


Abstract
The marine thermophilic archaeon Nanoarchaeum equitans possesses a monomeric primase encompassing the conserved domains of the small catalytic and the large regulatory subunits of archaeoeukaryotic heterodimeric primases in one protein chain. The recombinant protein primes on templates containing a triplet with a central thymidine, thus displaying a pronounced sequence specificity typically observed with bacterial type primases only. The N. equitans primase (NEQ395) is a highly active primase enzyme synthesizing short RNA primers. Termination occurs preferentially at about nine nucleotides, as determined by HPLC analysis and confirmed with mass spectrometry. Possibly, the compact monomeric primase NEQ395 represents the minimal archaeoeukaryotic primase and could serve as a functional and structural model of the heterodimeric archaeoeukaryotic primases, whose study is hindered by engagement in protein assemblies and rather low activity.
INTRODUCTION
In all cellular organisms, accurate copying of chromosomal DNA is required for a cell to divide successfully. DNA replication is the process by which DNA copies itself before cell division, utilizing a multiprotein machinery called the replisome. The DNA double helix is unwound by DNA helicases creating the replication fork, where the separated strands serve as templates for the synthesis of complementary DNA strands. Because of their opposite orientations and the fact that DNA polymerases only proceed in the 5′ to 3′ direction, the two strands are replicated differently. Upon initial priming, the so-called leading strand is continuously replicated by a processive DNA polymerase, whereas the lagging strand is discontinuously replicated, resulting in a more complex copying mechanism requiring a new primer for each Okazaki fragment.
Replicative DNA polymerases of cellular genomes are generally not able to synthesize new DNA strands de novo. Rather, they require a free 3′-OH of a primer already bound to the template strand to start the polymerization reaction. These primers are short RNA molecules synthesized by specialized RNA polymerases called DNA primases, which are capable of de novo synthesis of RNA chains on single-stranded DNA templates. Therefore, in the context of the fast-moving replisome, close cooperation between DNA primase and DNA polymerase on the lagging strand is required.
The basic organization of DNA replication is conserved over all domains of life, but the proteins of the replication machinery are not (1). Primases fall into two broad groups: bacterial primases and archaeoeukaryotic primases (AEPs), which do not share structural homology. Nevertheless, the unrelated protein folds of the enzymes perform the same function and probably share the same reaction mechanism for primer synthesis as highlighted by a similar arrangement of acidic catalytic residues in their active sites.
In bacteria, primases contain a characteristic catalytic domain displaying a topoisomerase-primase (TOPRIM) fold consisting of an α/β core with four conserved β-strands and three α-helices together with two conserved catalytic motifs (2). The monomeric cellular primase (DnaG) is typically associated with replicative DNA helicase (DnaB) and forms a complex that synthesizes a short RNA primer which can be extended by the bacterial replicative DNA polymerase (DnaE).
In contrast, archaeoeukaryotic primases involved in cellular genome replication are generally heterodimeric proteins consisting of one small catalytic subunit (PriS/AE_Prim_S) and one large accessory subunit (PriL). The catalytic core of PriS folds into a highly derived version of the RNA recognition motif (RRM) comprising four β-strands and two α-helices with three conserved catalytic motifs (3). In addition to the catalytic domain, the non-catalytic PriL appears to function as a regulatory subunit, that can be found in eukaryotic primases as well as many of the corresponding archaeal primases. PriL might act in different ways in the context of primer synthesis: Experimental evidence suggest that PriL could stabilize the primase-DNA complex (4), keeps the primase in the correct orientation for initiation (5) or determines the length of the primer (6–8).
PriL folds into a two-domain structure comprised of a larger, globular helical domain harboring a [4Fe–4S] cluster and a smaller mixed α/β domain that binds to PriS, indicating a significant conformational flexibility of the PriL subunit (9). The crystal structure of human primase revealed that the [4Fe–4S] cluster is buried inside the protein fulfilling a structural role (9). A switch of the iron oxidation state by charge transport along DNA might modulate the affinity of primases towards DNA as demonstrated for human as well as yeast primase (10,11). However, this hypothesis has been questioned (12) and further studies would be valuable to clarify the functional contribution of the [4Fe–4S] cluster towards primer synthesis. Of note, the [4Fe–4S] cluster itself does not appear to be directly involved in primer synthesis, as PriX, a variant of PriL observed in several orders of Crenarchaeota including Sulfolobus solfataricus (13), is devoid of a [4Fe–4S] cluster. A complex of PriS with a truncated fusion protein of PriX-PriL, which no longer contains a [4Fe–4S] cluster, is highly active (12). Thus, the exact contribution of the [4Fe–4S] cluster for primase activity remains unresolved. The cluster could have a purely structural role, but this alone is not easily reconciled with its high conservation in the domains of eukaryota and archaea. Possibly, the cluster could add an additional layer of primase regulation by taking advantage of the different charge states of the Fe ions.
In eukaryotic replication, PriS and PriL are tightly associated with DNA polymerase α (Pol α) and its B subunit (also called p70 subunit) constituting the primosome, a subcomplex of the replisome. Once a primer reaches its unit length of 8 to 12 nucleotides, the primer template is extended by Pol α with DNA to form a covalent DNA-RNA hybrid that is recognized by the replicative polymerase (6,14–16). Archaea do not possess orthologs of Pol α or the B subunit, indicating that the PriS-PriL heterodimer might be sufficient for the synthesis of primers which are then elongated by the replicative polymerase. In fact, for the well-studied archaea Pyrococcus furiosus, Pyrococcus abyssi, Archaeoglobus fulgidus and Haloferaxx volcanii the heterodimer PriS-PriL is probably the active primase (17–19). Notwithstanding, some orders of Crenarchaeota contain a third primase subunit PriX, which is a distant homologue of the C-terminal domain of eukaryotic PriL. In these organisms PriS appears to form a stable and highly active heterotrimer with PriL and PriX with a 1:1:1 stoichiometry. In the case of Sulfolobus solfataricus, it is proposed that the initiation, elongation and termination of primer synthesis is modulated by concerted interactions with the two non-catalytic subunits with PriS (13).
The predominantly heterodimeric organization of the primases into a small catalytic subunit and a large accessory subunit is typical for eukaryota and most archaea. In contrast to this, a fusion protein of both subunits is present in the archaeon N. equitans. It is not uncommon for functionally related proteins to fuse in some genomes. Systematic analysis of such gene fusions aids in the functional annotation of unknown genes and has been coined Rosetta stone (20).
Nanoarchaeaum equitans is a marine archaeon and lives as an obligate symbiont of the archaeal genus Ignicoccus (21,22). It grows optimally at about 80°C in slightly acidic environments around pH 6 and a salt concentration of 2%. The organism has a heavily reduced and compact genome of only 490 kilobasepairs. The reduction in genome size might have promoted the fusion leading to the single subunit archaeoeukaryotic primase (NEQ395, AAR39242.1). Remarkably, no other archaeoeukaryotic primase domain has been detected in the N. archaeaum genome (13,23,24), and furthermore, this primase gene fusion is also present in numerous other members of the archaeal superphylum DPANN. This underscores that monomeric primases such as NEQ395 perform their role as cellular replicative primase in Nanoarchaeaum equitans and other related archaea.
Monomeric archaeoeukaryotic primases have been reported before, but these primases are primarily responsible for the replication of extrachromosomal elements (e.g. repB’ from the bacterial plasmid RSF1010 (25), the replication protein from the archaeal plasmid pRN1 (26) and the polymerase protein from Nitratiruptor phage NrS-1 (27)). These primases are fusions of a PriS domain with helix bundle domains whose evolutionary relationship is unclear but may share functional homology with PriL (28). In contrast, the cellular primase NEQ395 from N. equitans is clearly a fusion of PriS and PriL and thus strongly resembles the cellular archaeoeukaryotic primases.
The superfamily of archaeoeukaryotic primases is structured into three major clades (3): The AEP proper clade including the NHEJ polymerase of bacteria and the name giving archaeoeukaryotic small subunit primases. The second clade is dominated by viral primases and the third clade, termed prim-pol clade, contains primases of various plasmids and phages as well as the prim-pol enzymes which are capable of priming and DNA polymerization (29). These enzymes mostly use dNTP as precursors (29–31). The long awaited structure of the initiation complex of primase, template and initiation as well as elongating nucleotide was finally determined with a member of the prim-pol clade (32). Representatives of the diverse superfamily of archaeoeukaryotic primases are therefore not only typical RNA primases, but also DNA primases and DNA polymerases with functions including repair, non-homologous end joining, trans-lesion synthesis or stalled replication fork recovery (33).
Bacterial and archaeoeukaryotic primases differ in terms of priming specificity: Prokarya typically initiate primer synthesis at a defined priming site, as shown for DnaG from Escherichia coli recognizing a 5′-CTG priming site (34), Aquifex aeolicus preferring 5′-CCC (35), Staphylococcus aureus recognizing 5′-CTA (36) or the T7 primase from Bacteriophage T7 using a 5′-GTC (37). In contrast, archaeoeukaryotic primases are considered to prime sequence-unspecifically although it is known that a purine is preferred as the 5′-terminal nucleotide of the primer (38,39). In general, the question whether archaeoeukaryotic primase activity is also sequence specific appears not to have been investigated systematically, possibly due to the low specific activities of these enzymes. Notwithstanding, we and other could demonstrate that in particular the monomeric archaeoeukaryotic primases of extrachromosomal elements display sequence specificity: RepB’ from the bacterial plasmid RSF1010 primes downstream of a hairpin (40). The structure of the RepB’ together with template DNA has been solved and shows that the hairpin structure is recognized by the catalytic domain of RepB’ (25). The phage NrS-1 polymerase primes most efficiently at 5′-NTTGPuPyPyA-3′ and strikingly an inverted repeat of these sequences is found downstream of the polymerase gene and could constitute the replication origin of this phage (27). The molecular basis of this pronounced sequence specificity has however not been elucidated yet, but more recent work (41) suggests that the priming specificity is more relaxed than previously reported. Our group demonstrated that the plasmidal archaeoeukaryotic primase from Sulfolobus islandicus has a strong preference for the motif 5′-GTG (42) and that this trinucleotide is specifically recognized by the helix bundle domain in the presence of ATP. We therefore suggested that the function of the helix bundle domain is to prepare dinucleotide formation by binding template and nucleotides (28).
In view of better understanding the primase reaction mechanism and in particular the interplay between the catalytic subunit (PriS) and the accessory domain (PriL) we recombinantly expressed and characterized the primase from N. equitans. Its compact organisation, stability and high activity make this protein more amenable to structural and functional studies compared to the more complex organisation of the eukaryotic primases while maintaining its core functionality and structure. For these reasons we consider the primase from N. equitans a suitable model enzyme of archaeoeukaryotic DNA primases.
MATERIALS AND METHODS
Materials
Synthetic DNA oligodeoxynucleotides used as primer-template substrates or ssDNA primer templates were synthesized and HPLC-purified by Microsynth AG (Table 1).
Table 1.
Oligodeoxynucleotides used for the following experiments. If present, the potential priming trinucleotide sites are printed in bold
| Primer extention assay | |
| Primer-template | 5′-FAM-TGTAAAACGACGGCCAGT |
| substrate | 3′- ACATTTTGCTGCCGGTCACGGTTCGAACGTACGGACGTCCAGCTGAGA |
| Primase assay | |
| t20 | 5′-GGAC ATA AATTG GTC ATTAC |
| I | 5′-GGAC ATA AAT |
| II | 5′- TG GTC ATTAC |
| III | 5′- TAAATTG GTC |
| IV | 5′-GGAC ATA AATTG GTC |
| V | 5′- TAAATTG GTC ATTAC |
| VI | 5′- GTC ATTAC |
| VII | 5′- ATTG GTC ATTAC |
| VIII | 5′- AATTG GTC ATTAC |
| IX | 5′- GTA AATTG GTC ATTA |
| t36 | 5′-AAAAGCGGCCGCTCATTT ATCATCATCATC TTTATA |
| t13 | 5′-AGTTG GTA ATTAC |
| Fid1 | 5′-AAAAAAAAACGATAAAAAAA |
| Fid2 | 5′-AAAAAAACGAAATAAAAAAA |
| Fid3 | 5′-AAAAACGAAAAATAAAAAAA |
| Fid4 | 5′-AAACGAAAAAAATAAAAAAA |
| OligoA | 5′-AAAAAAAAAAAAAAAAAAAA |
| OligoA+ATA | 5′-AAAAAAAAAAAATAAAAAAA |
| OligoA+CGATA | 5′-AAAAAAAAACGATAAAAAAA |
| OligoT | 5′-TTTTTTTTTTTTTTTTTTTT |
| OligoT+ATA | 5′-TTTTTTTTTTTATATTTTTT |
| OligoT+CGATA | 5′-TTTTTTTTTCGATATTTTTT |
| OligoC | 5′-CCCCCCCCCC |
| OligoC+ATA | 5′-CCCATACCCC |
| OligoC+AGATA | 5′-CAGATACCCC |
| t15 | 5′-TCTATAATGAGGATT |
| t15-ATA | 5′-TCTAGAATGAGGATT |
| t17 | 5′-CTCTACGCTATAATTAC |
| t17-ATA | 5′-CTCTACGCTTTTATTAC |
| t21 | 5′-TCGTTAGCTATATTCATGTTT |
| t21-ATA | 5′-TCGTTAGCTCTATTCATGTTT |
| DNA binding assay | |
| t9 | 5′-FAM-CTATACTCA |
| t9-ATA | 5′-FAM-CTGTGCTCA |
| t18 | 5′-FAM-TGTAAAACGACGGCCAGT |
| t18_ds | 5′-FAM-TGTAAAACGACGGCCAGT |
| 3′- ACATTTTGCTGCCGGTCA | |
Recombinant expression and purification of NEQ395
Wildtype NEQ395 (genbank accesssion AAR39242.1) as well as the point-mutants were expressed recombinantly in E. coli under the control of the T7 promoter. The gene sequence was synthesized codon-optimized (GeneScript Biotech) and the active site mutations were introduced using the QuickChange PCR protocol with two overlapping mutagenic oligonucleotide primers. After multiple cycle extension, parental DNA was digested by DpnI and successful mutations were verified by Sanger sequencing (Microsynth AG).
Expression plasmids incorporating a decahistidine tag at the C-termini of the proteins were transformed into E.coli BL21-CodonPlus (DE3)RIPL and cells then plated on LB agar containing 50 μg/mL kanamycin. Freshly transformed single colonies were inoculated into LB medium (50 μg/mL kanamycin, 200 μM ammonium iron (III) citrate and 30 μM l-methionine) and grown at 37°C until OD600 reached 0.6. Recombinant expression was induced by adding 1 mM isopropyl-beta-D-thiogalactoside (IPTG) and cultivation continued for 3 h at 30°C. Bacteria were sedimented (9000 g, 20 min) and the pellet resuspended in 25 mM Na-phosphate pH 8.0, 150 mM NaCl, 0.1% Triton X-100 (10 ml buffer per g of cells, supplemented by a cOmplete™ EDTA-free Protease Inhibitor Cocktail Tablet per 4 g of cells (Roche)). Cells were lysed by sonication (Branson Ultrasonics™ Sonifier S-250A; big needle, output level 8, 20% pulse, 2 × 15 min) followed by centrifugation (20 000 g, 4°C, 20 min) to clarify the whole cell lysate. From the supernatant, NEQ395 was purified by Co2+ Talon® affinity chromatography under N2 atmosphere to prevent the [4Fe–4S] cluster from oxidation. The clarified lysate was loaded onto the affinity column (5 mL) and bound protein washed with 5 column volumes (cv) of 25 mM Na-phosphate pH 8.0, 1 M NaCl to remove unspecifically interacting DNA and bacterial proteins followed by an imidazole step-gradient (5 cv—10 mM, 5 cv—20 mM, 3 cv—250 mM; in 25 mM Na-phosphate pH 8.0, 150 mM NaCl). Eluted protein was dialysed against 50 mM HEPES pH 6.5, 100 mM NaCl, 1 mM MnCl2, 1 mM DTT and finally concentrated by ultrafiltration using Amicon® Ultra centrifugal filters to approximately 500 μM. According to SDS-PAGE analysis, the protein preparation reached a purity of ∼95%. In order to avoid oxidation of the 4Fe–4S cluster no further purification steps were usually undertaken.
For some initial experiments the 250 mM imidazole fraction was concentrated to a volume of 2 mL and directly loaded with 25 mM HEPES pH 6.5, 150 mM NaCl onto a 150 mL Superdex 60/600 prep grade size exclusion chromatography column. Pooled fractions were dialysed against 50 mM HEPES pH 6.5, 100 mM NaCl, 1 mM MnCl2, 1 mM DTT and finally concentrated to approximately 500 μM. Protein concentrations were determined by using the theoretical extinction coefficient at 280 nm of 72 560 M−1cm−1. The yield of iron-sulfur cluster incorporation was calculated using an extinction coefficient at 410 nm of 16 000 M−1cm−1 (43).
Primer extension assay
Both strands of the primer-template substrate (see Table 1) were annealed in reaction buffer by the following temperature program: 98°C for 2 min, 70°C for 5 min, 50°C for 10 min, 40°C for 5 min, 25°C for 30 min. Reaction mixtures contained 1.0 μM enzyme, 100 nM primer-template substrate and 100 μM of the relevant rNTPs (or dNTPs) in 25 mM Tris/HCl pH 6.5, 10 mM MnCl2, 1 mM DTT. The polymerase assay was performed in a total volume of 10 μl. Reaction mixtures were incubated for 60 min at 50°C and the enzymatic reaction quenched by adding an equal volume of 20 mM EDTA, 0.25% (w/v) bromophenol and 0.25% (w/v) xylene cyanol in 95% (v/v) formamide. After heat treatment (90°C, 3 min), 10 μl of the reaction mixture was loaded onto 20% denaturing urea polyacrylamide gels (10 mM Tris/HCl pH 7.6, 10 mM boric acid, 200 μM ethylenediaminetetraacetic acid (EDTA), 7 M urea, acrylamide/bis (19:1)) and fluorescence images of the separated reaction products taken with an Fujifilm FLA-9000 image scanner. In case of the incorporation of 5-propargylamino-CTP-6-FAM (FAM-CTP), the rNTP composition was optimized to 100 μM ATP/GTP/UTP, 20 μM CTP and 1 μM FAM-CTP to achieve an efficient fluorophore incorporation. Likewise, in case of labelled UTP (5-(3-Aminoallyl)-UTP-6-FAM) the concentrations were adjusted as follows: 100 μM ATP/CTP/GTP, 20 μM UTP and 1 μM FAM-UTP.
Primase assay
Reactions were carried out in 25 mM Tris/HCl pH 6.5, 1 mM MnCl2, 1 mM DTT containing 1.0 μM enzyme, 5.0 μM ssDNA template and an rNTP composition optimized previously in the primer extension assay (100 μM ATP/GTP/UTP, 20 μM CTP and 1 μM FAM-CTP or 100 μM ATP/CTP/GTP, 20 μM UTP and 1 μM FAM-UTP). Assays were incubated at 70°C for 30 min, quenched, heat-denatured as described above and loaded onto a 30% denaturing urea polyacrylamide gel for separation and analysis.
In case of the HPLC-based assay, the fluorophore containing rNTP composition was replaced by 100 μM unlabeled rNTPs (each). Reactions were carried out in 15 μl and stopped by addition of 15 μl 15 mM EDTA pH 8.0. The resulting mixtures were centrifuged (5 min, 16 000 g) prior to analysis by HPLC (see below). The discrimination of NEQ395 between rNTPs and dNTPs at both the initiation and elongation site were tested by replacing an rNTP by the same concentration of a dNTP. For these assays we used the template Fid2 (Table 1).
To determine the apparent Michaelis-Menten constant Km and kcat, varying concentrations of template t13 (Table 1) or rNTPs were used in a primase assay with 0.75 μM enzyme. Varying concentrations of one of the substrates were added to the mix, while the other was kept constant at saturating concentration. Here, reaction mixtures were incubated for 20 min at 70°C and saturating concentrations of 0.5 mM template t13 and 0.25 mM rNTPs were used. The primer product consisting of seven nucleotides was quantified by HPLC and kinetic parameters obtained by using Michaelis-Menten kinetics for excess-substrate inhibition 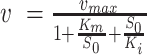 (44).
(44).
HPLC and mass spectrometry
20 μl of the primer synthesis reaction was analyzed by reverse phase (RP)-HPLC under thermally denaturing conditions at 55°C. The products of primase reactions were separated and analyzed on an Agilent 1100 Series HPLC system equipped with a photodiode array detector using an XBridge® C18 3.5 μm (3.0 mm inner diameter, 150 mm length) RP-HPLC column (Waters). A gradient elution was carried out using as solvent A 8 mM triethylamine and 200 mM hexafluoroisopropanol in water and as solvent B a mixture of 50% (v/v) A and 50% (v/v) methanol. De novo primer synthesis was monitored upon elution by measuring absorbance at 260 nm using a flow rate of 1 ml/min and a gradient from 5 to 50% B over 8 min. Amounts of primer products were quantified from the peak areas based on extinction coefficient and sequence.
In case of preparative RP-HPLC, the injection volume was increased to 100 μl and the appropriate peak fraction collected manually. After lyophilizing, the primer product was redissolved in water and identified by ESI mass spectrometry at the mass spectrometry facility of the University of Berne.
DNA binding assay
The DNA binding affinity of NEQ395 was determined by fluorescence anisotropy titration measurements. The experiments were performed with a Cary Eclipse Spectrophotometer by measuring the fluorescence at 20°C. Oligodeoxynucleotides (50 nM, Table 1) used for this assay were labelled with FAM at the 5′-end and binding isotherms obtained by reverse titration of NEQ395. Reactions were caried out in 25 mM Tris/HCl pH 6.5, 1 mM DTT and the protein concentration gradually decreased by replacing 37.5% of the mix in the cuvette with a solution of DNA in the same buffer (1.6x dilution). Fluorophores were excited at 490 nm (10 nm slit width) and the anisotropy was measured by monitoring the emission at 530 nm (10 nm slit width) and an integration time of 2 s. For the calculation of the dissociation constant (KD) the intensity of the bound and unbound labelled DNA was taken into account. At least three measurements per data point were collected and the data fitted with a single-site binding model using the following formula:  with
with 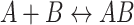 (45).
(45).
Structural modelling
The structure of NEQ395 was predicted with RoseTTAFold (46) using the webinterface provided through https://robetta.bakerlab.org/. The AlphaFold2 predictions (47) were performed with Jupyter Notebooks provided by Colabfold (48) using MMseqs2 (49) for homology detection. In both cases five models were generated with the standard settings. Structural superposition and visualization of the model was performed with Chimera (50).
RESULTS AND DISCUSSION
Domain structure of NEQ395
Archaea and eukaryota typically encode primases consisting of a small catalytic (PriS/AE_Prim_S) and a large accessory (PriL) subunit that are tightly associated as heterodimer. Here, we report a primase from Nanoarchaeum equitans (NEQ395, AAR39242.1) that is one of the rare examples of archaeoeukaryotic primases (AEPs) in which both primase subunits are expressed in a single polypeptide chain. Importantly, it is the only primase in N. equitans identified by analysis of the genome (AE017199.1). Furthermore the NEQ395 primase is detected in transcriptomic and proteomic studies underscoring its role as cellular primase for genome replication (23,24,51). The highly compact enzyme is smaller than the known heterodimeric AEPs but still shares important structural features. The shared core of the catalytic domain corresponds to an N-terminal (αβ)2 fold previously identified in human and archaeal primases (7,9,52,53) (annotated as α2β1 and α3β3 in human primase in Supplementary Figure S1A), and a domain that is considered as a derived version of the RNA recognition motif (RRM) with a β1α1β2β3α2β4 topology that forms a β-sheet packed against two α-helices (54) (Figure 1A and Supplementary Figure S1A). The catalytic core shared in all AEPs harbors three conserved motifs within the RRM fold including a highly conserved triad of acidic residues and a histidine residue (29,33): a hhhDhD/E motif (‘h’ is a hydrophobic residue) in strand 5, an sxH motif (‘s’ is a small residue, ‘x’ is any residue) in strand 7, and an hD/E motif in strand 8. Strand 8 can sometimes adopt a helical configuration like observed for human primase (9). The hhhDhD/E and hD/E motifs establish the coordination of divalent metal ions for catalysis, whilst the function of the sxH is less clear, potentially the histidine acts as a general base during the elongation reaction (52,55,32). Remarkably, the C-terminal domain of NEQ395 contains an arrangement that is typically documented in PriL of higher eukaryotes including a [4Fe–4S] cluster (56) (Figure 1A and Supplementary Figure S1B, coordinating cysteine residues in yellow).
Figure 1.

Domain structure of the monomeric DNA primase from Nanoarchaeum equitans and the heterodimeric primase from Homo sapiens and purification of NEQ395. (A) The conserved domain structure for the primases is given. Domains (numbers are bordering amino acid positions) with related function are colored similarly. For the catalytic domain of archaeoeukaryotic primases (AE_Prim_S) the four catalytic β-strands of the RRM fold containing the active site are highlighted with green bars (sequence alignment in Supplementary Figure S1A). The white box in the AE_Prim_S of the human primase is an unconserved helical insert. Likewise, in the large primase subunit (PriL) the positions of the coordinating cysteine residues of the [4Fe–4S] cluster are indicated by yellow bars (sequence alignment in Supplementary Fig. S1B). Sequence homology between the proteins was analyzed by HHpred (57). The extent of the binary alignment is represented by grey trapezoids and the alignment score S is given. The figure is drawn to scale. (B) SDS-PAGE of purified wildtype NEQ395 and investigated mutants. Recombinant His-tagged proteins were purified by Co2+ chelate chromatography under N2 atmosphere. (C) Appearance and UV-VIS spectra of the wildtype and a mutant devoid of the [4Fe–4S] cluster.
Due to the monomeric assembly and the similarity to human primase, particularly regarding the [4Fe–4S] cluster, NEQ395 could serve as an easily tractable model for eukaryotic primases, that are not only much larger, but also engaged in more complex protein assemblies.
In order to investigate the biochemical properties of the Nanoarchaeum equitans primase, NEQ395 (residues 1 to 393, 46.8 kDa) was recombinantly expressed in E. coli. A His10 tag was cloned C-terminally and used for an immobilized metal chelate-based affinity chromatography under N2 atmosphere to obtain a highly purified protein (49.3 kDa, Figure 1B). As expected for a protein containing an [4Fe–4S] cluster, the solution of the purified protein is brownish (Figure 1C top).
Next, we determined the quaternary structure of NEQ395 and found that the primase elutes as a monomer from an analytical gel permeation column (GPC) thus confirming its monomeric structure in solution (Supplementary Figure S2).
NEQ395 shows RNA polymerase activity
Classically, eukaryotic primases prefer ribonucleotides to prime single-stranded DNA in order to start the replication (29,58). However, the properties of DNA primases isolated from different archaea vary. For example, primases such as the plasmid-encoded pRN1 primase from Sulfolobus islandicus are known to synthesize mixed primers consisting of a ribonucleotide as the first nucleotide in the primer followed by dNTPs (42), or the primase from Pyrococcus furiosus that preferentially synthesizes DNA primers (19).
Lacking information on substrate requirements and reaction conditions for the primase activity of NEQ395, a primer extension assay to measure the inherent activity to elongate primers was the most practical approach to find suitable reaction conditions for a subsequent primase assay.
We therefore first investigated, if NEQ395 can extend a primer oligonucleotide hybridized to a template ssDNA with ribonucleotides or deoxynucleotides. Indeed, our initial characterization using denaturing urea polyacrylamide gel electrophoresis and fluorescent imaging by a fluorescein amidite (FAM) label at the 5′-end of the primer revealed that the monomeric NEQ395 is able to extend an artificial primer-template substrate (Table 1) up to the full length of the template strand in the presence of rNTPs but not dNTPs (Figure 2A). Thus, rNTPs are required and sufficient for the primer elongation activity of NEQ395.
Figure 2.

NEQ395 extends primer-template substrates. (A) NEQ395 (1.0 μM) was incubated with primer-template substrate (100 nM) and primer extended when incubated with rNTPs (100 μM each) and MnCl2 or MgCl2 (10 mM) at 50°C for 60 minutes. No elongation was observed using dNTPs (100 μM) or the NEQ395 mutants (D87A+D89A and H125A) confirming RNA polymerase activity of NEQ395. Replacing MnCl2 with MgCl2 dramatically reduced the elongation efficiency. (B) FAM-CTP is readily incorporated into the polymerization of the primer-template substrate when using an optimized rNTP composition (100 μM ATP/GTP/UTP, 20 μM CTP and 1.0 μM FAM-CTP). Similar results were observed for the incorporation of FAM-UTP. The same primer-template substrate as in Figure 2A was used but instead labelled with Cy5.5. Klenow fragment (large fragment of DNA polymerase I) was used as positive control for the primer extension assay.
Next, we optimized the reaction conditions using the primer extension assay in order to have a good starting point for the later investigation whether NEQ395 also has genuine primase activity. The polymerase activity required a slightly acidic environment with an optimum pH of 6.5 (Supplementary Figure S3A). Furthermore, NEQ395 required Mn2+ to extend a primer, whereas the efficiency was dramatically reduced when replacing Mn2+ with Mg2+ (Figure 2A). To exclude the possibility that other contaminating proteins were responsible for the observed extension activity, we replaced wildtype NEQ395 with the mutants D87A+D89A or H125A (Figure 2A). These mutations target the highly conserved catalytic residues of the primase active site. The active site mutants were devoid of elongation activity, as expected. Next, the temperature dependence and kinetics of the polymerase activity were assayed. NEQ395 showed highest polymerase activity at 50°C. Higher temperatures led to shorter polymerization products, probably due to a partially melted and/or degraded primer-template substrate (Supplementary Figure S3B). Although displaying robust and reproducible activity, the enzyme required nearly one hour to elongate the primer-template substrate under the reaction conditions (Supplementary Figure S3C) pointing towards rather slow elongation kinetics.
Next, we investigated and optimized the ability of NEQ395 to incorporate fluorophore-labeled rNTPs that were required to set up a non-radioactive electrophoresis-based primase assay. The fluorescent nucleotide analogue, 5-propargylamino-CTP-6-FAM (FAM-CTP) was added to the mixture of unlabeled rNTPs (Figure 2B). The fluorescence of incorporated FAM-labeled CTP was compared with the fluorescence of the 5′-Cy5.5-labeled DNA primer-template. To balance the competition between labeled to unlabeled nucleotides and to account for the reduced incorporation rate of modified nucleotides we assayed different ratios and found that optimal fluorophore incorporation was achieved with 100 μM ATP/GTP/UTP, 20 μM CTP and 1 μM FAM-CTP. Similar results have been gathered with aminoallyl-UTP-6-FAM (FAM-UTP). These experiments indicated the feasibility of investigating the primase activity using fluorescent nucleotides followed by gel electrophoresis.
NEQ395 is a primase
After having demonstrated that NEQ395 has primer elongation activity, we proceeded to test the ability of NEQ395 to prime on single-stranded DNA. Instead of a labeled DNA primer-template substrate, the enzyme was incubated with various single stranded oligodeoxynucleotide templates with lengths between 13 and 45 nucleotides (sequences given in Supplementary Figure S4A) in presence of the optimized unlabeled/labeled rNTP composition. When analyzing the results of this initial gel-based primase assay, we observed that not all the templates supported formation of oligonucleotide products containing the fluorophore, suggesting that template sequence and/or structure requirements exist for priming by the enzyme (Supplementary Figure S4B). It is also evident that most of the fluorescent reaction products have a length of less than 10 nucleotides. The ssDNA templates t20 and t36 were selected for further investigation because the relatively short template t20 (20 nucleotides) led to a distinct and intensive product band pattern, whereas NEQ395 did not appear to produce any primers on the longer template oligodeoxynucleotide t36 (36 nucleotides) (Figure 3, Supplementary Figure S4 and Table 1). No primer products were observed when the primase assays were repeated with the active site mutants D87A+D89A or H125A, suggesting that the wild-type NEQ395 enzyme has genuine primase activity. As an additional control, the experiments were repeated with FAM-UTP instead of FAM-CTP and similar results were observed in each reaction.
Figure 3.

NEQ395 efficiently primes on certain templates. When incubating NEQ395 (1.0 μM) with the previously optimized rNTP composition (100 μM ATP/GTP/UTP, 20 μM CTP and 1.0 μM FAM-CTP), MnCl2 (10 mM) and template t20 or t36 (5.0 μM, Supplementary Figure S4) respectively, only template t20 led to observable reaction products implicating a preference for a priming sequence that is absent in template t36. As expected, the mutants D87A+D89A and H125A did not synthesize any primers. Same observations were made using FAM-UTP.
Identification of the minimal template
To better define the sequence requirements of NEQ395 priming we selected template t20 as a starting point to elucidate the minimal template for priming. Different truncated variants of this 20-mer oligodeoxynucleotide were used as templates in subsequent experiments (Table 1 and Figure 4A). NEQ395 synthesized a distinct product of three nucleotides when the 3′-half (template II) of the oligodeoxynucleotide sequence t20 was present in the reaction mixture. In contrast, weak or no reaction was observed when incubating the enzyme with the 5′-half (template I) or the central part (template III). If the 5′-end of template II is further shortened, the primer synthesis is interrupted (see template VI). In contrast, the length of the primer product increases, if the 5′-end of template II is elongated beyond the original length (see template V, VII, VIII and IX). Note that template II does not support a measurable fluorescent primer production by NEQ395 when incubated with FAM-UTP due to the absence of the complementary adenine in the template strand (compare with FAM-CTP). Common to all functional template variants (Figure 4A, compare functional templates in green versus non-functional templates in red with the sequences shown in Table 1) is the sequence 5′-TGGTCATTA, suggesting that a recognition site for primer synthesis exist in this nine nucleotide long sequence.
Figure 4.

Identification of the minimal template substrate. (A) NEQ395 was incubated with different variants of ssDNA template t20 (Table 1). If the sequence 5′-TGGTCATTA (Template II) was present in the template, primers were detected. Templates leading to specific primer products are colored in green, whereas no synthesis was observed with the templates colored in red. (B) Temperature dependency and time course of the primase reaction was investigated with template t13 (see Table 1). NEQ395 functions over a broad temperature range with highest activity around 70°C and the enzyme showed a continuous product formation over a period of 120 min. (C) NEQ395 (1.0 μM), rNTPs (100 μM) and template t13 (5.0 μM) were incubated for 30 min at 70°C and analyzed by RP-HPLC. Unincorporated rNTPs as well as hydrolyzed rNTPs eluted between 0.5 and 2 min, whereas the 13-mer ssDNA template was detected at 6.98 min (black line). When adding NEQ395, a further sharp signal appeared at 5.15 min (red line). This retention time is expected for a primer of seven nucleotides and MS analysis identified the product as 5′-pppACCAACU (Supplementary Figure S5A). Addition of dNTPs instead of rNTPs did not lead to primer-product formation (blue line). (D) Variation of the 5′-end of the 13-mer ssDNA template showed that the primer synthesis stops at a primer length of 9 nucleotides (top). Furthermore, the presence of at least six bases 3′ of the priming site is necessary for efficient priming (bottom). Numbers in the chromatogram represent length of template and primer (with asterisk).
Having identified a suitable template which yields a clearly defined primer with either fluorescently labeled nucleotide, namely template VII, we next studied the temperature dependence and kinetics of primer formation (Figure 4B). The primase activity of NEQ395 is robustly observed over a broad temperature range from 37 to 90°C with an optimum at 70°C, in agreement with the organism's thermophilic growth optimum. In contrast, the synthesis of the fully elongated polymerization product shown before was less efficient at higher temperatures (Supplementary Figure S3B) possibly due to a melted primer-template substrate or because primer extension is only a side activity of the primase.
FAM-CTP and FAM-UTP are relatively bulky compared to the unlabeled nucleotide and, not surprisingly, are only reluctantly incorporated by NEQ395 (Figure 2B). Having established the basic substrate and nucleotide requirements of NEQ395 and in view of the robust activity of the primase, we therefore tempted to establish a label-free and quantitative HPLC-based assay. Reaction mixtures were analyzed by RP-HPLC under denaturing conditions at 55°C and primase products were detected upon elution by monitoring the UV absorbance at 260 nm. Unincorporated as well as hydrolyzed rNTPs eluted between 0.5 and 2 min, whereas the 13-mer ssDNA template t13 (see Table 1) was detected at a retention time of 6.98 min (Figure 4C, black line). When adding NEQ395, a further analyte eluted as a sharp signal at 5.15 min (Figure 4C, red line). The HPLC method separates oligonucleotides primarily by length. Based on the retention time of reference oligonucleotides (data not shown), the signal at 5.15 min represents a primer of about 7 nucleotides. MS analysis of the isolated primer confirmed that the primase product is indeed a 7-mer with the sequence 5′-pppACCAACU-OH-3′ (Supplementary Figure S5A). Thus, primer synthesis is initiated at thymidine at position 7 in template t13 (Figure 4C). As with the gel-based primase assay, activity was only observed in the presence of rNTPs but not dNTPs (Figure 4C, red vs. blue line).
Variation of the 5′ end of template t13 provides the primase with incrementally shorter and longer template sequences upstream of the priming site. The analysis of the different primase reactions clearly showed that the primase synthesizes run-off products until the 5′ end of the template as long as the template is shorter than 15 nucleotides (Figure 4D, traces until template length 15). However, as soon as the 5′ end upstream of the priming site reaches the length of 9 nucleotides the primer products length remains unchanged with a 9 nt long primer as the major reaction product regardless how long the 5′ extension is (Figure 4D, traces with template length 15–19). Thus, NEQ395 preferentially synthesized primers with a length of nine nucleotides and is capable of a rather precise termination at this primer length. However, precise primase termination is less evident from the gel-based primase assay (e.g. Figure 3 and Supplementary Figure S4) where we used longer templates and fluorescent nucleotides and observed diverse primer lengths, partly explained by run-off products and/or abortion due to fluorescent nucleotides.
Shortening of the 3′ end of the template indicates that the presence of approximately six bases 3′ of the priming site is necessary for efficient priming (Figure 4D bottom). This part of template might be required for the binding of the template by the primase. Taken together, these experiments show that N. equitans primase synthesizes primers starting from the T at position 7 of the template up to a length to approximately nine nucleotides, unless limited by shorter template strands in 5′ template direction.
We initially observed that Mn2+ is the preferred metal cofactor for the primer elongation activity. The titration of the divalent Mg2+, Mn2+ or Co2+ from 0 to 10 mM showed a strong primer synthesis on template t13 above 0.5 mM Mn2+ with highest primase activity around 3 to 4 mM (Supplementary Figure S5B). Unlike the majority of AEPs, NEQ395 requires Mn2+ as cofactor and is much less active when Mg2+ or Co2+ are offered as cofactor. Manganese is enriched in thermophile bacteria and archaea with concentrations ranging from the lower to the middle micromolar range (59,60). Manganese could act as antioxidant Mn2+ in these extremophilic organism thus preventing damage from ionizing radiation. It is also known that manganese can replace magnesium and zinc as cofactor in enzymatic reactions (61).
When titrating NEQ395 with an ssDNA template that enables primer termination (17-mer template from Figure 4D top), primer products could be observed by HPLC above 0.25 μM NEQ395 and the specific activity of NEQ395 plateaus at around 1.0 μM enzyme under these assay conditions (Supplementary Figure S5C). The quantitative label-free HPLC assay works optimally at a relatively high enzyme concentration (1 μM), but the reaction conditions assure a multi turn-over regime with 5 μM template and 100 μM rNTPs. In contrast, no primase activity was observed when only the catalytic domain PriS (see Figure 1A, NEQ395 residues 1–281) was incubated instead of wildtype NEQ395 under the same conditions. This experiment confirms the importance of PriL for primer synthesis although more recent report shows that primer formation is possible with the human PriS alone (32).
ATA is the preferred priming site
We next investigated which bases around the initiation site of a thymidine at position 7 (Figure 5A, top) constitute the recognition sequence of NEQ395 for primer initiation on the ssDNA template. Based on the 13-mer ssDNA template t13, possible recognition of the base positions 1 to 12 was tested by replacing each position with all the alternative bases. In a gel-based primase assay (Figure 5A), high primase activity was observed with each of the four bases at positions 1 to 5. At position 6, only adenosine or guanosine supported the initiation of a primer synthesis. A thymidine must follow at position 7 and an adenosine or cytosine at position 8 completes the recognized triplet. The bases at position 9–12 are not sequence-specifically recognized and were also not part of a template which is copied into the primer. The same set of templates was then used for a quantitative analysis by RP-HPLC. The relative activity of each base (given by the primer product peak area) at each position was then used to construct a position-specific scoring matrix (PSSM) and the result visualized as a logo (Figure 5B). While position 7 is invariantly a thymidine, position 6 and 8 allow for two nucleobases each. We next assessed all four combinations of the two neighboring bases and found that the template with the trinucleotide ATA is most active followed by GTA (Figure 5C). The activities of the four templates could be explained by single base specific contribution at each position; there was no indication that a particular combination of bases in position 6 and 8 is preferred.
Figure 5.

Detailed analysis of the primase priming site. (A) Based on template t13, priming was tested by replacing base positions 1 to 12 with all three other bases. The numbers above the template sequence indicates positions, that were mutated in the experiment. Primase reactions were assembled with NEQ395 (1.0 μM), ATP/GTP/UTP (100 μM each), CTP (20 μM) and FAM-CTP (1.0 μM) together with templates (5.0 μM) differing at positions 1 to 12. (B) The same templates (5.0 μM) were incubated with NEQ395 (1.0 μM) and rNTPs (100 μM each) and analyzed by HPLC. Integrated peak areas of the primer peak were summarized in a sequence LOGO. (C) Efficient primer synthesis requires the motif ATA. Possible further priming sequences show an activity order ATA > GTA > ATC > GTC. Peaks of the ssDNA templates were used as internal standard for product quantification.
The identified trinucleotide motif with the central thymidine was identified using the template t20 which we originally chose to investigate the priming specificity of NEQ395. We then tested whether this trinucleotide could also support priming in other contexts. We first chose homooligomeric templates. Priming activity is enhanced in the oligomeric context of deoxyadenosines, deoxythymidines and deoxycytidine as soon as the trinucleotide ATA is incorporated into the template (Supplementary Figure S6A). An oligodeoxyguanosine does not serve as template (data not shown) which we attribute to the formation of G-quadruplex structures which might interfere with template binding by NEQ395. In the case of oligodeoxycytidine template, primer formation was only observed when the template was shortened. For unknown reasons the longer homooligomeric template of deoxycytidine inhibited primer formation.
The homooligomeric templates will allow slippage of the primer-primase complex on the template. In fact, we observed primer products of different lengths using these templates. However, when the dinucleotide 5′-CG or 5′-AG is inserted upstream of the priming site 5′-ATA most of the primer products have a length of approximately 10 nucleotides. Next, we chose three short oligodeoxynucleotides devoid of the trinucleotide priming motif and introduced a priming site by mutating one or two bases. We could observe an increased albeit variable primer synthesis of the template carrying the mutation (Supplementary Figure S6B) again with a primer length of approximately 10 nucleotides.
It is possible that further priming sites might exist. However, due to the large variation in sequences of the assayed oligodeoxynucleotides (Supplementary Figure S4A) the existence of further strongly recognized priming motifs would likely have been detected.
Template binding and reaction kinetics of the primer synthesis
NEQ395 primes sequence-specifically. The sequence-specificity might be partially or completely caused by specific binding of the primase to templates containing a suitable priming site. We therefore analysed the binding equilibria of primase with templates using fluorescence anisotropy. Determining the dissociation constant of the binding equilibrium for the enzyme-template complex by reverse-titration of FAM-labeled templates (50 nM) with primase (Figure 6A) revealed that NEQ395 binds a nine nucleotide long single-stranded template devoid of the recognition triplet (t9-ATA) with a dissociation constant Kd of 2.87 μM (Figure 6A, black line). Introducing the ATA motif, which constitutes the best trinucleotide priming site in our prior analysis, into the same template (t9) increased the affinity slightly (Kd of 1.06 μM, Figure 6A, red line). Thus, we only observe a minor preference for sequence-specific interaction with templates containing the priming site. Template t9 serves as a substrate for NEQ395 in the HPLC-based primase assay, whereas the enzyme does not prime template t9-ATA lacking ATA (data not shown). It appears that NEQ395 has a small tendency to bind substrates which serve as templates with increased affinity. Nonetheless, binding to NEQ395 alone will not suffice for an oligonucleotide to be utilized as template. An additional discriminatory mechanism beyond binding is necessary to explain why t9-ATA is not used productively as template.
Figure 6.

DNA binding affinity and primer synthesis kinetics. (A) NEQ395 was titrated with single-stranded DNA of 9 nucleotides with the motif ATA (black) or without (red). Substrate t9 was bound with a dissociation constant of 1.1 μM, whereas substrate t9-ATA was bound with a 2.7 times lower affinity (KD = 2.9 μM). The binding affinity of t9 increases 1.8 fold in the presence of 250 μM ATP, but not with one of the other rNTPs. t9-ATA in contrast binds weaker to NEQ395 probably due to competition to the excessive rNTPs in solution. (B) The enzyme interacts with single-stranded DNA (t18, black) as well as with double-stranded DNA (t18_ds, red) showing no significant differences in the binding affinity. (C) The kinetic parameter of the reaction were investigated by using 0.75 μM enzyme, saturating template concentration (500 μM) and varying concentrations of rNTPs resulting in Km,rNTPs = 436 μM and kcat,rNTPs = 0.044 s−1 (top). When titrating template t13, rNTPs were kept at 250 μM and calculations led to Km,template = 5 μM and kcat,template = 0.016 s−1 (bottom). The reaction rate k refers to the primer formation rate. Data points are mean ± s.d. (n = 3).
The binding affinity of t9 increases in the presence of ATP, the cognate nucleotide for initiation (Kd of 0.58 μM, Figure 6A, table on the right) indicating the formation of a ternary complex consisting of NEQ395, ssDNA and ATP. In contrast, the affinity of the templates decreased when adding the non-cognate rNTPs as well as an rNTP mix.
Increasing the length of the template from 9 to 18 nucleotides (template t18) resulted in slightly higher anisotropy values of the unbound DNA and the complex of template and enzyme (Figure 6B, black line). In comparison with template t9, NEQ395 shows a slightly increased affinity to template t18 (Kd of 0.51 μM). t18 was annealed to its complement strand to test the ability of NEQ395 to interact with double-stranded DNA (t18_ds, Table 1). No large difference between the enzyme's affinity for single- versus double stranded DNA was observed (Figure 6B, 0.51 μM versus 0.65 μM). Compared to the DNA affinity of human PrimPol (Kd,DNA = 0.15 μM (62)), DNA affinity values measured with NEQ395 are in the same range. Possibly, NEQ395 can bind single- as well as double-stranded DNA in line with its primase and primer elongation activities.
Next, we performed a study of the enzyme kinetics of the primase activity of NEQ395. Kinetic data were collected for primer formation complementary to template t13, which yields a primer product with a length of seven nucleotides (see Figure 4C). For these experiments the concentrations of the substrate nucleoside triphosphates and of the template ssDNA were varied. The enzymatic activity was determined by analyzing the reaction mixture by RP-HPLC followed by integration of the product peak area for each experiment to quantify product yields. Plotting the enzymatic activity against the titrated component revealed that the simple Michaelis–Menten kinetic model did not fit the measured values. Instead, an adapted form of the Michaelis–Menten equation considering for excess-substrate inhibition was used to derive Km and kcat (see method section).
For the titration with rNTPs, NEQ395 (0.75 μM) was incubated with a saturating template concentration (500 μM) and the concentrations of the rNTP mix (containing equal amounts of each rNTP) was varied. Fitting the data revealed a Km, rNTPs of 436 μM with kcat, rNTPs of 0.044 s−1 (Figure 6C, top). NEQ395 appeared to be strongly sensitive to substrate inhibition (Ki = 245 μM) as the rate of incorporation began to decrease at >300 μM rNTPs. Reported Km values for rNTPs/dNTPs in reactions catalyzed by comparable eukaryotic and archaeal primases range from 8 to 250 μM (e.g. yeast primase: Km, ATP = 165 μM (63); mouse primase: Km, ATP = 250 μM (64); human PrimPol: Km, dTTP = 14 μM (62); Sulfolobus islandicus pRN1 primase: Km, dATP = 35 μM (42); Thermococcus kodakaraensis primase: Km, dATP = 30 μM (65)). Cellular concentrations of rNTPs and dNTPs have been documented in eukaryota and bacteria with average concentrations of approximately 0.3–3.1 mM rNTPs significantly exceeding the dNTP content of 5–37 μM (66,67), while the corresponding concentrations in archaea remain largely unknown. The apparent Km for ribonucleotides for NEQ395 is higher compared to values reported for other AEPs, but within the same order of magnitude. Under physiological intracellular conditions the rNTP concentrations can be expected to be saturating based on our data.
For the titration of the template, the enzymatic reaction was carried out with NEQ395 (0.75 μM) and ‘optimal’ rNTP concentration (0.25 mM) before onset of inhibition (see above). Titration of the template resulted in an apparent Km of 5 μM with a kcat of 0.016 s−1 (Figure 6C bottom). Again, NEQ395 was sensitive to substrate inhibition (Ki = 322 μM) as the rate of incorporation started to decrease at >50 μM template. The Michaelis-Menten constant for the template of NEQ395 primase was similar to that of other eukaryotic primases, as shown for mammalian primase (Km, Oligo(dT4) (human placenta) = 5.8 μM, Km, Oligo(dT4) (calf-tymus) = 0.8 μM) (68), Km, poly(dT290) (mouse) = 4.1 μM (64) and human PrimPol (Km,DNA = 3.6 μM (62)).
Overall, both titration experiments yielded a maximum reaction constant k of about 0.012 s−1. This activity is comparable to the synthesis rate observed for the trimeric archaeal primase from Sulfolobus solfataricus where a kcat of 0.016 s−1 has been reported for the primer synthesis (13) and for the mouse primase (kcat ∼ 0.04) (69).
Fidelity of primase activity
DNA polymerase activity in presence of the metal cofactor Mn2+ is linked to reduced fidelity (70,71). We therefore investigated whether NEQ395 incorporates the cognate base during primer elongation. The fidelity of the primase reaction was assayed with ssDNA templates in which a CG was incorporated into a polyadenine sequence upstream of the ATA priming triplet (Table 1, templates Fid1–4). Using different rNTP mixes (Supplementary Figure S7), the primer synthesis can be specifically abrogated when a cognate nucleotide is missing, and the primase is reluctant to proceed in the absence of the correct nucleotide. The templates chosen by us allow to hinder the elongation when the cognate ribonucleotides GTP and CTP are absent in the reaction.
Depending on the distance between the CG and the priming triplet and in the presence of ATP and UTP primers of 4, 6 or 8 nt were synthesized (Supplementary Figure S7, blue traces) confirming the start of the reaction opposite the T of the triplet ATA and a faithful termination because of lack of the ribonucleotide CTP. In case of the template used in Supplementary Figure S7A, the expected dinucleotide is not well resolved and probably elutes together with the unprocessed rNTPs and further buffer components at the beginning of the chromatography run (tR < 2 min). Addition of CTP to the respective reaction mixtures allowed the efficient synthesis of a primer of 3, 5, 7 or 9 nt (Supplementary Figure S7, red traces), thus of exactly one additional base in agreement with a faithful termination opposite to C since GTP is still missing. Incubating the reaction mixtures with the rNTP mixture containing all four bases enabled NEQ395 to synthesize run-off primer products of 9 –11 nt (Supplementary Figure S7, traces in black). Noteworthy, using an rNTP mix without ATP (the first cognate base needed for synthesis) did not initiate primer synthesis confirming the importance of the central T in the ATA priming site (Supplementary Figure S7, gray traces). The templates contain longer homopolymeric stretches and primase slippage is observed that leads to more heterogenous primer products. This effect is most prominent when the primase is synthesizing a primer opposite to the homoadenine stretch as in the reactions shown in Supplementary Figure S7C and D. Nevertheless, NEQ395 strongly prefers to incorporate only the cognate ribonucleotides.
Next, we investigated how strict NEQ395 is in incorporating ribonucleotides versus deoxynucleotides. Primer formation is dependent on ribonucleotides, deoxynucleotides alone do not allow for primer synthesis as shown in Figure 4C. Nevertheless, it is possible that deoxynucleotides are accepted as substrates either as initiating nucleotide or as elongating nucleotide with low efficiency. Using the templates of the previous experiment we can address specifically the requirements of the initiating nucleotide by using dATP or ATP. As shown in Supplementary Figure S7E, dATP is poorly accepted with only about 10% of the primer product compared to ATP (Supplementary Figure S7E, blue line). We analogously used the pair GTP and dGTP to investigate the suitability of a deoxynucleotide as elongating nucleotide. In presence of GTP, extension to the full-length primers occurs. In contrast, when dGTP is present, the primer extension is stalled at the 5 nucleotide long primer (Supplementary Figure S7E, black and grey lines). Thus, elongation using a deoxynucleotide is very inefficient. In fact, the reaction products are comparable with the reaction in absence of GTP (Supplementary Figure S7B, red line). We thus conclude, that NEQ395 strongly prefers rNTPs as initiating and elongating nucleotides. The structural basis for this specificity is not known and we attempted to identify residues which could be responsible for the specificity (see below).
Model structure of the monomeric primase NEQ395
NEQ395 encompasses two well conserved and structurally characterized domains (see Figure 1A). We therefore attempted to build model structures using two state of the art deep learning modelling techniques, namely AlphaFold2 and RoseTTAFold (46,47). Five models were generated each and compared. The ensemble of AlphaFold2 was more diverse whereas the RoseTTAFold ensemble had an overall RMSD of only 1.4 Å. The analysis showed that most parts of the models agreed very well, but that the structures differed considerably in the relative orientations of the PriS and PriL domain (Figure 7, Supplementary Figure S8). All ten models had a consistent structure of the N-terminal PriS domain and the first part of the helical linker connecting the PriS and the PriL domain (residues 1–266, overall average RMSD 1.4 Å). Likewise, the PriL domain alone (residues 285–393) also displayed a consistent structure over all models (RMSD 1.1 Å). The two conserved and consistently modelled domains are connected by a long alpha-helix (residues 251–280) which is straight in most models but kinked past Q266 in three AlphaFold2 models. The models in the ensemble differ mainly by this kink in the middle of the helix and as well as in the loop connecting the linker helix with the first conserved helix of the PriL domain, i.e. between residues P281 to P288. The different conformations confined to these two short amino acid stretches result in different orientations of PriS relative to PriL and make a superposition of all models across the entire polypeptide chain impossible. Noteworthy, the overall structures of the models are reminiscent of the crystal structure of RepB’ from RSF1010, where the catalytic PriS-like domain and the regulatory helix bundle domain are also connected by a long helix of 30 amino acids.
Figure 7.

Representative AlphaFold2 model structure of the Nanoarchaeum equitans primase. PriS and PriL domain are predicted to be linked with a long alpha-helix. The backbone is colored according to conservation with other PriS-PriL fusions (red: highly conserved). Some residues mutated in the study are highlighted with atomic resolution. Mutations are situated in PriS at the active site and the binding pocket for the elongating nucleotide. In PriL the mutations are in the central helix which could participate in template and initiating nucleotide binding. The models generated by AlphaFold2 and RoseTTAfold differ mainly in the conformation of the helix linking PriS and PriL and in particular at the transition from the end of the linking helix to the beginning of PriL. This part of the protein is also the region of preferential proteolysis. The cysteines forming the [4Fe–4S] cluster in PriL are in close neighborhood, but the FeS cluster itself is not explicitly modeled by Alphafold2.
The model ensemble gave hints on the conformational flexibility of the NEQ395 fusion protein. A certain conformational flexibility between the PriS and PriL domain is expected for the cooperation of the PriS with the PriL domain for initiation and termination of primer synthesis. We can even expect that a certain amount of conformational flexibility is required in case of fusion proteins with covalent linkage of domains which usually exist as separate polypeptide chains. Unstructured and flexible amino acid stretches can be more susceptible to proteolysis. We therefore performed a limited proteolysis experiment to locate less structured and more flexible regions in the primase. Limited proteolysis of the primase yielded two fragments of approximately 11 and 35 kDa (Supplementary Figure S9A). Tryptic digestions of the fragments followed by mass-spectrometry suggests that limited proteolysis occurs predominantly between amino acid K282 and I290. Peptides terminating up to K282 (C-terminal tryptic end) were much more abundant in the protein fragment encompassing PriS and the connecting alpha-helix (∼35 kDa) than in the PriL fragment (∼11 kDa). Accordingly, peptides starting at I290 and N292 could only be detected in the PriL fragment. Noteworthy, these latter peptides do not have an N-terminal tryptic end suggesting that limited proteolysis by proteinase K yielded the N-termini of these peptides.
Taken together, the model ensemble and the limited proteolysis suggest that the PriS domain together with the connecting linker is well-structured. The linker could have a largely helical conformation. In contrast, the highest amount of conformational flexibility can be expected to be in the loop from the C-terminus of the predicted linker helix to the first conserved helix of the PriL domain.
Functional dissection of the two domains of NEQ395
We aimed to analyse the constituting domains as separate proteins and constructed a pair of deletion constructs. Based on the information from structural modelling and limited proteolysis, we decided to engineer a deletion mutant up to P281 encompassing PriS and the connecting helix as well as a deletion mutant starting at K282.
The catalytic domain could be expressed as a soluble recombinant protein in E. coli with good yields (∼15 mg/l culture, Supplementary Figure S10A, 35.8 kDa). In contrast, the regulatory domain was only poorly soluble when overexpressed in E. coli (Supplementary Figure S10A, 16.2 kDa). In an attempt to purify the regulatory domain through metal chelate affinity chromatography, we obtained an impure protein preparation with a slightly brownish appearance suggesting the presence of a small amount of correctly folded regulatory domain with an intact [4Fe–4S] cluster.
Nonetheless, both deletion mutants were analysed in the RNA polymerase and primase assay (Supplementary Figure S10B). PriS successfully elongates the applied primer-template substrate, but with reduced efficiency compared to wildtype NEQ395. The reduction might derive from the deletion of the PriL domain that could stabilize the arrangement of PriS with its template and/or the rNTPs during polymerisation. As expected, PriL does not show polymerase nor primase activity. The impurity of the preparation however rendered a better characterisation of PriL impossible. We tested two further deletion constructs of the regulatory domain, but the alternative deletion mutants were also poorly soluble.
In order to understand better the contribution of the domains for primer synthesis we then constructed several point mutations of conserved amino acids which should inactivate either the PriS or PriL domain (Table 2).
Table 2.
Activity profile of deletion and point mutants of the N. equitans primase. Relative changes are classified as follows: +++: unchanged activity relative to wildtype NEQ395, ++: ∼ 50% of activity, +: ∼10% of activity, -: inactive, *: poor protein preparation
| Mutant | Polymerase activity | Primase activity | Predicted function | Remarks |
|---|---|---|---|---|
| Wildtype | +++ | +++ | ||
| Catalytic domain | ||||
| 1–281 | ++ | - | RNA polymerase activity | PriS domain |
| H58A | - | - | Binding of elongating nucleotide | |
| H58Y | - | - | Binding of elongating nucleotide | |
| D87A+D89A | - | - | Active site residues | |
| S119A | - | + | Binding of elongating nucleotide | |
| K122A | ++ | ++ | Binding of elongating nucleotide | Less conserved residue |
| H125A | - | - | Active site residues | |
| D194A | - | - | Binding of initiating nucleotide | |
| D194E | - | - | Binding of initiating nucleotide | |
| H202A | - | - | Binding of elongating nucleotide | |
| H202K | - | - | Binding of elongating nucleotide | |
| R205A | - | - | Binding of elongating nucleotide | |
| Regulatory domain | ||||
| 282–393* | - | - | Initiation of primase activity | PriL domain |
| R301A | +++ | + | Binding of initiating nucleotide or 5′ primer end | |
| R303A | +++ | + | Binding of initiating nucleotide or 5′ primer end | |
| R301A+R303A | +++ | - | Binding of initiating nucleotide or 5′ primer end | |
| F306A | +++ | + | Primer/template binding | |
| C289A | +++ | +++ | [4Fe–4S] cluster | |
| C364A | +++ | +++ | [4Fe–4S] cluster | |
| C376A | +++ | ++++ | [4Fe–4S] cluster | Increased primase activity |
| C384A | +++ | ++++ | [4Fe–4S] cluster | Increased primase activity |
Based on sequence alignments and structural information of the homologous domains we selected highly conserved and surface exposed residues. In particular, we chose S119, K122 and R205 in PriS. These amino acids should participate at the binding of the nucleotide at the elongating position. In fact, they are homologous to T220, R223 and K277 in the CRISPR-associated primase-polymerases from Marinitoga piezophile, another AEP. A structure of this primase has recently been solved in complex with template and two nucleotides thus representing the long awaited initiation complex of archaeoeukaryotic primases (32). In this structure (pdb: 7P9J) T220, R223 and K277 bind to the phosphates of dATP which is in the elongation site.
For the PriL domain, we chose R301 and R303. Both arginines are at the C-terminal end of the second helix in PriL and could be implicated in binding the nucleotide at the initiating position or the phosphorylated 5′ primer end, respectively (see also Supplementary Figure S11B). We also selected F306 which is a nearly invariant amino acid in the close homologues of NEQ395. Based on the structure of the human primase bound to a primer/template (pdb: 5F0S (72)), an amino acid at this position could stabilize the transition of double to single-stranded DNA at the 5′ primer end (see Supplementary Figure S11B). Moreover, we single-mutated each cysteine participating in the formation of the [4Fe–4S] cluster (C289, C364, C376 and C384).
All selected amino acids were mutated to alanine and the respective mutant proteins were assayed towards primase and RNA polymerase activity (Table 2 and Supplementary Figure S10C and D). All the mutants in the PriS domain can neither synthesize a primer nor elongate a primer/template. These findings are in line with the expected critical role of these amino acids for the polymerization reaction.
The mutants R301, R303 and F306 in the PriL domain continue to extend a primer/template pair but fail to efficiently synthesize a primer further underscoring the contribution of PriL for the initiation of primer synthesis. These mutants show a similar behavior as the complete deletion of the regulatory domain emphasizing again the importance of the regulatory domain for the initiation of primer synthesis (Supplementary Figure S10D). A comparable contribution of the regulatory domain is seen in other archaeoeukaryotic primases, e.g. primases from S. solfataricus, P. furiosus and also for the pRN1 replication enzyme.
Surprisingly, the cysteine mutants which disrupt the [4Fe–4S] cluster formation, as can be seen by the lack of coloration of the protein solution and the lack of extinction at 410 nm (Figure 1C), are still active in primase as well as elongation activity. Thus, the [4Fe–4S] cluster per se is not required for primer synthesis under these in vitro conditions. A similar observation has already been made with the heterotrimeric primase of Sulfolobus solfataricus. A fusion protein of PriX with a truncated PriL, which does not longer contain a [4Fe–4S] cluster, forms a highly active complex with PriS (12).
On the other hand in an in vivo context carried out in yeast, double mutants of the [4Fe–4S] cluster in PriL led to a growth defect (73) whereas single mutants display unaffected growth as long as C434 (homologous to C384 in NEQ395) is not mutated. Thus, the contribution of the highly conserved [4Fe–4S] cluster in archaeoeukaryotic primases towards primer synthesis and its possible regulation by charge transfer remains enigmatic.
We note that two of the cysteine point mutants display a somewhat higher primase activity. Therefore, it is possible that an intact [4Fe–4S] cluster could exert a slight inhibitory activity. The inhibition might be amplified or suppressed by other proteins in the context of the replisome and therefore offer an additional layer of regulation of the primase.
With the availability of point mutants either inactive in catalysis (PriS) or defect in the regulatory subunit (PriL) we could test whether an active primase can be restored by complex formation of two inactive primases. We therefore mixed the active site mutants D87A+D89A or H125A with the PriL mutants R301A or F306A and performed the HPLC-based activity assay (Figure 8). A distinct primer product was not observed in the mixtures excluding the possibility of activation in trans. Thus, in accordance with monomeric quaternary structure determined via gel permeation chromatography (Supplementary Figure S2), NEQ395 is functional as a monomer.
Figure 8.

NEQ395 is not activated in trans. Mixing 1.0 μM of PriL mutant (R301A or F306A) with 1.0 μM of PriS mutant (D87A+D89A or H125A) did not restore primase activity (compare Supplementary Figure S10D). Reaction condition as detailed in Figure 4, template t13 was used as substrate.
The two domains of NEQ395 do not interact in trans, implying that covalent binding between them is required for primer synthesis. We next investigated how the linker length could influence the primase activity. We systematically changed the linker length by constructing mutants with a deletion and insertion of 2, 4, 6, 8 and 10 amino acids. The length changes were carried out at amino acid position I277, thus in the C-terminal non-conserved part of the linker connecting both domains close to the determined proteolytic cleavage site (Supplementary Figure S9). Elongating the linker (+2, +4, +6, +8 and + 10 mutants) reduced the activity of NEQ395 while the length distribution of the primer products remained unchanged (Figure 9). The lower primase activity could be due to the reduced local concentration of the domains and is in line with the finding, that both domains do not act in trans.
Figure 9.

Variation of linker length. 1.0 μM of the proteins were assayed with the label-free primase assays using the 17-mer template from Figure 4D top. Shortening the linker led to a higher proportion of aborted primer products, whereas the elongated linkers did not alter the primer length distribution.
Shortening the linker (−2, −4, −6, −8 and −10 mutants) led to a changed primer length distribution. Already the mutant with a linker shortened by two amino acids (mutant −2) has a strong increase of prematurely aborted primer products with the length of 4–5 nucleotides. Shortening the linker further (mutants −4, −6, −8 and −10) progressively increases the amounts of aborted primers at the expense of the full-length primers.
Primases usually have a rather narrow distribution of primer lengths, which precludes a purely stochastic termination. In this case termination would occur at each extension step with the same probability regardless of the length of the primer already synthesized.
In the caliper model (8), the primase would sense the primer length by conformational restrains, which impedes primer elongation at a specific length of the already synthesized primer. The caliper model can however not fully explain the results presented here, as this model would predict that primer length increases when the conformational restrains are released by elongating the linker with additional amino acids. The mutants with shorter linkers however can be explained with the caliper model. The shortened linker renders the primer elongation less probable and leads to a higher proportion of aborted primer products.
The third hypothesis brought forward to explain a narrow distribution of primer lengths is the steric clash model (72). It predicts that primer termination occurs when a certain primer length is reached which in turn leads to a clash primarily caused by the rotational movement imposed through synthesis of double-stranded DNA.
The data on NEQ395 cannot be explained fully by these models. We suggest that the wildtype protein terminates primer synthesis either due to a steric clash between the PriS and the PriL domains or because a favourable interaction between both domains is weakened when the primer reaches a length of about nine nucleotides. The mechanistic basis of this termination is currently unclear and would require additional structural investigation of NEQ395 in complex with a primer/template.
Specificity towards ribonucleotides
NEQ395 has a strong preference for ribonucleotides as initiating and elongating nucleotides. Thus, there shall be specific interactions with the hydroxyl group at position 2′ present in ribonucleotides but absent in deoxynucleotides. Using the structure of CRISPR-associated primase-polymerases from Marinitoga piezophile in the precatalytic conformation before dinucleotide synthesis as a guide, we identified D194, H58 and H202 as residues which could potentially differentiate between deoxy- and ribonucleotide (Supplementary Figure S11).
D194 was selected for the initiating position. This amino acid is homologous to E260 in Marinitoga sp. CRISPR-associated primase-polymerase where it is crucial for hydrogen bonding with the initiating ribonucleotide as well as coordinating one of the catalytic metal ions (32). An acidic residue at this position is highly conserved within the AEP and in fact a mutation to alanine destroyed the priming and elongation activity of NEQ395 using alternatively ribonucleotides and deoxynucleotides. Next, we substituted D194 with glutamic acid, but this mutant proved also to be inactive. We therefore concluded that the sugar selectivity at the initiation site cannot be altered by mutating this position although this amino acid is closest to the 2′ position of the ribose of the initiating nucleotide.
For the elongating position we selected H58 and H202. H202 could establish a favourable hydrogen bond with the 2′ oxygen at the elongating position (Supplementary Figure S11). An alanine mutation at this residue destroyed the primase and elongation activity using alternatively ribonucleotides and deoxynucleotides. H202 might be needed to bind and position the elongating ribonucleotide and without this favourable interaction, the enzyme is rendered inactive. We next chose to replace H202 with lysine since this amino acid is present in some homologous proteins. Lysine is bulkier than histidine and might prevent the binding of a ribonucleotide. Again, the activity of the enzyme was destroyed by this mutation.
H58 is more distant than H202 to the sugar of the nucleotide at the elongating position. We nevertheless investigated this position as it is homologous to Y100 in the human PrimPol (pdb: 5L2X). Y100 plays a crucial role in selecting deoxynucleotides at the elongation site. Mutating the tyrosine into histidine relaxed the specificity towards ribonucleotides with an overall only minor reduction of polymerase activity (74). Unfortunately, we were not able to change the sugar specificity by mutating H58. The mutants H58A and H58Y were both inactive.
Our attempts to identify the molecular basis of the sugar selectivity were not successful. The mutant protein with amino acids changed in proximity of the sugars of the nucleotides were inactive. It seems that the wild-type amino acids make critical favourable interactions with the nucleotides, which are lost in the mutants. A change in sugar specificity might require more subtle changes and might require additional compensating mutations of further residues in the neighbourhood of the nucleotide binding pockets.
CONCLUSIONS
We here present the characterization of a remarkable archaeoeukaryotic primase from the archaeon Nanoarchaeaum equitans. The monomeric enzyme arises from a gene fusion between the genes of the small catalytic and large regulatory primase subunits or might represents an ancestral primordial form. Although the monomeric Nanoarchaeaum equitans primase is even smaller than the typical large primase subunits of archaea and eukaryotes the enzyme is as active as other archaeoeukaryotic primases. Sequence alignment and structural modelling demonstrated that apart from the linker the whole protein is highly conserved to either the small or large primase subunit. Possibly, this monomeric enzyme represents the most compact and minimal archaeoeukaryotic primase.
The N. equitans primase displays a preference for initiating primer synthesis at sites in the templates containing triplets with a central thymidine. Considering the high sequence conservation of the N. equitans primase with related archaeoeukaryotic primases, it appears possible that other primases might also prime sequence-specifically. This however has not been systematically investigated so far.
This primase also reconfirms that the regulatory PriL domain is needed for efficient primer synthesis since mutant proteins at selected amino acids in PriL destroy the primase activity. Most probably the PriL domains are required to bind the phosphates of the initiating nucleotide which becomes the 5′ primer end (6,12,28,72). The primase of N. equitans terminates primer synthesis preferentially at about nine nucleotides.
Given its small size, its high specific activity and the lack of quaternary structure, this primase could serve as model to investigate more closely the reaction mechanism of archaeoeukaryotic primases, in particular the interplay between the catalytic and the regulatory subdomains and the conformational changes during the reaction cycle from initiation to termination of primer synthesis.
DATA AVAILABILITY
The data underlying this article are available in the article and in its online supplementary material.
Supplementary Material
ACKNOWLEDGEMENTS
The services of Urs Kämpfer at the mass spectrometry facility at University of Berne are gratefully acknowledged.
Contributor Information
Andy Schneider, Institute of Chemistry and Bioanalytics, University of Applied Sciences Northwestern Switzerland, 4132 Muttenz, Switzerland.
Jan Bergsch, Institute of Chemistry and Bioanalytics, University of Applied Sciences Northwestern Switzerland, 4132 Muttenz, Switzerland.
Georg Lipps, Institute of Chemistry and Bioanalytics, University of Applied Sciences Northwestern Switzerland, 4132 Muttenz, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
SNF [310030_185252]. Funding for open access charge: SNF and/or Universtiy.
Conflict of interest statement. None declared.
REFERENCES
- 1. Yao N.Y., O’Donnell M.E. Evolution of replication machines. Crit. Rev. Biochem. Mol. Biol. 2016; 51:135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keck J.L., Roche D.D., Lynch A.S., Berger J.M.. Structure of the RNA polymerase domain of E. coli primase. Science. 2000; 287:2482–2486. [DOI] [PubMed] [Google Scholar]
- 3. Iyer L.M., Koonin E.V., Leipe D.D., Aravind L.. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005; 33:3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stadlbauer F., Brueckner A., Rehfuess C., Eckerskorn C., Lottspeich F., Forster V., Tseng B.Y., Nasheuer H.-P.. DNA replication in vitro by recombinant DNA-polymerase-alpha-primase. Eur. J. Biochem. 1994; 222:781–793. [DOI] [PubMed] [Google Scholar]
- 5. Kilkenny M.L., Longo M.A., Perera R.L., Pellegrini L.. Structures of human primase reveal design of nucleotide elongation site and mode of Pol alpha tethering. Proc.Natl.Acad.Sci.U.S.A. 2013; 110:15961–15966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baranovskiy A.G., Lisova A.E., Morstadt L.M., Babayeva N.D., Tahirov T.H.. Insight into RNA–DNA primer length counting by human primosome. Nucleic Acids Res. 2022; 50:6264–6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lao-Sirieix S.H., Nookala R.K., Roversi P., Bell S.D., Pellegrini L.. Structure of the heterodimeric core primase. Nat Struct Mol Biol. 2005; 12:1137–1144. [DOI] [PubMed] [Google Scholar]
- 8. Yan J., Holzer S., Pellegrini L., Bell S.D.. An archaeal primase functions as a nanoscale caliper to define primer length. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:6697–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baranovskiy A.G., Zhang Y., Suwa Y., Babayeva N.D., Gu J., Pavlov Y.I., Tahirov T.H.. Crystal structure of the human primase. J. Biol. Chem. 2015; 290:5635–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O’Brien E., Salay L.E., Epum E.A., Friedman K.L., Chazin W.J., Barton J.K.. Yeast require redox switching in DNA primase. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:13186–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O’Brien E., Holt M.E., Thompson M.K., Salay L.E., Ehlinger A.C., Chazin W.J., Barton J.K.. The (4Fe4S) cluster of human DNA primase functions as a redox switch using DNA charge transport. Science. 2017; 355:eaag1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holzer S., Yan J., Kilkenny M.L., Bell S.D., Pellegrini L.. Primer synthesis by a eukaryotic-like archaeal primase is independent of its Fe-S cluster. Nat. Commun. 2017; 8:1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu B., Ouyang S., Makarova K.S., Xia Q., Zhu Y., Li Z., Guo L., Koonin E.V., Liu Z.-J., Huang L.. A primase subunit essential for efficient primer synthesis by an archaeal eukaryotic-type primase. Nat. Commun. 2015; 6:7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baranovskiy A.G., Zhang Y., Suwa Y., Gu J., Babayeva N.D., Pavlov Y.I., Tahirov T.H.. Insight into the Human DNA primase interaction with template-primer. J. Biol. Chem. 2016; 291:4793–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pellegrini L. MacNeill S. The pol α-primase complex. The Eukaryotic Replisome: a Guide to Protein Structure and Function, Subcellular Biochemistry. 2012; 62:Netherlands, Dordrecht: Springer; 157–169. [Google Scholar]
- 16. Urban M., Joubert N., Purse B.W., Hocek M., Kuchta R.D.. Mechanisms by which human DNA primase chooses to polymerize a nucleoside triphosphate. Biochemistry. 2010; 49:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jozwiakowski S.K., Gholami F.B., Doherty A.J.. Archaeal replicative primases can perform translesion DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E633–E638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le Breton M., Henneke G., Norais C., Flament D., Myllykallio H., Querellou J., Raffin J.-P.. The heterodimeric primase from the euryarchaeon pyrococcus abyssi: a multifunctional enzyme for initiation and repair?. J. Mol. Biol. 2007; 374:1172–1185. [DOI] [PubMed] [Google Scholar]
- 19. Liu L., Komori K., Ishino S., Bocquier A.A., Cann I.K.O., Kohda D., Ishino Y.. The Archaeal DNA primase: biochemical characterization of the p41-p46 complex Frompyrococcus furiosus. J. Biol. Chem. 2001; 276:45484–45490. [DOI] [PubMed] [Google Scholar]
- 20. Marcotte E.M., Pellegrini M., Thompson M.J., Yeates T.O., Eisenberg D.. A combined algorithm for genome-wide prediction of protein function. Nature. 1999; 402:83–86. [DOI] [PubMed] [Google Scholar]
- 21. Huber H., Hohn M.J., Rachel R., Fuchs T., Wimmer V.C., Stetter K.O.. A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature. 2002; 417:63–67. [DOI] [PubMed] [Google Scholar]
- 22. Huber H., Hohn M.J., Stetter K.O., Rachel R.. The phylum nanoarchaeota: present knowledge and future perspectives of a unique form of life. Res. Microbiol. 2003; 154:165–171. [DOI] [PubMed] [Google Scholar]
- 23. Randau L. RNA processing in the minimal organism nanoarchaeum equitans. Genome Biol. 2012; 13:R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waters E., Hohn M.J., Ahel I., Graham D.E., Adams M.D., Barnstead M., Beeson K.Y., Bibbs L., Bolanos R., Keller M.et al.. The genome of nanoarchaeum equitans: insights into early archaeal evolution and derived parasitism. Proc. Natl. Acad. Sci. 2003; 100:12984–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geibel S., Banchenko S., Engel M., Lanka E., Saenger W.. Structure and function of primase RepB’ encoded by broad-host-range plasmid RSF1010 that replicates exclusively in leading-strand mode. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:7810–7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lipps G., Rother S., Hart C., Krauss G.. A novel type of replicative enzyme harbouring atpase, primase and DNA polymerase activity. EMBO J. 2003; 22:2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu B., Wang L., Mitsunobu H., Lu X., Hernandez A.J., Yoshida-Takashima Y., Nunoura T., Tabor S., Richardson C.C.. Deep-sea vent phage DNA polymerase specifically initiates DNA synthesis in the absence of primers. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boudet J., Devillier J.-C., Wiegand T., Salmon L., Meier B.H., Lipps G., Allain F.H.-T.. A small helical bundle prepares primer synthesis by binding two nucleotides that enhance sequence-specific recognition of the DNA template. Cell. 2019; 276:154–166. [DOI] [PubMed] [Google Scholar]
- 29. Iyer L.M. Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res. 2005; 33:3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blanco L., Calvo P.A., Diaz-Talavera A., Carvalho G., Calero N., Martínez-Carrón A., Velázquez-Ruiz C., Villadangos S., Guerra S., Martínez-Jiménez M.I.. Mechanism of DNA primer synthesis by human PrimPol. The Enzymes. 2019; 45:289–310. [DOI] [PubMed] [Google Scholar]
- 31. Guilliam T.A., Jozwiakowski S.K., Ehlinger A., Barnes R.P., Rudd S.G., Bailey L.J., Skehel J.M., Eckert K.A., Chazin W.J., Doherty A.J.. Human PrimPol is a highly error-prone polymerase regulated by single-stranded DNA binding proteins. Nucleic Acids Res. 2015; 43:1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li A.W.H., Zabrady K., Bainbridge L.J., Zabrady M., Naseem-Khan S., Berger M.B., Kolesar P., Cisneros G.A., Doherty A.J.. Molecular basis for the initiation of DNA primer synthesis. Nature. 2022; 605:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guilliam T.A., Keen B.A., Brissett N.C., Doherty A.J.. Primase-polymerases are a functionally diverse superfamily of replication and repair enzymes. Nucleic Acids Res. 2015; 43:6651–6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoda K., Okazaki T.. Specificity of recognition sequence forEscherichia coli primase. Mol. Gen. Genet. MGG. 1991; 227:1–8. [DOI] [PubMed] [Google Scholar]
- 35. Larson M.A., Bressani R., Sayood K., Corn J.E., Berger J.M., Griep M.A., Hinrichs S.H.. Hyperthermophilic Aquifex aeolicus initiates primer synthesis on a limited set of trinucleotides comprised of cytosines and guanines. Nucleic Acids Res. 2008; 36:5260–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koepsell S.A., Larson M.A., Griep M.A., Hinrichs S.H.. Staphylococcus aureus helicase but not Escherichia coli helicase stimulates S. aureus primase activity and maintains initiation specificity. J. Bacteriol. 2006; 188:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frick D.N., Kumar S., Richardson C.C.. Interaction of ribonucleoside triphosphates with the gene 4 primase of bacteriophage T7. J. Biol. Chem. 1999; 274:35899–35907. [DOI] [PubMed] [Google Scholar]
- 38. Davey S.K., Faust E.A.. Murine DNA polymerase.Alpha-primase initiates RNA-primed DNA synthesis preferentially upstream of a 3′-CC(C/A)-5′ motif. J. Biol. Chem. 1990; 265:3611–3614. [PubMed] [Google Scholar]
- 39. Grosse F., Krauss G.. The primase activity of DNA polymerase alpha from calf thymus. J. Biol. Chem. 1985; 260:1881–1888. [PubMed] [Google Scholar]
- 40. Banchenko S., Weise C., Lanka E., Saenger W., Geibel S.. Helix bundle domain of primase RepB’ is required for dinucleotide formation and extension. ACS Omega. 2021; 6:28903–28911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang F., Lu X., Yu C., Sliz P., Wang L., Zhu B.. Molecular dissection of the primase and polymerase activities of deep-sea phage NrS-1 primase-polymerase. Front. Microbiol. 2021; 12:3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beck K., Lipps G.. Properties of an unusual DNA primase from an archaeal plasmid. Nucleic Acids Res. 2007; 35:5635–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sweeney W.V., Rabinowitz J.C.. Proteins containing 4Fe–4S clusters: an overview. Annu. Rev. Biochem. 1980; 49:139–161. [DOI] [PubMed] [Google Scholar]
- 44. Ignesti G. Equations of substrate-inhibition kinetics applied to pig kidney diamine oxidase (DAO, E.C. 1.4.3.6). J. Enzyme Inhib. Med. Chem. 2003; 18:463–473. [DOI] [PubMed] [Google Scholar]
- 45. Kernchen U., Lipps G.. Thermodynamic analysis of the single-stranded DNA binding activity of the archaeal replication protein A (RPA) from Sulfolobus solfataricus. Biochemistry. 2006; 45:594–603. [DOI] [PubMed] [Google Scholar]
- 46. Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D.et al.. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021; 373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M.. ColabFold: making protein folding accessible to all. Nat. Methods. 2022; 19:679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Steinegger M., Söding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:nbt.3988. [DOI] [PubMed] [Google Scholar]
- 50. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J.Comput.Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 51. Giannone R.J., Huber H., Karpinets T., Heimerl T., Küper U., Rachel R., Keller M., Hettich R.L., Podar M.. Proteomic characterization of cellular and molecular processes that enable the nanoarchaeum equitans-ignicoccus hospitalis relationship. PLoS One. 2011; 6:e22942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Augustin M.A., Huber R., Kaiser J.T.. Crystal structure of a DNA-dependent RNA polymerase (DNA primase). Nat Struct Biol. 2001; 8:57–61. [DOI] [PubMed] [Google Scholar]
- 53. Ito N., Nureki O., Shirouzu M., Yokoyama S., Hanaoka F.. Crystal structure of the pyrococcus horikoshii DNA primase-UTP complex: implications for the mechanism of primer synthesis. Genes Cells. 2003; 8:913–923. [DOI] [PubMed] [Google Scholar]
- 54. Cléry A., Blatter M., Allain F.H.. RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 2008; 18:290–298. [DOI] [PubMed] [Google Scholar]
- 55. Lipps G., Weinzierl A.O., von Scheven G., Buchen C., Cramer P.. Structure of a bifunctional DNA primase-polymerase. Nat. Struct. Mol. Biol. 2004; 11:157–162. [DOI] [PubMed] [Google Scholar]
- 56. Holt M.E., Salay L.E., O’Brien E., Barton J.K., Chazin W.J.. Functional and structural similarity of human DNA primase (4Fe4S) cluster domain constructs. PLOS ONE. 2018; 13:e0209345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meier A., Söding J.. Automatic prediction of protein 3D structures by probabilistic multi-template homology modeling. PLoS Comput. Biol. 2015; 11:e1004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arezi B., Kuchta R.D.. Eukaryotic DNA primase. Trends Biochem Sci. 2000; 25:572–576. [DOI] [PubMed] [Google Scholar]
- 59. Sharma A., Gaidamakova E.K., Grichenko O., Matrosova V.Y., Hoeke V., Klimenkova P., Conze I.H., Volpe R.P., Tkavc R., Gostinčar C.et al.. Across the tree of life, radiation resistance is governed by antioxidant Mn 2+, gauged by paramagnetic resonance. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E9253–E9260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Webb K.M., DiRuggiero J.. Role of Mn 2+ and compatible solutes in the radiation resistance of thermophilic bacteria and archaea. Archaea. 2012; 2012:845756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bock C.W., Katz A.K., Markham G.D., Glusker J.P.. Manganese as a replacement for magnesium and zinc: functional comparison of the divalent ions. J. Am. Chem. Soc. 1999; 121:7360–7372. [Google Scholar]
- 62. Xu W., Zhao W., Morehouse N., Tree M.O., Zhao L.. Divalent cations alter the rate-limiting step of PrimPol-catalyzed DNA elongation. J. Mol. Biol. 2019; 431:673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brooks M., Dumas L.B.. DNA primase isolated from the yeast DNA primase-DNA polymerase complex. J. Biol. Chem. 1989; 264:3602–3610. [PubMed] [Google Scholar]
- 64. Copeland W.C., Wang T.S.. Enzymatic characterization of the individual mammalian primase subunits reveals a biphasic mechanism for initiation of DNA replication. J. Biol. Chem. 1993; 268:26179–26189. [PubMed] [Google Scholar]
- 65. Chemnitz Galal W., Pan M., Kelman Z., Hurwitz J.. Characterization of DNA primase complex isolated from the archaeon, Thermococcus kodakaraensis. J. Biol. Chem. 2012; 287:16209–16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994; 140:1–22. [DOI] [PubMed] [Google Scholar]
- 67. Nick McElhinny S.A., Watts B.E., Kumar D., Watt D.L., Lundstrom E.-B., Burgers P.M.J., Johansson E., Chabes A., Kunkel T.A.. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:4949–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Anarbaev R.O., Vladimirova O.V., Lavrik O.I.. The interaction of synthetic templates with eukaryotic DNA primase. Eur. J. Biochem. 1995; 228:60–67. [PubMed] [Google Scholar]
- 69. Copeland W.C., Tan X.. Active site mapping of the catalytic mouse primase subunit by Alanine Scanning Mutagenesis. J. Biol. Chem. 1995; 270:3905–3913. [DOI] [PubMed] [Google Scholar]
- 70. Calvo P.A., Sastre-Moreno G., Perpiñá C., Guerra S., Martínez-Jiménez M.I., Blanco L.. The invariant glutamate of human PrimPol DxE motif is critical for its Mn2+-dependent distinctive activities. DNA Repair. 2019; 77:65–75. [DOI] [PubMed] [Google Scholar]
- 71. Kirk B.W., Kuchta R.D.. Human DNA primase: Anion inhibition, manganese stimulation, and their effects on In vitro start-site selection. Biochemistry. 1999; 38:10126–10134. [DOI] [PubMed] [Google Scholar]
- 72. Baranovskiy A.G., Babayeva N.D., Zhang Y., Gu J., Suwa Y., Pavlov Y.I., Tahirov T.H.. Mechanism of concerted rna-dna primer synthesis by the HUMAN primosome. J.Biol.Chem. 2016; 291:10006–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu L., Huang M.. Essential role of the iron-sulfur cluster binding domain of the primase regulatory subunit Pri2 in DNA replication initiation. Protein Cell. 2015; 6:194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Díaz-Talavera A., Calvo P.A., González-Acosta D., Díaz M., Sastre-Moreno G., Blanco-Franco L., Guerra S., Martínez-Jiménez M.I., Méndez J., Blanco L.. A cancer-associated point mutation disables the steric gate of human PrimPol. Sci. Rep. 2019; 9:1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.