


Abstract
RNA-binding proteins (RBPs) form highly diverse and dynamic ribonucleoprotein complexes, whose functions determine the molecular fate of the bound RNA. In the model organism Sacchromyces cerevisiae, the number of proteins identified as RBPs has greatly increased over the last decade. However, the cellular function of most of these novel RBPs remains largely unexplored. We used mass spectrometry-based quantitative proteomics to systematically identify protein–protein interactions (PPIs) and RNA-dependent interactions (RDIs) to create a novel dataset for 40 RBPs that are associated with the mRNA life cycle. Domain, functional and pathway enrichment analyses revealed an over-representation of RNA functionalities among the enriched interactors. Using our extensive PPI and RDI networks, we revealed putative new members of RNA-associated pathways, and highlighted potential new roles for several RBPs. Our RBP interactome resource is available through an online interactive platform as a community tool to guide further in-depth functional studies and RBP network analysis (https://www.butterlab.org/RINE).
Graphical Abstract
Graphical Abstract.
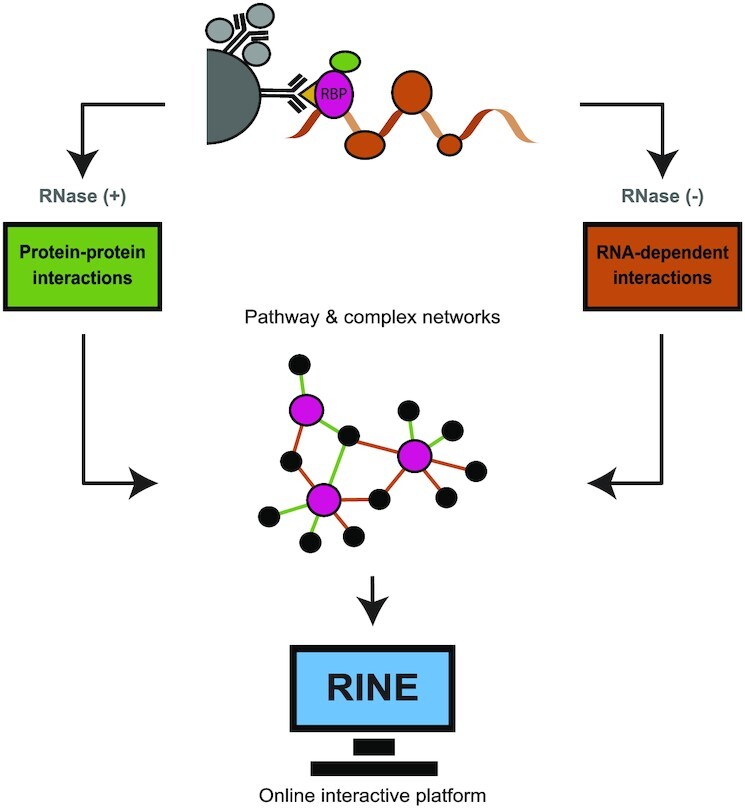
RNA-dependent interactome allows network-based assignment of RNA-binding protein function.
INTRODUCTION
Throughout their life cycle, mRNAs are bound by different RNA-binding proteins (RBPs) forming transient ribonucleoprotein (RNP) complexes. RNP complexes are critical to determine the downstream effects of the bound mRNAs (1). These downstream effects involve the initial mRNA processing, export from the nucleus, transport and localization within the cytoplasm and ultimate translation and degradation of the mRNA. mRNA processing mechanisms, including the initial steps of capping, splicing and polyadenylation, typically require RBP-mediated modifications of the mRNA. These modifications are later recognized by additional RBPs that trigger further coupled processes (2–4). Then, transmembrane RBPs play a critical role in facilitating the passage of mRNA from the nucleus into the cytoplasm (5,6). Once the mRNA has reached the cytoplasm, RNP complexes are again assembled in a sequential and contemporaneous manner to regulate mRNA cellular fate, such as localization, translation or degradation within a large interconnected network (7).
This web of connections is facilitated by interactions with a unique combination of RBPs, as either core or regulatory factors. Core factors are the central players in RNA processes and can be found to interact with a plethora of RNA species. For example, Pab1 is a critical core factor playing a central role in several steps of mRNA processing and metabolism (8,9). Regulatory factors, on the other hand, are more specific; they include, among others, the post-translational regulators interacting with specific sequences or structures of untranslated regions in mRNA (10). There is an interplay between core and regulatory factors, and the recruitment of the latter may result in the assembly of complexes that dictate exchange amid the core factors (11). Thus, the RBP combination of an mRNA determines its cellular fate.
Several large-scale approaches have been applied to discover RBPs in Saccharomyces cerevisiae. Previously, RBPs were identified by using protein arrays in which the capability of each arrayed protein to capture fluorescently labelled RNAs was measured (12,13). Additionally, mass spectrometry (MS) proteomics techniques were developed, including oligo(dT) capture (12) and in vivo RNA interactome capture (RIC), either with conventional cross-linking (14,15) or with photoactivatable ribonucleoside-enhanced cross-linking (16). Furthermore, MS-based techniques have been applied to identify RBPs by validation of a short RNA remnant fragment after cross-linking (17). Over the last decade, these studies have been consolidated into a census of 1273 proteins annotated as RBPs in S. cerevisiae (18). Within this census, there are a large number of proteins lacking canonical RNA-binding domains (RBDs), such as the eukaryotic RNA-recognition motif (RRM) or the heterogeneous nuclear RNP K homology (KH) domain. Of the seven studies included in the S. cerevisiae RBP census, the two largest contributors only reported 7% and 34% of proteins containing classical RBDs (15,16).
Among these proteins without canonical RBDs are a high number of metabolic enzymes. In recent years, there have been an increasing number of studies on the presence and evolutionary conservation of metabolic proteins having a secondary role as RBPs (19,20). In S. cerevisiae, 10% of the RBPs in the census are classified as metabolic enzymes (18). For instance, glyceraldehyde-3-phosphate dehydrogenase (21) and cytosolic aconitase (22,23) are well characterized to function as RBPs. This recurrent evidence showing metabolic enzymes acting as RBPs suggests an extensive enzyme activity regulation network acting through RNAs.
To better understand these RBPs, we need to accompany the RBP catalogue expansion with functional characterization of these proteins (24). Thus, it becomes paramount to correctly identify functional roles for the plethora of RBPs. One strategy used previously is to connect RBPs with a specific RNA sequence or structure to facilitate functional studies (25–27). Another strategy is to use the interconnection of RBPs binding to specific subsets of RNAs to disentangle functionality (28). To implement this experimentally, RBPs can be immunoprecipitated in the presence or absence of RNA to describe concurrent interactions with other RBPs. This can be used to suggest the involvement of the bait RBP in functional pathways (29). Indeed, RNA-dependent protein interactors are more likely to be RBPs themselves, which has been used to predict RBPs from protein–protein interaction (PPI) networks (30). We reasoned that with sufficient data, this strategy can be extended to identify functional associations for previously uncharacterized RBPs. Thus, we immunoprecipitated 40 S. cerevisiae RBPs involved in different RNA pathways and identified their concurrent RNA-dependent and -independent interactors by quantitative proteomics. We further quantified proteome-wide protein expression level changes upon knockout of 13 of these RBPs. Integration of these data with pathway and protein complex annotations revealed new associations and functions of selected RBPs within core RNA maturation and regulation pathways, such as splicing.
MATERIALS AND METHODS
Yeast culture and lysis
Saccharomyces cerevisiae tandem affinity purification (TAP)-tagged strains (31) or knockout (KO) strains (32) (Horizon discovery) were grown for 2 days at 30°C on YPD agar plates. The resulting isolated colonies were inoculated on 15 ml of YPD medium and grown at 30°C and 180 rpm until saturation. Saturated cultures were spiked into 500 ml (RBP interactome screen) or 100 ml (KO screen) of YPD and grown (30°C and 180 rpm) until exponential growth (OD600 between 0.8 and 1.0 absorbance units), when cells were harvested at 3000 g for 5 min. Pelleted cells were suspended in 200 μl of Buffer 1 [50 mM Tris pH 7.5, 150 mM NaCl, 5 mM MgCl2 and freshly added 1 μg/ml pepstatin and leupeptin, 1 mM phenylmethylsulphonyl fluoride (PMSF) and 0.5 mM dithiothreitol (DTT)] and transferred to a 2 ml screw lid tube containing 0.5 mm diameter zirconia/silica beads (Roth). Cells were lysed on a FasPrep-24 (MP Biomedicals) with two 30 s cycles at 6.5 m/s, allowing the samples to cool on ice in between. Cell lysates were topped up with 800 μl of Buffer 2 (Buffer 1 + 0.2% IGEPAL), vortexed and transferred into a new tube, leaving the beads behind. Cell lysates were centrifuged at 15 g twice for 5 min at 4°C, and the supernatant was transferred into a clean tube after each cycle. Finally, the protein concentration was measured with a Bradford assay (Protein Assay Dye Reagent, Bio-Rad).
Immunoprecipitation
Protein G magnetic Dynabeads (30 mg/ml, Invitrogen) were separated with a magnetic rack and washed twice with 1 ml of Buffer 2 (see ‘Yeast culture and lysis’). Beads [20 μl/immunoprecipitation (IP)] were coupled with 1 μg/IP anti-TAP antibody (0.5 mg/ml, GenScript Biotech) in 500 μl of Buffer 2 for 30 min on a rotating wheel at room temperature. Then, the beads were washed twice with 200 μl of Buffer 2 and suspended in 100 μl of Buffer 2. For each immunoprecipitation, 12 mg of protein lysate was combined with the 100 μl of suspended beads and incubated for 3 h on a rotating wheel at 4°C. Then, the beads were washed with 1 ml of Buffer 2 and split into two groups. One group was washed three times with 200 μl of Buffer 3 (Buffer 2 + 10% glycerol) containing 50 μg/IP RNase A from bovine pancreas (Sigma-Aldrich); the other was washed three times with 200 μl of Buffer 3 containing 0.5 μl/ml Ribolock RNase inhibitor (40 U/μl, Fisher Scientific). Finally, the beads were spun down and eluted with 30 μl of lithium dodecylsulphate (LDS) + 10 mM DTT.
MS sample preparation
Both RBP interactome screen immunoprecipitation elution products and KO screen protein lysates (100 μg in 30 μl LDS + 10 mM DTT) were heated for 10 min at 70°C. Proteins were then each separated on either a 4–12% (RBP interactome screen) or a 10% (KO screen) NOVEX gradient SDS gel (Thermo Scientific) for 8 min at 180 V in 1× MES buffer (Thermo Scientific). Proteins were fixed and stained with a Coomassie solution [0.25% Coomassie Blue G-250 (Biozym), 10% acetic acid, 43% ethanol]. The gel lane was cut into slices, minced and destained with a 50% ethanol/50 mM ammonium bicarbonate pH 8.0 solution. Proteins were reduced in 10 mM DTT for 1 h at 56°C and then alkylated with 50 mM iodoacetamide for 45 min at room temperature, in the dark. Proteins were digested with LysC (Wako Chemicals) overnight at 37°C. Peptides were extracted from the gel twice using a mixture of acetonitrile (30%) and 50 mM ammonium bicarbonate pH 8.0 solution, and three times with pure acetonitrile, which was subsequently evaporated in a concentrator (Eppendorf) and loaded on activated C18 material (Empore) StageTips as previously described (33).
MS data acquisition and analysis
Peptides were separated on a 25 cm self-packed column (New Objective) with a 75 μm inner diameter filled with ReproSil-Pur 120 C18-AQ (Dr. Maisch GmbH) with reverse-phase chromatography. The EASY-nLC 1000 (Thermo Fisher) was mounted on a Q-Exactive plus mass spectrometer (Thermo Fisher) and peptides were eluted from the column in an optimized 90 min (RBP interactome screen) or 4 h (KO screen) gradient from 2% to 40% MS-grade acetonitrile/0.1% formic acid solution at a flow rate of 200 nl/min. The mass spectrometer was used in data-dependent acquisition mode with one MS full scan and up to 10 MS/MS scans using HCD fragmentation. Raw MS data were searched using the Andromeda search engine (34) integrated into MaxQuant software suite 1.6.5.0 (35) using the S288C_Genome_Release_64-2-1_orf_trans_all.fasta protein sequences from the Saccharomyces Genome Database (SGD) (36). For the analysis, carbamidomethylation at cysteine was set as a fixed modification and methionine oxidation and protein N-acetylation were variable modifications. The match between runs option was activated.
Knockout library validation
For 18 of our 40 investigated RBPs, deletion strains are available in the S. cerevisiae KO collection (32). For the 18 available strains, we were able to validate the respective RBP knockout on the proteome level for 10 strains, visible as a strong down-regulation due to imputation of missing LFQ (label-free quantitation) values. In four cases, knockout validation was not possible because the target RBP was not detected in the wild type (WT). For the cdc2-Δ strain, Cdc2 expression levels in the KO strain were equal to those in the WT strain. We thus decided to check all strains by colony polymerase chain reaction (PCR) targeting the respective open reading frame (ORF) and the presence of an incorporated kanamycin resistance marker, which should be present in all KO clones.
KO and WT strains were streaked on YPD agar plates and grown for 3 days at 30°C. A single colony was resuspended in 10 μl of fresh 0.02 N NaOH, and incubated in PCR tubes at 99°C for 10 min. The supernatant was transferred to a fresh tube, and chilled on ice for 10 min. To amplify the samples, OneTaq (NEB, M0480S) was used according to the manufacturer’s instructions, containing 2 μl of yeast lysis supernatant in a 25 μl reaction. Horizon discovery ‘YKO Primers from SGDP’ were used. A_confirmation_primer and B_confirmation_primer were used to detect the WT allele, and A_confirmation_primer and kanB were used to detect the KO allele. The annealing temperature was set at 50°C and used with a 90 s elongation cycle. Samples were separated on a 1% agarose gel and imaged with Gel Doc™ XR+ (Bio-Rad) with a 1 s exposure.
This validated 13 KO strains—the 10 previously validated with proteome evidence and another three previously not detected by MS. The two strains without validation (cdc2-Δ and msl1-Δ) were excluded from further downstream analysis.
Bioinformatics analysis
For protein quantification, contaminants, reverse database hits, protein groups only identified by site and protein groups with <2 peptides (at least one of them classified as unique) were removed by filtering from the MaxQuant proteinGroups.txt file. Missing values were imputed by shifting a beta distribution, obtained from the LFQ intensity values, to the limit of quantitation. Further analysis and graphical representation were performed on an R framework (37) incorporating ggplot2 (38), for visualization, among other packages. For the RBP interactome screen, the protein enrichment threshold was set to a P-value <0.05 (Welch t-test) and fold change >2, c = 0.05. Enriched proteins were overlapped with all interactors with physical evidence reported at The Biological General Repository for Interaction Datasets (BioGRID) (39) and with proteins in the RBP census (18). For the KO screen, the protein enrichment threshold was set to a P-value <0.05 (Welch t-test) and to abs(fold change) >2, c = 0.05.
For the protein signature enrichment analysis, domains were queried with InterProScan version 5.50-84.0 (40), and hits in the Pfam (41) and SUPERFAMILY (42) databases were selected for downstream analysis. Signatures for a particular group of enriched proteins were tested for over-representation (P-value <0.01; Fisher’s exact test) against all signatures found in the background (defined as all quantified proteins in the comparison, enriched or not). Signatures found to be over-represented in at least two bait RBPs were selected for graphical representation.
For the molecular function enrichment analysis, terms were queried in the Gene Ontology (GO) database (43) with the ClusterProfiler R package (44). Terms for a particular group of enriched proteins were tested for over-representation {adjusted P-value [false discovery rate (FDR)] <0.05; Fisher’s exact test} against all terms found in the background (defined as all quantified proteins in the comparison, enriched or not). The top five most found terms per group, which were over-represented in at least two bait RBPs, were selected for graphical representation.
For the pathway enrichment analysis, terms were queried in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (45) and Reactome (46) databases with the ClusterProfiler R package (44). Terms for a particular group of enriched proteins were tested for over-representation [adjusted P-value (FDR) <0.05; Fisher’s exact test] against all terms found in the background, defined as all other quantified proteins in the comparison.
For the protein complex analysis, enriched proteins were overlapped with the manually curated heteromeric protein complexes included in the CYC2008 data (47). A ratio for each complex was calculated by dividing the number of proteins overlapping with a particular complex by the total number of proteins in that complex. For the RBP interactome screen data, protein complexes with a ratio >0.5 were selected for graphical representation. Meanwhile, for the KO screen data, the threshold was set at a ratio ≥0.5.
Finally, protein networks were generated with in-house scripts based on an R framework incorporating igraph (48), with the Fruchterman–Reingold force-directed layout algorithm implementation, among other packages. All networks were drawn with the spoke model. For the PPI and RNA-dependent interaction (RDI) global networks, bait RBPs with an associated KEGG term and their prey were selected as nodes. For the functional subnetworks, bait RBPs and their prey were included as nodes when associated with a particular functionality via KEGG analysis, even when associated with multiple terms. Preys were then coloured in grey tones when reported to BioGRID (to any of its interacting baits) and blue tones when not, and with darker tones when reported at the RBP census and lighter tones when not. For the complex subnetworks, bait RBPs and their prey were selected as nodes when interacting or being part of a particular complex. The network explorer interactive platform was developed with the Shiny R package (49).
RESULTS AND DISCUSSION
Quantitative interactomics screen identifies protein–protein and RNA-dependent interactions
We selected RBPs from pathways that span the RNA life cycle, including less characterized RBPs from recent RIC studies (14–16). These RBPs are involved in seven major RNA-associated processes: (i) capping; (ii) splicing; (iii) cleavage; (iv) polyadenylation; (v) nuclear export; (vi) transport and localization; and (vii) degradation (Figure 1A). To select the individual RBPs for the interactome screen, we queried the RBP census (18) for proteins annotated with these roles in the KEGG and Reactome databases (Supplementary Figure S1). Notably, we included 40 RBPs covering various stages of the mRNA life cycle either with specialized roles in particular pathways or with broader functional descriptions (highlighted with an asterisk in Figure 1A). Of these 40 RBPs, two (Spt5 and Sto1) are involved in capping, 14 in splicing, three (Cft1, Mpe1 and Rna14) in cleavage, Pab1 in polyadenylation and Puf3 in transport/localization. Furthermore, two RBPs (Ndc1 and Mex1) were selected for nuclear export and 17 RBPs are associated with RNA degradation.
Figure 1.

RBP interactome screen. (A) The 40 selected bait RBPs are listed with a schematic drawing of their RNA biological processes. Proteins highlighted with an asterisk are associated with multiple processes. (B) Schematic representation of the experimental design to screen for protein–protein interactors and RNA-dependent ineractors in parallel.
We performed a quantitative label-free proteomics screen with the 40 chosen TAP-tagged RBPs (31) using two different conditions and a WT (Figure 1B), which allowed us to differentiate between protein–protein associations/interactions (PPIs) and RNA-dependent associations/interactions (RDIs). To identify the PPIs, we compared immunoprecipitated tagged RBP against WT lysate, both treated with RNase A to digest the RNA, similar to previous large-scale yeast PPI screens (50,51). We also included a second condition where we immunoprecipitated the tagged RBP, but omitted the RNase A treatment, which reveals the RDIs that are only observable in the presence of RNA. Each condition comprised multiple replicate IP experiments that were prepared in parallel and measured on the mass spectrometer as a set applying LFQ.
This study design allowed for quality control benchmarking within each IP set using the bait RBP and RNase A treatment. In the case of RBP-IP compared with the WT lysate, the tagged RBP was expected to be enriched (P-value <0.05 and fold change >2, c = 0.05) (Figure 2A). Indeed, 38 of 40 bait RBPs showed strong enrichment, between 3.3- and 14.1-fold (Supplementary Figure S2). The remaining two tagged RBPs, Ndc1 and Spt5, also showed enrichment of 2.4- and 2.1-fold, respectively, despite slightly less statistical significance of P-value = 0.07 and P-value = 0.10. Additionally, when comparing the RBP IPs with and without RNase treatment, the tagged RBP is expected to be equally abundant (Figure 2B). Again, this was the case for almost all experiments (39 of 40), with only Sub2 showing a slight offset (Supplementary Figure S3). Similarly, we clearly see that RNase A is found in the non-enriched background cloud of proteins, when comparing the RBP IPs with RNase treatment [IP RNase (+) versus WT]. Meanwhile, we observed the RNase A enriched, with a negative fold change, when comparing the non-treated with the treated RBP IPs [IP RNase (–) versus IP RNase (+)] (Figure 2A, B; Supplementary Figures S2 and S3).
Figure 2.

RBP interactome screen reveals different PPIs and overlapping RDIs among bait RBPs. (A) Volcano plot of PPIs for Pab1 comparing enriched proteins of the Pab1-TAP IPs digested with RNase A (n = 4) or WT (n = 4) determined by label-free quantitative proteomics. The enrichment threshold (dotted line) is set to P-value <0.05 (Welch t-test) and fold change >2, c = 0.05. Each dot represents a protein; enriched proteins are shown in black. Pab1-TAP (red) and RNaseA (orange) are indicated. (B) Volcano plot of RDIs for Pab1 comparing enriched proteins of Pab1-TAP IPs (n = 4), with or without RNase A digestion, determined by label-free quantitative proteomics. The enrichment threshold (dotted line) is set to P-value <0.05 (Welch t-test) and fold change >2, c = 0.05. Each dot represents a protein; enriched proteins are shown in black. Pab1-TAP and RNaseA are not enriched (orange). (C and D) Bar plot of PPIs (C) and RDIs (D) for the 40 bait RBPs. Each bar represents the number of enriched proteins [P-value <0.05 (Welch t-test) and fold change >2, c = 0.05]. Each bar is mirrored to show the protein’s overlap with reported interactors at the BioGRID database (left side, dark grey) and with the RBP census (right side, black). Proteins not contained in either are coloured in light grey. (E) Bar plot depicting all enriched interactors of the 40 bait RBPs: unique PPI (green), unique RDI (orange) and shared interactors (grey).
We obtained valuable information for the PPIs (Figure 2C; Supplementary Table S1) and RDIs (Figure 2D; Supplementary Table S2) for the 40 chosen RBPs. For the PPI group, the number of enriched proteins ranged from 4 (Dbp2) to 112 (Mpe1) (Figure 2C), while for the RDI group, the number of enriched proteins ranged from 5 (Upf3) to 143 (Nam7) (Figure 2D). We then used BioGRID, a database of established protein interactions, to check how many of the identified interactions had been previously reported with physical experimental evidence (light grey, left side, Figure 2C and D). The ratio of reported interactions was dependent on the bait RBP, ranging from 0 (for Dbp2, among others) to 80% (for Dhh1) for PPIs and from 0 (for Dbp2, among others) to 82% (for Dhh1) for RDIs. As expected, the percentage of previously identified interactions in the BioGRID database was overall higher for PPIs than for RDIs. For 11 RBPs, our PPIs included at least half of the previously described interactions, while for our RDIs, this was the case for only two bait RBPs (Supplementary Figure S4A, B). Additionally, for 26 of our investigated RBPs, BioGRID classifies more than half of the reported interactions as ‘Affinity Capture-MS’, confirming the experimental results of our approach (Supplementary Figure S4C). Irrespective of PPIs or RDIs, we found high overlap with RBPs included in the published RBP census (18) (black, right side, Figure 2C, D). However, the fraction of RBP-annotated proteins was higher within RDIs compared with PPIs. While the RDI partners of 34 bait RBPs consisted of >70% of RBPs, only 14 bait RBPs showed this high fraction for their PPI partners (Supplementary Figure S4D). Finally, when we checked the overlap of the interactors among the PPI and RDI groups of each bait RBP, we observed that 0 (for Dbp1, among others) to only 22% (for Sto1) are identical (dark grey, Figure 2E), clearly showing that they are two specific subsets of interactors.
Overall, our label-free quantitative RBP interactome screen resulted in two distinct groups of enriched interactors among the PPI and RDI datasets. In the case of RDIs, the majority of protein interactors were included in the RBP census, outlining that our approach is able to uncover hitherto unknown RDIs among a large set of RBPs. With this, we provide complementary information to the previous large-scale screens in yeast that were designed to only report RNA-independent PPIs.
RNA-related functionalities are over-represented among enriched interactors
We wanted to investigate whether RNA functionalities were over-represented and shared among our enriched interactors, from the structural protein domain to the functional pathway level. Thus, we queried for protein signatures among the enriched interactors with InterProScan. For this analysis, we noted that the descriptors ‘RNA recognition motif domain’ and ‘RNA-binding’ (Figure 3A) were significantly over-represented (P-value <0.01) among multiple bait RBPs for both PPIs and RDIs in the structural domain databases Pfam and SUPERFAMILY. Within the PPIs, there were three baits with enrichment of these terms (green, Figure 3A; Supplementary Table S3). We then applied the same protein signature analysis for the enriched RDIs. RNA binding-related domains were most prevalent among the over-represented protein domains (orange, Figure 3A; Supplementary Table S4). For instance, ‘RRM’ (Pfam) and ‘RNA-binding domain’ (SUPERFAMILY) were over-represented among the interactors of 17 and 18 of our 40 bait RBPs, respectively. Overall, this shows a general trend of the RDIs having a larger amount of canonical RBDs.
Figure 3.

Computational analysis links the enriched interactors with RNA-related functionalities. (A) Bar plot of PPI (green) and RDI (orange) protein signatures. Each bar represents the number of bait RBPs with the over-represented signature (P-value <0.01; Fisher’s exact test) found for at least two different bait RBPs. RNA binding-related signatures are in bold. (B) Bar plot of the PPI (green) and RDI (orange) GO molecular function terms. Each bar represents the number of bait RBPs with the over-represented term [adjusted (FDR) P-value <0.05; Fisher’s exact test] for at least two bait RBPs. The top five terms per group are shown. RNA binding-related signatures are in bold. (C) Heat map of the PPI (green) and RDI (orange) KEGG analysis. Each row contains an over-represented KEGG term [adjusted (FDR) P-value <0.05; Fisher’s exact test], with a blue colour gradient representing the gene ratio. The second horizontal bar represents the bait RBP functional selection criterion. The vertical bar represents the global function of the KEGG terms associated with the interactors.
Further interrogation of the enriched PPI set using GO revealed an over-representation [adjusted (FDR) P-value <0.05] of molecular function GO terms such as ‘nucleic acid binding’ among the interactors of nine bait RBPs and ‘RNA-binding’ among the interactors of six bait RBPs (Figure 3B; Supplementary Table S5). Over-represented RNA-related GO molecular function terms were also identified for the enriched RDI set and were more predominant than in the PPI set (Figure 3B; Supplementary Table S6). In particular, the term ‘mRNA binding’ was over-represented among the enriched RDIs for 26 of our 40 bait RBPs. Despite the domain and GO molecular function analysis revealing an enrichment for RNA binding functionalities, especially for RDI partners, the number of canonical RBDs among the bait RBPs’ interactors is low (Supplementary Tables S3 and S4) albeit still significant for the RRM domain (Figure 3A; Supplementary Figure S5). This highlights that among our enriched interactors, RBPs lacking canonical RBDs might be abundant and thus may have less studied functions in the context of RNA biology.
To investigate shared functionalities among our interactors, we queried the KEGG for over-represented pathways among interactors in the PPI and RDI datasets [adjusted (FDR) P-value <0.05; Supplementary Tables S7 and S8, respectively]. As expected, for both PPIs and RDIs, we obtained over-represented KEGG terms associated with known bait RBP functionality in several cases (Figure 3C). In particular, within the 14 selected splicing-associated RBPs, the KEGG term ‘spliceosome’ was over-represented among PPIs of the bait RBPs Snp1, Ist3 and Hsh49, as well as for the RDIs of the bait RBP Msl1. For Cbc2 and Lsm2, the ‘spliceosome’ term was over-represented for both PPIs and RDIs. The degradation-associated RBPs Rrp40, Dhh1, Dis3, Sup35 and Hbs1 had RNA degradation-associated KEGG terms over-represented among their PPIs, while Sgn1 and Tif4631 showed this among their RDIs. We also enriched interactors related to ‘ribosome’ and ‘ribosome biogenesis in eukaryotes’ in four RDI (Cbc2, Mtr4, Mud2 and Tif4631) and two PPI (Gbp2 and Hsh49) datasets. Additionally, while not among our selection criteria for the bait RBPs, we observed a strong over-representation of metabolic and synthesis pathways among the interactors for 7 and 13 baits of the PPI and RDI groups, respectively. There were 525 metabolism-related proteins within the combined PPI and RDI datasets, which included 70 of the 154 metabolic RBPs described in the RBP census. Previously, metabolic proteins have been characterized to have unusual RNA binding function, which coincides with the lack of enrichment of canonical RBDs across all RBPs (18). Through our unbiased, global analysis, our dataset adds more evidence to the growing field of metabolic enzymes with RBP functionalities (18,20).
Overall, despite a high number of interactors without canonical RBDs, we confirm that interactors of both PPI and RDI groups are involved in RNA biology through association with structural domains, molecular functions and biological pathways. This is in agreement with the SONAR dataset, where they already used this observation on a smaller set of baits for the prediction of hitherto unknown RBPs (30).
RBP knockouts reveal possible processes regulated by these RBPs
RBPs that interact with a certain set of mRNAs have sometimes been associated with the post-transcriptional regulation of genes belonging to a specific biological process or protein complex (10,52). To further investigate the downstream biological processes that are likely to be regulated by the action of our bait RBPs, we aimed to identify proteins that are differentially expressed at the protein level upon knockout of our selected RBPs. For 18 of our 40 investigated RBPs, deletion strains were available in the S. cerevisiae KO collection (32). Of the 22 unavailable KO strains, 21 correspond to essential genes and one is not included in the library. Further experimental validation of the 18 available strains resulted in the confirmation of RBP knockout in 10 strains at the proteomic level, and an additional 3 strains at the genomic level (Supplementary Figures S6 and S7A, B; see also the Materials and Methods). Thus, 13 RBP strains were utilized for further experimental investigations; these included five RBPs involved in mRNA nuclear processing, one in RNA transport and localization, and seven in degradation (Supplementary Figure S1A).
We measured the proteomes of WT and individual KO clones by MS and performed label-free quantification with multiple replicates (Figure 4A). Per knockout, we quantified between 2854 and 3184 proteins (Supplementary Table S9), approximately two-thirds of the expressed yeast proteome (53). To determine significant protein expression changes, we compared the WT with the KOs, setting a threshold at P-value <0.05 and abs(fold change) >2, c = 0.05 (Figure 4B). For the 13 RBP KOs, significant protein expression changes ranged from 7 to 230 proteins, representing on average a higher number of differentially expressed proteins than observed for a genome-wide KO screen performed in Schizosaccharomyces pombe (54) and being on a par with expression changes observed for their S. pombe homologues (Figure 4C; Supplementary Figures S6 and S8; Supplementary Table S10). This shows that the knockout of the selected RBPs led on average to more profound expression changes than knockout of other genes. As expected, we observed that differentially expressed proteins overlapped little with prey identified in the PPI and RDI datasets of each respective bait RBP; ranging from 0 (for gbp2-Δ, among others) to 4.4% (for sto1-Δ) (Figure 4C). This supports our initial hypothesis that the differentially expressed proteins in the KO strains point to downstream processes regulated by the RBPs and are thus different from the RBP-associated proteins. However, differential expression of some proteins can also be the result of functional compensation mechanisms.
Figure 4.

Protein expression changes among the 13 RBP KO strains. (A) Schematic representation of the KO screen experimental design. (B) Volcano plot of ski2-Δ comparing its proteome with that of the WT by label-free quantitative proteomics (n = 4). The enrichment threshold (dotted line) is set to P-value <0.05 (Welch t-test) and abs(fold change) >2, c = 0.05. Each dot represents a protein; enriched proteins are shown in black with Ski2 highlighted (red). (C) Bar plot of the altered proteins in the 13 RBP KOs. Each bar shows the number of altered proteins with highlighted overlap for PPI (green) and RDI (orange) with the RBP interactome screen. (D) Heat map of the KEGG analysis for the 13 RBP KOs. Each row contains an over-represented KEGG term [adjusted (FDR) P-value <0.05; Fisher’s exact test] with a blue colour gradient for the gene ratio. The horizontal bar delineates up- and down-regulated proteins. (E) Network of the protein complexes with at least half of their subunits included among the differentially expressed proteins in the RBP KOs. Nodes are RBP KOs (purple) and protein complexes (blue gradient). Edges are highlighted for up-regulated (red) or down-regulated (blue) proteins.
To further characterize the biological pathways that are affected and likely to be regulated by the knocked out RBPs, we tested for KEGG pathway enrichment among the up- and down-regulated proteins separately in each strain (Figure 4D; Supplementary Tables S11 and S12). Only three pathways related to RNA were over-represented in three KO strains, ‘aminoacyl-tRNA biosynthesis’, ‘mRNA surveillance pathway’ and ‘ribosome’ (Figure 4D). Interestingly, amino acid and nucleic acid synthesis pathways as well as various metabolic pathways were enriched among proteins that were down-regulated in KOs of RBPs related to splicing (ist3-Δ and gbp2-Δ) and to degradation (sgn1-Δ, mip6-Δ, ski2-Δ, dhh1-Δ, upf3-Δ and hbs1-Δ). However, these do not overlap with the over-represented metabolic KEGG pathways from the RBP interactome screen.
To further interrogate cellular processes that are likely to be regulated by the selected RBPs, we identified known yeast protein complexes for which at least half of all subunits were either up- or down-regulated in the individual KO strains (Figure 4E). We primarily obtained dimeric complexes (27 of 31) across the 13 selected RBPs. These complexes covered a wide range of functionalities. Some were expected, such as the down-regulation of the PAN complex that is directly related to the knockout of one of its subunits, Pan3. Others echoed with the over-represented KEGG pathways. For instance, we obtained several metabolism-related complexes that were up-regulated in dhh1-Δ. Similarly, mip6-Δ and gbp2-Δ, both having KEGG metabolic pathways down-regulated, had a subunit of the Pmt3p/Pmt5p complex down-regulated. Additionally, we obtained further insights into RBPs when examining the up- and down-regulated complex members. In dhh1-Δ we found a down-regulation of mismatch repair proteins. This is in line with the involvement of Dhh1 in DNA repair (55). Dhh1 has also been shown to be critical to G1/S phase cell cycle progression, and its deletion causes ionizing radiation sensitivity (56). There are emerging studies that have linked RBPs with DNA repair processes (57,58). Taken together, this KO dataset could lead to the further association of RBPs with metabolism and DNA repair.
These results collectively point to interesting hypotheses for possible downstream processes regulated by the selected RBPs and can guide future experimental investigations.
Network analysis identifies putative new members of RNA pathways
To connect information from the individual experiments and visualize shared interactors among the RBPs, we built an extensive interaction network for the PPI (Figure 5A; Supplementary Table S13) and RDI (Figure 5B; Supplementary Table S14) datasets. Within each network, the bait RBPs and their interactors were included when enriched for a KEGG term (Figure 3C). These KEGG terms were grouped as degradation, export, metabolism, ribosome, splicing, synthesis or multiple. Each of these individual categories was used to create specific subnetworks. To gain further insights into which complexes are captured within our subnetworks, we annotated our proteins with the manually curated heteromeric protein complexes included in the CYC2008 dataset (47). We calculated for each complex a coverage ratio by dividing the number of subunits included in the network by the total number of complex members, and those with a ratio >0.5 were included in the downstream analysis. Within all splicing, export, ribosome, synthesis, metabolism and degradation subcomplexes, 56 unique complexes were identified. These combined networks as well as individual subnetworks are available online for each enriched biological process within the RBP interactome network explorer (RINE) at https://www.butterlab.org/RINE.
Figure 5.

PPI and RDI networks link novel interactors to RNA-related functionalities. (A and B) PPI network (A) and RDI network (B) with all bait RBPs labelled. Bait RBP nodes and edges are coloured according to single (colour indicated in the key) or multiple (purple) higher order KEGG-associated functions. The nodes of the RBP prey are light grey and node sizes are determined by the number of interactors. The networks are drawn with the spoke model.
We wanted to check the presence of expected and unexpected complexes as well as cross-talk between these complexes in an exemplary RNA process. Thus, we further examined the PPI and RDI subnetworks of the baits enriched for splicing-related KEGG terms, which have been widely studied in S. cerevisiae (59). Within the interactions, there were seven baits included in the PPI subnetwork (Ist3, Lsm2, Cbc2, Dhh1, Snp1, Sto1 and Hsh49) (Figure 6A; Supplementary Table S15), and there were four bait RBPs included in the RDI subnetwork (Ist3, Cbc2, Msl1 and Lsm2) (Figure 6B; Supplementary Table S16). We had 15 and 20 previously unreported interactions in the PPIs and RDIs, respectively, when compared with BioGRID and RBP census data (Figure 6A, B). Among these PPIs, there were three uncharacterized interactors, namely YHR214C-B and YMR315W with bait RBP Hsh49, and YPL225W with bait RBP Lsm2. We additionally integrated both the PPI and RDI interactomes into one combined subnetwork (Figure 6C; Supplementary Table S17).
Figure 6.

Splicing subnetworks link bait RBPs with protein complexes. (A and B) PPI splicing (A) and RDI splicing (B) subnetworks, with bait RBPs possessing interactors, with KEGG-associated splicing pathways highlighted in pink. Interactors reported at BioGRID (light grey), at the RBP census (dark blue) or at both (dark grey) are indicated. Interactors not found in BioGRID or the RBP census are indicated in light blue. The node size of the bait RBPs is determined by the number of interactors. The networks are drawn with the spoke model. (C) Combined (PPI and RDI) network, with bait RBPs having interactors with KEGG-associated splicing pathways in pink. Edges are highlighted for unique PPI (green), unique RDI (orange) or shared interactors (grey). The network is drawn with the spoke model. (D) Protein complex Cleveland dot plot. Protein complexes with a coverage ratio >0.5 are represented. The number of proteins in the network (purple) and in the complex (orange) are shown as dots. (E) Heat map of the protein complexes. Each row names a protein complex, with a blue colour gradient for the coverage ratio. The horizontal bar indicates the bait RBP functional selection criterion.
Using the protein complexes from the CYC2008 dataset, we established complexes present at a ratio >0.5 within the combined RDI and PPI subnetwork (Figure 6D; Supplementary Table S18). There were 19 complexes within the PPIs and RDIs with over-represented splicing-related KEGG pathways that surpassed this threshold. These complexes contained the anticipated spliceosome components as well as other complexes that are involved in the mRNA life cycle. The complexes that were not spliceosome components function in capping, degradation, nuclear export and translation (Figure 6D). For example, the transcription export (TREX) complex, which contains the THO complex, were both found among the interactors. These complexes are critical for the nuclear export of mRNA (60). There were six baits with interactors that are components of the TREX complex (Figure 6E). Of these, Msl1, Snp1 and Hsh49 are considered splicing-associated proteins, while Dhh1 is considered to be degradation associated. This shows the interconnectedness of mRNA splicing, export and degradation. An unexpected complex was the RENT complex, which is responsible for rDNA silencing in S. cerevisiae. The bait RBPs Ist3 and Msl1, which are both splicing proteins, had members of the RENT complex among their PPI interactors. This association had not been previously reported.
While there were complexes with a wide range of functionalities in the splicing subnetwork, most identified subunits were part of the spliceosome (Figure 6E). We built a network for each annotated spliceosome complex shared by more than one bait (Figure 7; Supplementary Table S19). Within these baits, 16 of the 21 members of the commitment complex (or E complex), were detected (61). The three baits with the largest number of E complex subunits were Snp1, Sto1 and Cbc2. Snp1 is a portion of U1 small nuclear RNP (snRNP), and Sto1 and Cbc2 are capping proteins, and are all part of the S. cerevisiae commitment complex (59,62). This demonstrates that our approach is able to identify known complexes. The U2-associated complex SF3a/b had a high average coverage ratio across many baits (ratio = 0.67 and 0.83, respectively). However, the other U2 snRNP-associated complex, called RES, was exclusively found in the interactome of its complex member and bait RBP, Ist3.
Figure 7.

Spliceosome protein complex subnetworks reveal novel bait RBP functionalities. Spliceosome complexes shared by more than one bait RBP and with a coverage ratio >0.5 are shown. Bait RBPs being part of (dark orange) or associated with (purple) a protein complex are shown. Prey being part of a protein complex (light orange) are shown. Edges are coloured for PPI (green), RDI (orange) or both (grey). The networks are drawn with the spoke model.
The U4/U6 × U5 tri-snRNP complex (ratio = 0.82) joins splicing complex A (including the U1 and U2 snRNP complex) to form the preB splicing complex. However, despite the thorough U6 coverage (ratio = 1.0), its complex members were found only with Lsm2 and Dhh1. Lsm2 is a well known complex member of U6 snRNP, whereas Dhh1 has not been characterized in detail as a U6 snRNP-associated protein. Dhh1 facilitates decapping and inhibits translation (63,64). Nevertheless, there has been a yeast two-hybrid study confirming the Lsm2 and Dhh1 interaction (65), and there have been studies associating Lsm4 and Dhh1 with P-granules, which in turn are associated with inhibition of translation (66,67). We noticed that Dhh1 association was limited to snRNPs that join the spliceosome later during the splicing process.
Another unexpected interaction occurred between the RES complex subunit Ist3 and the U5 snRNP complex. The RES complex is critical for the successful formation of the pre-spliceosome complex via its interaction with U2 snRNP (3). Within the diverse roles of the RES complex, no association with the U5 complex apart from Prp8 was described (63,64). However, within our network analysis we found several PPIs between Ist3 and the U5 snRNP complex members Prp8, Brr2 and Snu114. These data suggest that there might be more U5 members serving as a bridge to the RES complex.
In summary, we found complexes that were critical to the overall mRNA life cycle, with particular enrichment of complexes needed for the assembly and catalytic activity of the spliceosome with putative new roles for RBPs based on concurrent binding patterns.
CONCLUSIONS
Here, we provide an extensive S. cerevisiae RBP interactome network to systematically map both PPIs and RDIs. The approach to study RDIs at a larger scale gives the unique opportunity to group RBPs by concurrent binding patterns and thus provides suggestions for functions for the RBPs themselves as well as for their interaction partners. An additional integration of the RINE resource with next-generation sequencing data containing information about the RNAs bound to RBPs would provide further insights into the functionalities of both the RBPs and their associated RNAs. By providing interactive and visual access to the data of this study, the RINE resource (https://www.butterlab.org/RINE) can serve as a starting point for further data analysis and exploration of individual candidates.
DATA AVAILABILITY
The R scripts required for the data statistical analysis and its visualization are available at Zenodo (https://doi.org/10.5281/zenodo.7753608). The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (68) partner repository with the dataset identifiers PXD035979 (RBP interactome screen) and PXD035971 (KO screen). The protein interactions from this publication have been submitted to the IMEx (http://www.imexconsortium.org) consortium through IntAct (69) and assigned the identifier IM-29638.
Supplementary Material
ACKNOWLEDGEMENTS
Assistance by the Proteomics Core Facility and the Media lab at IMB, and critical reading of the manuscript by Julian König is gratefully acknowledged.
Contributor Information
Albert Fradera-Sola, Quantitative Proteomics, Institute of Molecular Biology, D-55128 Mainz, Germany.
Emily Nischwitz, Quantitative Proteomics, Institute of Molecular Biology, D-55128 Mainz, Germany.
Marie Elisabeth Bayer, Quantitative Proteomics, Institute of Molecular Biology, D-55128 Mainz, Germany.
Katja Luck, Integrative Systems Biology, Institute of Molecular Biology, D-55128 Mainz, Germany.
Falk Butter, Quantitative Proteomics, Institute of Molecular Biology, D-55128 Mainz, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The work was supported by the Deutsche Forschungsgemeinschaft [BU 2996/7-1 (SPP1935: ‘Deciphering the mRNP code: RNA-bound Determinants of Post-Transcriptional Gene Regulation’) and project number 439669440 (TRR319 RMaP TP C03) to F.B. and LU 2568/1-1 to K.L.]. Funding for open access charge: DFG 439669440 [TRR319 RMaP].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dreyfuss G., Kim V.N., Kataoka N.. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 2002; 3:195–205. [DOI] [PubMed] [Google Scholar]
- 2. Ramanathan A., Robb G.B., Chan S.-H.. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016; 44:7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilkinson M.E., Charenton C., Nagai K.. RNA splicing by the spliceosome. Annu. Rev. Biochem. 2020; 89:359–388. [DOI] [PubMed] [Google Scholar]
- 4. Neve J., Patel R., Wang Z., Louey A., Furger A.M.. Cleavage and polyadenylation: ending the message expands gene regulation. RNA Biol. 2017; 14:865–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tutucci E., Stutz F.. Keeping mRNPs in check during assembly and nuclear export. Nat. Rev. Mol. Cell Biol. 2011; 12:377–384. [DOI] [PubMed] [Google Scholar]
- 6. Xie Y., Ren Y.. Mechanisms of nuclear mRNA export: a structural perspective. Traffic. 2019; 20:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Licatalosi D.D., Darnell R.B.. RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 2010; 11:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mangus D.A., Evans M.C., Jacobson A.. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 2003; 4:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kühn U., Wahle E.. Structure and function of poly(A) binding proteins. Biochim. Biophys. Acta. 2004; 1678:67–84. [DOI] [PubMed] [Google Scholar]
- 10. Glisovic T., Bachorik J.L., Yong J., Dreyfuss G.. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008; 582:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rissland O.S. The organization and regulation of mRNA–protein complexes. Wiley Interdiscip. Rev. RNA. 2017; 8:e1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsvetanova N.G., Klass D.M., Salzman J., Brown P.O.. Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS One. 2010; 5:e12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scherrer T., Mittal N., Janga S.C., Gerber A.P.. A screen for RNA-binding proteins in yeast indicates dual functions for many enzymes. PLoS One. 2010; 5:e15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitchell S.F., Jain S., She M., Parker R.. Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 2013; 20:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matia-González A.M., Laing E.E., Gerber A.P.. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 2015; 22:1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beckmann B.M., Horos R., Fischer B., Castello A., Eichelbaum K., Alleaume A.-M., Schwarzl T., Curk T., Foehr S., Huber W.et al.. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 2015; 6:10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kramer K., Sachsenberg T., Beckmann B.M., Qamar S., Boon K.-L., Hentze M.W., Kohlbacher O., Urlaub H.. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods. 2014; 11:1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 19. Hentze M.W., Preiss T.. The REM phase of gene regulation. Trend. Biochem. Sci. 2010; 35:423–426. [DOI] [PubMed] [Google Scholar]
- 20. Curtis N.J., Jeffery C.J.. The expanding world of metabolic enzymes moonlighting as RNA binding proteins. Biochem. Soc. Trans. 2021; 49:1099–1108. [DOI] [PubMed] [Google Scholar]
- 21. Singh R., Green M.R.. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science. 1993; 259:365–368. [DOI] [PubMed] [Google Scholar]
- 22. Hentze M.W., Argos P.. Homology between IRE-BP, a regulatory RNA-binding protein, aconitase, and isopropylmalate isomerase. Nucleic Acids Res. 1991; 19:1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rouault T.A., Stout C., Kaptain S., Harford J.B., Klausner R.D.. Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications. Cell. 1991; 64:881–883. [DOI] [PubMed] [Google Scholar]
- 24. Kilchert C., Sträßer K., Kunetsky V., Änkö M.-L.. From parts lists to functional significance—RNA–protein interactions in gene regulation. Wiley Interdiscip. Rev. RNA. 2020; 11:e1582. [DOI] [PubMed] [Google Scholar]
- 25. Butter F., Scheibe M., Mörl M., Mann M.. Unbiased RNA–protein interaction screen by quantitative proteomics. Proc. Natl Acad. Sci. USA. 2009; 106:10626–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scheibe M., Butter F., Hafner M., Tuschl T., Mann M.. Quantitative mass spectrometry and PAR-CLIP to identify RNA–protein interactions. Nucleic Acids Res. 2012; 40:9897–9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Casas-Vila N., Sayols S., Pérez-Martínez L., Scheibe M., Butter F.. The RNA fold interactome of evolutionary conserved RNA structures in S. cerevisiae. Nat. Commun. 2020; 11:2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hogan D.J., Riordan D.P., Gerber A.P., Herschlag D., Brown P.O.. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008; 6:e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klass D.M., Scheibe M., Butter F., Hogan G.J., Mann M., Brown P.O.. Quantitative proteomic analysis reveals concurrent RNA–protein interactions and identifies new RNA-binding proteins in Saccharomycescerevisiae. Genome Res. 2013; 23:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brannan K.W., Jin W., Huelga S.C., Banks C.A., Gilmore J.M., Florens L., Washburn M.P., Van Nostrand E.L., Pratt G.A., Schwinn M.K.et al.. SONAR discovers RNA-binding proteins from analysis of large-scale protein–protein interactomes. Mol. Cell. 2016; 64:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghaemmaghami S., Huh W.-K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S.. Global analysis of protein expression in yeast. Nature. 2003; 425:737–741. [DOI] [PubMed] [Google Scholar]
- 32. Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H.et al.. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999; 285:901–906. [DOI] [PubMed] [Google Scholar]
- 33. Rappsilber J., Mann M., Ishihama Y.. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using stagetips. Nat. Protoc. 2007; 2:1896–1906. [DOI] [PubMed] [Google Scholar]
- 34. Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M.. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011; 10:1794–1805. [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 36. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R.et al.. Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. R Core Team R: A Language and Environment for Statistical Computing. 2021; Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 38. Wickham H. Ggplot2—Elegant Graphics for Data Analysis. 2016; Cham: Springer International Publishing. [Google Scholar]
- 39. Oughtred R., Rust J., Chang C., Breitkreutz B.-J., Stark C., Willems A., Boucher L., Leung G., Kolas N., Zhang F.et al.. The BioGRID Database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Prot. Sci. 2021; 30:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blum M., Chang H.-Y., Chuguransky S., Grego T., Kandasaamy S., Mitchell A., Nuka G., Paysan-Lafosse T., Qureshi M., Raj S.et al.. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021; 49:D344–D354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J.et al.. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilson D., Pethica R., Zhou Y., Talbot C., Vogel C., Madera M., Chothia C., Gough J.. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009; 37:D380–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. The Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021; 49:D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L.et al.. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021; 2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Prot. Sci. 2019; 28:1947–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R.et al.. The reactome pathway knowledgebase. Nucleic Acids Res. 2020; 48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pu S., Wong J., Turner B., Cho E., Wodak S.J.. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res. 2009; 37:825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Csardi G., Nepusz T.. The igraph software package for complex network research. InterJournal, Complex Systems. 2006; 1695:1–9. [Google Scholar]
- 49. Chang W., Cheng J., Allaire J., Sievert C., Schloerke B., Xie Y., Allen J., McPherson J., Dipert A., Borges B.. shiny: Web Application Framework for R. 2023; R package version 1.7.4.9002https://shiny.rstudio.com/.
- 50. Ho Y., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.-L., Millar A., Taylor P., Bennett K., Boutilier K.et al.. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002; 415:4. [DOI] [PubMed] [Google Scholar]
- 51. Krogan N.J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P.et al.. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006; 440:637–643. [DOI] [PubMed] [Google Scholar]
- 52. Corley M., Burns M.C., Yeo G.W.. How RNA-binding proteins interact with RNA: molecules and mechanisms. Mol. Cell. 2020; 78:9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Godoy L.M.F., Olsen J.V., Cox J., Nielsen M.L., Hubner N.C., Fröhlich F., Walther T.C., Mann M.. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008; 455:1251–1254. [DOI] [PubMed] [Google Scholar]
- 54. Öztürk M., Freiwald A., Cartano J., Schmitt R., Dejung M., Luck K., Al-Sady B., Braun S., Levin M., Butter F.. Proteome effects of genome-wide single gene perturbations. Nat. Commun. 2022; 13:6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bergkessel M., Reese J.C.. An essential role for the Saccharomyces cerevisiae DEAD-Box Helicase DHH1 in G1/S DNA-damage checkpoint recovery. Genetics. 2004; 167:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Westmoreland T., Olson J., Saito W., Huper G., Marks J., Bennett C.. Dhh1 regulates the G1/S-checkpoint following DNA damage or BRCA1 expression in yeast1. J. Surg. Res. 2003; 113:62–73. [DOI] [PubMed] [Google Scholar]
- 57. Dutertre M., Lambert S., Carreira A., Amor-Guéret M., Vagner S.. DNA damage: RNA-binding proteins protect from near and far. Trend. Biochem. Sci. 2014; 39:141–149. [DOI] [PubMed] [Google Scholar]
- 58. Klaric J.A., Wüst S., Panier S.. New faces of old friends: emerging new roles of RNA-binding proteins in the DNA double-strand break response. Front. Mol. Biosci. 2021; 8:668821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Plaschka C., Newman A.J., Nagai K.. Structural basis of nuclear pre-mRNA splicing: lessons from yeast. Cold Spring Harb. Perspect. Biol. 2019; 11:a032391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Katahira J. mRNA export and the TREX complex. Biochim. Biophys. Acta. 2012; 1819:507–513. [DOI] [PubMed] [Google Scholar]
- 61. Larson J.D., Hoskins A.A.. Dynamics and consequences of spliceosome E complex formation. eLife. 2017; 6:e27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gonatopoulos-Pournatzis T., Cowling V.H.. Cap-binding complex (CBC). Biochem. J. 2014; 457:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sweet T., Kovalak C., Coller J.. The DEAD-box protein dhh1 promotes decapping by slowing ribosome movement. PLoS Biol. 2012; 10:e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Coller J.M., Tucker M., Sheth U., Valencia-Sanchez M.A., Parker R.. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA. 2001; 7:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Uetz P., Giot L., Cagney G., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P.et al.. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000; 403:623–627. [DOI] [PubMed] [Google Scholar]
- 66. Cary G.A., Vinh D. B.N., May P., Kuestner R., Dudley A.M.. Proteomic analysis of Dhh1 complexes reveals a role for Hsp40 chaperone Ydj1 in yeast P-body assembly. G3 Genes|Genomes|Genetics. 2015; 5:2497–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rao B.S., Parker R.. Numerous interactions act redundantly to assemble a tunable size of P bodies in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 2017; 114:E9569–E9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Perez-Riverol Y., Bai J., Ban dla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M.et al.. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022; 50:D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N.et al.. The MIntAct Project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014; 42:D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R scripts required for the data statistical analysis and its visualization are available at Zenodo (https://doi.org/10.5281/zenodo.7753608). The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (68) partner repository with the dataset identifiers PXD035979 (RBP interactome screen) and PXD035971 (KO screen). The protein interactions from this publication have been submitted to the IMEx (http://www.imexconsortium.org) consortium through IntAct (69) and assigned the identifier IM-29638.