


Abstract
Blockage of replication forks can have deleterious consequences for the cell as it may prompt premature termination of DNA replication. Moreover, the blocked replication intermediate (RI) could be particularly sensitive to recombination processes. We analysed the different populations of RIs generated in vivo in the bacterial plasmid pPI21 after pausing of replication forks at the inversely oriented ColE1 origin. To achieve this goal, a new method was developed based on two-dimensional agarose gel electrophoresis. This method allows the isolation of specific RIs, even when they were rather scarce, from the total DNA. Here we describe the occurrence of RI restriction fragments containing reversed forks. These Holliday-like structures have been postulated but never observed before.
INTRODUCTION
The replication machinery, despite its high accuracy and processivity, does not always progress in a smooth way along the DNA template. Structural elements such as hairpins, triplexes and quadruplexes, or DNA modifications induced by radiation or chemical agents, may cause replication forks to slow down, pause or even arrest (reviewed in 1). DNA-binding proteins can also lead to blockage of replication forks. In the chromosomal terminus region of Escherichia coli and Bacillus subtilis, replication termination proteins interact with specific DNA sequences causing a polar replication fork arrest (reviewed in 2). Protein-mediated replication pause sites have also been reported in eukaryotes. The replication initiator protein EBNA-1 binds to the replication origin of the Epstein–Barr virus blocking progression of DNA replication in one direction. This leads to unidirectional replication of the viral genome (3). Additionally, replication forks pause at yeast centromeres, as a result of the protein–DNA complexes required for centromeric function in Saccharomyces cerevisiae (4). Finally, recent reports indicate that replication fork barriers in the ribosomal RNA repeats require the presence of a specific protein: Fob1 in S.cerevisiae (5) and the transcription terminator factor TTF-1 in the mouse (6,7).
The consequences of replication fork blockage are of particular interest. Premature termination of DNA replication may lead to double-stranded breaks in recBC mutants (8). Moreover, it was proposed that it could induce DNA replication fork reversal in E.coli (9). The presence of DNA ends and single-stranded regions at a blocked fork can also easily trigger genome rearrangements, as these structures are more prone to DNA recombination (1).
Replication arrest leads to the accumulation of a specific population of partially replicated intermediates in the cell. To further understand the consequences of replication arrest it is necessary to analyse the accumulated replication intermediate (RI) by mapping the leading and lagging strands at the arrested fork and, in this way, characterise the mechanism of the replication blockage. This implies isolation of the accumulated, but still scarce, partially replicated intermediate from the total DNA.
We have proposed that plasmid multimers containing two inversely oriented ColE1 origins do not occur in nature because these plasmids cannot complete their replication due to polar blockage of the replication fork at the silent and inversely oriented ColE1 origin (10). In the present study, our goal was to detect all kinds of RIs generated in vivo as a consequence of replication fork blockage. The system used was the plasmid pPI21, which contains two inversely oriented ColE1 replication origins separated by 1.4 kb. Although both origins are equally competent to initiate replication, only one initiation event occurs per molecule (10,11). The silent ColE1 origin blocks replication fork progression in an orientation-dependent manner (10,12) and consequently leads to the accumulation in the cell of a specific RI containing an internal bubble spanning the region between both origins (the accumulated bubble). For the isolation of such a specific population of RIs and other less abundant species that might be related to the pausing of replication forks we developed a new method consisting of two successive agarose gel electrophoreses. This method is a modification of the neutral/neutral 2-dimensional (2D) agarose gel electrophoresis technique for the analysis of branched RIs (13,14).
The procedure allowed us to characterise by electron microscopy the RIs containing an internal bubble, the sizes of parental and nascent strands by neutral/alkaline 2D agarose gels (10) and fine mapping of the replication fork pausing site at the silent ColE1 origin by primer extension (D.Santamaría, P.Hernández, M.Martínez-Robles, D.Krimer and J.Schvartzman, submitted for publication). Furthermore, it allowed the identification of a subpopulation of RIs containing knots with different numbers of nodes in their replicated portion (10,15). In the present study we report that this procedure also led us to detect another subpopulation of molecules containing reversed forks. Molecules containing reversed forks have been postulated (9,16) but never observed before.
MATERIALS AND METHODS
Strains and plasmids
Escherichia coli DH5αF′ cells were transformed with plasmid pPI21. Cells were grown in LB medium containing 50 µg/ml ampicillin.
Isolation of plasmid DNA
Plasmid DNA was isolated from an exponentially growing culture, as described in Santamaría et al. (12). Basically, cells from a fresh culture were grown in 1 litre LB medium at 37°C until the OD reached 0.4–0.6, quickly chilled, centrifuged and washed with 20 ml of STE buffer (0.1 M NaCl, 10 mM Tris–HCl pH 8.0, and 1 mM EDTA, pH 8.0). Cells were collected by centrifugation and resuspended in 5 ml of 25% sucrose in 0.25 M Tris–HCl pH 8.0. Lysozyme (10 mg/ml) and RNase A (0.1 mg/ml) were added and the suspension was maintained on ice for 5 min. Afterwards, 2 ml of 0.25 M EDTA, pH 8.0, were added and the suspension was kept on ice for another 5 min. Cell lysis was achieved by adding 8 ml of lysis buffer (1% Brij-58, 0.4% sodium deoxycholate, 63 mM EDTA pH 8.0, and 50 mM Tris–HCl pH 8.0) and incubating for 1 h on ice. Plasmid DNA was recovered from the supernatant after centrifugation at 26 000 g for 60 min at 4°C, precipitated by adding a 2/3 vol of 25% polyethylene glycol 6000 and 1.25 M NaCl in TE (10 mM Tris–HCl pH 8.0, and 1 mM EDTA) and kept overnight at 4°C. The precipitated DNA was pelleted by centrifugation at 6000 g for 15 min at 4°C and the pellet resuspended in 5 ml of digestion buffer (100 µg/ml proteinase K in 1 M NaCl, 10 mM Tris–HCl pH 9.0, 1 mM EDTA and 0.1% SDS) and incubated at 65°C for 30 min. Proteins were extracted twice with phenol:chloroform:isoamyl alcohol (25:24:1) and once with chloroform:isoamyl alcohol (24:1). The DNA was precipitated overnight at –20°C with 2.5 vol of absolute ethanol and resuspended in TE. The DNA was digested with restriction endonucleases (Roche Molecular Biochemicals) as recommended by the manufacturer in the presence of 100 µg/ml RNase A.
Isolation of specific replication intermediates
Analysis of RIs by standard 2D agarose gel electrophoresis was performed as described (11). For the isolation of a specific RI, two successive agarose electrophoreses were performed, as depicted in Figure 1. After restriction digestion, the preparative DNA sample was loaded in a 10 cm well in a 0.4% agarose gel; this allows loading of as much as 100 µg of total plasmid DNA without distorting DNA migration. The first electrophoresis was run under the same conditions as those employed in the first dimension of a neutral/neutral 2D agarose gel to minimise the effect of molecular shape [0.4% agarose gel in TBE buffer (85 mM Tris base, 89 mM borate and 2.5 mM EDTA) at 0.6 V/cm at room temperature for 37 h]. An aliquot of the preparative sample was run in a separate single lane and used as a control (control sample). After the first dimension, this control lane was cut out, stained with ethidium bromide and examined under UV light. In this way, we calculated the distance migrated by the accumulated bubbles. This distance was then extrapolated to the preparative sample (see Fig. 1A). A segment of the preparative gel from the position of the accumulated bubbles to a little beyond the linear molecules was excised from the gel and placed on a new gel in the same orientation as it had in the first dimension (see Fig. 1B).
Figure 1.

Schematic representation of the procedure used to isolate specific RIs. (A) First gel electrophoresis. The digested DNA sample (preparative sample) is loaded in a long well. In this way, 200 times the amount of total DNA generally used in a classical 2D agarose gel could be loaded. This would further increase the yield of recovery of specific RIs. An aliquot of this sample was used as a control (control sample). The conditions for the first dimension electrophoresis separate molecules mainly according to their mass. Dotted lines represent the region of the gel cut for the second electrophoresis. Thick lines indicate the migration position of the linear unreplicated molecules. The grey background in the control sample indicates the window selected for isolation of specific RIs from the preparative sample. (B) Second gel electrophoresis. The excised region corresponding to the preparative sample was placed in a new gel that was run in the same orientation as the first electrophoresis. In this way, non-replicated linear molecules, much more abundant than RIs, were selectively eliminated. The lane corresponding to the control sample was excised, rotated 90° and transferred to the same second gel. Low melting point agarose (1%) was then poured. Second electrophoresis conditions emphasised the effect of molecular shape. Horizontal dotted lines in the preparative sample represent the seven regions excised and analysed further. The diagrammatic representation of the control sample indicates the 2D gel pattern for the fragment under study, used as a reference. This procedure allows discrimination between RIs containing different branched structures that co-migrated in the first dimension because they have similar masses.
On the same gel, a similar but not UV-irradiated control lane was cut, rotated 90° and aligned together with the preparative gel slab as in the classic 2D agarose gel technique. This second control allowed the proper recognition of different RIs in the preparative sample, as the final distance migrated by each molecular species from the start of the gel should be roughly the same for the control and preparative samples. Thus, knowing the position of the accumulated bubbles in the control sample let us know that at approximately the same ‘height’ we should have accumulated bubbles in the preparative sample. Low melting point agarose (1%) containing 0.5 µg/ml ethidium bromide was then poured around the excised lanes. A second electrophoresis was then run in TBE buffer containing 0.5 µg/ml ethidium bromide at 6 V/cm for 7 h at 4°C to maximise the effect of molecular shape. Part of the gel, corresponding to the control sample, was cut, transferred to a nitrocellulose membrane (BAS85®; Schleicher and Schuell), hybridised with a specific probe and the 2D agarose gel patterns analysed using a phosphorimager (Molecular Dynamics). Meanwhile, the remains of the gel containing the preparative sample were kept at 4°C in the running buffer. The region of the gel with the accumulated bubbles was cut out horizontally in a rectangle 0.3 cm wide (Fig. 1B, sample 1). Parallel pieces of the gel containing molecules migrating between the accumulated bubble and the linear molecules were also cut in the same way (Fig. 1B, samples 2–7).
DNA was extracted after melting the agarose at 60°C for 10 min and digested with 1 U β-agarase (New England Biolabs) per 100 µl at 40°C for 2 h. Remaining insoluble products were eliminated by centrifugation. Digests were gently treated with phenol:chloroform:isoamyl alcohol (25:24:1) and precipitated overnight at –20°C in 0.3 M sodium acetate with 3 vol 100% ethanol. After centrifugation and air drying, DNA samples were resuspended in TE. Electroelution of DNA is a cheaper alternative for the extraction of RIs from the gel that could avoid denaturation by heat.
Electron microscopy
DNA samples enriched for specific RIs were filtered by passage through Sephadex LH60 and prepared for electron microscopy by cytochrome c spreading in 50% formamide and carbonate buffer on a water hypophase. The spreading film was picked up with Parlodion-coated cooper grids, the DNA shadowed with platinum/iridium (80:20) and micrographs were recorded using a Phillips EM400 electron microscope (17).
RESULTS
Replication of the E.coli plasmid pPI21, which contains two oppositely oriented ColE1 origins, leads to the accumulation of a population of RIs containing an internal bubble. Conventional methods, such as sedimentation by centrifugation through CsCl gradients (18) or extraction from an agarose gel (19,20), did not allow us to isolate this specific population of RIs or other less abundant subpopulations. Therefore, we developed a new method for the isolation of large amounts of a specific RI from the total DNA sample by means of two successive agarose gel electrophoreses. This procedure allowed recovery, after the second electrophoresis, of seven samples corresponding to different molecular species that migrated between the unreplicated linear molecules and the accumulated RI containing an internal bubble (Fig. 1).
To verify the quality of the enrichment, an aliquot of each of these seven samples was analysed by standard gel electrophoresis. In order to enhance separation of molecules according to their shape, the gel was run under the same conditions as those used in the second dimension of a 2D agarose gel. Figure 2 shows an autoradiogram corresponding to the analysis of the different samples. A prominent uniform spot, migrating at the most retarded position in the gel, was detected in sample 1 (Fig. 2, lane 1). This spot corresponds precisely to the migration position expected for the specific PstI restriction fragment containing an internal bubble spanning between both ColE1 origins. This RI is generated when the replication fork pauses at the silent origin. Another less intense intermediate migrating band was also detected in this sample that corresponds to broken accumulated bubbles. If a break occurs at one of the two forks of the RI containing an internal bubble, the shape of the molecule changes from a bubble to an asymmetric fork. This broken molecule migrates faster in an agarose gel. Samples 2–4 (Fig. 2, lanes 2–4) contained several discrete bands migrating faster than the accumulated bubbles. As previously mentioned, during the replication of plasmids containing two inversely oriented ColE1 origins, a specific RI accumulates that contains an internal bubble. These bubbles are frequently knotted in vivo. Knotted bubbles with different numbers of nodes migrate differently, leading to the discrete bands migrating faster than the accumulated bubbles (10,15). For the restriction fragment analysed, these knotted RIs were expected to migrate ahead of the accumulated bubbles. Finally, samples 5–7 (Fig. 2, lanes 5–7) generated a more diffuse pattern containing molecules migrating between the knotted molecules and the unreplicated linear forms.
Figure 2.
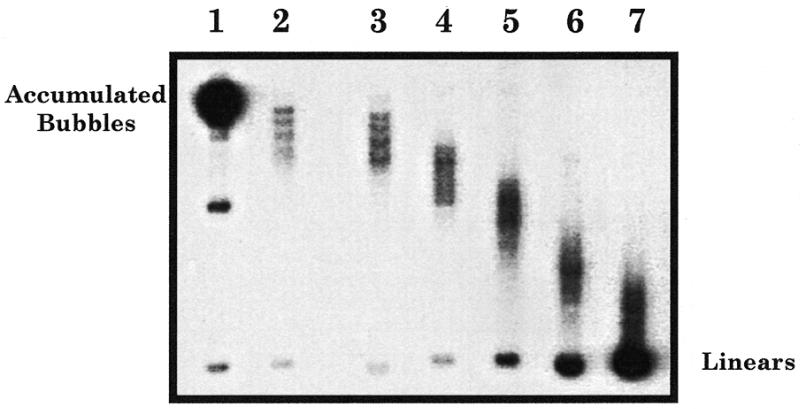
Analysis of the samples enriched for the specific RIs by agarose gel electrophoresis. An aliquot of the seven samples selected from the preparative gel was loaded (lanes 1–7). To enhance separation of molecules according to their shape, the gel was run under the same electrophoresis conditions as those used for the second dimension of a neutral/neutral 2D gel (see Materials and Methods). After electrophoresis, the gel was transferred and hybridised with the 0.5 kb PvuII fragment of pPI21, used as probe.
Selected molecules from the seven enriched DNA samples visualised under the electron microscope are shown in Figure 3. Seventy-nine of the 95 molecules analysed (83%) from lane 1 consisted of RIs containing an internal bubble (Fig. 3A). The size of the two branches and the bubble corresponded exactly to the sizes expected for the PstI restriction fragment containing an internal bubble spanning the region between both replication origins (schematised in Fig. 4, top). A minor population of molecules (2%) with unequal size branches was found that probably corresponded to broken accumulated bubbles that were induced during the extraction procedure (not shown). Interestingly, in sample 1 we observed nine molecules, corresponding to 10% of the molecules analysed, containing an internal bubble with a particular structure (Fig. 3B–K). These RIs contained one additional branch of variable length at one of the replication forks. In addition, the length of this third branch increased when the size of the bubble decreased (compare Fig. 3B and K). A blocked replication fork contains two newly synthesised strands that are complementary and consequently could anneal to each other (see Fig. 4, middle). We propose that these molecules were arrested RIs containing a reversed fork formed by annealing of the newly synthesised strands. The resultant structure would resemble a Holliday junction that might be able to branch migrate (Fig. 4, bottom). Samples 2–4 (Fig. 2) were enriched in partially replicated knotted RIs, molecules containing an internal bubble with a knot showing different numbers of nodes in the replicated region (Fig. 3M1 and M2). These molecules have been studied in detail by Sogo et al. (15). RIs containing a bubble smaller than the accumulated one were observed in samples 5 and 6, as expected for molecules migrating in this region of the gel (Fig. 3L). These growing bubbles corresponded to transient RIs.
Figure 3.
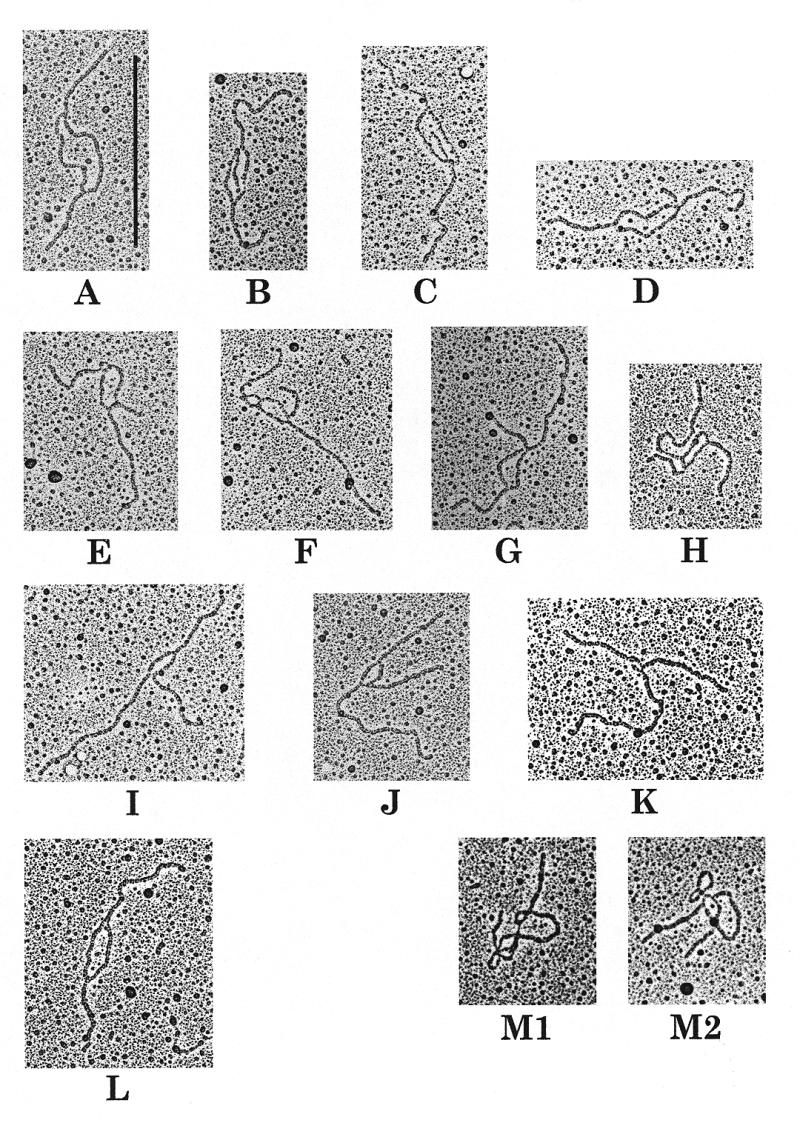
Electron microscopy visualisation of the plasmid RIs from the enriched samples. (A–K) Selected molecules obtained from sample 1. Eighty-five percent (79 of 93) of the molecules analysed contained an internal bubble (A). Ten percent (10 molecules obtained from two independent experiments are shown) contained a third additional branch (B–K). (L) From sample 5, consisting of transient RIs containing an internal bubble smaller than the accumulated one. (M1 and M2) From samples 2–4, corresponding to knotted bubbles. The vertical bar in (A) corresponds to 0.5 µm.
Figure 4.

Pausing of the replication fork leads to the generation of reversed fork molecules. (Top) Schematic representation of the 3.6 kb PstI–PstI restriction fragment of pPI21 containing both origins. DnaB helicase is blocked by the RNAII–DNA hybrid at the silent ColE1 origin and a RI containing an internal bubble is accumulated. The ends of the newly synthesised nascent strands at the silent origin separate from their corresponding parental strands and anneal to each other, forming a reversal of the fork. (Bottom) This structure resembles a Holliday junction, able to branch migrate. A similar scheme would apply if fork reversal occurred at the active origin.
DISCUSSION
The technique we developed allowed the isolation of specific RIs that accumulated in the cell (Fig. 2, lanes 1–4) as well as other less abundant transient species (Fig. 2, lanes 5 and 6). The enriched samples could be used for further analyses (10,15). We believe that this technique could also be used to purify other naturally occurring DNA molecules with higher complexity than linear ones, such as recombination intermediates or D-loops, as well as to characterise the structure of replication forks blocked by various elements in either prokaryotic or eukaryotic systems.
Among the population of RIs accumulated in the cell, a subpopulation of DNA molecules was shown to contain a fork with an additional arm. We propose that these structures arise due to reversion of the replication fork. Formation of reversed fork molecules has been proposed to arise as a consequence of replication fork arrest and presumably is implicated in different cellular processes. Reversion of replication forks in E.coli generates a Holliday junction that might be stabilised by binding of the RuvAB proteins. Processing of these Holliday junctions by recombination proteins was proposed to allow recombination-dependent replication restart (9). In the same way, it was suggested that the Fob1-dependent blockage of replication forks in the rDNA locus of S.cerevisiae could form a similar structure that, upon the action of a nuclease, might generate a double-strand break (DSB). Intrachromosomal recombination of these DSBs could lead to the accumulation of extrachromosomal rDNA circles, a possible cause for ageing in yeast (16).
How could these reversed fork molecules have formed? We recently showed that pausing of replication forks at an inversely oriented ColE1 origin is caused by the inability of DnaB helicase to resolve a RNAII–DNA hybrid at the silent origin (12), leading to the accumulation of a RI containing an internal bubble. Blockage of the helicase could lead to dissociation of the replisome, generating nascent DNA ends that would be free to anneal. No molecules containing two reversed forks were detected. We ignore, however, whether the active origin or the pausing one was more prone to result in this particular structure. To address this question, asymmetrically cleaved DNA molecules should be studied.
We think the molecules containing reversed forks depicted in Figure 3 were formed in vitro: in 2D gels a bubble with a reversed fork should migrate at a different position than accumulated bubbles with no reversed fork. However, in our case they were isolated from the same position in the gel. No effect of ethidium bromide on supercoiling should be expected in this case, as these molecules were exposed to the drug after restriction digestion. The reversed forks we observed could have formed during the extraction of DNA from low melting point agarose gels. As described in Materials and Methods, DNA was extracted from the gels after agarose melting and β-agarase digestion. It is well known that in short DNA stretches heating may prompt the complete extrusion of nascent double-stranded DNA molecules (21).
Nevertheless, it is interesting to speculate about the possibility for fork reversal to occur in vivo. Evidence for the existence of these molecules has been obtained by genetic means in E.coli (9). Formation of reversed fork molecules might be triggered by the topological stress generated in arrested RIs, as originally proposed by Postow et al. (22). The stress produced by positive supercoiling could be partially removed by the annealing of newly synthesised strands at the arrested fork. However, it was recently shown that in vivo RIs are negatively supercoiled (15,23,24) and presumably preventing replication fork reversal is another function of negative supercoiling. The possible role of topoisomerases should also be taken into account (e.g. gyrase might be required to ‘retract’ the reversed fork in vivo) and the exonuclease activity of E.coli RecBCD complex, acting on linear double-stranded DNA, could specifically degrade these molecules.
Regardless of the origin of reversed fork molecules, the present data demonstrate that fork reversal is possible and that the technique we developed allows the isolation of scarce RIs from total DNA. Analysis in different genetic backgrounds is required for further characterisation of this phenomenon in vivo.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to A. Boistov for plasmid pPI21, B. Michel, D. Santamaría, D. Canceill, E. Lechatelier, V. Bidnenko and M. A. Petit for critical reading of the manuscript, and M. L. Martínez-Robles, P. Robles and Beate Rückert for technical assistance. We would also like to acknowledge J. C. Alonso for encouraging us to study the replication of plasmids containing two inversely oriented ColE1 origins. This work was partially supported by grants 93/0161, 96/0470 and 99/0850 from the Spanish Fondo de Investigación Sanitaria, grant 08.6/0016/1997 from the Comunidad Autónoma de Madrid, and grant PM97-0138 from the Spanish Comisión Interministerial de Ciencia y Tecnología (CICYT).
REFERENCES
- 1.Bierne H. and Michel,B. (1994) Mol. Microbiol., 13, 17–23. [DOI] [PubMed] [Google Scholar]
- 2.Bussiere D.E. and Bastia,D. (1999) Mol. Microbiol., 31, 1611–1618. [DOI] [PubMed] [Google Scholar]
- 3.Gahn T.A. and Schildkraut,C.L. (1989) Cell, 58, 527–535. [DOI] [PubMed] [Google Scholar]
- 4.Greenfeder S.A. and Newlon,C.S. (1992) Mol. Cell. Biol., 12, 4056–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi T., Heck,D.J., Nomura,M. and Horiuchi,T. (1998) Genes Dev., 12, 3821–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerber J.K., Gogel,E., Berger,C., Wallisch,M., Muller,F., Grummt,I. and Grummt,F. (1997) Cell, 90, 559–567. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Estraño C., Schvartzman,J.B., Krimer,D.B. and Hernández,P. (1998) J. Mol. Biol., 277, 249–256. [DOI] [PubMed] [Google Scholar]
- 8.Michel B., Ehrlich,S.D. and Uzest,M. (1997) EMBO J., 16, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seigneur M., Bidnenko,V., Ehrlich,S.D. and Michel,B. (1998) Cell, 95, 419–430. [DOI] [PubMed] [Google Scholar]
- 10.Viguera E., Hernández,P., Krimer,D.B., Boistov,A.S., Lurz,R., Alonso,J.C. and Schvartzman,J.B. (1996) J. Biol. Chem., 271, 22414–22421. [DOI] [PubMed] [Google Scholar]
- 11.Martín-Parras L., Hernández,P., Martínez-Robles,M.L. and Schvartzman,J.B. (1991) J. Mol. Biol., 220, 843–853. [DOI] [PubMed] [Google Scholar]
- 12.Santamaría D., de la Cueva,G., Martínez-Robles,M.L., Krimer,D.B., Hernández,P. and Schvartzman,J.B. (1998) J. Biol. Chem., 273, 33386–33396. [DOI] [PubMed] [Google Scholar]
- 13.Brewer B.J. and Fangman,W.L. (1987) Cell, 51, 463–471. [DOI] [PubMed] [Google Scholar]
- 14.Friedman K.L. and Brewer,B.J. (1995) Methods Enzymol., 262, 613–627. [DOI] [PubMed] [Google Scholar]
- 15.Sogo J.M., Stasiak,A., Martínez-Robles,M.L., Krimer,D.B., Hernández,P. and Schvartzman,J.B. (1999) J. Mol. Biol., 286, 637–643. [DOI] [PubMed] [Google Scholar]
- 16.Defossez P.A., Prusty,R., Kaeberlein,M., Lin,S.J., Ferrigno,P., Silver,P.A., Keil,R.L. and Guarente,L. (1999) Mol. Cell, 3, 447–455. [DOI] [PubMed] [Google Scholar]
- 17.Seufert W., Dobrinski,B., Lurz,R. and Messer,W. (1988) J. Biol. Chem., 263, 2719–2723. [PubMed] [Google Scholar]
- 18.Bruand C., Ehrlich,S.D. and Janniere,L. (1991) EMBO J., 10, 2171–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucchini R. and Sogo,J.M. (1995) Nature, 374, 276–280. [DOI] [PubMed] [Google Scholar]
- 20.Kuzminov A., Schabtach,E. and Stahl,F.W. (1997) J. Mol. Biol., 268, 1–7. [DOI] [PubMed] [Google Scholar]
- 21.Zannis-Hadjopoulus M., Persico,M. and Martin,R.G. (1981) Cell, 27, 155–163. [DOI] [PubMed] [Google Scholar]
- 22.Postow L., Ullsperger,C. and Cozzarelli,N. (1999) In Hurwitz,J. and Kowalczykowski,S.C. (eds), Molecular Mechanisms in DNA Replication and Recombination. Keystone Symposia on Molecular and Cellular Biology. Taos, NM, p. 76.
- 23.Peter B.J., Ullsperger,C., Hiasa,H., Marians,K.J. and Cozzarelli,N.R. (1998) Cell, 94, 819–827. [DOI] [PubMed] [Google Scholar]
- 24.Postow L., Peter,B.J. and Cozzarelli,N.R. (1999) Bioessays, 21, 805–808. [DOI] [PubMed] [Google Scholar]