work has shown that 5′P-MicC is rapidly degraded by RNase E in vitro, whereas the 5′PPP-MicC variant is largely protected (40). RNase E cleavage sites in MicC were mapped to positions +9 and +24, the former of which is preferred in 5′P-MicC and cleavage at this site likely initiates decay. To learn whether processing at position +9 initiates decay of 5′P-MicC in vivo, we exchanged the uridines at position +11 and +12 with cytidine residues (Figure 2A). These mutations should eliminate cleavage at position +9. However, they also abolish base-pairing of position +11 in MicC.
Accordingly, we compared steady state levels of wild-type (wt) MicC and mutated MicC (designated MicCmut), produced either as 5′PPP or as 5′P variants (Supplementary Figure S3). Indeed, the slightly shorter MicC species (designated MicC−9) disappeared in case of MicCmut, regardless whether produced in 5′PPP or in 5′P state (Supplementary Figure S3, cf. lanes 4, 5 with 6, 7). Simultaneously, the levels of the full-length sRNAs increased, which was in particular pronounced for the 5′P variant. Nonetheless, its amount remained below the level of 5′PPP-MicC. This is likely the result of cleavage at position + 24, reflecting that a corresponding species also accumulated when 5′P-MicC was produced (Supplementary Figure S3, cf. lanes 5, 7 with 4, 6). These data indicate that following production in vivo, 5′P-MicC is rapidly cleaved at positions +9 and +24 and degraded. This high intrinsic instability explains its weak regulatory strength as compared to 5′PPP-MicC.
5′P-RyhB is highly unstable in vivo and instantly turned over contrarily to 5′PPP-RyhB
RyhB is a post-transcriptional regulator of >50 mRNAs involved in iron metabolism and related functions (56). One of the best characterized targets is sodB mRNA in E. coli, which is regulated by RyhB via two mechanisms. Assisted by Hfq, RyhB binds to the translation initiation region (TIR) of sodB and represses translation (Figure 4A; (57,58)). Additionally, RyhB recruits the RNA degradosome to stimulate degradation of sodB by RNase E, which cleaves 350 nt downstream of the base-pairing site (7,8,34).
Figure 4.

Analysis of sodB mRNA repression caused by 5′PPP- and 5′P-RyhB. (A) RNA duplex formed by RyhB with the sodB start region (58). RNase E cleavage of sodB mRNA downstream in the coding region is indicated. (B) Northern blot experiment addressing sodB mRNA steady state levels in presence of the GlmZ’-gfp control fusion, 5′PPP-RyhB and 5′P-RyhB, respectively. Strain Z1013 (ΔryhB ΔglmY ΔglmZ ΔrapZ) containing plasmid pYG205 (rapZ), carried additionally either plasmid pYG215 (glmZ’-gfp, lane 5, 6), pYG275 (ryhB, lanes 7, 8) or plasmid pYG274 (glmZ’-ryhB, lanes 9, 10). IPTG (1 mM) was added to induce expression of corresponding RNA constructs, while expression of rapZ from pYG205 was induced with 0.2% arabinose where indicated (+). The empty ancestor strains S4197 (wild-type), Z956 (ΔglmY ΔglmZ ΔrapZ), Z1013 and JW1648 (ΔsodB) were included as additional controls (lanes 1–4). Total RNAs were isolated and 1.5 μg each were separated on two denaturing 8% PAA gels. After blotting, the membranes were either hybridized with probes detecting sodB and GlmZ (first and second panel from top), or probed for RyhB (fourth panel from top). Blots were re-probed for 5S rRNA to obtain loading controls. (C) Quantification of Northern blots. Bar graphs presenting the average normalized signal intensities and SD of two independent experiments. sodB mRNA signal intensities, in presence of RyhB (light grey) and GlmZ’-RyhB (dark grey), were compared to sodB signal intensity obtained in presence of GlmZ’-gfp (lane 5 and 6; =100%) and normalized to the corresponding 5S rRNA loading controls.
Production of RyhB in 5′PPP state led to accumulation of the sRNA and decreased sodB mRNA levels concomitantly, compared to the glmZ’-gfp control RNA (Figure 4B, cf. lanes 7, 8 with 5, 6; Figure 4C), as expected. Surprisingly, 5′P-RyhB remained undetectable upon induction of transcription and cleavage of the glmZ’-ryhB fusion RNA, albeit the GlmZ’ 5′ cleavage product readily accumulated reflecting successful cleavage (Figure 4B, lanes 9 and 10). Across several experiments, we reiteratively observed decreased sodB levels although 5′P-RyhB itself became never detectable (Figure 4; Supplementary Figure S4, lanes 6, 7; Supplementary Figure S5; data not shown). Next, we studied sodB and RyhB decay (Supplementary Figure S5; Table 1). Somewhat lower sodB mRNA levels were detectable at the time of rifampicin addition when RyhB was produced in 5′PPP state as compared to 5′P-RyhB, reflecting a somewhat stronger repression in the former strain. Over time, sodB levels decreased with comparable rates in presence of 5′PPP-RyhB (t1/2 = 8.4 ± 2.6 min) or when releasing 5′P-RyhB from the glmZ’-ryhB precursor RNA (t1/2 = 8.9 ± 3.4 min). 5′P-RyhB remained undetectable, whereas 5′PPP-RyhB decayed only slowly (t1/2 = 15.2 ± 2.1 min). Consequently, RyhB must be 5′ tri- (or di-) phosphorylated in the latter case (Supplementary Figure S5; Figure 4B, lanes 7 and 8). We conclude that 5′P-RyhB is highly unstable and instantly turned over either together with its target (34), when not base-paired or via a so far unknown mechanism. However, our data indicate that both RyhB variants repress sodB mRNA with comparable efficiency.
Small RNAs carrying 5′P ends naturally are stable but do not perform better in target RNA destabilization than their 5′PPP variants
Additionally, we tested sRNAs, whose biogenesis involves processing from parental transcripts catalyzed by RNase E (13,59). Therefore, these sRNAs naturally carry a 5′P group. A well-studied example is sRNA CpxQ, which is produced from the 3 ′UTR of the cpxP mRNA (43). In Salmonella, CpxQ targets mRNA nhaB via two base-pairing sites. Base-pairing with the site located at the TIR inhibits ribosome binding. Interaction with a second site located ∼300 nt downstream in the CDS, stimulates nhaB decay by RNase E in vitro. Again, 5′P-CpxQ stimulates faster nhaB decay as compared to 5′PPP-CpxQ in such assays, emphasizing a stimulatory role of the 5′P group (43).
The CpxQ base-pairing site at the TIR is not conserved, but the base-pairing site in the CDS exists also in E. coli, except that the nhaB A312/U7 CpxQ base-pairing in Salmonella is replaced with a G:U pairing (Figure 5A). Consequently, we studied regulation of the 1542 nt long (Supplementary Figure S6) endogenous nhaB RNA by CpxQ in E. coli using the two-plasmid system. Addition of increasing amounts of IPTG elicited accumulation of both CpxQ variants and downregulated nhaB mRNA concomitantly, confirming that regulation is conserved (Figure 5B). 5′PPP- and 5′P-CpxQ repressed nhaB with similar efficiency. For a thorough comparison, precise CpxQ levels were considered. According to signal quantifications, induction of the 5′PPP-CpxQ construct with 0.01 – 0.05 mM IPTG generated sRNA levels that were comparable to those produced by the 5′P-CpxQ construct when induced with 1 mM IPTG (Figure 5B, lanes 5 and 6 and 13; Figure 5C, bottom graph). Under these conditions, the two sRNA variants achieved a comparable repression of nhaB mRNA (Figure 5C, top graph).
Figure 5.

Effect of different CpxQ variants on nhaB mRNA regulation. (A) RNA duplex likely formed by CpxQ with the nhaB CDS in E. coli as deduced from the base-pairing occurring in Salmonella (43). (B) Northern blot experiment addressing nhaB steady state levels in presence of the GlmZ’-gfp control fusion, 5′PPP-CpxQ and 5′P-CpxQ, respectively. Strain Z1012 (ΔcpxQ ΔglmY ΔglmZ ΔrapZ), containing plasmid pYG205, carried additionally either plasmid pYG215 (glmZ’-gfp, lanes 2, 3), pYG273 (cpxQ, lanes 4–8) or plasmid pYG272 (glmZ’-cpxQ, lanes 9–13). Strain JW1175 (ΔnhaB, lane 1) shows nhaB probe specificity. IPTG (10 μM, 50 μM, 100 μM or 1 mM) was added to induce expression of corresponding RNA constructs. Expression of rapZ from pYG205 was induced using 0.2% arabinose where indicated (+). To detect nhaB mRNA (first panel from top), 3.5 μg of total RNA was separated on an 1% agarose gel and subsequently transferred to the membrane via vacuum blotting. EtBr stained 23S and 16S rRNAs were imaged after blotting serving as loading controls (second panel from top). For detection of CpxQ (third panel from top) and GlmZ (fifth panel from top), 3 μg of total RNA was separated on two denaturing PAA gels containing 8% or 5% acrylamide. The RNA was transferred to the membranes via semi-dry electroblotting. After hybridization with either probes detecting CpxQ or GlmZ, both membranes were re-probed for 5S rRNA. (C) Quantification of Northern blots. Bar graphs presenting the average normalized signal intensities and SD of 3–7 independent experiments. nhaB mRNA signal intensities, in presence of CpxQ (light grey) and GlmZ’-CpxQ (dark grey), were compared to nhaB signal intensities obtained in presence of GlmZ’-gfp (lanes 2, 3; = 100%) and normalized to the corresponding 23S rRNA loading controls (top graph). CpxQ signal intensities were compared to the strongest signal obtained for CpxQ (lane 8; = 100%) and normalized to the 5S rRNA loading controls (bottom graph).
Closer inspection of the blot indicated somewhat different migration velocities for 5′P-CpxQ and 5′PPP-CpxQ. Side by side comparison with endogenously produced CpxQ revealed a slightly increased length for the latter variant (Supplementary Figure S7), suggesting that transcription starts few nucleotides upstream to avoid usage of U as start nucleotide. In any case, this CpxQ variant is fully active in regulation.
RNA half-life determinations showed that decay of nhaB mRNA in presence of both CpxQ variants was comparable (glmZ’-cpxQ: t1/2nhaB = 2.6 ± 1.3 min; cpxQ: t1/2nhaB = 2.4 ± 0.6 min) (Figure 6; Table 1). Interestingly, both CpxQ variants were fairly stable (t1/2 > 16 min) indicating that the 5′ phosphorylation state has no impact on CpxQ decay.
Figure 6.

Impact of 5′PPP- and 5′P-CpxQ on nhaB mRNA decay rates and comparison of their stabilities. (A) Northern blot assessing nhaB decay rates in presence 5′PPP-CpxQ and 5′P-CpxQ and determination of CpxQ stability. Transcription was stopped with rifampicin (t0) and samples were harvested at indicated times for RNA extraction and Northern blotting. Strain Z1012 (ΔcpxQ ΔglmY ΔglmZ ΔrapZ) carried plasmid pYG205 (rapZ) and additionally either plasmid pYG273 (cpxQ) or plasmid pYG272 (glmZ’-cpxQ). The transformants were grown in presence of 1 mM IPTG to induce transcription of cpxQ and the glmZ’ fusion genes. Arabinose (0.2%) was additionally added to trigger cleavage of the fusion RNAs. To detect nhaB mRNA (first panel from top), 3.5 μg of total RNA was separated on an 1% agarose gel and subsequently transferred to the membrane via vacuum blotting. EtBr stained 23S and 16S rRNAs were imaged after blotting serving as loading controls (second panel from top). For detection of CpxQ (fifth panel from top) and GlmZ (third panel from top), 3 μg of total RNA was separated on two denaturing PAA gels containing 8% and 5% acrylamide. The RNA was transferred to the membranes via semi-dry electroblotting. After hybridization with either probes detecting CpxQ or GlmZ, both membranes were re-probed for 5S rRNA. (B) Semi-logarithmic plots of nhaB and CpxQ signal intensities for half-life determination. In the semi-logarithmic plots data are presented as mean, n = 2–4. Half-lives values are presented as mean ± SD.
The ∼160 nt long sRNA SroC represents another naturally 5′ monophosphorylated sRNA that is generated by RNase E through processing of the gltI mRNA (60). SroC uses two short base-pairing regions to sponge sRNA GcvB and induce its degradation by RNase E (Figure 7A). We triggered production of both, 5′PPP-SroC and 5′P-SroC by using incremental IPTG concentrations and followed the fate of GcvB (Supplementary Figure S8). Interestingly, both SroC variants were detectable in the Northern blot in absence of IPTG and their levels increased further upon IPTG addition. This may be explained by the exceptional stability of SroC (t1/2 > 32 min, (61)), allowing SroC to accumulate even when produced at low levels as resulting from promoter leakiness in absence of the inducers. Consequently, GcvB levels decreased in the absence of inducers and declined further with incremental IPTG concentrations. Importantly, the two SroC variants exhibited no major differences regarding repression of GcvB. Determination of sRNA decay rates confirmed this result (glmZ’-sroC: t1/2 GcvB = 3.8 ± 0.9 min; sroC: t1/2 GcvB = 3.8 ± 1.3 min) (Figure 7; Table 1). SroC itself remained stable over time, regardless of its 5′ end phosphorylation state (t1/2 > 16 min).
Figure 7.

GcvB decay rates triggered by 5′PPP- and 5′P-SroC and determination of SroC stability. (A) SroC-GcvB base-pairing as shown in Miyakoshi et al. 2015 (60). Base-pairing occurs at two regions that are in close proximity within SroC, but 137 nt apart in GcvB. (B) Northern blot for determination of GcvB decay rates in presence of 5′PPP- SroC and 5′P-SroC and analysis of SroC stability. Rifampicin was added to stop transcription (t0) and samples were removed at indicated times for RNA extraction and Northern blotting. Strain Z1014 (ΔsroC ΔglmY ΔglmZ ΔrapZ) carried plasmid pYG205 (rapZ) and additionally either plasmid pYG277 (sroC) or plasmid pYG276 (glmZ’-sroC). The empty strains S4197 (wild-type), Z1014 and JVS06081 (ΔgcvB) were included as controls (lanes 1–3). The transformants were grown in presence of 0.01 mM IPTG to induce transcription of sroC and glmZ’ fusion genes. Arabinose (0.2%) was added to trigger RapZ-mediated cleavage of the fusion RNAs. Total RNAs were separated on two denaturing 8% PAA gels (1.5 μg each) and subsequently blotted. One blot was probed for GcvB and SroC (first and second panel from top), while the other blot was probed for GlmZ (fourth panel from top). Blots were re-probed for 5S rRNA to obtain loading controls. (C) Semi-logarithmic plots of GcvB and SroC signal intensities for half-life determination. The data are presented as mean, n = 3. Half-lives values are presented as mean ± SD.
These results show that the naturally 5′ monophosphorylated sRNAs CpxQ and SroC retain their regulatory potential when produced in 5′PPP form. Interestingly, the 5′ phosphorylation state has no role for CpxQ and SroC stabilities, which is in remarkable contrast to sRNAs MicC and RyhB.
Pyrophosphohydrolase RppH has little impact on the regulatory performance 5′PPP-sRNAs
Our results could not detect any major difference in target repression strength exerted by 5′ monophosphorylated sRNAs or their 5′ triphosphorylated counterparts, at least when compared at similar steady state levels. However, it cannot be ruled out completely that the 5′PPP-sRNA species undergo rapid dephosphorylation leading to a fraction of 5′ monophosphorylated species that was actually responsible for the observed regulation. It is also conceivable that dephosphorylation could take place after the annealing step. The stable 5′PPP-sRNA variants might be able to bind their targets, which could then be followed by their dephosphorylation, allowing the sRNA to boost RNase E activity.
To address these possibilities, we performed experiments in a ΔrppH strain background. The results did not deviate significantly from those obtained in the rppH+ strains. Again, 5′P-CpxQ and 5′PPP-CpxQ repressed nhaB with similar efficiency when assessed at comparable steady state levels (Supplementary Figure S9, cf. conditions shown in lanes 7 and 12). Likewise, both SroC variants reduced GcvB levels to a comparable extent when transcription was induced using ≤ 0.01 mM IPTG (Supplementary Figure S10, lanes 4–13). Surprisingly, repression of GcvB by 5′P-SroC was relieved again when using higher IPTG concentrations for induction of glmZ’-sroC transcription (Supplementary Figure S10, lanes 20–23). This phenomenon was also observable in the rppH+ strain, albeit less pronounced (i.e. only at 1 mM IPTG; Supplementary Figure S8, lane 22 and 23). Concomitantly, an exceptional strong accumulation of the parental GlmZ’-SroC RNA could also be observed. We assume that the high GlmZ’-SroC levels (promoted though the notorious stability of SroC) may sequester RNase E, thereby limiting degradation of GcvB. Absence of RppH might exacerbate limitation of RNase E as it may prolong its retention time on 5′ triphosphorylated substrates, i.e. the latter could be bound by RNase E but their cleavage is impaired due to absence of 5′P groups (62). Therefore, we used only 0.01 mM IPTG for sRNA induction when studying GcvB decay rates (Figure 7; Supplementary Figures S11; Table 1). Under these conditions, both SroC variants destabilized GcvB with similar efficiency. Finally, also the 5′PPP-MicC variant retained the ability to promote degradation of ompD in the ΔrppH background (Supplementary Figure S12; Table 1). Collectively, these results are not in agreement with the hypothesis that a 5′ monophosphorylated fraction of the triphosphorylated sRNAs was responsible for the observed regulatory effects.
MicC does not require the 5′P sensing pocket of RNase E to promote ompD decay
Our experiments, using the GlmZ aptamer, did not yield any evidence that a 5′P group would allow sRNAs to boost RNase E activity towards their targets. For further insight, we explored regulation of ompD mRNA by MicC in a Salmonella strain carrying the rneR169K allele encoding a variant of RNase E that is defective in 5′P sensing (40,60). In vitro, the R169K substitution reduces the ability of RNase E to cleave ompD in a MicC-guided manner >5-fold (28). The chromosomal micC copy was deleted in this strain, allowing us to study the effect of plasmid-encoded 5′PPP-MicC, independent of endogenous transcriptional control mechanisms. The bacteria were cultivated in absence or presence of various IPTG concentrations for induction of micC transcription, allowing for thorough comparison.
The 5′P sensing defect of the Salmonella rneR169K mutant was evident from the accumulation of the 5S rRNA precursor as reported previously (Figure 8A; (60)). Quantification of ompD signal intensities over multiple replicates and 5S loading control normalization showed that MicC repressed ompD with comparable or even somewhat improved efficiency when RNase E was defect in 5′P sensing (Figure 8A and B). Interestingly, MicC accumulated to much higher levels in the rneR169K mutant as compared to the wt rne strain (Figure 8A, cf. lane 6 with 12; Figure 8B, bottom graph). At lower IPTG concentrations, only the MicC variant cleaved at position + 9 was detectable in the wt rne strain and this variant likewise accumulated in the rneR169K strain (Figure 8A, cf. lanes 4–6 and 10–12). Additional longer transcripts arising above the MicC signal in the rneR169K mutant presumably represent read-through transcripts from upstream located start sites, which might undergo rapid degradation via the RNase E 5′P pathway in the wt rne strain.
Figure 8.

MicC does not require the 5′P sensory pocket of RNase E for downregulation of ompD in vivo. (A) Northern blot experiment addressing ompD steady state levels in presence of MicC in Salmonella Typhimurium strains Z1266 (wild-type rne, ΔmicC) and Z1267 (rneR169K, ΔmicC), respectively. The strains carried either plasmid pYG215 (glmZ’-gfp, lanes 1, 2, 7, 8) or plasmid pYG314 (micC, lanes 3–6 and 9–12). Transformants were grown in absence or presence of IPTG, 0.05, 0.1 and 1 mM IPTG were used in case of micC and 1 mM in case of glmZ’-gfp, to induce transcription of RNA genes. Total RNAs (1.5 μg each) were separated on two denaturing PAA gels, containing 5% or 8% acrylamide, and were subsequently blotted. The blot resulting from the 5% PAA gel was hybridized with a probe detecting ompD (first panel from top), while the other blot was probed for MicC (third panel from top). Membranes were re-probed for 5S rRNA to obtain loading controls. Full-length (FL) MicC and its decay products are indicated with arrows. (B) Quantification of Northern blots. Bar graphs presenting the average normalized signal intensities and SD of 2–6 independent experiments. ompD mRNA signal intensities, in presence of wild-type (WT) rne (light grey) and rneR169K (dark grey), were compared to ompD signal intensities obtained in presence of GlmZ’-gfp in the respective strain background (lanes 1, 2 or 7, 8; = 100%) and normalized to the corresponding 5S rRNA loading controls (top graph). MicC signal intensities, in presence of wild-type (WT) rne (light grey) and rneR169K (dark grey) were compared to the strongest signal obtained for MicC (lane 12; = 100%) and normalized to the 5S rRNA loading controls (bottom graph). In the rneR169K strain, signal intensities of the 5S signal and the 5S rRNA precursor were summed up for normalization.
Examination of RNA decay indicated a somewhat faster degradation of ompD in the rneR169K mutant as in the wt rne strain (rneR169K: t1/2ompD = 3.4 ± 1.1 min, wt rne: t1/2ompD = 5.6 ± 0.2 min; Supplementary Figure S13; Table 1), whereas the opposite was the case for MicC. Both, full-length MicC and its shorter MicC-9 variant were stabilized in the rneR169K strain. These data suggest that degradation of MicC occurs at least partially via the RNase E 5′P pathway. Most importantly, MicC efficiently promotes degradation of ompD even in the rneR169K mutant, indicating that the 5′P sensory pocket in RNase E plays no major role for this process.
The C-terminal half of RNase E is indispensable for regulation of ompD by MicC
An alternative mechanism was proposed, in which sRNAs employ Hfq to recruit RNase E in order to stimulate target decay (7,31,39). In light of our finding that the 5′P group of MicC and the corresponding binding pocket in RNase E play no direct role for destabilization of ompD, we wondered whether the RNase E-CTH might be critical instead. To this end, we tested regulation of ompD by MicC in an E. coli strain carrying the RNase1-598 variant lacking the CTH. Once again, we introduced the two plasmids allowing us to produce MicC in 5′PPP state or with a 5′P group via processing from the GlmZ’-MicC precursor. For comparison, we included corresponding transformants of the isogenic strain carrying full-length rne. As observed before (Figure 2), full induction of 5′PPP-MicC caused strong repression of ompD in the latter strain, whereas repression by 5′P-MicC was weaker (Figure 9A, lanes 1–7; Figure 9B). Strikingly, regulation of ompD was strongly relieved in the strain synthesizing the truncated RNase E1–598 variant (Figure 9A, lanes 8–14; Figure 9B). Both MicC variants largely failed to stimulate ompD decay in this strain (Supplementary Figure S14; Table 1). We conclude that the CTH of RNase E is essential for destabilization of ompD by 5′P-MicC or 5′PPP-MicC.
Figure 9.
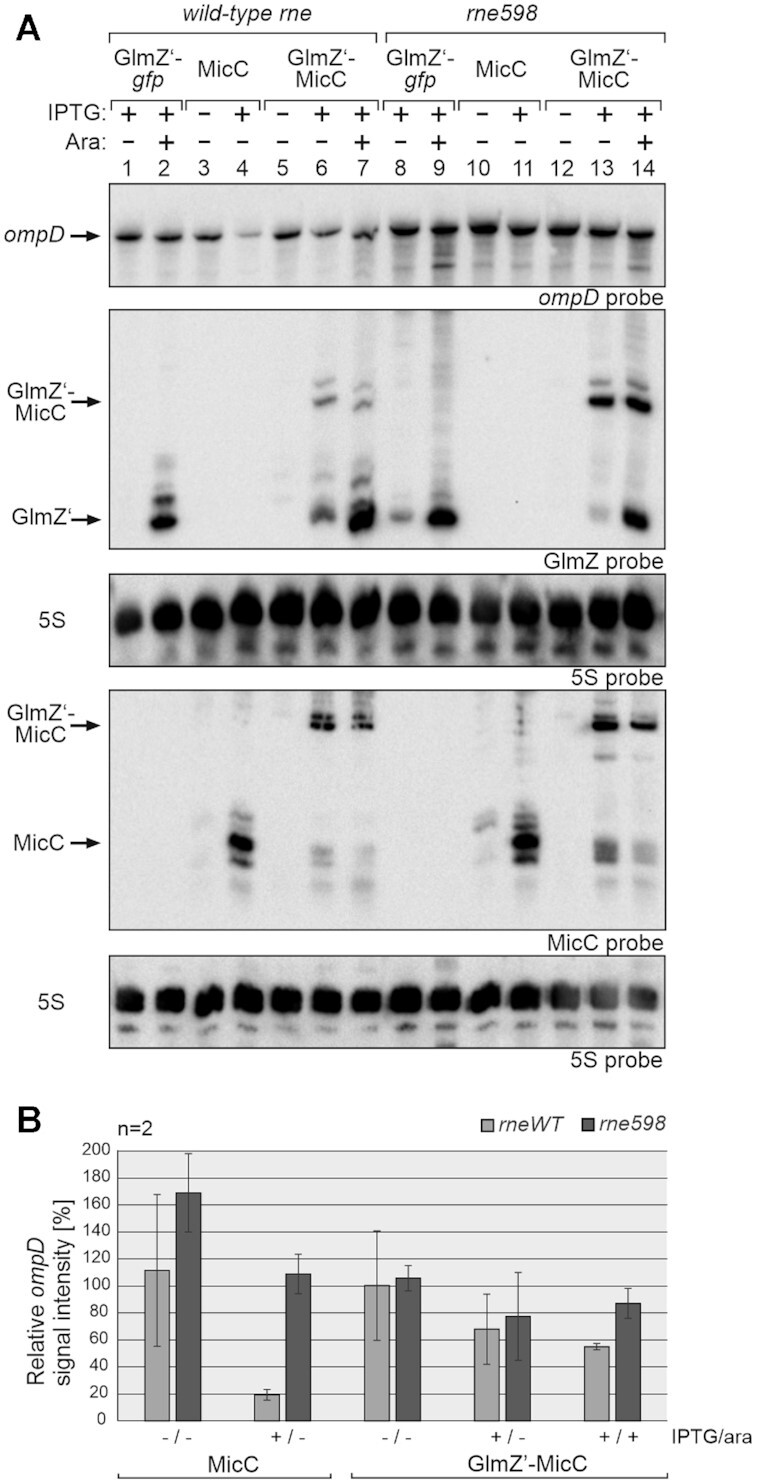
The C-terminal half of RNase E is essential for regulation of ompD by MicC. (A) Northern blot experiment comparing regulation of ompD mRNA by MicC in strains expressing wild-type rne (Z1129, lanes 1–7) or the truncated rne598 variant (Z1245, lanes 8–14). The strains carried plasmid pYG205 (rapZ) and additionally either control plasmid pYG215 (glmZ’-gfp, lanes 1, 2, 8, 9), pYG314 (micC, lanes 3, 4, 10, 11) or pYG313 (glmZ’-micC, lanes 5–7, 12–14). 1 mM IPTG and 0.2% arabinose were added as indicated. Total RNAs were extracted and separated (1.5 μg each) on two denaturing PAA gels containing 5% or 8% acrylamide, respectively, and subsequently blotted. The blot resulting from the 5% PAA gel was consecutively hybridized with probes detecting ompD and GlmZ (first and second panel from top), while the other blot was probed for MicC (fourth panel from top). Membranes were re-probed for 5S rRNA to obtain loading controls. (B) Quantification of Northern blots. Bar graphs presenting the average normalized signal intensities and SD of 2 independent experiments. ompD mRNA signal intensities, in wild-type (WT) rne (light grey) and rne598 (dark grey) strain background, in presence of either MicC or GlmZ’-MicC, were compared to ompD signal intensities obtained in presence of GlmZ’-gfp (lanes 1, 2 and 8, 9; =100%) in the respective strains and normalized to the corresponding 5S rRNA loading controls.
DISCUSSION
In this work, we examined the impact of 5′P ends on the stability and regulatory performance of sRNAs in the E. coli cell. This was possible through usage of the GlmZ aptamer, which together with protein RapZ allows the controllable release of 5′P sRNAs from precursor transcripts in vivo (Figure 1; (44)). Generated 5′P sRNAs were directly compared to their 5′PPP counterparts, which were expressed to comparable levels from isogenic plasmids. The results do not indicate a major role of the 5′P group of sRNAs for stimulation of target RNA decay by RNase E, as previously proposed (40,43). In contrast, they emphasize the importance of the RNase E-CTH (7,31), as it is essential for the regulatory activity of MicC sRNA towards ompD mRNA. Our results also show that a 5′P end marks MicC and RyhB for rapid degradation, either in a coupled process with their target or when not base-paired (Figure 10). In contrast, this does not apply to sRNAs, which are generated through processing (i.e. CpxQ and SroC), indicating that they are protected. In this respect, GlmZ/RapZ provides a suitable tool to determine whether a particular transcript is degraded via the RNase E 5′P-dependent pathway in vivo.
Figure 10.

Model depicting decay pathways and regulatory activities of 5′P- and 5′PPP- sRNA variants as revealed in this study. (A) 5′PPP-MicC is highly stable and regulatory active. It can base pair with its target mRNA ompD and induce RNase E-mediated degradation. These variants may act catalytically until they become dephosphorylated and degraded. In contrast, 5′P-MicC is highly unstable and its major fraction is rapidly degraded before base-pairing can occur (40). Only a minor fraction might be able to find its target to induce mRNA decay (grey dotted arrow). Consequently, MicC displays only a weak regulatory activity in vivo when monophosphorylated. (B) Like MicC, the 5′PPP variant of RyhB is perfectly stable and regulatory active. After base-pairing, RyhB undergoes coupled degradation with its targets (34). The 5′P variant of RyhB is extremely unstable. However, a major fraction of 5′P-RyhB might be able to find its targets and undergoes (coupled) degradation. A minor fraction might be degraded rapidly before base-pairing can occur (grey dotted arrow). (C) sRNAs CpxQ and SroC are produced by RNase E-catalyzed processing from the 3′ UTRs of mRNAs and therefore equipped with 5′P groups. Nonetheless, both sRNAs escape the 5′P-dependent decay pathway of RNase E and are exceptionally stable, regardless of their 5′ phosphorylation state, suggesting that they act in a catalytic manner on their targets. A 5′P for stimulation of target destabilization by RNase E is not required, at least not for sRNAs CpxQ and SroC. A stimulatory effect of the 5′P variants of MicC and RyhB was not observed, but cannot be excluded entirely. sRNA-mediated stimulation of target degradation by RNase E is indicated by a yellow flash.
A major group of Hfq-binding sRNAs are released from the 3′ UTRs of mRNAs through processing (43,60,63–68). This biogenesis pathway equips sRNAs with 5′P groups, which could be used to destabilize cognate targets through interaction with the 5′ monophosphate sensing pocket of RNase E (13,41,43). However, we observe that the 5′ phosphorylation state has no major role for the regulatory activities of CpxQ and SroC. Both, 5′P and 5′PPP variants of these sRNAs regulate their targets with comparable efficiency (Figures 5–7; Supplementary Figure S8). The same effect was observed in absence of the pyrophosphohydrolase RppH (Supplementary Figures S9–S11), which could dephosphorylate the initially 5′PPP sRNAs shifting their regulatory potential. Our findings for CpxQ/nhaB are reinforced by observations made recently for the CpxQ/cfa mRNA pair. Similar to nhaB, CpxQ destabilizes cfa through recruitment of RNase E, but apparently with little impact of its 5′ end phosphorylation state (69). It appears conceivable that 5′ end phosphorylation of CpxQ is irrelevant for target RNA destabilization in general, extending the concept beyond the target studied herein. Interestingly, both CpxQ and SroC are exceptionally stable, regardless of their 5′ phosphorylation state (t1/2 >16 min; Figures 6 and 7). Features such as elimination of potential RNase E cleavage sites or obstacles hindering substrate scanning by RNase E (70), may prevent CpxQ and SroC from entering the 5′P-dependent decay pathway. However, the underlying characteristics that determine the molecular stability of these sRNAs remain to be disassembled.
sRNAs MicC and RyhB are stable when tri- or (di-) phosphorylated (Figure 3; Supplementary Figure S5). However, when produced in monophosphorylated form, MicC was rapidly degraded and RyhB was not detectable at all, indicating that they are eliminated through the 5′P-dependent decay pathway of RNase E. Mutational analysis of MicC indicates that RNase E cleavage at position + 9 initiates this process (Supplementary Figure S3), which is in agreement with previous in vitro analysis (40). Instability of 5′P-MicC relieved target repression compared to 5′PPP-MicC, but under conditions where the steady state levels of both sRNA variants were comparable, they regulated ompD with similar efficiency (Figures 2).
MicC is degraded when not protected through base-pairing with its targets (40). Therefore, most of the 5′P-MicC molecules produced from the GlmZ’-MicC aptamer were likely removed by RNase E before base-pairing could occur (Figure 10). The small fraction of 5′P-MicC becoming detectable, may represent those molecules that escaped degradation via base-pairing (Figure 2; Supplementary Figures S2 and S3). As MicC has a small target spectrum, the number of protecting molecules is limited, which may explain why the monophosphorylated fraction could not be increased above a certain threshold (Supplementary Figure 2, cf. lanes 7 and 8). In agreement with this observation, 5′P-MicC levels could be elevated by producing additional ompD molecules (data not shown). Rapid elimination of free 5′P-MicC via the RNase E 5′P sensing pathway is also supported by the accumulation of MicC in the presence of the RNase E-R169K variant affected in 5′ sensing (Figure 8). Clearly, an intact RNase E 5′P sensing pocket is required to destabilize MicC, but not ompD.
In contrast to MicC, RyhB was shown to be degraded in a coupled process together with its targets (34). We observed reiteratively in all experiments that both RyhB variants repressed sodB mRNA with similar efficiency (Figure 4; Supplementary Figure S4 and S5). Opposite to 5′P-MicC, the major fraction of 5′P-RyhB might be able to find and bind its targets, but is then eliminated through co-degradation. However, since monophosphorylated RyhB could not be detected at all and the amounts of ‘free- and co-degraded’ 5′P-RyhB are not known, the regulatory potential of the RyhB variants towards sodB mRNA may differ potentially (Figure 10). RyhBs broad target spectrum and other regulatory elements, e.g. the AspX sRNA sponge repressing RyhB that was recently identified, 3′ETSLeuZ or poly (A) polymerase (PAP I) (71,72), might add additional layers of complexity to this in vivo analysis. Moreover, PNPase, which is part of the RNA degradosome, can affect stability and activity of some sRNAs including RyhB (73,74). Furthermore, it was shown that, besides RNase E, RNase III is another player controlling RyhB induced degradation of sodB mRNA (75). Altered secondary structures of the sRNA variants could potentially impact target RNA binding and interaction with the RNA degradosome. Finally, it cannot be excluded that one RyhB variant might primarily cause translational block of sodB, while the other variant actively induces sodB decay far downstream of the ribosome binding site by recruiting RNase E (8). Overall, 5′P-RyhBs detailed mode of action is still ambiguous and remains to be elucidated.
In light of our findings, previous in vitro results, suggesting a stimulatory role of the sRNA 5′P group for target RNA decay (40,43), appear debatable. Presumably, these in vitro assays are not yet suited to recapitulate all facets of the complexity of sRNA function in the cell. 5′P variants of sRNAs like MicC are too short-lived to achieve efficient regulation (Figure 3). This obstacle is not reflected by in vitro assays, in which base-pairing of the RNAs is allowed, providing sRNA protection, before RNase E is added in the next step (28,40,43). Often, only fragments of the target RNAs are used in vitro (40,43), which could bind differently to RNase E or lack secondary structures important for RNase E activity (28,70,76). It appears also unclear whether the base-paired sRNA is identical with the molecule interacting with the RNase E 5′ sensing pocket in such in vitro assays. Activation by an additional 5′P-RNA molecule appears also possible. However, several limitations apply to our own study. We performed our experiments in the absence of conditions and stimuli known to naturally activate the studied sRNAs such as iron limitation or envelope stress. Consequently, our results do not account for the possible impact of co-regulated RNAs and proteins on the sRNA-induced target decay mechanism under the physiological stress condition. Second, off-target effects generated through RapZ or GlmZ’ cannot be excluded. Overproduction of RapZ affects the transcriptome significantly and the recently identified RNA-binding mode of RapZ suggests potential recognition of similar structured RNAs beyond GlmY/GlmZ (27,54). For instance, we cannot exclude that RapZ regulates an RNA species or another factor, which could affect the stability of a RyhB variant.
Structural work has suggested different modes of recognition of RNA duplexes by RNase E. Crystal structures detected a hairpin RNA mimicking the MicC/ompD duplex in the RNase E catalytic center, while the sRNA 5′P group concomitantly binds the 5′ sensing pocket (17,28). Other RNA duplexes bind to a surface composed by the RNase H and the small domains in the principal RNase E dimer (28). It remains unclear how these models could account for interaction of RNase E with other sRNA/target pairs, e.g. the GcvB/SroC base-pairing, which involves two duplexes in close proximity (60), or RyhB/sodB, which leads to cleavage of sodB far downstream of the duplex (8). Likely, the CTH of RNase E with its additional RNA binding sites and protein binding partners such as Hfq plays a role. Whether Hfq hands over the RNAs, or has an active role beyond, remains to be clarified. There is rising evidence that association of Hfq with the RNase E-CTH likely occurs in presence of RNA (7,16,33,39,77,78). A new model proposes, that complex double-stranded RNA substrates could be recognized by both, Hfq and RNase E, forming complexes analogous as demonstrated in vitro for the RNaseE-NTD/RapZ/GlmZ complex (54). But how this model could involve the CTD of RNase E, which was shown in this study to be mandatory for regulation of ompD by MicC in vivo, remains unclear.
However, the precise molecular mechanism of Hfq-mRNA-sRNA-RNase E complex formation in vivo, for the individual RNA pairs, is still unknown. Finally, it is also possible that specialized RNase E complexes cooperate with sRNAs to silence transcripts in vivo. A recent study showed that RNase E multimerization can be stimulated by protein interaction partners (76). Notably, increased multimerization enhances cleavage of substrates exceeding 100 nt regardless of their 5′ phosphorylation state, whereas this does not apply to shorter RNAs containing the same cleaved sequence.
In summary, our data clearly show that a 5′P group is not important for target RNA destabilization by the processing-derived sRNAs CpxQ and SroC. Both sRNAs are remarkably stable in the cell, even with a 5′P monophosphate group. In contrast, 5′ phosphorylation was observed to be crucial for the stability of MicC and RyhB, sRNAs which are originally equipped with a 5′ triphosphate. Our experiments indicate that the phosphorylation state of MicC and RyhB plays no direct role for target RNA destabilization. However, the data cannot rule out completely that 5′P-MicC is more effective than 5′PPP-MicC in triggering degradation of ompD, once it is base-paired. The same could be the case for RyhB. Nevertheless, we show that the RNase E 5′ sensing pocket is not required for MicC-mediated regulation of ompD, while the importance of the RNase CTD is highlighted instead. Finally, we present a new genetic tool, which provides an interesting device for future research as it allows the generation of small RNAs of choice in a 5′ monophosphorylated state in vivo.
DATA AVAILABILITY
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yvonne Göpel for support in the initial stage of the project and Florian Sikora for help with construction of plasmids. We are grateful to Udo Bläsi for discussion and comments. We thank Jörg Vogel for providing strains.
Contributor Information
Alexandra Schilder, Department of Microbiology, Immunobiology and Genetics, Max Perutz Labs, University of Vienna, Vienna Biocenter (VBC), 1030 Vienna, Austria; Doctoral School in Microbiology and Environmental Science, University of Vienna, Vienna, Austria.
Boris Görke, Department of Microbiology, Immunobiology and Genetics, Max Perutz Labs, University of Vienna, Vienna Biocenter (VBC), 1030 Vienna, Austria.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Austrian Science fund through a stand-alone [P32410 to B.G.]; Doktoratskolleg RNA Biology [W1207-B09]. Funding for open access charge: Austrian Science Fund [P32410].
Conflict of interest statement. None declared.
REFERENCES
- 1. Santiago-Frangos A., Woodson S.A.. Hfq chaperone brings speed dating to bacterial sRNA. Wiley Interdiscip. Rev. RNA. 2018; 9:e1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hör J., Matera G., Vogel J., Gottesman S., Storz G.. Trans-acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica. EcoSal Plus. 2020; 9: 10.1128/ecosalplus.esp-0030-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quendera A.P., Seixas A.F., Dos Santos R.F., Santos I., Silva J.P.N., Arraiano C.M., Andrade J.M. RNA-binding proteins driving the regulatory activity of small non-coding RNAs in bacteria. Front. Mol. Biosci. 2020; 7:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lalaouna D., Simoneau-Roy M., Lafontaine D., Massé E.. Regulatory RNAs and target mRNA decay in prokaryotes. Biochim. Biophys. Acta. 2013; 1829:742–747. [DOI] [PubMed] [Google Scholar]
- 5. Laalami S., Zig L., Putzer H.. Initiation of mRNA decay in bacteria. Cell. Mol. Life Sci. 2014; 71:1799–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfeiffer V., Papenfort K., Lucchini S., Hinton J.C., Vogel J.. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 2009; 16:840–846. [DOI] [PubMed] [Google Scholar]
- 7. Morita T., Maki K., Aiba H.. RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 2005; 19:2176–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prévost K., Desnoyers G., Jacques J.F., Lavoie F., Massé E.. Small RNA-induced mRNA degradation achieved through both translation block and activated cleavage. Genes Dev. 2011; 25:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miczak A., Kaberdin V.R., Wei C.L., Lin-Chao S.. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leroy A., Vanzo N.F., Sousa S., Dreyfus M., Carpousis A.J.. Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA. Mol. Microbiol. 2002; 45:1231–1243. [DOI] [PubMed] [Google Scholar]
- 11. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 12. Hui M.P., Foley P.L., Belasco J.G.. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014; 48:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chao Y., Li L., Girodat D., Forstner K.U., Said N., Corcoran C., Smiga M., Papenfort K., Reinhardt R., Wieden H.J.et al.. In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell. 2017; 65:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ali N., Gowrishankar J.. Cross-subunit catalysis and a new phenomenon of recessive resurrection in Escherichia coli RNase E. Nucleic Acids Res. 2020; 48:847–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strahl H., Turlan C., Khalid S., Bond P.J., Kebalo J.M., Peyron P., Poljak L., Bouvier M., Hamoen L., Luisi B.F.et al.. Membrane recognition and dynamics of the RNA degradosome. PLoS Genet. 2015; 11:e1004961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bruce H.A., Du D., Matak-Vinkovic D., Bandyra K.J., Broadhurst R.W., Martin E., Sobott F., Shkumatov A.V., Luisi B.F.. Analysis of the natively unstructured RNA/protein-recognition core in the Escherichia coli RNA degradosome and its interactions with regulatory RNA/Hfq complexes. Nucleic Acids Res. 2018; 46:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Callaghan A.J., Marcaida M.J., Stead J.A., McDowall K.J., Scott W.G., Luisi B.F.. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005; 437:1187–1191. [DOI] [PubMed] [Google Scholar]
- 18. Bandyra K.J., Luisi B.F.. RNase E and the high-fidelity orchestration of RNA metabolism. Microbiol Spectr. 2018; 6: 10.1128/microbiolspec.RWR-0008-2017. [DOI] [PubMed] [Google Scholar]
- 19. Koslover D.J., Callaghan A.J., Marcaida M.J., Garman E.F., Martick M., Scott W.G., Luisi B.F.. The crystal structure of the Escherichia coli RNase E apoprotein and a mechanism for RNA degradation. Structure. 2008; 16:1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Callaghan A.J., Redko Y., Murphy L.M., Grossmann J.G., Yates D., Garman E., Ilag L.L., Robinson C.V., Symmons M.F., McDowall K.J.et al.. Zn-link”: a metal-sharing interface that organizes the quaternary structure and catalytic site of the endoribonuclease, RNase E. Biochemistry. 2005; 44:4667–4675. [DOI] [PubMed] [Google Scholar]
- 21. Jiang X., Belasco J.G.. Catalytic activation of multimeric RNase E and RNase G by 5′-monophosphorylated RNA. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:9211–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mackie G.A. Ribonuclease E is a 5′-end-dependent endonuclease. Nature. 1998; 395:720–723. [DOI] [PubMed] [Google Scholar]
- 23. Deana A., Celesnik H., Belasco J.G.. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008; 451:355–358. [DOI] [PubMed] [Google Scholar]
- 24. Luciano D.J., Vasilyev N., Richards J., Serganov A., Belasco J.G.. A novel RNA phosphorylation state enables 5′ end-dependent degradation in Escherichia coli. Mol. Cell. 2017; 67:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clarke J.E., Kime L., Romero A.D., McDowall K.J.. Direct entry by RNase E is a major pathway for the degradation and processing of RNA in Escherichia coli. Nucleic Acids Res. 2014; 42:11733–11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Del Campo C., Bartholomaus A., Fedyunin I., Ignatova Z.. Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLoS Genet. 2015; 11:e1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Durica-Mitic S., Göpel Y., Amman F., Görke B.. Adaptor protein RapZ activates endoribonuclease RNase E by protein-protein interaction to cleave a small regulatory RNA. RNA. 2020; 26:1198–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bandyra K.J., Wandzik J.M., Luisi B.F.. Substrate recognition and autoinhibition in the central ribonuclease RNase E. Mol. Cell. 2018; 72:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Updegrove T.B., Kouse A.B., Bandyra K.J., Storz G.. Stem-loops direct precise processing of 3′ UTR-derived small RNA MicL. Nucleic Acids Res. 2019; 47:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anupama K., Leela J.K., Gowrishankar J.. Two pathways for RNase E action in Escherichia coli in vivo and bypass of its essentiality in mutants defective for rho-dependent transcription termination. Mol. Microbiol. 2011; 82:1330–1348. [DOI] [PubMed] [Google Scholar]
- 31. Ikeda Y., Yagi M., Morita T., Aiba H.. Hfq binding at RhlB-recognition region of RNase E is crucial for the rapid degradation of target mRNAs mediated by sRNAs in Escherichia coli. Mol. Microbiol. 2011; 79:419–432. [DOI] [PubMed] [Google Scholar]
- 32. Caillet J., Baron B., Boni I.V., Caillet-Saguy C., Hajnsdorf E.. Identification of protein-protein and ribonucleoprotein complexes containing Hfq. Sci. Rep. 2019; 9:14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Worrall J.A., Gorna M., Crump N.T., Phillips L.G., Tuck A.C., Price A.J., Bavro V.N., Luisi B.F.. Reconstitution and analysis of the multienzyme Escherichia coli RNA degradosome. J. Mol. Biol. 2008; 382:870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Massé E., Escorcia F.E., Gottesman S.. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003; 17:2374–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fei J., Singh D., Zhang Q., Park S., Balasubramanian D., Golding I., Vanderpool C.K., Ha T.. RNA biochemistry. Determination of in vivo target search kinetics of regulatory noncoding RNA. Science. 2015; 347:1371–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tree J.J., Granneman S., McAteer S.P., Tollervey D., Gally D.L.. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell. 2014; 55:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Waters S.A., McAteer S.P., Kudla G., Pang I., Deshpande N.P., Amos T.G., Leong K.W., Wilkins M.R., Strugnell R., Gally D.L.et al.. Small RNA interactome of pathogenic E.coli revealed through crosslinking of RNase E. EMBO J. 2017; 36:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dendooven T., Luisi B.F.. RNA search engines empower the bacterial intranet. Biochem. Soc. Trans. 2017; 45:987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sinha D., De Lay N.R.. Target recognition by RNase E RNA-binding domain AR2 drives sRNA decay in the absence of PNPase. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2208022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bandyra K.J., Said N., Pfeiffer V., Gorna M.W., Vogel J., Luisi B.F.. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol. Cell. 2012; 47:943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Belasco J.G. Ribonuclease E: chopping knife and sculpting tool. Mol. Cell. 2017; 65:3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bandyra K.J., Luisi B.F.. Licensing and due process in the turnover of bacterial RNA. RNA Biol. 2013; 10:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chao Y., Vogel J.. A 3′ UTR-derived small RNA provides the regulatory noncoding arm of the inner membrane stress response. Mol. Cell. 2016; 61:352–363. [DOI] [PubMed] [Google Scholar]
- 44. Göpel Y., Khan M.A., Görke B.. Domain swapping between homologous bacterial small RNAs dissects processing and Hfq binding determinants and uncovers an aptamer for conditional RNase E cleavage. Nucleic Acids Res. 2016; 44:824–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wilson G.G., Young K.Y., Edlin G.J., Konigsberg W.. High-frequency generalised transduction by bacteriophage T4. Nature. 1979; 280:80–82. [DOI] [PubMed] [Google Scholar]
- 46. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uzzau S., Figueroa-Bossi N., Rubino S., Bossi L.. Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:15264–15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diederich L., Rasmussen L.J., Messer W.. New cloning vectors for integration in the lambda attachment site attB of the Escherichia coli chromosome. Plasmid. 1992; 28:14–24. [DOI] [PubMed] [Google Scholar]
- 49. Guillier M., Gottesman S.. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol. Microbiol. 2006; 59:231–247. [DOI] [PubMed] [Google Scholar]
- 50. Göpel Y., Khan M.A., Görke B.. Ménage à trois: post-transcriptional control of the key enzyme for cell envelope synthesis by a base-pairing small RNA, an RNase adaptor protein and a small RNA mimic. RNA Biol. 2014; 11:433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Urban J.H., Vogel J.. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol. 2008; 6:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Göpel Y., Papenfort K., Reichenbach B., Vogel J., Görke B.. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev. 2013; 27:552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gonzalez G.M., Durica-Mitic S., Hardwick S.W., Moncrieffe M.C., Resch M., Neumann P., Ficner R., Görke B., Luisi B.F.. Structural insights into RapZ-mediated regulation of bacterial amino-sugar metabolism. Nucleic Acids Res. 2017; 45:10845–10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Islam M.S., Hardwick S.W., Quell L., Durica-Mitic S., Chirgadze D.Y., Görke B., Luisi B.F.. Structure of a bacterial ribonucleoprotein complex central to the control of cell envelope biogenesis. EMBO J. 2023; 42:e112574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen S., Zhang A., Blyn L.B., Storz G.. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J. Bacteriol. 2004; 186:6689–6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chareyre S., Mandin P.. Bacterial iron homeostasis regulation by sRNAs. Microbiol. Spectr. 2018; 6:e01730-18. [DOI] [PubMed] [Google Scholar]
- 57. Morita T., Mochizuki Y., Aiba H.. Translational repression is sufficient for gene silencing by bacterial small noncoding RNAs in the absence of mRNA destruction. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:4858–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Geissmann T.A., Touati D. Hfq, a new chaperoning role: binding to messenger RNA determines access for small RNA regulator. EMBO J. 2004; 23:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dar D., Sorek R.. Bacterial noncoding RNAs excised from within protein-coding transcripts. mBio. 2018; 9:e01730-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miyakoshi M., Chao Y., Vogel J.. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015; 34:1478–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vogel J., Bartels V., Tang T.H., Churakov G., Slagter-Jäger J.G., Hüttenhofer A., Wagner E.G.. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 2003; 31:6435–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kime L., Jourdan S.S., Stead J.A., Hidalgo-Sastre A., McDowall K.J.. Rapid cleavage of RNA by RNase E in the absence of 5′ monophosphate stimulation. Mol. Microbiol. 2010; 76:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang C., Chao Y., Matera G., Gao Q., Vogel J.. The conserved 3′ UTR-derived small RNA NarS mediates mRNA crossregulation during nitrate respiration. Nucleic Acids Res. 2020; 48:2126–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Miyakoshi M., Matera G., Maki K., Sone Y., Vogel J.. Functional expansion of a TCA cycle operon mRNA by a 3′ end-derived small RNA. Nucleic Acids Res. 2019; 47:2075–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. De Mets F., Van Melderen L., Gottesman S.. Regulation of acetate metabolism and coordination with the TCA cycle via a processed small RNA. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Melamed S., Peer A., Faigenbaum-Romm R., Gatt Y.E., Reiss N., Bar A., Altuvia Y., Argaman L., Margalit H.. Global mapping of small RNA-target interactions in bacteria. Mol. Cell. 2016; 63:884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Walling L.R., Kouse A.B., Shabalina S.A., Zhang H., Storz G.. A 3′ UTR-derived small RNA connecting nitrogen and carbon metabolism in enteric bacteria. Nucleic Acids Res. 2022; 50:10093–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ponath F., Hor J., Vogel J.. An overview of gene regulation in bacteria by small RNAs derived from mRNA 3′ ends. FEMS Microbiol. Rev. 2022; 46:fuac017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bianco C.M., Fröhlich K.S., Vanderpool C.K.. Bacterial cyclopropane fatty acid synthase mRNA is targeted by activating and repressing small RNAs. J. Bacteriol. 2019; 201:e00461-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Richards J., Belasco J.G.. Obstacles to scanning by RNase E govern bacterial mRNA lifetimes by hindering access to distal cleavage sites. Mol. Cell. 2019; 74:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen J., To L., de Mets F., Luo X., Majdalani N., Tai C.H., Gottesman S.. A fluorescence-based genetic screen reveals diverse mechanisms silencing small RNA signaling in E. coli. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2106964118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sinha D., Matz L.M., Cameron T.A., De Lay N.R.. Poly(A) polymerase is required for RyhB sRNA stability and function in Escherichia coli. RNA. 2018; 24:1496–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cameron T.A., De Lay N.R.. The phosphorolytic exoribonucleases polynucleotide phosphorylase and RNase PH stabilize sRNAs and facilitate regulation of their mRNA targets. J. Bacteriol. 2016; 198:3309–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dendooven T., Sinha D., Roeselova A., Cameron T.A., De Lay N.R., Luisi B.F., Bandyra K.J.. A cooperative PNPase-Hfq-RNA carrier complex facilitates bacterial riboregulation. Mol. Cell. 2021; 81:2901–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Afonyushkin T., Vecerek B., Moll I., Bläsi U., Kaberdin V.R.. Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res. 2005; 33:1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moore C.J., Go H., Shin E., Ha H.J., Song S., Ha N.C., Kim Y.H., Cohen S.N., Lee K.. 2021) Substrate-dependent effects of quaternary structure on RNase E activity. Genes Dev. 35:286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Park S., Prévost K., Heideman E.M., Carrier M.C., Azam M.S., Reyer M.A., Liu W., Massé E., Fei J.. Dynamic interactions between the RNA chaperone Hfq, small regulatory RNAs, and mRNAs in live bacterial cells. Elife. 2021; 10:e64207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lalaouna D., Prévost K., Park S., Chenard T., Bouchard M.P., Caron M.P., Vanderpool C.K., Fei J., Massé E.. Binding of the RNA chaperone Hfq on target mRNAs promotes the small RNA RyhB-induced degradation in Escherichia coli. Noncoding RNAs. 2021; 7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.