


Abstract
Intrinsically disordered regions (IDRs) direct transcription factors (TFs) towards selected genomic occurrences of their binding motif, as exemplified by budding yeast's Msn2. However, the sequence basis of IDR-directed TF binding selectivity remains unknown. To reveal this sequence grammar, we analyze the genomic localizations of >100 designed IDR mutants, each carrying up to 122 mutations within this 567-AA region. Our data points at multivalent interactions, carried by hydrophobic—mostly aliphatic—residues dispersed within a disordered environment and independent of linear sequence motifs, as the key determinants of Msn2 genomic localization. The implications of our results for the mechanistic basis of IDR-based TF binding preferences are discussed.
INTRODUCTION
Transcription factors (TFs) contain DNA binding domains (DBDs) that bind specifically to short DNA sequence motifs. Most DBDs belong to families of known 3D folds. In eukaryotes, however, those structured domains occupy only a portion of TF sequences with much of the remaining sequences being of low complexity and devoid of a stable 3D structure (1–5). Intrinsically disordered regions (IDRs) within TFs include activation domains (ADs) that recruit co-activators (6–8) and, in some cases, incorporate TFs into transcription condensates (9). Whether these roles fully explain the enrichment of long IDRs within TFs is unclear, as most IDRs remain poorly characterized.
The short DNA motifs recognized by DBDs are highly abundant in genomes but, when measured inside cells, most motif occurrences remain unbound (10–13). This common observation presents a fundamental question: how do TFs distinguish between the various motif occurrences, binding to only a selected subset of sites? Despite immense interest, the basis of TF binding selectivity, at the genomic scale, remains unclear (14–21).
We recently reported that IDRs can direct TF binding along the genome (22). Msn2 and Yap1 are budding yeast TFs whose long (>500 residues) non-DBDs consist mostly of IDRs. We have shown that both TFs locate their binding sites using a multiplicity of weak determinants distributed along their IDRs. While conserved in function across considerable evolutionary distances, the IDR sequence displayed rapid sequence divergence (22), similar to other functional IDRs (23–27), rendering it incompatible with alignment-based comparative analysis. Newly devised approaches for comparative IDR analysis are now available, and go beyond sequence alignment to detect poorly aligned features such as short linear motifs (SLiMs) or sequence composition (25,27–37), yet those are still limited in defining the unknown sequence grammar within long IDRs of hundreds of residues.
Here, we tested a range of compositional or linear sequence features that could form the basis for the sequence code, or grammar, employed by the IDR in directing genomic binding preferences. For this, we followed previously employed guidelines (38–41), to design over 100 IDR mutants each carrying dozens of sequence changes spread across 567 residues, and mapped their binding locations across the full genome. Our results point at specific interactions carried by individual, bulky hydrophobic residues as the key determinants of genomic preferences. The flexible and disordered sequence context is needed for exposing those hydrophobic residues, as well as limiting deleterious (self)-interactions leading to loss of IDR activity, reduced expression, or cytoplasmic retention. Those design features are notably similar to features implicated in other IDR functions, including recruitment of co-activator by ADs (42–46) and bio-molecular condensate formation (39,40,47–49). We discuss the similarities and differences between the IDR sequence grammar identified here and those associated with other functions, and the implications of our results for the mechanism through which IDRs directs TF binding preferences along the genome.
MATERIALS AND METHODS
Generating Msn2 mutants
All Msn2 sequence mutants were designed in silico, codon-optimized for yeast and ordered from Twist Bioscience (see supplementary Table S2 for a full list of mutants and sequences). Mutant DNA sequences were PCR-amplified and transformed into the endogenous MNAse or YFP-tagged MSN2 locus replacing the native residues 2–567 using CRISPR, as described in (50).
Yeast transformation
All yeast transformations were performed using LiAc-PEG high-efficiency transformation as described by (51). After confirming successful transformations with PCR and Sanger DNA sequencing, CRISPR plasmids were lost by growing the cells for ∼20 generations in liquid YPD without selection, plating individual colonies and then selecting those clones w/o resistance. See supplementary Table S1 for a list of all strains used in this study.
ChEC-seq experiments
ChEC-seq experiments were performed as described in (52) with small modifications described in (53). For absolute quantification of the binding signal, a fixed amount of calibration spike-in (0.5 ml of OD4 BY4741 with MNAse-tagged Aro80) was added to every sample just before the first wash of the ChEC-seq procedure.
Next-generation-sequencing library generation and sequencing
Next-generation sequencing libraries were prepared as described in (53).
NGS data processing
After sequencing, raw reads from ChEC-seq libraries were demultiplexed using bcl2fastq (Illumina). Adaptor dimers and short reads were filtered out using cutAdapt (54) with parameters: ‘–O 10 –pairfilter = any –max-n 0.8 –action = mask’. Filtered reads were subsequently aligned to the S. cerevisiae genome R64-1-1 using Bowtie2 (55) with the options ‘‐‐end-to-end ‐‐trim-to 40 ‐‐verysensitive’. The genome coverage of fully aligned read pairs was calculated with GenomeCoverage from BEDTools (56) using the parameters ‘-d -5 –fs 1’. MATLAB was used for further processing of genome coverage files. For all samples, the genome coverage, i.e. ChEC signal, was normalized so that the average read density along the nuclear genome, excluding ribosomal RNA genes and CUP1-1/2, is one read/ base. After normalization, biological repeats were combined into a mean profile, which is used for all analyses (except Supplementary Figure S2A, B; see Figure S2B and Table S1 for number of biological repeats p. strain).
Bioinformatics analysis
Promoter definition and promoter binding signal (Figures 3B, 3G, 4C, 6B, S1C,E, S3)
Figure 3.
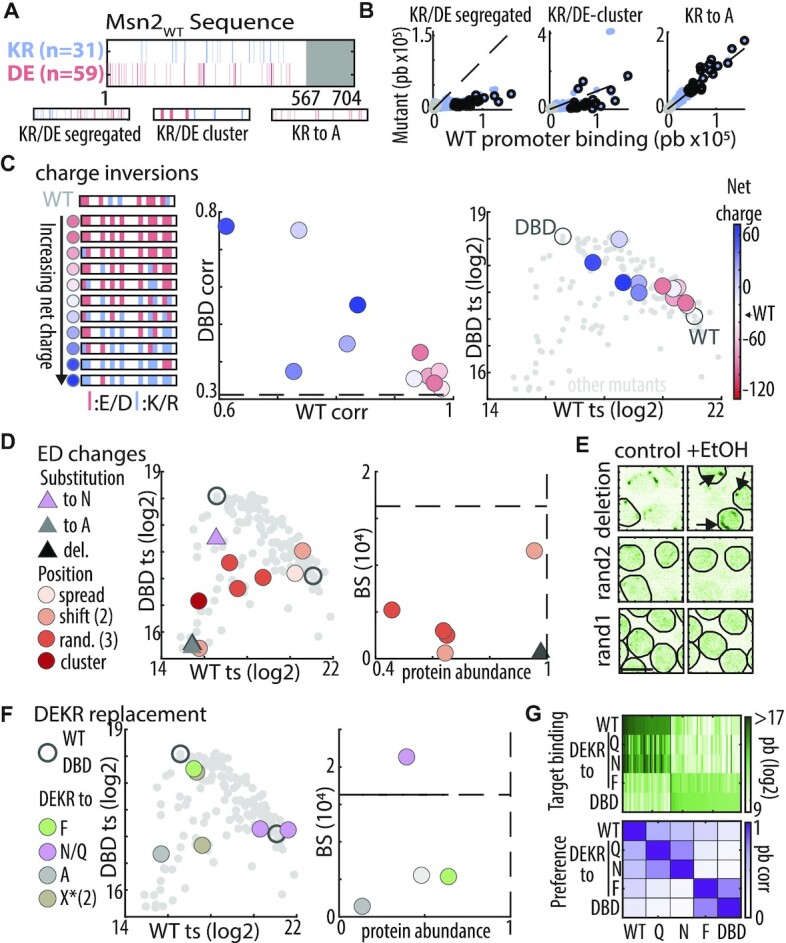
Charged residues do not explain the IDR-based DNA binding: (A) Distribution of basic and acidic residues within Msn2 nonDBD: shown are the locations of basic (R or K, blue) and acidic (D or E, red) residues within Msn2 IDR (top) and in three indicated mutants (bottom). (B) IDR function is sensitive to clustering of charged residues but insensitive to removal of positive residues: the plots compare promoter preferences. Each dot is a promoter, located according to binding signal of Msn2 and the indicated mutant. Black and grey dots correspond to Msn2WT and Msn2DBD unique promoters, respectively (see Supplementary Figure S3 for KR to E/D mutants). (C) Increasing IDR charge reduces binding at Msn2WT promoters: acidic and basic residues at different ratios were distributed randomly within the 90-charged locations of the tested IDR (left). Shown are similarity (correlation) of promoter preferences between mutants and Msn2WT or Msn2DBD (middle), and binding signal at Msn2WT and Msn2DBD unique targets (right). Color-coding indicates net charge (Msn2WT: -28). For comparison, dashed black line indicates the Msn2WT–Msn2DBD correlation (promoter preference plot), or small grey dots indicate the position of all mutants and empty circles Msn2WT and Msn2DBD position (target signal plot). (D, E) Acidic residues may contribute to IDR-based binding: shown in (D) are the binding signals at Msn2DBD versus Msn2WT promoters (middle) and the absolute motif binding vs. protein abundance relative to Msn2WT (right) for the indicated mutants. Dashed black lines indicate the relative protein abundance (= 1) and motif binding of Msn2WT. Nuclear localization before and after EtOH addition is shown in (E). Black arrows indicate cytoplasmic Msn2 clusters (scale bar corresponds to 5 μm). (F, G) Asparagine and glutamine can compensate for acidic residues: shown in (F) are binding signals at Msn2DBD vs. Msn2WT promoters (left) and the absolute motif binding versus relative protein abundance relative to Msn2WT (right) for the indicated mutants. The consequences of charge→polar replacements are shown also in (G), displaying promoter binding across all target promoters (top, Msn2WT promoters on the left, Msn2DBD promoters on the right) and correlation of promoter preferences between the indicated mutants (bottom). Note that DEKR replacement by either Q or N maintains binding to WT targets and avoids DBD targets, but changes the ordering among those targets in a similar way.
Figure 4.

Asparagine provides the disordered environment required for DNA binding. (A) Distribution of disorder promoting residues within Msn2 nonDBD: shown are the locations and number of the indicated residues within Msn2 IDR. (B, C) Replacing N + Q residues with alanine strongly reduces binding strength and nuclear localization but not binding preferences: binding phenotypes of the indicated mutants are summarized as in Figure 3C (B). Also shown is the similarity of promoter binding preferences between Msn2WT and the N + Q→A and NQ deletion mutant (scatter plots in C, left) and their effect on nuclear localization after EtOH exposure (C, right). (D) Disorder promoting residues retrieve N contribution to IDR-based binding: binding phenotypes of the indicated mutants are summarized as in Figure 3C above (D). Also shown is the absolute motif binding vs. relative protein abundance (right) of the same mutants. Dashed black line indicates Msn2WT to Msn2DBD correlation (left) or Msn2WT abundance and motif binding (right). The NQ to A mutant is indicated with *. (E, F) Acidic residues bias binding towards Msn2DBD promoters: binding phenotypes of the indicated mutants are summarized as in Figure 3C above (E). Change in binding to Msn2DBD targets, measured as fold-change (FC), is also compared between control substitution of N-neighboring and the respective N-to-X substitution (F).
Figure 6.
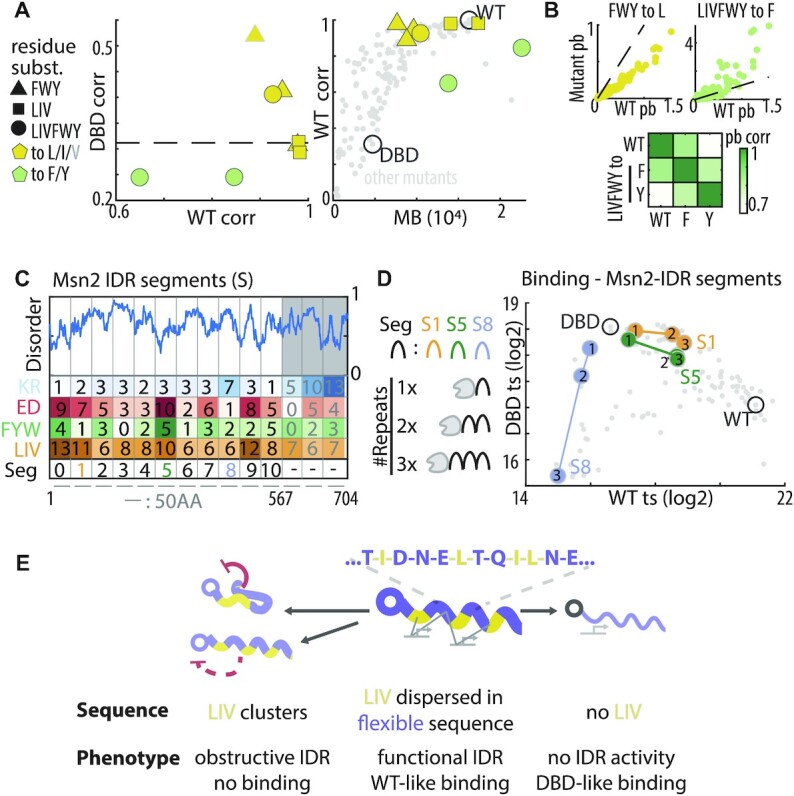
IDR based binding preferences remain invariant to the replacement of aromatic by aliphatic residues: (A, B) Aliphatic but not aromatic residues are critical for promoter preferences. Shown in (A) are the phenotypes of the indicated mutants, summarized as in Figure 3C above (left). Also shown is a comparison between the promoter preference similarity of each mutant to Msn2WT as a function of its motif binding strengths (right, each dot is a mutant with indicated mutants highlighted). Grey outline indicates the valine replacement which did not provide full rescue. Also shown are the scatter plots comparing promoter binding by the two indicated exemplary mutants and Msn2WT (top, line indicates equal 1:1 binding) and a heatmap showing the respective pairwise correlations (bottom). (C, D) Predicting the phenotype of tandem Msn2 segments: all 50-AA Msn2 IDR segments include multiple aliphatic residues and most are negatively charged (C, left). In total, 28 tandem repeat constructs of different segments were created (C, right) and profiled. The binding phenotype of tandem repeats for three representative segments is shown, one of which only contains one aromatic residue (S1) and one is positively charged (S8) (D, see Supplementary Figure S6 for all segment tandem repeats). (E) Model for IDR-based binding: hydrophobic and in particular aliphatic residues provide the specific interactions mediating IDR-based TF targeting. This role requires dispersion of these residues within a disordered environment, in the absence of which the same residues abolish all binding at both Msn2WT and Msn2DBD promoters, either via direct DBD inhibition or by overruling the NLS and preventing its nuclear localization.
If available, for each gene, we defined a consensus transcription start (TSS) site by comparing three different TSS datasets (57–59). The promoter of each gene was then defined as the distance between the start codon of a gene and the closest upstream verified ORF or at least 700 bp upstream of the TSS, or Start Codon if TSS not was available. For each mutant, the binding signal on every promoter, i.e. promoter binding ‘pb’, was then calculated as the cumulative, normalized ChEC-signal along this promoter. If not noted otherwise the axis scales are of the magnitude 105 normalized reads.
Target promoter selection (Figure 2B)
Figure 2.

IDR sequence mutants span a range of binding profiles. (A) Msn2 mutant types- a scheme: Engineered mutations included (1) removing same-identity residues by substituting with other residues or by deletion, and (2) changing locations of same-identity residues (‘shift’, ‘cluster’ or ‘random’). Red lines indicate mutated residues. (B) Binding phenotypes of Msn2 mutants: The 509 promoters bound strongly by at least one of the Msn2 mutants were selected. Heatmap shows the relative binding of each mutant to each promoter, ordered by clustering. Note the three general patterns: Msn2WT-like (cluster 1), Msn2DBD-like (cluster 2), and loss of binding (cluster 3). The two bottom rows indicate the identity of the Msn2WT (WT) and Msn2DBD (DBD) unique target promoters (Supplementary Figure S1B, and materials and methods). Bar graphs on the right show the correlation of binding preferences of each mutant with the Msn2WT and Msn2DBD, and its overall binding to the Msn2 motif (MB). (C−G) Msn2 mutants span a range of binding correlations and binding strengths: Shown in (C) are the similarity (correlation) in promoter binding preferences between mutants and Msn2WT or Msn2DBD, as a function of binding signal at Msn2WT or Msn2DBD unique target promoters, as indicated (target signal, ts). Binding signals at those target sets are compared in (D−G), color-coded by mutant-Msn2WT correlation (D), mutant-Msn2DBD correlation (E), mutant protein abundance (F), and mutants subcellular localization (G). Examples for differential localization is shown in G, with scale bar corresponding to 5 μm (see Supplementary Figure S2D for full frames). In all plots, mutants with missing data are indicated as small grey dots. In (D) dot size indicates total motif binding (MB). Of note, target signal is highly reproducible with 80% of mutants showing <25% in WT ts and <13% variance in DBD ts between biological repeats (i.e. Supplementary Figure S2A-C).
For each mutant, the target promoters were selected based on Z-scores. After Z-score normalizing the binding signal of a TF at each promoter against the promoter binding signal of this TF across all promoters, target promoters were defined as promoters with a Z-score >3.5. Shown are all promoters crossing this threshold in at least one mutant. Unique Msn2WT and Msn2DBD targets were selected based on a stricter Z-score threshold (5 and 4, see Supplementary Figure S1C) and also needed to be excluded from targets of Msn2DBD and Msn2WT respectively.
Relative promoter binding (Figures 2B, S1C)
To compare the binding of one mutant to that of a different mutant (or the median mutant) to the same promoter, we calculate the relative binding as:
Δlog2(pbA,X) = log2(pbA,X + 700) – log2(pbB,X + 700), where pbA,X (pbB,X) is the binding of TF A (B) to promoter X. The pseudo count of 700 is added to repress noise from weakly bound promoters. In Figure 2B, each mutant is compared against the median TF. In Supplementary Figure S1C, Msn2WT is compared against Msn2DBD and the relative binding at each promoter subsequently correlated with the enrichment of each dinucleotide.
Target signal and binding preferences (Figures 2–6, S2–S6)
The binding phenotype of each mutant was defined as the sum of the binding signal on a set of unique target promoters for Msn2WT and Msn2DBD, respectively (see above for definition and Table S3 for a full list of target promoters). In all figures, the log2-transformed value is shown. To compare the binding preference of each mutant with Msn2WT and Msn2DBD, we used the Pearson's correlation across the binding signal over all non-telomeric promoters (n = 5358).
Motif binding and 7mer enrichment (Figure S1B)
In order to verify the functional binding of Msn2WT and Msn2DBD, two separate analysis were performed. (i) We first determined the occurrences of all in-vitro Msn2 motifs from CISBP (20) (M00036_2.00) in intergenic regions using FIMO (n = 394) and then calculated the mean binding profile of of Msn2WT and Msn2DBD around these motifs, and compared it to a mean profile, of Msn2WT and Msn2DBD, across the same number of randomly chosen promoter positions (rdn). For better comparison the mean profiles were normalized by the average occupancy of a intergenic position in of Msn2WT and Msn2DBD, respectively. This analysis is shown in Supplementary Figure S1B on the left. (ii) We then calculated the mean occupancy (±15 bp) across all occurrences of each possible 7mer (n = 8192, not differentiating between forward and reverse) in intergenic regions, these mean occupancies are then normalized against the mean occupancy across all intergenic regions, i.e. enrichment. The enrichment values of all possible 8192 7mers in the Msn2WT and Msn2DBD profile is shown in Supplementary Figure S1 on the right and 7mers containing AGGGG or its reverse CCCCT, e.g. TTAGGGG or GCCCCTA, are highlighted.
Motif binding score (MB) (Figures 2–6, S1F, S5A)
As a proxy for the absolute binding strength of each mutant, we calculated the motif binding score. Therefore, we calculated the sum of the mean signal around (±25 bp) around each motif occurrence (see above).
Cluster score (Figure S5A)
To assess the clustering of individual amino acids or amino acids with similar biophysical properties in Msn2 and the mutants, we first used a window of 17 residues to calculate the relative density of each amino acid around each residue in the Msn2 IDR. The Gini coefficient of this density distribution was then calculated and compared to the Gini coefficient of 104 random sequences with the same amino acid composition as this IDR mutant. The cluster score shown is the Z-score normalized Gini coefficient of the Msn2 mutant when compared to the 104 random sequences.
Amino acid abundance and enrichment in the IDRs of Msn2 and its orthologs (Figure 1B)
Figure 1.
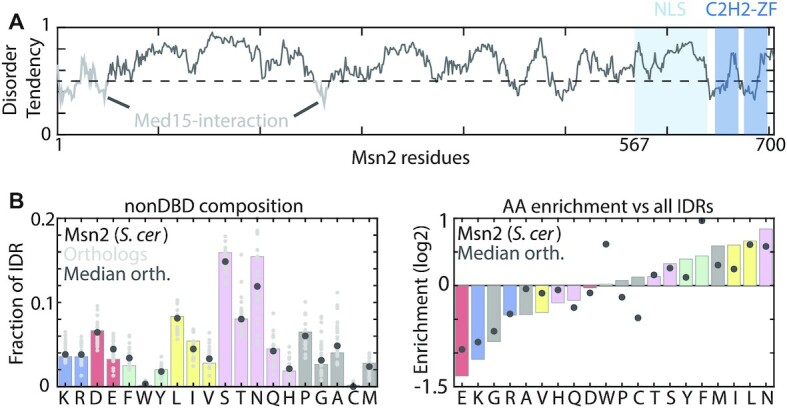
Msn2 IDR enrichment signature is conserved across yeast species. (A) The Msn2 protein: Shown is the predicted disorder tendency along the Msn2 sequence. The DBD and NLS (64) are indicated (light and dark blue). Mutations were restricted to the 567 AA N-terminus (white). (B) The sequence composition of Msn2: Shown is the frequency of each amino acid within the Msn2 nonDBD (left), and their enrichment relative to other IDRs in the budding yeast proteome (right). Enrichment was examined for Msn2 of S. cerevisiae and for 25 homologs from 17 yeast species.
The amino acid sequences of Msn2 homologs from 17 yeast species were obtained from YGOB (60), i.e. all except S. mikatae, S. kudriavzevii and S. bayanus. For better comparison and without knowing the NLS sequence of Msn2 homologs, we first defined the IDR of each Msn2 homolog as the sequence between the first residue and the first PFAM-defined C2H2 zinc finger and calculated the relative abundance of each amino acid in this region. To calculate the corresponding amino acid enrichment compared to other IDRs, we first downloaded the complete proteome of each yeast species and determined all IDRs using IUPred. After determining the relative abundance of each AA in each proteome, we calculated the enrichment of each AA in the Msn2 IDR compared to the total IDR-ome as: log2(fracMsn2) − log2(fracIDRome)
Dinucleotide enrichment in promoters (Figure S1D)
For each intergenic region upstream of a gene, we calculated the relative abundance of each Dinucleotide, 10 Dinucleotides combining forward and reverse complement, and compared it to the relative abundance of each dinucleotide in all intergenic regions: Enr(NN in Promoter X) = log2(fracNN-PromX) −log2(fracNN-All).
Flow cytometry based protein abundance measurements of msn2 IDR mutants (Figures 2–6)
To assess the expression level of each Msn2 mutant, we used yeast strains where the Msn2 mutants were tagged with YFP instead of MNAse and grew them overnight to saturation in SC + glucose media. We then diluted the cells to an OD of 0.02 and grew them for around three cell cycles to reach early exponential phase. The YFP level in these exponentially growing cells was then determined on a BD LSRII system with an excitation laser of 488 nm and an emission filter at 525 ± 25 nm. The relative abundance of each mutant is then calculated as:
(F(Msn2Var) − F(Blank))/(F(Msn2WT) − F(Blank)), where F(Msn2Var), F(Msn2WT) and F(Blank) are the median fluorescence levels of the Msn2 mutant, Msn2WT and non-tagged control cells, respectively.
Live imaging of Msn2 mutants and nuclear localization (Figures 2G, 3E, 4C, 5C, S2D, S4A)
Figure 5.

Dispersion of aliphatic and aromatic residues is required for IDR-based binding. (A) Distribution of hydrophobic residues within the Msn2 nonDBD: shown are the locations of the indicated hydrophobic residues within the Msn2 IDR. (B, C) Clustering of hydrophobic residues perturbs genomic binding and nuclear localization. Binding phenotypes of the indicated mutants are summarized as in Figures 3C above (Color indicates target residue and shape mutation type). Also shown is the localization of LIV cluster mutants after EtOH exposure (C, arrows indicate possible aggregates, scale bar corresponds to 5 μm). Note that binding preferences remain invariant to random or shifted positioning of the hydrophobic residues, while clustering leads to complete loss of binding and nuclear localization. (D) Deletion of hydrophobic residues or non-similar replacement biases binding towards Msn2DBD promoters: binding phenotypes of the indicated mutants are summarized as in Figures 3C above (Shape indicates target residue and color indicates mutation type, see also Supplementary Figure S5B, for target signal and Supplementary Figure S5C for alanine replacements). (E) Genomic preferences depend on the additive contribution of hydrophobic residues located throughout the IDR. Shown are the effects of sequential deletion of hydrophobic residues from either end of the IDR on promoter preference similarity with Msn2WT (left), Msn2DBD (middle) and absolute motif binding (right). The effects of sequential IDR truncation are also shown for comparison (Data from (22)). Note that removing only the hydrophobic residues has a similar effect to deleting the corresponding IDR region, and that the effect only depends on the number of the removed residues not on their position, i.e. N- or C-terminal.
Strains carrying YFP-tagged Msn2 mutants, were grown overnight in SC + glucose (SD) to full saturation and then diluted to an OD of 0.02 and grown for ∼3 cell cycles as described above. The cell cultures were then transferred to ConA-coated imaging slides (Ibidi), incubated, and gently washed 3 times with clean SD medium and imaged as a control. To trigger nuclear localization, we added 5% EtOH to each culture, waited for 5 and 10 min, respectively, and performed another imaging session. Images were either acquired on an Axio Observer Z1 widefield (Zeiss) or DragonFly confocal (Andor) microscope. After segmenting yeast cells based on the bright field (Zeiss) or fluorescence (Andor) channel using YeastSpotter (61), we used MATLAB to determine the nuclear localization. For this, we first defined the 2.5% brightest pixel (YFP channel) in each cell and then calculated the hull area spanned by these 2.5% brightest pixels. Bright top pixels in a small area indicate nuclear localization, and dim, dispersed pixels indicate cytoplasmic localization.
RESULTS
Designing and profiling IDR mutants
Msn2 is a 704 amino acid (AA)-long TF that regulates the budding yeast stress response (62,63). Its 64-AA C2H2 zinc-finger DBD is located at its C-terminus, flanked by a 73-AA region including the nuclear localization signal (NLS) (64) (Figure 1A). The remaining 567 AAs (80% of its sequence) include two short structured segments required for recruiting the Med15 co-activator (65), but are otherwise mostly IDR (as predicted by e.g IUPred (66)). Relative to other disordered proteins, the IDR of Msn2 is enriched with the hydrophobic residues leucine (L) and phenylalanine (F), depleted of charged residues and lacks apparent AA clusters or recurring motifs (Figure 1B). The most abundant residues, asparagine (N, 16.5%), serine (S, 15.5%) and leucine (L, 8.8%), are distributed throughout the sequence at median distances of 4, 4 and 8, respectively. This composition is similar among Msn2 orthologues (Figure 1B).
Defining protein sequence grammar requires mutating domains of functional relevance. Our previous study revealed that removal of any < 200AA region within the Msn2’s non-DBD has no detectible effect on Msn2 binding profile (22), and, in preliminary analysis, we found that no 50AA-segment is sufficient, by itself, for directing binding preferences (Supplementary Figure S1A). We therefore selected a long region of 567 non-DBD residues, containing most of the IDR but excluding the NLS (64). We designed mutations that spread across this full sequence, following established frameworks for testing IDR features of potential functional relevance, including sequence composition, short sequence motifs, or AA clustering (25,27–37,40,42–46,67) (Figure 2A). In particular, our mutants either changed the AA composition (e.g. deleting or replacing same-type residues) or preserved it (e.g. random AA distributions or clustering) and included local shifts designed to abrogate potential short sequence motifs.
Overall, we analyzed 106 mutants, each changing 2%-22% of hethe tested sequence (12–122 AAs). The mutated sequences were integrated into the genome, replacing the nonDBD of the endogenous Msn2 sequence, and their binding profiles were mapped using ChEC-seq (52). The intact Msn2WT and a mutant lacking the mutated region (denoted as Msn2DBD), served as controls. As we reported previously (22), both Msn2WT and Msn2DBD localized to their known motif (AGGGG, Supplementary Figure S1B), yet selected different subsets of motif occurrences, leading to distinct promoter preferences (correlation c = 0.3, Supplementary Figure S1C).
IDR mutants span a continuum of genomic binding profiles
To obtain a global view on the span of binding profiles displayed by the designed IDR mutants, we clustered the mutants based on similarity (correlation) in promoter preferences, with promoter preference defined as the total binding signal mapped to promoter sequence. This analysis distinguished three groups: mutants that retained Msn2WT preferences, mutants that became Msn2DBD-like, and mutants that lost binding to both promoter classes (Figure 2B). Of note, while mutants of the second group have lost IDR activity but retained binding to Msn2DBD targets, mutants of the third cluster showed little DBD-dependent binding, as quantified by the fraction of ChEC-signal localizing to AGGGG motif sites and verified using external controls (Supplementary Figure S1E, F). Therefore, in these third-cluster mutants, the mutated non-DBD, inhibited, rather than directed DNA binding.
To examine the mutants’ binding phenotypes at a higher resolution, we assembled two sets of promoters bound uniquely by either Msn2WT or Msn2DBD (Supplementary Figure S1C, see Materials and Methods). For each individual mutant, binding signal localizing to each promoter group provided a measure of binding strength, complementing the correlation-based measure of promoter preferences. The IDR mutants displayed a continuum of binding strengths that spread across the ‘Msn2WT−Msn2DBD−None’ binding space (Figure 2C–G, Supplementary Figure S1C). Therefore, mutating the IDR quantitatively tunes Msn2 binding, consistent with multivalent interactions contributing to this phenotype (22).
Several features of the mutants’ binding phenotypes are notable. First, compared to the spread of binding strengths, changes in promoter preferences were limited (Figure 2C, D). In fact, mutants that retained Msn2WT preferences (c > 0.9) still displayed over 3-fold differences in binding strengths at Msn2WT targets (Figure 2C). Second, as promoter preferences shifted towards that of Msn2DBD, overall binding to all AGGGG sites decreased (Figure 2B–E). This apparent decrease in DBD-DNA binding is notable, as the Msn2DBD mutant was of increased abundance at the protein level and constitutively nuclear (Figure 2F, G). Finally, mutants that lost binding to both Msn2WT and Msn2DBD targets were generally of low abundance or poor nuclear localization. Consistently, none of our mutants gained a nonDBD-like profile (Supplementary Figure S1G). We conclude that mutations can reduce IDR activity, but could also render it inhibitory for DBD–DNA binding, by limiting its abundance, nuclear localization or, perhaps, through direct IDR–DBD interactions as described for p53 (68).
The Msn2 IDR can direct binding preferences independent of electrostatic interactions
Within IDRs, charged residues influence the conformation ensembles (69–73) through their overall charges (70–72,74) or patterning of oppositely charged residues (74–77). In our context of TF-DNA association, charges appear particularly relevant, first because the DNA is negatively charged, and second because acidic residues promote co-activator recruitment by ADs (42–46). The Msn2 IDR contains 31 positively charged (lysine K, and arginine R) and 59 negatively charged (16 E, and 43 D) residues (Figure 3A). These charged residues distribute largely uniformly along the sequence, showing some bias for the N-terminus and the middle of the IDR, regions that also show some AD function (46). This dispersion of charges might be important, as segregating acidic and basic residues to opposite sides led to a loss of binding and, when additionally clustered, to changes in promoter preferences (Figure 3B).
We tested the role of net IDR charge through systematic charge inversions (Figure 3C). Contrasting our expectations, decreasing the net charge, or even replacing all 31 K/R with either aspartate or glutamate (D/E) had little, if any, effect on promoter preferences or binding strengths, as did their replacement with the neutral alanine (Figure 3B, C and Supplementary Figure S3; correlations with Msn2WTc = 0.98, 0.97, and 0.94, respectively). By contrast, increasing the net charge by replacing acidic residues by alanine (A) or deleting them led to a full loss of binding while systematically replacing acidic residues with basic ones, or asparagine (N) shifted promoter preferences gradually away from Msn2WT and towards Msn2DBD (Figure 3C, D). Therefore, positive charges are not required within the IDR, but negative charges might be needed for IDR activity, at least in the presence of positively charged ones.
We noted that the effects of charge inversions on promoter preferences were not monotonic (Figure 3C), and this was further emphasized when examining binding strength, which was reduced also in acidic mutants retaining Msn2WT-like preferences (Figure 3D). Since the tested mutants differed not only in net charge but also in the (randomly selected) charge-inverted locations (Figure 3C), we examined whether acidic residues at particular positions contribute through short sequence motifs (Figure 3D). Random dispersion or clustering of the available acidic residues generally reduced binding signal, protein abundance or nuclear localization, indicating changes in the conformation ensemble that, perhaps, occluded the NLS or promoted other interactions (Figure 3D, E). Other, similar, perturbations, however, had no effects, including locally shifting acidic residues or dispersing them evenly within the sequence (Figure 3D). As these perturbations abrogated any potential short motifs, we conclude that the IDR directs promoter preferences independent of short sequence motifs containing those residues.
The variable sensitivity of the IDR to the re-distributions of acidic residues, and the non-monotonic effects of charge inversion, could reflect the loss acidic residues from particular regions, or inhibitory effects caused by other residues affected by those changes. Examples include exposure of hydrophobic residues that could induce hydrophobic collapse or aggregation. Consistent with the later, deleting all acidic residues led to a full loss of binding and cytoplasmic aggregation (Figure 3D, E). To examine this further, while avoiding inhibitory effects of net positive charge, we replaced all charged residues with phenylalanine (F), alanine (A), asparagine (N), glutamine (Q) or ‘random’ residues retaining the uncharged IDR composition (Figure 3F). F replacement, as well as one of the composition-preserving replacements, led to Msn2DBD-like binding, indicating a loss of IDR activity perhaps through hydrophobic collapse. Similarly, charge-to-A replacement, as well as a second composition-preserving replacement led to a complete loss of binding, at least in part through reduced abundance and cytoplasmic retention (Figure 3F). Most notably, replacement of all charged residues by N or Q retained strong binding to Msn2WT promoters, although modulating relative preferences within this promoter set (c = 0.56, 0.65; Figure 3G). Together, those results refute a dominant role of electrostatics in the specific interactions directing Msn2 binding preference.
High disorder content is required for nonDBD function, but residues of low hydrophobicity/high flexibility do not explain the specific interactions guiding promoter preferences
The ability to replace the charged residues using the polar N or Q (but not, e.g. A or F), while retaining binding to Msn2WT promoters, suggests some shared property of functional relevance. Polar and charged residues are similar in being of low hydrophobicity and high flexibility, based on the Kyte–Doolittle hydrophobicity and Vihinen's flexibility scales, respectively (78,79). As the IDR contains additional residues of similar properties, we examined the roles of those using four replacement mutants: N + Q→A (121 residues), P→A (37), S→A (88) and G→A (12) (Figure 4A, B). The last two were of little effect, while P→A increased binding to Msn2WT promoters, causing a slight shift in preferences (c = 0.81, Figure 4B). Also of limited effects were local shifts of P-G, N-Q or S, as well as local clustering of N or S into small groups, refuting a role for short sequence motifs involving those flexible residues (Figure 4B).
By contrast, the most extensive, N + Q→ A replacement (121 residues), which reduced nuclear localization (Figure 4C, Supplementary Figure S4A), reduced binding strength, as did deleting all N/Q residues (Figure 4B, C). Notably, both mutants still showed strong preference for Msn2WT promoters (c = 0.78 and 0.85, respectively; Figure 4B, C). Further, replacing N by high-flexibility residues provided full (Q, G) or partial compensation (D, E, T, H) (Figure 4D; Note the increased binding strength and slight change in relative preference of the N→H replacements (Supplementary Figure S4B). By contrast, replacing N with the aromatic tyrosine (Y) or with arginine (R) reduced binding overall and also, in the case of tyrosine, shifted preferences towards Msn2DBD, indicating loss of IDR activity, potentially driven by hydrophobic collapse (Figure 4D). Finally, N→S (serine) replacement reduced nuclear localization (Supplementary Figure S4A), which may result from the generation of new phosphorylation sites, as S phosphorylation limits Msn2 nuclear translocation (64), and N←→S swapping was of no effect (Figure 4D).
N replacements, therefore, led to a range of phenotypes, with high flexibility residues mostly capable of full rescue, while others shifted binding preferences or decreased overall binding at least in part through reduced protein abundance or nuclear localization. Notably, replacing N-neighboring residues (n = 82), rather than the N residues themselves, led to a similar range of effects (Figure 4E, F): S, T and Q fully compensated, E/D shifted binding towards Msn2DBD promoters, and Y or R fully abolished binding (Figure 4E, F). Together with the N/Q deletion mutant retaining preferred binding to Msn2WT promoter (despite reduced abundance), we conclude that the high enrichment of polar residues within the Msn2 IDR provides a necessary disordered environment, but is not sufficient for explaining the IDR-based interactions directing binding preferences.
Hydrophobic residues direct binding towards Msn2 promoters
Four of the five top-enriched residues within the IDR sequence were hydrophobic: the aliphatic residues leucine and isoleucine (L, I), which together comprise 82 (14.4%) residues, and the aromatic residues phenylalanine and tyrosine (F, Y), which comprise additional 27 (4.6%) residues (Figure 1B). The spread of those residues was largely even, showing a higher dispersion than expected by chance (Figure 5A, Supplementary Figure S5A). This dispersion is relevant, as clustering those residues led to a loss of DNA binding and cytoplasmic aggregation, indicating emerging intra-molecular interactions (Figure 5B, C). Of note, aliphatic clusters emerged also in mutants testing the dispersion of acidic residues (e.g. Figure 3 and Supplementary Figure S5A), likely explaining their differential effects.
Locally shifting the hydrophobic residues, or dispersing them randomly within the IDR sequence had no effect for most realizations, refuting the use of short linear motifs (Figure 5B). By contrast, deleting the three aliphatic residues (L/I/V), or the three aromatic residues (F/Y/W), shifted binding preferences away from Msn2WT and towards Msn2DBD promoters (Figure 5D). Further, removing all six residues (L/I/V/F/Y/W) (n = 122, 22%) caused an almost complete shift in preference, increasing similarity with Msn2DBD to ∼80% correlation (Figure 5D). Polar or acidic residues could not compensate for the hydrophobic AAs, as demonstrated by testing N, Q or D (Figure 5D). Therefore, the hydrophobic residues are required for IDR activity, but their role is independent of linear motifs.
To examine whether the hydrophobic residues could explain the multiplicity of specificity determinants spread throughout the IDR (22), we sequentially eliminated the hydrophobic content from segments of increasing sizes, in steps of 20 L/I/V/F/W/Y residues deleted from either side of the IDR (Figure 5E). As predicted, promoter preferences followed the hydrophobic content, gradually shifting away from Msn2WT and towards the Msn2DBD, similar to IDR truncations (22) (Figure 6B). We conclude that hydrophobic residues located throughout the IDR are essential for its role in directing promoter preferences.
A sequence-based model of Msn2 binding specificity predicts the effect of IDR segments
Our results suggest that the IDR directs promoter preferences through multiple interactions carried by hydrophobic residues dispersed within a flexible sequence environment. This general pattern was indeed realized by all short (50 AA) protein segments used in our previous truncation experiments (Figures 5A, 6C and Supplementary Figure S1A), consistent with the gradual effects of those truncations on promoter preferences (22) (Figure 5E). We noted, however, that while obtaining a full DBD-like promoter preference required removal of both aliphatic and aromatic residues, some of the 50-AA segments contained only aliphatic residues but lacked aromatic ones (Figure 6C).
To test whether aliphatic residues, by themselves, are sufficient for directing binding preferences, we replaced all aromatic residues by different aliphatic ones. Binding preferences were maintained when using the most abundant leucine (L) (Figure 6A) (c = 0.98), although binding strength was somewhat reduced (Figure 6B). Good compensation was also achieved by isoleucine (c = 0.95), but less so by the least abundant valine (c = 0.88). Consistently, replacement of both aliphatic and aromatic residues by the aromatic phenylalanine (F) or tyrosine (Y) increased binding at the Msn2WT promoters, but also shifted their relative preferences in a similar manner (Figure 6B). Therefore, while both aliphatic (L, I) and aromatic residues (F,Y) contribute to binding at Msn2WT promoters, their binding preferences differ with the wild type one depending more on the prominent aliphatic (14.4%) than the low aromatic (4.6%) content.
To finally probe the redundancy of the code, we considered the 50-AA segments used in the previous truncation experiments (22). As mentioned, none of those was sufficient, on its own, for shifting preference away from Msn2DBD (Supplementary Figure S1A). Yet, the additive nature of the interactions predicted that tandem repeats will succeed in biasing promoter preferences towards Msn2WT (Figure 6C,D). To test this, we fused 1–3 tandem repeats of each segment to Msn2DBD (Figure 6C, Supplementary Figure S6). As predicted, increasing the number of repeats led to a gradual change in binding profiles, increasing similarity with Msn2WT. The only exception was segment #8, (AA 401 to 450) which gradually inhibited binding also to Msn2DBD promoters, and this correlated with its unique basic net-charge, which we found to be inhibitory for IDR activity (Figure 6C, D). Of note, two of the segments had no (#3: AA 151–200) or a single (#1: 51–100) aromatic residue, but still biased binding towards Msn2WT promoters (Figure 6C, Supplementary Figure S6). We conclude the IDR of Msn2 directs promoter preferences through multivalent interactions carried by hydrophobic - mostly aliphatic- residues spread within a disordered environment (Figure 6E).
DISCUSSION
Protein regions lacking a stable 3D structure can still interact with other biomolecules in at least two ways. First, short linear sequence motifs (SLiMs) within IDRs can provide structural or chemical complementarities associated with specific molecular recognition. Second, multivalent interactions can arise from the chemical composition characterizing the low complexity sequences of disordered domains (32,36,74,80–82). In this work, we examine the role of IDRs in directing TF binding towards a particular subset of promoters. Given the need for specific recognition, we expected promoter binding preferences to depend on short sequence motifs. This, however, was refuted by our data, as Msn2 binding pattern remained largely insensitive to mutations designed to abrogate such motifs. Our data further refuted a dominant role of electrostatics in guiding promoter preference, as Msn2 retained strong target binding (albeit with some changing preferences) when replacing charged residues with polar ones. Positive charges, in fact, inhibited IDR activity and could be tolerated only when dispersed within negative ones.
Rather, our data suggest that the IDR of Msn2 directs promoter recognition using multivalent interactions carried by hydrophobic AAs separated by flexible (hydrophilic) residues (Figure 6E). The hydrophobic residues include a majority of aliphatic (leucine and isoleucine) and a minority of aromatic ones, while the interspacing residues are mostly polar and a minority charged. Perturbing this design led to a range of phenotypes. First, removal of hydrophobic AAs abrogated IDR activity, shifting binding preferences towards DBD-bound promoters. Second, grouping hydrophobic residues together caused cytoplasmic aggregation and loss of nuclear localization and all DNA binding, indicating emerging intra-molecular interactions. Third, increasing the aromatic content (Y or F) by replacing flexible residues inhibited IDR activity, perhaps through hydrophobic collapse. Finally, multiple mutations including deletion of polar or acidic residues led to a decrease in protein abundance and cytoplasmic retention, indicating changes in conformation assembly that promote degradation or occlude the NLS. IDR function therefore depends on the balancing of hydrophobic residues, which provide the specific interactions required for promoter recognition, and flexible residues, which keep those residues exposed (Figure 6E). At a finer resolution, a balance between charged vs. polar residues and between aliphatic and aromatic ones, tunes relative preferences at the various target promoters.
The inferred sequence grammar may shed light on the mechanisms through which the IDR directs promoter preferences. It is notable that its central features resemble the design used by ADs in co-factor recruitment (42–46): a hydrophobic motif surrounded by negative charge and intrinsic disorder that keep it exposed and prevent its collapse (43). This similarity might suggest a shared mechanism, yet several differences are notable. First, in our context of binding preferences, the residues carrying the interactions are aliphatic (leucine and isoleucine), while ADs require aromatic residues (42,44–46), with leucine (but not isoleucine) contributing in human but not in yeast (43). Second, acidic residues are essential in ADs, but their replacement by the polar N or Q retains strong binding at target promoters. Finally, while short, 13–30AA regions are sufficient for activating expression when fused to an endogenous DBD, a larger region of hundreds of residues is required for shifting binding preferences.
Our model is also aligned with a ‘stickers-and-spacers’ model explaining the formation of bio-molecular condensates (34,39,83). Within this model, uniform spacing of stickers separated by flexible spacers (e.g. hydrophobic residues in a hydrophilic sequence) allows cross interactions leading to condensate formation. This similarity raises the possibility that the IDR incorporates Msn2 into transcription condensates. We disfavor this possibility for two reasons. First, self-assembly into condensates was mostly observed for low-complexity sequences with clustered aromatic residues (39,40,47–49), whereas aliphatic residues, central to our case, were shown to inhibit condensate formation, at least in some contexts (84). Furthermore, we observed Msn2-YFP to be diffused in cells, consistent with previous failure to detect Msn2 clustering (85). In fact, aggregates did arise, but only in mutants that, e.g. grouped hydrophobic residues together. Aggregation of the intact protein is therefore limited, perhaps by a balance favoring flexibility over hydrophobic interactions.
A third possibility is that hydrophobic residues direct specific interactions with other TFs, in which case specificity would result from the co-binding of interacting TFs to the associated composite motifs. This is perhaps the most probable possibility, which we are actively pursuing in parallel projects.
Finally, the IDR could recognize its promoters through direct DNA interactions. Since long IDRs are absent from crystal structures, IDR–DNA interactions are sparse in existing databases. Still, several literature evidences might point to the possibility of specific IDR–DNA interactions: First, short IDRs flanking the DBD are observed in available structures, and those often insert within the minor groove (reviewed in (5)). Consequently, IDRs could provide specificity through indirect readouts of DNA shape. Second, analysis of base-contacts within the minor groove revealed preferences for hydrophobic (aliphatic) residues, specifically for A-form DNA (86). Asparagine is also preferred in these interactions, but requires closer association with DNA, while the hydrophobic residues can act at intermediate distances (86) and are therefore more compatible with transient interactions that, perhaps, do not require overcoming large entropic barriers. Third, transition to A-form DNA is more common in the presence of GG and GC di-nucleotides (87–89), and those dinucleotides are enriched within Msn2WT-bound, but not the Msn2DBD-bound promoters (Supplementary Figure S1B, C). Further studies are required to test these and other possibilities.
DATA AVAILABILITY
All raw sequencing reads and genomic profiles are available on GEO (GSE212466). Additional sequencing data for previously profiled mutants (22) were taken from Bioproject: PRJNA573518. All Matlab scripts to analyze the data are available on GITHUB (https://github.com/barkailab/Carmi2022) and on Zenodo (DOI: 10.5281/zenodo.7677339).
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Barkai Lab for careful reading of the manuscript, their comments, and scientific discussions. We also like to thank Divya Kumar for sharing the Aro80 strain and profile.
Author contributions: F.J., M.C., B.K. and N.B. conceived the study and designed the experiments. M.C. and B.K. performed most of the experiments with the help of J.S., F.J. analyzed the data. All authors wrote the manuscript.
Contributor Information
Felix Jonas, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
Miri Carmi, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
Beniamin Krupkin, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
Joseph Steinberger, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
Sagie Brodsky, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
Tamar Jana, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
Naama Barkai, Department of Molecular Genetics, Weizmann Institute of Science, Rehovot 76100, Israel.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Israel Science Foundation, Horizon Europe (European Research Council); Minerva Foundation. Funding for open access charge: Internal Grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Liu J., Perumal N.B., Oldfield C.J., Su E.W., Uversky V.N., Dunker A.K.. Intrinsic disorder in transcription factors. Biochemistry. 2006; 45:6873–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang C., Uversky V.N., Kurgan L.. Disordered nucleiome: abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics. 2016; 16:1486–1498. [DOI] [PubMed] [Google Scholar]
- 3. Minezaki Y., Homma K., Kinjo A.R., Nishikawa K.. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006; 359:1137–1149. [DOI] [PubMed] [Google Scholar]
- 4. Ward J.J., Sodhi J.S., McGuffin L.J., Buxton B.F., Jones D.T.. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004; 337:635–645. [DOI] [PubMed] [Google Scholar]
- 5. Brodsky S., Jana T., Barkai N.. Order through disorder: the role of intrinsically disordered regions in transcription factor binding specificity. Curr. Opin. Struct. Biol. 2021; 71:110–115. [DOI] [PubMed] [Google Scholar]
- 6. Kornberg R.D. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 2005; 30:235–239. [DOI] [PubMed] [Google Scholar]
- 7. Hope I.A., Struhl K.. Functional dissection of a eukaryotic transcriptional activator protein, GCN4 of yeast. Cell. 1986; 46:885–894. [DOI] [PubMed] [Google Scholar]
- 8. Hope I.A., Mahadevan S., Struhl K.. Structural and functional characterization of the short acidic transcriptional activation region of yeast GCN4 protein. Nature. 1988; 333:635–640. [DOI] [PubMed] [Google Scholar]
- 9. Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A.. A phase separation model for transcriptional control. Cell. 2017; 169:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jana T., Brodsky S., Barkai N.. Speed-specificity trade-offs in the transcription factors search for their genomic binding sites. Trends Genet. 2021; 37:421–432. [DOI] [PubMed] [Google Scholar]
- 11. Krieger G., Lupo O., Wittkopp P., Barkai N.. Evolution of transcription factor binding through sequence variations and turnover of binding sites. Genome Res. 2022; 32:1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., Thurman R.E., John S., Sandstrom R., Johnson A.K.et al.. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012; 489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y.et al.. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inukai S., Kock K.H., Bulyk M.L.. Transcription factor-DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev. 2017; 43:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Todeschini A.L., Georges A., Veitia R.A.. Transcription factors: specific DNA binding and specific gene regulation. Trends Genet. 2014; 30:211–219. [DOI] [PubMed] [Google Scholar]
- 16. Pan Y., Tsai C.-J., Ma B., Nussinov R.. Mechanisms of transcription factor selectivity. Trends Genet. 2010; 26:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dror I., Rohs R., Mandel-Gutfreund Y.. How motif environment influences transcription factor search dynamics: finding a needle in a haystack. BioEssays. 2016; 38:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kribelbauer J.F., Rastogi C., Bussemaker H.J., Mann R.S.. Low-affinity binding sites and the transcription factor specificity paradox in eukaryotes. Annu. Rev. Cell Dev. Biol. 2019; 35:357–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X.et al.. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K.et al.. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wunderlich Z., Mirny L.A.. Spatial effects on the speed and reliability of protein–DNA search. Nucleic Acids Res. 2008; 36:3570–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brodsky S., Jana T., Mittelman K., Chapal M., Kumar D.K., Carmi M., Barkai N.. Intrinsically disordered regions direct transcription factor in vivo binding specificity. Mol. Cell. 2020; 79:459–471. [DOI] [PubMed] [Google Scholar]
- 23. Brown C.J., Johnson A.K., Daughdrill G.W.. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol. 2010; 27:609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown C.J., Takayama S., Campen A.M., Vise P., Marshall T.W., Oldfield C.J., Williams C.J., Keith Dunker A.. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002; 55:104–110. [DOI] [PubMed] [Google Scholar]
- 25. Cohan M.C., Shinn M.K., Lalmansingh J.M., Pappu R.V.. Uncovering non-random binary patterns within sequences of intrinsically disordered proteins. J. Mol. Biol. 2022; 434:167373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen Ba A.N., Yeh B.J., Van Dyk D., Davidson A.R., Andrews B.J., Weiss E.L., Moses A.M.. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci. Signal. 2012; 5:rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zarin T., Strome B., Nguyen Ba A.N., Alberti S., Forman-Kay J.D., Moses A.M.. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. Elife. 2019; 8:e46883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benz C., Ali M., Krystkowiak I., Simonetti L., Sayadi A., Mihalic F., Kliche J., Andersson E., Jemth P., Davey N.E.. Proteome-scale mapping of binding sites in the unstructured regions of the human proteome. Mol. Syst. Biol. 2022; 18:e10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davey N.E., Shields D.C., Edwards R.J.. Masking residues using context-specific evolutionary conservation significantly improves short linear motif discovery. Bioinformatics. 2009; 25:443–450. [DOI] [PubMed] [Google Scholar]
- 30. Davey N.E., Van Roey K., Weatheritt R.J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., Gibson T.J.. Attributes of short linear motifs. Mol. Biosyst. 2012; 8:268–281. [DOI] [PubMed] [Google Scholar]
- 31. Tompa P., Fuxreiter M.. Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci. 2008; 33:2–8. [DOI] [PubMed] [Google Scholar]
- 32. Bugge K., Brakti I., Fernandes C.B., Dreier J.E., Lundsgaard J.E., Olsen J.G., Skriver K., Kragelund B.B.. Interactions by disorder–a matter of context. Front. Mol. Biosci. 2020; 7:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amin A.N., Lin Y.-H., Das S., Chan H.S.. Analytical theory for sequence-specific binary fuzzy complexes of charged intrinsically disordered proteins. J. Phys. Chem. B. 2020; 124:6709–6720. [DOI] [PubMed] [Google Scholar]
- 34. Choi J.-M., Holehouse A.S., Pappu R.V.. Physical principles underlying the complex biology of intracellular phase transitions. Annu. Rev. Biophys. 2020; 49:107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borg M., Mittag T., Pawson T., Tyers M., Forman-Kay J.D., Chan H.S.. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:9650–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Riback J.A., Katanski C.D., Kear-Scott J.L., Pilipenko E.V., Rojek A.E., Sosnick T.R., Drummond D.A.. Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell. 2017; 168:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langstein-Skora I., Schmid A., Emenecker R.J., Richardson M.O.G., Götz M.J., Payer S.K., Korber P., Holehouse A.S.. Sequence- and chemical specificity define the functional landscape of intrinsically disordered regions. 2022; bioRxiv doi:11 February 2022, preprint: not peer reviewed 10.1101/2022.02.10.480018. [DOI]
- 38. Zarin T., Strome B., Peng G., Pritišanac I., Forman-Kay J.D., Moses A.M.. Identifying molecular features that are associated with biological function of intrinsically disordered protein regions. Elife. 2021; 10:e60220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin E.W., Holehouse A.S., Peran I., Farag M., Incicco J.J., Bremer A., Grace C.R., Soranno A., Pappu R.V., Mittag T.. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. 2020; 367:694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang J., Choi J.M., Holehouse A.S., Lee H.O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D.et al.. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell. 2018; 174:688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zarin T., Tsai C.N., Nguyen Ba A.N., Moses A.M.. Selection maintains signaling function of a highly diverged intrinsically disordered region. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E1450–E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Staller M.V., Holehouse A.S., Swain-Lenz D., Das R.K., Pappu R.V., Cohen B.A.. A high-throughput mutational scan of an intrinsically disordered acidic transcriptional activation domain. Cell Syst. 2018; 6:444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Staller M.V., Ramirez E., Kotha S.R., Holehouse A.S., Pappu R.V., Cohen B.A.. Directed mutational scanning reveals a balance between acidic and hydrophobic residues in strong human activation domains. Cell Syst. 2022; 13:334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ravarani C.N., Erkina T.Y., De Baets G., Dudman D.C., Erkine A.M., Babu M.M.. High-throughput discovery of functional disordered regions: investigation of transactivation domains. Mol. Syst. Biol. 2018; 14:e8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Erijman A., Kozlowski L., Sohrabi-Jahromi S., Fishburn J., Warfield L., Schreiber J., Noble W.S., Söding J., Hahn S.. A high-throughput screen for transcription activation domains reveals their sequence features and permits prediction by deep learning. Mol. Cell. 2020; 78:890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sanborn A.L., Yeh B.T., Feigerle J.T., Hao C.V., Townshend R.J., Lieberman Aiden E., Dror R.O., Kornberg R.D.. Simple biochemical features underlie transcriptional activation domain diversity and dynamic, fuzzy binding to Mediator. Elife. 2021; 10:e68068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmidt H.B., Barreau A., Rohatgi R.. Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun. 2019; 10:4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Holehouse A.S., Ginell G.M., Griffith D., Böke E.. Clustering of aromatic residues in prion-like domains can tune the formation, state, and organization of biomolecular condensates: published as part of the biochemistry virtual special issue “protein condensates. Biochemistry. 2021; 60:3566–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boke E., Ruer M., Wühr M., Coughlin M., Lemaitre R., Gygi S.P., Alberti S., Drechsel D., Hyman A.A., Mitchison T.J.. Amyloid-like self-assembly of a cellular compartment. Cell. 2016; 166:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anand R., Memisoglu G., Haber J.. Cas9-mediated gene editing in Saccharomyces cerevisiae. 2017; https://protocolexchange.researchsquare.com/article/nprot-5791/v1.
- 51. Gietz R.D., Schiestl R.H.. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007; 2:31–34. [DOI] [PubMed] [Google Scholar]
- 52. Zentner G.E., Kasinathan S., Xin B., Rohs R., Henikoff S.. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat. Commun. 2015; 6:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gera T., Jonas F., More R., Barkai N.. Evolution of binding preferences among whole-genome duplicated transcription factors. Elife. 2022; 11:e73225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011; 17:10–12. [Google Scholar]
- 55. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pelechano V., Wei W., Steinmetz L.M.. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013; 497:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park D., Morris A.R., Battenhouse A., Iyer V.R.. Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res. 2014; 42:3736–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Policastro R.A., Raborn R.T., Brendel V.P., Zentner G.E.. Simple and efficient profiling of transcription initiation and transcript levels with STRIPE-seq. Genome Res. 2020; 30:910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Byrne K.P., Wolfe K.H.. The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 2005; 15:1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu A.X., Zarin T., Hsu I.S., Moses A.M.. YeastSpotter: accurate and parameter-free web segmentation for microscopy images of yeast cells. Bioinformatics. 2019; 35:4525–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schmitt A.P., McEntee K.. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Martínez-Pastor M.T., Marchler G., Schüller C., Marchler-Bauer A., Ruis H., Estruch F.. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 1996; 15:2227–2235. [PMC free article] [PubMed] [Google Scholar]
- 64. Görner W., Durchschlag E., Martinez-Pastor M.T., Estruch F., Ammerer G., Hamilton B., Ruis H., Schüller C.. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998; 12:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sadeh A., Baran D., Volokh M., Aharoni A.. Conserved motifs in the Msn2-activating domain are important for Msn2-mediated yeast stress response. J. Cell Sci. 2012; 125:3333–3342. [DOI] [PubMed] [Google Scholar]
- 66. Mészáros B., Erdős G., Dosztányi Z.. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018; 46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pak C.W., Kosno M., Holehouse A.S., Padrick S.B., Mittal A., Ali R., Yunus A.A., Liu D.R., Pappu R.V., Rosen M.K. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol. Cell. 2016; 63:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. He F., Borcherds W., Song T., Wei X., Das M., Chen L., Daughdrill G.W., Chen J.. Interaction between p53 N terminus and core domain regulates specific and nonspecific DNA binding. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8859–8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeng X., Ruff K.M., Pappu R.V.. Competing interactions give rise to two-state behavior and switch-like transitions in charge-rich intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2200559119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mao A.H., Crick S.L., Vitalis A., Chicoine C.L., Pappu R.V.. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:8183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Müller-Späth S., Soranno A., Hirschfeld V., Hofmann H., Rüegger S., Reymond L., Nettels D., Schuler B.. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14609–14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Marsh J.A., Forman-Kay J.D.. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010; 98:2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wiggers F., Wohl S., Dubovetskyi A., Rosenblum G., Zheng W., Hofmann H.. Diffusion of a disordered protein on its folded ligand. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2106690118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chong S., Mir M.. Towards decoding the sequence-based grammar governing the functions of intrinsically disordered protein regions. J. Mol. Biol. 2021; 433:166724. [DOI] [PubMed] [Google Scholar]
- 75. Srivastava D., Muthukumar M.. Sequence dependence of conformations of polyampholytes. Macromolecules. 1996; 29:2324–2326. [Google Scholar]
- 76. Sawle L., Ghosh K.. A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J. Chem. Phys. 2015; 143:085101. [DOI] [PubMed] [Google Scholar]
- 77. Das R.K., Pappu R.V.. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vihinen M., Torkkila E., Riikonen P.. Accuracy of protein flexibility predictions. Proteins. 1994; 19:141–149. [DOI] [PubMed] [Google Scholar]
- 79. DeForte S., Uversky V.N.. Order, disorder, and everything in between. Molecules. 2016; 21:1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Borgia A., Borgia M.B., Bugge K., Kissling V.M., Heidarsson P.O., Fernandes C.B., Sottini A., Soranno A., Buholzer K.J., Nettels D. Extreme disorder in an ultrahigh-affinity protein complex. Nature. 2018; 555:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Davey N.E. The functional importance of structure in unstructured protein regions. Curr. Opin. Struct. Biol. 2019; 56:155–163. [DOI] [PubMed] [Google Scholar]
- 82. Van Roey K., Gibson T.J., Davey N.E.. Motif switches: decision-making in cell regulation. Curr. Opin. Struct. Biol. 2012; 22:378–385. [DOI] [PubMed] [Google Scholar]
- 83. Bremer A., Farag M., Borcherds W.M., Peran I., Martin E.W., Pappu R.V., Mittag T.. Deciphering how naturally occurring sequence features impact the phase behaviours of disordered prion-like domains. Nat. Chem. 2022; 14:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lin Y., Currie S.L., Rosen M.K.. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 2017; 292:19110–19120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chowdhary S., Kainth A.S., Pincus D., Gross D.S.. Heat shock factor 1 drives intergenic association of its target gene loci upon heat shock. Cell Rep. 2019; 26:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tolstorukov M.Y., Jernigan R.L., Zhurkin V.B.. Protein–DNA hydrophobic recognition in the minor groove is facilitated by sugar switching. J. Mol. Biol. 2004; 337:65–76. [DOI] [PubMed] [Google Scholar]
- 87. Gupta A., Kulkarni M., Mukherjee A.. Accurate prediction of B-form/A-form DNA conformation propensity from primary sequence: a machine learning and free energy handshake. Patterns. 2021; 2:100329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Basham B., Schroth G.P., Ho P.S.. An A-DNA triplet code: thermodynamic rules for predicting A- and B-DNA. Proc. Natl. Acad. Sci. 1995; 92:6464–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tolstorukov M.Y., Ivanov V.I., Malenkov G.G., Jernigan R.L., Zhurkin V.B.. Sequence-dependent B↔A transition in DNA evaluated with dimeric and trimeric scales. Biophys. J. 2001; 81:3409–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing reads and genomic profiles are available on GEO (GSE212466). Additional sequencing data for previously profiled mutants (22) were taken from Bioproject: PRJNA573518. All Matlab scripts to analyze the data are available on GITHUB (https://github.com/barkailab/Carmi2022) and on Zenodo (DOI: 10.5281/zenodo.7677339).