

Abstract
Acute respiratory distress syndrome (ARDS) is a highly morbid lung pathology induced by exposure to chemical warfare agents, including vesicants, phosgene, chlorine, and ricin. In this review, we describe the pathology associated with the development of ARDS in humans and experimental models of acute lung injury following animal exposure to these high-priority threat agents. Potential future approaches to disease-modifying treatment used in preclinical animal studies, including antioxidants, anti-inflammatories, biologics, and mesenchymal stem cells, are also described. As respiratory pathologies, including ARDS, are the major cause of morbidity and mortality following exposure to chemical threat agents, understanding mechanisms of disease pathogenesis is key to the development of efficacious therapeutics beyond the primary intervention principle, which remains mechanical ventilation.
Keywords: acute respiratory distress syndrome, chemical warfare agents, mustards, inflammation, oxidative stress
Introduction
Acute respiratory distress syndrome (ARDS) is the most severe clinical correlate of acute lung injury (ALI). It is defined as impaired oxygenation and respiratory failure that is characterized by bilateral pulmonary infiltrates not fully explained by cardiac failure or fluid overload and that develops within 1 week of a known clinical insult.1,2 Potential etiologies of ARDS include exposure to chemical warfare agents, such as vesicants, phosgene, chlorine, and ricin. ARDS is a seriously morbid condition with associated mortality estimated to be as high as 40%.3 Moreover, survivors of ARDS often suffer long-term debilitating pulmonary and systemic disease.4
There are only a few treatment modalities that improve case fatality rates in patients with ARDS, including limiting the tidal volumes delivered with mechanically controlled breaths and prone position ventilation in severely hypoxemic ARDS.5,6 However, as insight into the pathophysiology of ARDS grows, there is the potential for development of targeted therapies to treat this lethal condition. One of the primary goals of the National Institutes of Health Countermeasures Against Chemical Threats (CounterACT) program is to prevent and treat acute conditions caused by chemical threat agents. Thus, given the severity of ARDS, it is important to review chemical agents that cause ALI, how they do so, and some potential approaches for treating specific agent-induced ARDS.
Pathophysiology of ARDS
The major functions of the lung are to facilitate the transport of oxygen from ambient air into the systemic circulation to supply other organs, excrete carbon dioxide, and maintain a homeostatic acid–base balance. Hypoxemia can develop due to five pathophysiologic processes: low ambient oxygen content (e.g., high altitude and smoke inhalation), hypoventilation, ventilation/perfusion (V/Q) mismatch, shunting of deoxygenated blood, and impaired diffusion from the alveoli to the pulmonary capillaries. Hypercapnia develops when there is either a decrease in total minute ventilation (respiratory rate × tidal volume), a disproportionate increase in dead space ventilation (increased ventilation in areas of the lung that are poorly perfused), and/or loss of lung elastic recoil. As hydrogen ion concentration in the plasma is directly proportional to the partial pressure of arterial CO2, hypercapnia results in decreased plasma pH, otherwise termed acidemia.
In ARDS, hypoxemia develops from a combination of shunting deoxygenated blood due to diffuse alveolar filling/edema, V/Q mismatch from pulmonary microthrombi, and diffusion impairment across a thickened alveolar septum. Hypercapnia develops because of a combination of increased dead space ventilation (pulmonary microthrombi and increased blood viscosity) and decreased total ventilation owing to respiratory muscle fatigue. ARDS can develop as a consequence of direct or indirect lung injury. The etiologies of direct lung injury are pneumonia, gastric content aspiration, pulmonary contusion, fat emboli, near-drowning, and inhalational injury.4 Etiologies of indirect lung injury are sepsis, trauma, hemorrhagic shock, cardiopulmonary bypass, drug overdose, acute pancreatitis, transfusions of blood products, and surgical reperfusion edema.4
ARDS occurs in three, often overlapping, phases. The first phase is the exudative phase (days 1–6).7 In response to injury, inflammatory lung macrophages develop a proinflammatory/cytotoxic (M1) phenotype, releasing proinflammatory cytokines (tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1) and chemokines (IL-8, CCL7, and CCL2), and promoting further accumulation of monocytes and neutrophils in the alveolar space.4 Neutrophils release additional inflammatory mediators, reactive oxygen species (ROS), and proteinases, which degrade the basement membrane and epithelial-endothelial barrier.4 Neutrophils also release extracellular traps containing injurious histones and proteases that stimulate the release of more proinflammatory cytokines via the NLRP3 inflammasome.8 Inactivation and loss of surfactant results in reduced alveolar hysteresis and alveolar collapse.9 Loss of plasma volume entering the lung results in hemoconcentration and increased blood viscosity, further impairing gas exchange.10 TNF-mediated expression of tissue factor leads to dysregulation of intravascular and intra-alveolar coagulation, microthrombi formation, and hyaline membrane formation along denuded basement membranes (Fig. 1).4 The combination of these pathologic processes results in increased dead space ventilation and significant intrapulmonary shunting of blood culminating in hypoxemia that is refractory to the administration of supplemental oxygen, and ultimately respiratory failure (Fig. 2). Different chemical agent exposures induce a unique mix of these physiologic derangements, ultimately leading to pathology similar to the exudative phase of ARDS.
Figure 1.
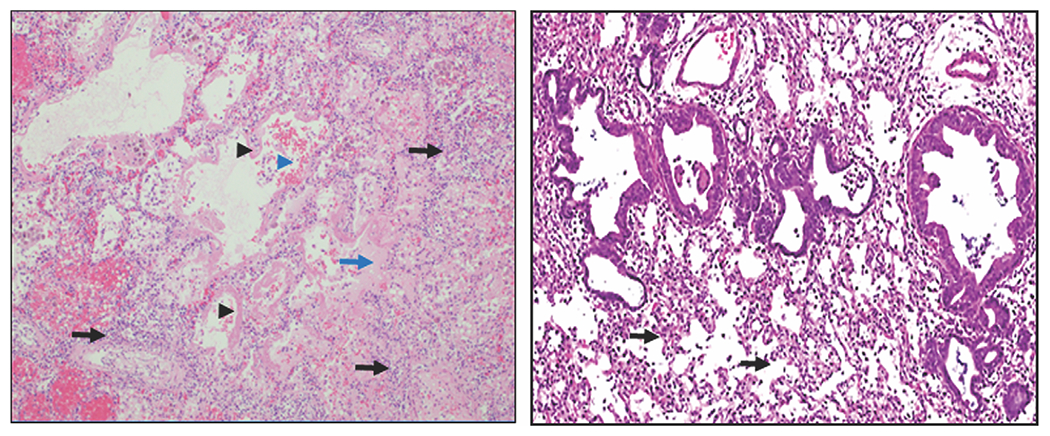
ARDS histopathology in humans and rats. Left panel: Human diffuse alveolar damage (original magnification, 100×). Note thickened alveolar interstitium with inflammatory cell infiltrate (black arrows), hyaline membrane formation (black arrowheads), proteinaceous fluid accumulation in alveoli (blue arrow), and alveolar hemorrhage (blue arrowhead). Right panel: Rat ALI 3 days following NM exposure (original magnification, 40×). Note thickened alveolar interstitium with inflammatory cell infiltrate (black arrows).
Figure 2.
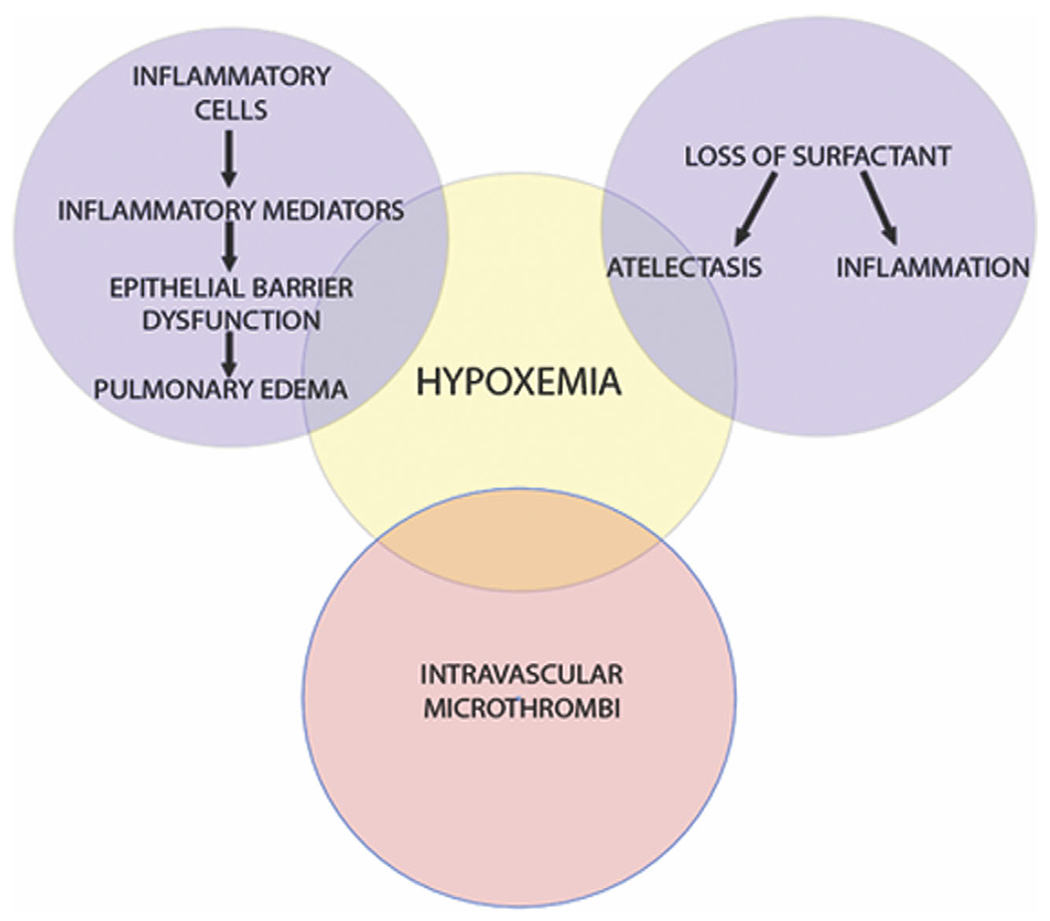
Pathophysiology of hypoxemia in ARDS. Pictured is a Venn diagram displaying the development of hypoxemia. The top left and top right circles represent processes resulting in shunting of deoxygenated blood. The bottom circle represents processes increasing dead space ventilation.
Pulmonary edema develops due to imbalance in Starling forces, namely, changes in the difference between capillary and interstitial hydrostatic pressure, as well as plasma and interstitial oncotic pressures. Increases in capillary hydrostatic pressure relative to interstitial hydrostatic pressure and/or increases in interstitial oncotic pressures relative to plasma oncotic pressures culminate in alveolar edema development. Moreover, surfactant dysfunction increases alveolar interstitial pressures via the law of Laplace, promoting edema development.11 Excessive inflammation, as described above, compromises vascular integrity, augmenting the leakage of colloids from the plasma to the interstitium, increasing interstitial oncotic pressure.12 As such, patients with noncardiogenic pulmonary edema have greater extravascular lung water at a given left ventricular end-diastolic pressure compared with those having cardiogenic pulmonary edema.13 However, the relative role of cardiovascular changes and resultant aberrations in hydrostatic pressure gradients in the development of edema in ARDS cannot be understated. While ARDS is defined by a low clinical suspicion of cardiac failure, studies have shown elevations of left ventricular end-diastolic pressure are common in ARDS patients, and cardiac dysfunction may be present depending on the individual inhalation injury.14,15
The second, proliferative phase (days 7–14) is characterized by the resolution of inflammation and initiation of wound repair.4,7 This is mediated, in part, by anti-inflammatory/reparative M2 macrophages, which release anti-inflammatory cytokines (e.g., IL-10, IL-4, and IL-13), resolvins, lipoxins, and growth factors that promote epithelial and endothelial repair, macrophage phagocytosis of proinflammatory apoptotic neutrophils, and release matrix metallopeptidases (MMPs) that cleave chemokines.4 Alveolar type II cells begin to proliferate in part due to Wnt/β-catenin signaling and differentiate into type I cells, replacing the damaged alveolar epithelial barrier while increasing the production of anti-inflammatory proteins.16,17 With restoration of the alveolar epithelium, there is reestablishment of tight junctions and increased expression of alveolar ion and aquaporin channels, leading to resorption of alveolar edema.4 Thus, this phase of ARDS is essential in the resolution of the disease.
The third, fibrotic phase (post-14 days) does not occur in all patients, as it is thought to be due to aberrant resolution of inflammation. During this phase, there is an excessive fibrogenic response driven by differentiation of resident fibroblasts into myofibroblasts following exposure to profibrotic mediators, such as platelet-derived growth factor (PDGF), transforming growth factor-beta (TGF-β), and insulin-like growth factor (IGF-1) released by M2 macrophages.4 The result is persistent and debilitating pulmonary fibrosis characterized by continued hypoxemia due to regional ventilation-perfusion mismatch and oxygen diffusion impairment through a thickened alveolar interstitium.
General supportive treatment of ARDS
The majority of patients with ARDS require invasive mechanical ventilation. While this is a lifesaving therapy, it is an unnatural form of oxygenation and ventilation. Mechanical ventilation with large tidal volumes leads to lung injury, referred to as ventilator-induced lung injury. This results from alveolar stress due to barotrauma (overdistention of lung), volutrauma (excessive lung stretching), atelectrauma (repetitive opening and closing of alveoli with ensuing epithelial sloughing, pulmonary edema, and hyaline membrane formation), or biotrauma (epithelial microtears culminating in translocation of inflammatory mediators into the systemic circulation).18 All of these forms of trauma have the potential to promote inflammation and exacerbate ARDS progression. Thus, the most successful mechanical treatment for ARDS is low tidal volume ventilation, in which tidal volumes are limited to 6–8 mL/predicted body weight.5 This has been shown to modestly decrease absolute ARDS case fatality rate from 40% to 31%.5 As such, the use of low tidal volume ventilation is strongly recommended by major critical care societies.19
In addition to ventilating the lung with low tidal volumes, providing mechanical ventilation to patients in the prone versus supine position has been demonstrated to reduce mortality in patients with severe ARDS.6,20 Prone ventilation increases recruitment of the dorsal part of the lung (greater anatomical mass than ventral lung), which is compressed during supine ventilation, resulting in more homogenous ventilation and oxygenation and less lung injury.20,21 A large multicenter randomized controlled trial and a meta-analysis of earlier studies confirm that prone position ventilation improves 28-day mortality from 32.8% (supine) to 16% (prone).6,20 Thus, like low tidal volume ventilation, prone positioning is strongly recommended for use in patients with severe ARDS.19
Pulmonary edema decreases respiratory system static compliance, increases shunt-like hypoxia, and impairs the function of surfactant.22,23 Furthermore, pulmonary edema and fluid retention have been shown to be associated with increased mortality.24 Thus, a common treatment strategy used to prevent pulmonary edema exacerbation in ARDS is to limit fluid administration and induce diuresis as needed for relative circulatory overload (central venous pressure >9 mmHg or pulmonary artery occlusion pressure >13 mmHg with a cardiac index ≥2.5 L/min/m2).25 The largest study examining this strategy demonstrated improved oxygenation, pulmonary mechanics, increased oncotic pressure, and shortened duration of mechanical ventilation, but there was no improvement in 60-day mortality.25
Chemical threat agent–induced ALI
Specific chemical threat agents that cause ALI include vesicants, phosgene, chlorine, and ricin. Treatment generally includes airway management, mechanical lung protection strategies, aggressive pulmonary toilet, and avoidance of circulatory volume overload.26 These strategies, especially lung protective ventilation, are paramount to improve mortality but biochemical therapeutics targeted to different etiologies have the potential to improve outcomes. Hence, there is a clear need for disease-modifying therapies based on the underlying agent-specific ALI pathogenesis.
In animals, B2 agonists, exogenous surfactant and surfactant protein C, omega 3 fatty acids, neutrophil elastase inhibitors, statins, granulocyte-macrophage colony-stimulating factor (GMCSF), activated protein C, steroids, and N-acetylcysteine (NAC) have shown some benefits in animal models of ALI; however, they have failed to demonstrate significant efficacy in humans.27–39 ARDS is a syndrome with increasingly recognized phenotypic variation, with differential response to treatment.40 As such, certain therapies like steroids and NAC may in fact be useful for treating specific chemical injuries. Herein, we review the specific pathologies of ALI caused by chemical threat agents and highlight some biochemical treatment strategies used in preclinical, mechanistic animal models with the potential for human use if developed further (Table 1).
Table 1.
Summary of disease-modifying agents used in vivo to treat chemical agent-induced ARDS
| Medication | Mechanism of action | Route | Species | Refs. |
|---|---|---|---|---|
| Mustard vesicants | ||||
| Anti-TNF-α Ab | Block binding of TNF-α to TNF-α receptor | IV | Rat | 55 |
| Pentoxyphilline | Downregulate TNF-α production | IP | Rat | 62 |
| Aminoguanidine | iNOS inhibition | IP | Rat | 55 |
| Melatonin | iNOS inhibition | IP | Rat | 64 |
| NAC | Antioxidant | Oral, IT | Guinea pig | 65 |
| A | Pig | 66 | ||
| Valproic acid | Histone deacetylase inhibition | IP | Rat | 68 |
| MSCs | Increased M2/M1 ratio | IP | Mouse | 71, 72 |
| IV | ||||
|
| ||||
| Phosgene | ||||
| Ibuprofen | COX-2 inhibition | IP | Rat | 82 |
| NAC | Upregulation of Nrf2 | IP | Rat | 83 |
| 1400w | Inhibition of Nos2-mediated RNS production | N | Mouse | 84 |
| MSCs | Increased Wnt/β-catenin signaling | IV | Rat | 87 |
| MSC-CXCl7 | Increased MSC honing and differentiation in type II pneumocytes | IV | Rat | 89 |
| MSC-derived exosomes | Proinflammatory cytokine reduction | IT | Rat | 90 |
| Angiopoetin-1 | Reduced NLRP3 inflammasome, NF-κB, and p38 MAPK signaling | IV | Rat | 93 |
| Melatonin with ulinastatin | Increased Wnt/β-catenin signaling | IP | Rat | 88 |
|
| ||||
| Chlorine | ||||
| Ascorbic acid | Antioxidant | IM | Mouse | 112 |
| IP | Rat | |||
| Deferoxamine | Antioxidant | IM | Mouse | 112 |
| IP | Rat | |||
| NAC | Antioxidant | IP | Rat | 112 |
| Nitrite | Antioxidant | IM | Rat | 114 |
| Trehalose | Autophagy stimulation | A | Mouse | 115 |
| TPRV4 | Decreased pulmonary edema development | IP | Mouse | 118 |
| IM | ||||
| Rolipram | Type 4 phosphodiesterase inhibitor; clearance of pulmonary edema | IP | Mouse | 120 |
| IM, IN | ||||
| Arformoterol | Beta-agonist; clearance of pulmonary edema | IN | Mouse | 121 |
| Budesonide | Steroidal anti-inflammatory | IM | Mouse | 122 |
| Mometasone | Steroidal anti-inflammatory | IP | Mouse | 122 |
| Dexamethasone + NAC | Steroidal anti-inflammatory + antioxidant | IP | Mouse | 123 |
| Triptolide | Nonsteroidal anti-inflammatory | IM | Mouse | 97 |
| Heparin | Endothelial anticoagulation | A | Mouse | 106 |
|
| ||||
| Ricin | ||||
| Monoclonal anti-ricin antibody | Ricin competitive binding | IV | Mouse | 130 |
| OA | ||||
| Polyclonal anti-ricin antibody | Ricin competitive binding | A | Mouse | 131 |
| Doxycycline | Anti-inflammatory; increased VE cadherin expression | IP | Mouse | 126 |
| Anakinra | IL-1 receptor blockade | A | Mouse | 133 |
| Ricin vaccine | Immunogenic protection | IM | Rhesus macaque | 138 |
| Mouse | 136 | |||
| Rats | 137 | |||
Abbreviations: A, aerosolized; IN, intranasal; IP, intraperitoneal; IV, intravenous; IT, intratracheal; IM, intramuscular; MSC, mesenchymal stem cell; NAC, N-acetylcysteine; N, nebulized; OA, oropharyngeal aspiration.
Vesicants
Vesicants (blistering agents) recognized by the U.S. Department of Homeland Security (DHS) include the mustard agents: sulfur mustard (SM, bis[2-chloroethyl]sulfide; DHS chemical access service (CAS) no. 505-60-2) and nitrogen mustard (NM, bis[2-chloroethyl]methylamine hydrochloride; DHS CAS no. 55-86-7). SM is an oily, yellow–brown liquid at room temperature, whereas NM is pale amber, clear, or yellow colored. SM was first used by Germany as a chemical warfare agent in World War I, coining it the nickname “King of the Battle Gases.”41,42 It was later utilized as a chemical agent in several conflicts, including the Iran–Iraq war.42,43 At least a dozen countries have been known to possess stockpiles (13,839 reported tons), as it is easy and inexpensive to manufacture.42 Most recently, SM was identified as causative agent of the death of civilians in the 2016 Syria/Iraq conflict.44 Although NM and related derivatives were never used in warfare, they were produced in the 1920s and 1930s as chemical threat agents and stockpiled. As such, these agents are of major concern and are listed as DHS chemical agents of interest.
Pathophysiology.
Mustard agents are bifunctional DNA alkylating chemicals with low water solubility, allowing them to penetrate the lower respiratory tract.45 There, they react with lipids, proteins, and DNA, forming monoadducts and intra and intermolecular crosslinks.46 Mustards induce cellular damage by a variety of mechanisms, including arresting cells in the cell cycle and activating chromosomal poly(ADP-ribose)polymerase, which reduces intracellular oxidized nicotinamide adenosine dinucleotide, inhibiting glycolysis.42 This leads to activation of the hexose monophosphate shunt, which releases proteases that damage structural proteins, inducing inflammation and ultimately cell death.47 Mustard agents also alkylate thiol groups, depleting cellular glutathione, inducing an accumulation of ROS that can react with phospholipids to damage cell membranes.47
Within 24 h of SM exposure in humans, pulmonary edema and pulmonary failure with sloughing of the respiratory epithelium and loss of pulmonary surfactant consistent with ARDS develops.48–50 As observed in ARDS, survivors of SM exposure develop permanent pulmonary fibrosis.48 Experimental studies from our laboratory have demonstrated similar pathology and disease progression in rodents following exposure to SM or NM. Thus, within 1–3 days, there is thickening of the alveolar septa and inflammatory cell infiltrates, consistent with an experimental classification of ALI (Fig. 1).51–53 The human respiratory LD50 for mustard agents is estimated at 1500 mg-min/m3, although NM is less potent than SM.47,54
As in the exudative phase of human ARDS, experimental models also showed that early in mustard-induced ALI, there is an accumulation of proinflammatory/cytotoxic macrophages, which express inducible nitric oxide synthase (iNOS), cyclooxygenase (COX)-2, and TNF-α.52,53,55,56 By day 28 postexposure, rodents display numerous areas of fibrosis in the airways and bronchioles, similar to pathology observed in Iran–Iraq war veterans 20 years after exposure to SM.57 Fibrosis develops in part due to accumulation of profibrotic macrophages in the lung, which produce TGF-β, mirroring the fibrotic phase of ARDS.55,58
Potential treatment strategies.
One potential targeted treatment for mustard-induced ALI is anti-TNF-α therapy. As described earlier, TNF-α is an important proinflammatory mediator produced mainly by alveolar macrophages during the exudative phase of ARDS. Therapeutics using monoclonal antibodies to target TNF-α were the first in the class of rapidly growing biologics.59 Anti-TNF antibodies are currently approved for use in chronic inflammatory diseases, such as rheumatoid arthritis, Crohn’s disease, psoriasis, and ankylosing spondylitis.59 Our group has demonstrated that anti-TNF-α antibody attenuates NM-induced ALI and fibrosis, as evidenced by decreased interstitial thickening, inflammation, epithelial barrier dysfunction, and collagen deposition.55 A similar therapeutic response has recently been observed in a rat model of inhaled SM pulmonary toxicity (unpublished data). This is accompanied by decreased expression of alveolar macrophage proinflammatory markers (iNOS, COX-2, and TNF-α) 3–7 days post-NM exposure and reduced fibrosis at 28 days postexposure.55 These results suggest that using anti-TNF-α agents to target proinflammatory macrophages could be an effective therapeutic for humans with mustard-induced ALI. However, there are no prior human trials specifically using anti-TNF antibodies to treat ARDS and the benefit of such therapy will need to be weighed against the risk of increased bacterial and nosocomial infections.
Instead of blocking the downstream effects of TNF-α, medications can target TNF-α production. Pentoxifylline is a methyl xanthine phosphodiesterase inhibitor that downregulates the production of TNF-α in humans.60 Pentoxifylline is FDA approved as an anti-TNF therapy and has been used to treat rheumatoid arthritis.61 Treatment of rats with pentoxifylline 15 min after NM exposure reduced epithelial barrier dysfunction, neutrophil infiltration into the lung, histologic evidence of lung injury, and levels of proinflammatory macrophages while increasing levels of anti-inflammatory macrophages.62 No large studies have examined pentoxifylline use for the treatment of ARDS, but a small prospective randomized study of 30 cancer patients with ARDS demonstrated decreased TNF-α levels and improved 30-day mortality posttreatment.63
Oxidative stress is an important mechanism leading to lung injury following mustard exposure. Nitric oxide is produced by inflammatory macrophages via the enzyme iNOS; nitric oxide and its oxidation products cause oxidative stress and tissue damage.55 Our group has demonstrated that blocking iNOS activity with aminoguanidine decreases oxidative and nitrosative stress, inflammation, epithelial barrier dysfunction, and lung injury induced by NM in rats.55 Similarly, treatment of animals with melatonin, which also inhibits iNOS, reduces levels of reactive oxygen and nitrogen species and attenuates mustard-induced lung injury.64 Future studies evaluating the utility of iNOS inhibitors to treat ARDS must weigh these benefits against a potential added risk for infection as nitric oxide is critical for bacterial killing. Lung injury, inflammation, oxidative stress, and pulmonary function induced by SM and 2-chloroethyl ethyl sulfide (half mustard), which is a less potent analog of SM, have also been reported to be mitigated in guinea pigs and pigs by NAC, a thiol-mediated free-radical scavenger.65–67
Valproic acid (VPA) is a histone deacetylase inhibitor used clinically as a mood stabilizer and anticonvulsant. Evidence suggests that histone deacetylases are important epigenetic regulators of proinflammatory leukocyte activation.68 As such, they may be an attractive immunomodulator. Human monocytes treated in vitro with VPA secrete less IL-6 and TNF-α in response to lipopolysaccharide, and neutrophils from patients chronically treated with VPA have reduced chemotaxis.69 Our laboratory has demonstrated that treatment of rats with VPA reduces NM-induced increases in bronchoalveolar lavage (BAL) cell numbers and proinflammatory M1 macrophages, and increases anti-inflammatory/wound repair macrophages 3 days postexposure.68 While the treatments described above are potentially interesting, it should be noted that, based on the time of onset of SM-induced ALI in humans, they would theoretically need to be started within 24 h of exposure in order to expect efficacy.
Alternative treatments using mesenchymal stem cells (MSCs) to promote wound healing have been tested in the later, proliferative phase of injury. MSCs can be derived from bone marrow, adipose tissue, pancreas, placenta, and umbilical cord.70 They have been reported to facilitate the resolution of lung injury with excellent safety profiles.70 This is thought to be due to their ability to polarize macrophages toward an anti-inflammatory/wound healing phenotype, reduce proinflammatory cytokine production, and restore epithelial barrier function.70 As such, they have become a potential ARDS treatment strategy. Mice treated with adipose-derived MSCs after half mustard exposure display attenuated histological evidence of ALI, increased M2/M1 macrophage ratios, and decreased lung IL-1β and TNF-α levels.71 Similarly, bone marrow–derived MSCs administered to mice 24 h after exposure to SM effectively decreased pulmonary edema/epithelial barrier dysfunction and increased survival, a response associated with decreases in serum and lung IL-6, IL-1β, and TNF-α levels and with increases in M2/M1 macrophage ratios and levels of IL-10 and tissue repair growth factors (epidermal growth factor (EGF), fibroblast growth factor (FGF), and PDGF).72 Early phase clinical trials of MSCs in human ARDS have demonstrated safety but a lack of clinical efficacy, possibly due to variations in viability of the MSCs delivered.73
Phosgene
Phosgene (carbonic dichloride; DHS CAS no. 75-44-5) is a highly reactive, colorless gas that was used as a chemical warfare agent in World War I.42 Additionally, it was used by Egypt in the North Yemen Civil War (1963–1967).74 Contemporarily, 5 million metric tons of phosgene are used globally as an intermediate in the manufacture of plastics, polyurethanes, polycarbonates, dyestuffs, pharmaceuticals, and agrochemicals.75
Pathophysiology.
Phosgene is an acylating agent with low water solubility, enabling it to penetrate and damage the lower respiratory tract.45 As it has negligible scrubbing of the airway, its toxic dose in the lower respiratory system is mainly derived from the time inhaled as opposed to concentration inhaled.15 Human phosgene exposures result in acute bilateral pulmonary infiltrates, pulmonary edema, and increased blood coagulation, culminating in severe/deadly hypoxemia, consistent with ARDS.76–78 At high levels of exposure, animal lungs are characterized by alveolar and interstitial edema, inflammatory cell infiltrates, fibrin, hemorrhage, and necrosis.75 The human LC0 and LC50 of phosgene is ~1200 (similar to that required to induce clinically relevant pulmonary edema) and ~2000 mg/m3 × min, respectively.75,79 The onset of pulmonary edema in humans has been described 6–24 h after exposure.80
Animal models have demonstrated that initiation of ALI by phosgene is considerably different than that by mustard vesicants. The reaction of phosgene with water is slower than its reaction with nucleophilic moieties of proteins and phospholipids present in the alveoli.15 This enables phosgene-induced acylation of surfactant, causing surfactant dysfunction, increased alveolar surface tension, and resultant atelectasis.15,75 Phosgene’s chemical reaction with thiol groups depletes and oxidizes glutathione reserves, reducing the antioxidant buffering capacity of the lung.15,75 Phosgene also oxidizes red blood cell membrane structural proteins, leading to the release of free heme into the plasma, which itself is an oxidant and proinflammatory mediator.81 Lastly, phosgene induces additional free radicals, which damage endothelial, epithelial, and innate immune cells, resulting in inflammation and epithelial barrier dysfunction.15,81
The development of pulmonary edema post-phosgene exposure is also influenced by changes in cardiovascular function. Phosgene damages vagal C-fiber nerve endings, causing a loss of neurovascular control, resulting in cardiovascular disturbances and pulmonary edema.15 Phosgene exposure is also associated with bradycardia, diminished cardiac output, and systemic vasoconstriction, further promoting plasma leakage into the lung.15 As alluded to earlier, this translocation of plasma causes hemoconcentration and increased blood viscosity, further exacerbating deficits caused by pulmonary edema.15
Potential treatment strategies.
To counteract oxidative damage induced by phosgene exposure, antioxidants have been evaluated experimentally.82,83 The transcription factor Nrf2 regulates production of antioxidant proteins, including phase 2 detoxifying enzymes and glutathione-regenerating enzymes.83 The antioxidant NAC has been reported to attenuate ALI from phosgene exposure via upregulation of Nrf2-mediated increases in glutathione reductase.83 Ibuprofen, a COX-2 inhibitor with antioxidant activity, has also been reported to reduce pulmonary edema in rats induced by phosgene.82 Reactive nitrogen species (RNS) also play an important role in phosgene-induced ALI.84 As indicated above, RNS are generated in macrophages via iNOS.85 Selective inhibition of iNOS with 1400W [N-(3-(aminomethyl)benzyl)acetamidine] was found to reduce ALI and preserve epithelial barrier integrity in phosgene-exposed mice.84 Conversely, inhaled nitric oxide exacerbated phosgene-induced pulmonary edema.86
As observed in mustard-induced ALI, MSCs have been reported to promote the resolution of ALI induced by phosgene. MSCs administered to rats with phosgene-induced ALI upregulated Wnt/β-catenin signaling and reduced epithelial barrier dysfunction.87 Similarly, the combination of melatonin and ulinastatin, a urinary trypsin inhibitor, administered after phosgene exposure attenuated ALI via activation of Wnt/β-catenin signaling in rats.88 MSCs in combination with CXCR7 have been used in phosgene-induced ALI. In this rodent study, MSC honing to the lung and MSC differentiation into type II alveolar cells was increased and ALI was blunted.89 MSC-derived exosomes administered to rats with phosgene-induced ALI were also found to reduce levels of TNF-α, IL-6, IL-8, and MMP9.90 Angiopoietin-1 (Ang1) is a growth factor known to inhibit leukocyte vascular permeability and cytokine production; it has also been used in animal models of ALI.91,92 Ang1 administered to rats after phosgene exposure decreased ALI and reduced inflammatory cytokine levels via disruption of the NLRP3 inflammasome.92 In another rat exposure study, Ang1 was found to suppress TNF-α, IL-6, and IL-8 production post-phosgene exposure via the NF-κB and p38 MAPK pathways.93 Additionally, MSCs overexpressing Ang1 exhibited increased homing to the lung, upregulating IL-10 expression while decreasing ALI and IL-1β production following phosgene exposure.94
Chlorine
Chlorine (DHS CAS no.7782-50-5) is a halogen gas first used as a chemical agent in World War I and more recently by insurgents in Iraq. It is thought to be one of the most commonly used chemical weapons in the Levant region of the Middle East.95,96 DHS estimates that chlorine release in an urban area could produce as many as 100,000 respiratory injuries requiring hospitalization.97 Thus, DHS designates chlorine as a chemical agent of interest.
Pathophysiology.
Unlike mustard vesicants and phosgene, chlorine has intermediate water solubility and the principle area of absorption is the upper airway.26,98 Therefore, the presence of ALI is accompanied by extensive airway damage, and, in contrast to phosgene, chlorine’s effects on the lower lung are highly dependent on concentrations of exposure >15 ppm, with chemical pneumonitis developing at concentrations >50 ppm.15,98,99 The LC50 for a 10-min exposure is 364 and 210 ppm in populations exposed for 10 and 30 min, respectively.100 ARDS has been described in World War I soldiers and civilians with high-level chlorine exposures from cylinder leaks and accidents.101 More recently, a train derailment in 2005 released 42–60 tons of chlorine gas, resulting in significant pulmonary toxicity in exposed individuals. Approximately 58% of those hospitalized due to chlorine inhalation met the clinical criteria of ARDS within 24 h of admission.102 Animals exposed to chlorine develop pulmonary edema and alveolar inflammation characterized by neutrophil recruitment and the appearance of foamy macrophages, alveolar damage with epithelial barrier dysfunction, capillary microthrombi, and fibrin deposition.103–106 Similarly, inhalation of high doses of chlorine results in human ALI characterized by pneumonitis, pulmonary edema, and decrements in lung function.102
Animal exposure models suggest that much of the damage induced by chlorine can be attributed to oxidative stress.99 When added to an aqueous environment, chlorine reacts to produce hydrochloric and hypochlorous acid, which further reacts with oxygen and nitrogen dioxide to form ROS and RNS.99,107 Chlorine also damages the red blood cell cytoskeleton, resulting in the previously described oxidizing, cell-free heme.108 These oxidative reactions alter surfactant function, increasing alveolar surface tension and lung elastance.107 Resultant pulmonary edema develops due to epithelial barrier injury and increased capillary hydraulic conductance.109 The presence of cardiomegaly and increased pulmonary vascular resistance in chlorine-exposed animals suggest an additional cardiogenic component to the edema.99
Potential treatment strategies.
Most preclinical treatment strategies have targeted chlorine-induced oxidative stress. Following chlorine exposure, increases in nitrotyrosine residues in proteins have been observed, along with an accumulation of 8-isoprostane and decreased ascorbate and glutathione in the lung, indicating oxidative stress.110–112 Low molecular weight antioxidants (ascorbic acid, deferoxamine, and NAC) administered to rats after chlorine exposure attenuate epithelial barrier dysfunction and neutrophilic inflammation, which is associated with improved gas exchange.112,113 Nitrite, an oxidation product of nitric oxide, administered to rats after chlorine exposure, decreases neutrophil recruitment.114
Chlorine also increases superoxide anion production in damaged alveolar type II cell mitochondria, promoting inflammation in vitro.115 As damaged cellular proteins and organelles are targeted for removal by the autophagy–lysosomal pathway, pretreatment of mice with the autophagy inducer trehalose has been reported to decrease epithelial barrier dysfunction and neutrophil influx, depending on the timing and method of delivery.115 This suggests that, if demonstrated effective in humans, trehalose could be trialed as a prophylactic therapy for those at high risk of chlorine exposure, in order to decrease the incidence of ALI. Additionally, in a rodent model, treatment with hemopexin, a heme protein scavenger, postexposure to the halogen gas bromine, reduces epithelial barrier dysfunction, lung collagen levels, and improves lung mechanics.116 These data suggest that hemopexin may be effective in treating chlorine gas exposure.
Another potential approach for the treatment of chlorine-induced ALI is the reduction of pulmonary edema. Evidence suggests that calcium transit through transient receptor potential vanilloid 4 (TPRV4) located on lung epithelial and endothelial cells mediates the development of pulmonary edema in ARDS.117 Treatment of mice with TRLV4 inhibitors after chlorine exposure reduces macrophage and neutrophil counts and improves gas exchange, pulmonary mechanics, epithelial barrier function, and histologic lung injury scores.118 As an alternative for reducing pulmonary edema, drugs can also be used to promote fluid removal. Rolipram is a type 4 phosphodiesterase inhibitor with the potential to increase alveolar fluid clearance and decrease pulmonary edema via increased cAMP levels in alveolar epithelial cells.119,120 Administration of rolipram to mice following chlorine exposure decreases pulmonary edema.120 Beta-agonists are another commonly used class of medications that facilitate alveolar fluid removal via cAMP upregulation. The beta-agonist arformoterol applied to nares of mice treated with chlorine was found to increase sodium-dependent alveolar fluid clearance.121
As inflammation promotes tissue damage in ARDS, anti-inflammatory therapy may be of benefit in treating chemical-induced ALI. The anti-inflammatory steroids budesonide and mometasone have been used in murine models of chlorine-induced ALI.97,122 Budesonide was successful in reducing chlorine-induced neutrophil influx into the lung by 90%.97 Similarly, mometasone or budesonide administered after chlorine exposure blunted neutrophil influx and pulmonary edema.122 Aerosolized heparin administered after chlorine exposure decreases microthrombi formation and epithelial barrier dysfunction, with the added benefit of reducing lung neutrophilia.106 Additionally, triptolide, a plant diterpenoid and nonsteroidal anti-inflammatory agent, decreased chlorine-induced neutrophil recruitment into the lung by up to 82%.97 Moreover, the use of anti-inflammatory therapies with anti-oxidants may have synergistic benefit as dexamethasone in combination with NAC decreases BAL neutrophil counts while improving maximum peripheral tissue resistance compared with steroids or NAC alone.123
Ricin
Ricin differs from the chemical agents described above, as it is a plant-derived toxin from the seeds of castor beans. One million tons of castor beans are processed into castor oil per year and the waste produces is ~5% ricin by weight.42 Due to this high availability and ease of production, ricin has been considered as a chemical warfare agent since 1918 and has been used in attempted terrorist acts.124 The United States and Iraq have manufactured and tested weapons-grade ricin in animal experiments and in the field.124 Thus, ricin remains an important agent of concern.
Pathophysiology.
Ricin is directly cytotoxic to cells via inhibition of ribosome-mediated protein synthesis.125 It also increases proinflammatory signaling through activation of NF-κB and p38 MAPK and the NALP3 inflammasome.125 Ricin particles less than 5 μm can deposit in the lower airways, and postmortem examination of lungs from non-human primates exposed to ricin shows ARDS-like pathology.124 The primary targets of inhaled ricin cytotoxicity in this model are types I and II pneumocytes.124 In mice, there is disruption of epithelial barrier function due to loss of the junction proteins VE-cadherin, claudin 5, and connexin 43, and a rapid influx of neutrophils.126 Pigs exposed to ricin develop inflammatory pulmonary edema and histological evidence of diffuse alveolar damage.127 The LD50 for inhaled ricin is 3–5 μg/kg in mice.128 In humans, the LD50 for oral ingestion is 30 mg/kg.124 While there is a lack of reports of inhaled ricin in humans, monkeys exposed by this route develop pulmonary edema within 36–48 hours.129
Potential treatment strategies.
Antibodies represent a major treatment approach that appear to have some beneficial activity in ricin-induced ALI. In this context, a monoclonal antibody used in an oropharyngeal aspiration model of ricin poisoning in mice was reported to be effective in reducing lung edema, alveolar inflammation, necrosis, and thickening of the alveolar septum.130 Likewise, improved epithelial barrier function and reduced neutrophil recruitment to the lung have been observed in mice treated with an equine anti-ricin antibody.126 Additionally, mice exposed to aerosolized ricin and treated with aerosolized polyclonal anti-ricin antibody exhibit reduced pulmonary edema and alveolar necrosis.131
Efforts have also focused on targeting the proinflammatory cascade triggered during ricin-induced ALI. Anakinra, an IL-1 receptor antagonist, is a biologic approved for use in rheumatoid arthritis.132 Mice treated with aerosolized anakinra at the time of ricin exposure showed decreases in vascular congestion, alveolar destruction, and neutrophil recruitment to the lung.133 Decreases in pulmonary inflammatory cells and epithelial barrier dysfunction were also observed in ricin-exposed mice treated with ciprofloxacin, an antibiotic with immunomodulatory properties.134,135 Doxycycline has also been found to improve ALI in ricin-treated mice via improved barrier function with increased VE-cadherin expression and a reduction in inflammatory cytokines and oxidative stress markers.126 Additionally, the combination of an anti-ricin antibody and doxycycline restored barrier integrity in ricin-exposed mice.126
An ideal strategy to combat ricin use as a chemical agent would be to vaccinate individuals at high risk of exposure. Mice treated with an anti-ricin vaccine display increased survival and reduced alveolar necrosis or edema.136 Rats vaccinated via two intratracheal liposomes 3 weeks apart also developed less epithelial barrier dysfunction and lung neutrophilia.137 In a very promising translational study, a recombinant ricin vaccine was produced and administered as part of a monthly three-injection protocol in rhesus macaques.138 Eleven out of 12 macaques that received the vaccine survived challenge with ricin and did not demonstrate the diffuse alveolar damage exhibited postmortem in nonvaccinated animals.138
Summary and conclusions
A number of highly toxic chemical warfare agents, including vesicants, phosgene, chlorine, and ricin, have been identified that target the respiratory tract. Although their mechanisms of action are distinct, they each induce ARDS-like pathology and disease in humans and animals. As pulmonary toxicity underlies most of the morbidity and mortality in exposed victims, identification of mechanistic targets and the development of therapeutics are essential. At present, antioxidants and anti-inflammatories, including targeted biologics, along with MSCs are among the most promising approaches in preclinical development to mitigate chemical threat agent pulmonary toxicity. It may be that combinations of these approaches will be required to suppress the development of ARDS and other chronic lung diseases in exposed victims.
Acknowledgments
The authors thank Christine Minerowicz for the human ARDS photomicrograph. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Grant/Award number: U54AR055073) and the National Institute of Environmental Health Sciences (Grant/Award numbers: P30ES005022 and R01ES004738).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, et al. 2012. Acute respiratory distress syndrome: the Berlin Definition. JAMA 307: 2526–2533. [DOI] [PubMed] [Google Scholar]
- 2.Walkey AJ, Summer R, Ho V, et al. 2012. Acute respiratory distress syndrome: epidemiology and management approaches. Clin. Epidemiol 4: 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellani G, Laffey JG, Pham T, et al. 2016. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315: 788–800. [DOI] [PubMed] [Google Scholar]
- 4.Thompson BT, Chambers RC & Liu KD. 2017. Acute respiratory distress syndrome. N. Engl. J. Med 377: 562–572. [DOI] [PubMed] [Google Scholar]
- 5.Acute Respiratory Distress Syndrome Network, Brower RG, Matthay MA, et al. 2000. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med 342: 1301–1308. [DOI] [PubMed] [Google Scholar]
- 6.Guérin C, Reignier J, Richard J-C, et al. 2013. Prone positioning in severe acute respiratory distress syndrome. N. Engl. J. Med 368: 2159–2168. [DOI] [PubMed] [Google Scholar]
- 7.Rezoagli E, Fumagalli R & Bellani G. 2017. Definition and epidemiology of acute respiratory distress syndrome. Ann. Transl. Med 5: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Standiford TJ & Ward PA. 2016. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl. Res 167: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wheeler AP & Bernard GR. 2007. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 369: 1553–1564. [DOI] [PubMed] [Google Scholar]
- 10.Lukey BJ, Romero JA Jr. & Salem H. 2019. Chemical Warfare Agents: Biomedical and Psychological Effects, Medical Countermeasures, and Emergency Response. 3rd ed. Boca Raton, FL: CRC Press. [Google Scholar]
- 11.Zucker AR, Holm BA, Crawford GP, et al. 1992. PEEP is necessary for exogenous surfactant to reduce pulmonary edema in canine aspiration pneumonitis. J. Appl. Physiol 73: 679–686. [DOI] [PubMed] [Google Scholar]
- 12.Chelazzi C, Villa G, Mancinelli P, et al. 2015. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit. Care 19: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibbald WJ, Short AK, Warshawski FJ, et al. 1985. Thermal dye measurements of extravascular lung water in critically ill patients. Intravascular Starling forces and extravascular lung water in the adult respiratory distress syndrome. Chest 87: 585–592. [DOI] [PubMed] [Google Scholar]
- 14.Casey JD, Semler MW & Rice TW. 2019. Fluid management in acute respiratory distress syndrome. Semin. Respir. Crit. Care Med 40: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W & Pauluhn J. 2017. Phosgene-induced acute lung injury (ALI): differences from chlorine-induced ALI and attempts to translate toxicology to clinical medicine. Clin. Transl. Med 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillamat-Prats R, Puig F, Camprubí-Rimblas M, et al. 2018. Intratracheal instillation of alveolar type II cells enhances recovery from acute lung injury in rats. J. Heart Lung Transplant 37: 782–791. [DOI] [PubMed] [Google Scholar]
- 17.Tanjore H, Degryse AL, Crossno PF, et al. 2013. B-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am. J. Respir. Crit. Care Med 187: 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slutsky AS & Ranieri VM. 2013. Ventilator-induced lung injury. N. Engl. J. Med 369: 2126–2136. [DOI] [PubMed] [Google Scholar]
- 19.Fan E, Del Sorbo L, Goligher EC, et al. 2017. An Official American Thoracic Society/European Society of Intensive Care Medicine/Society of Critical Care Medicine Clinical Practice Guideline: mechanical ventilation in adult patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med 195: 1253–1263. [DOI] [PubMed] [Google Scholar]
- 20.Gattinoni L, Carlesso E, Taccone P, et al. 2010. Prone positioning improves survival in severe ARDS: a pathophysiologic review and individual patient meta-analysis. Minerva Anestesiol. 76: 448–454. [PubMed] [Google Scholar]
- 21.Gattinoni L, Taccone P, Carlesso E, et al. 2013. Prone position in acute respiratory distress syndrome. Rationale, indications, and limits. Am. J. Respir. Crit. Care Med 188: 1286–1293. [DOI] [PubMed] [Google Scholar]
- 22.Ware LB & Matthay MA. 2000. The acute respiratory distress syndrome. N. Engl. J. Med 342: 1334–1349. [DOI] [PubMed] [Google Scholar]
- 23.Roch A, Guervilly C & Papazian L. 2011. Fluid management in acute lung injury and ARDS. Ann. Intensive Care 1: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakr Y, Vincent J-L, Reinhart K, et al. 2005. High tidal volume and positive fluid balance are associated with worse outcome in acute lung injury. Chest 128: 3098–3108. [DOI] [PubMed] [Google Scholar]
- 25.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Wiedemann HP, Wheeler AP, et al. 2006. Comparison of two fluid-management strategies in acute lung injury. N. Engl. J. Med 354: 2564–2575. [DOI] [PubMed] [Google Scholar]
- 26.Saeed O, Boyer NL, Pamplin JC, et al. 2018. Inhalation injury and toxic industrial chemical exposure. Mil. Med 183: 130–132. [DOI] [PubMed] [Google Scholar]
- 27.Gao Smith F, Perkins GD, Gates S, et al. 2012. Effect of intravenous β-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet 379: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Matthay MA, Brower RG, et al. 2011. Randomized, placebo-controlled clinical trial of an aerosolized β2-agonist for treatment of acute lung injury. Am. J. Respir. Crit. Care Med 184: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kesecioglu J, Beale R, Stewart TE, et al. 2009. Exogenous natural surfactant for treatment of acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med 180: 989–994. [DOI] [PubMed] [Google Scholar]
- 30.Spragg RG, Lewis JF, Walmrath H-D, et al. 2004. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N. Engl. J. Med 351: 884–892. [DOI] [PubMed] [Google Scholar]
- 31.Rice TW, Wheeler AP, Thompson BT, et al. 2011. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 306: 1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeiher BG, Artigas A, Vincent J-L, et al. 2004. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit. Care Med 32: 1695–1702. [DOI] [PubMed] [Google Scholar]
- 33.McAuley DF, Laffey JG, O’Kane CM, et al. 2014. Simvastatin in the acute respiratory distress syndrome. N. Engl. J. Med 371: 1695–1703. [DOI] [PubMed] [Google Scholar]
- 34.National Heart, Lung, and Blood Institute ARDS Clinical Trials Network, Truwit JD, Bernard GR, et al. 2014. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N. Engl. J. Med 370: 2191–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paine R, Standiford TJ, Dechert RE, et al. 2012. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit. Care Med 40: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu KD, Levitt J, Zhuo H, et al. 2008. Randomized clinical trial of activated protein C for the treatment of acute lung injury. Am. J. Respir. Crit Care Med 178: 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor RW, Zimmerman JL, Dellinger RP, et al. 2004. Low-dose inhaled nitric oxide in patients with acute lung injury: a randomized controlled trial. JAMA 291: 1603–1609. [DOI] [PubMed] [Google Scholar]
- 38.Steinberg KP, Hudson LD, Goodman RB, et al. 2006. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N. Engl. J. Med 354: 1671–1684. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Ding S, Li C, et al. 2017. Effects of N-acetylcysteine treatment in acute respiratory distress syndrome: a meta-analysis. Exp. Ther. Med 14: 2863–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reilly JP, Calfee CS & Christie JD. 2019. Acute respiratory distress syndrome phenotypes. Semin. Respir. Crit. Care Med 40: 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geraci MJ 2008. Mustard gas: imminent danger or eminent threat? Ann. Pharmacother 42: 237–246. [DOI] [PubMed] [Google Scholar]
- 42.Bismuth C, Borron SW, Baud FJ, et al. 2004. Chemical weapons: documented use and compounds on the horizon. Toxicol. Lett 149: 11–18. [DOI] [PubMed] [Google Scholar]
- 43.Mosayebzadeh M, Ghazanfari T, Delshad A, et al. 2016. Evaluation of apoptosis in the lung tissue of sulfur mustard-exposed individuals. Iran J. Allergy Asthma Immunol 15: 283–288. [PubMed] [Google Scholar]
- 44.Report of the OPCW fact-finding mission in Syria regarding the incident of 16 September as reported in the note verbale of the Syrian Arab Republic number 113 Dated 29 November 2016. May 1, 2018. https://www.opcw.org/sites/default/files/documents/Fact_Finding_Mission/s-1491-2017_e_.pdf.
- 45.Gorguner M & Akgun M. 2010. Acute inhalation injury. Eurasian J. Med 42: 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shakarjian MP, Heck DE, Gray JP, et al. 2010. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol. Sci 114: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balali-Mood M & Abdollahi M, Eds. 2015. Basic and Clinical Toxicology of Mustard Compounds. Cham: Springer International Publishing. [Google Scholar]
- 48.Graham JS & Schoneboom BA. 2013. Historical perspective on effects and treatment of sulfur mustard injuries. Chem. Biol. Interact 206: 512–522. [DOI] [PubMed] [Google Scholar]
- 49.Ghabili K, Agutter PS, Ghanei M, et al. 2010. Mustard gas toxicity: the acute and chronic pathological effects. J. Appl. Toxicol 30: 627–643. [DOI] [PubMed] [Google Scholar]
- 50.World Health Organization mustard gas fact sheet. Accessed June 4, 2020. http://www.emro.who.int/ceha/information-resources/mustard-gas-fact-sheet.html.
- 51.Matute-Bello G, Frevert CW & Martin TR. 2008. Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol 295: L379–L399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunil VR, Patel KJ, Shen J, et al. 2011. Functional and inflammatory alterations in the lung following exposure of rats to nitrogen mustard. Toxicol. Appl. Pharmacol 250: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sunil VR, Vayas KN, Abramova EV, et al. 2020. Lung injury, oxidative stress and fibrosis in mice following exposure to nitrogen mustard. Toxicol. Appl. Pharmacol 387. 10.1016/j.taap.2019.114798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balali-Mood M & Hefazi M. 2005. The pharmacology, toxicology, and medical treatment of sulphur mustard poisoning. Fundam. Clin. Pharmacol 19: 297–315. [DOI] [PubMed] [Google Scholar]
- 55.Malaviya R, Sunil VR, Venosa A, et al. 2015. Attenuation of nitrogen mustard-induced pulmonary injury and fibrosis by anti-tumor necrosis factor-α antibody. Toxicol. Sci 148: 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malaviya R, Sunil VR, Cervelli J, et al. 2010. Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol. Appl. Pharmacol 248: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malaviya R, Sunil VR, Venosa A, et al. 2016. Macrophages and inflammatory mediators in pulmonary injury induced by mustard vesicants. Ann. N.Y. Acad. Sci 1374: 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Venosa A, Malaviya R, Choi H, et al. 2016. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol 54: 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Monaco C, Nanchahal J, Taylor P, et al. 2015. Anti-TNF therapy: past, present and future. Int. Immunol 27: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.González-Espinoza L, Rojas-Campos E, Medina-Pérez M, et al. 2012. Pentoxifylline decreases serum levels of tumor necrosis factor alpha, interleukin 6 and C-reactive protein in hemodialysis patients: results of a randomized double-blind, controlled clinical trial. Nephrol. Dial. Transplant 27: 2023–2028. [DOI] [PubMed] [Google Scholar]
- 61.Raza A 2000. Anti-TNF therapies in rheumatoid arthritis, Crohn’s disease, sepsis, and myelodysplastic syndromes. Microsc. Res. Tech 50: 229–235. [DOI] [PubMed] [Google Scholar]
- 62.Sunil VR, Vayas KN, Cervelli JA, et al. 2014. Pentoxifylline attenuates nitrogen mustard-induced acute lung injury, oxidative stress and inflammation. Exp. Mol. Pathol 97: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ardizzoia A, Lissoni P, Tancini G, et al. 1993. Respiratory distress syndrome in patients with advanced cancer treated with pentoxifylline: a randomized study. Support. Care Cancer 1: 331–333. [DOI] [PubMed] [Google Scholar]
- 64.Macit E, Yaren H, Aydin I, et al. 2013. The protective effect of melatonin and S-methylisothiourea treatments in nitrogen mustard induced lung toxicity in rats. Environ. Toxicol. Pharmacol 36: 1283–1290. [DOI] [PubMed] [Google Scholar]
- 65.Das SK, Mukherjee S, Smith MG, et al. 2003. Prophylactic protection by N-acetylcysteine against the pulmonary injury induced by 2-chloroethyl ethyl sulfide, a mustard analogue. J. Biochem. Mol. Toxicol 17: 177–184. [DOI] [PubMed] [Google Scholar]
- 66.Jugg B, Fairhall S, Smith A, et al. 2013. N-acetyl-L-cysteine protects against inhaled sulfur mustard poisoning in the large swine. Clin. Toxicol. (Phila.) 51: 216–224. [DOI] [PubMed] [Google Scholar]
- 67.Mukherjee S, Stone WL, Yang H, et al. 2009. Protection of half sulfur mustard gas-induced lung injury in guinea pigs by antioxidant liposomes. J. Biochem. Mol. Toxicol 23: 143–153. [DOI] [PubMed] [Google Scholar]
- 68.Venosa A, Gow JG, Hall L, et al. 2017. Regulation of nitrogen mustard-induced lung macrophage activation by valproic acid, a histone deacetylase inhibitor. Toxicol. Sci 157: 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soria-Castro R, Schcolnik-Cabrera A, Rodríguez-López G, et al. 2019. Exploring the drug repurposing versatility of valproic acid as a multifunctional regulator of innate and adaptive immune cells. J. Immunol. Res 2019. 10.1155/2019/967809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laffey JG & Matthay MA. 2017. Fifty years of research in ARDS. Cell-based therapy for acute respiratory distress syndrome. Biology and potential therapeutic value. Am. J. Respir. Crit. Care Med 196: 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sadeghi S, Mosaffa N, Hashemi SM, et al. 2020. The immunomodulatory effects of mesenchymal stem cells on long term pulmonary complications in an animal model exposed to a sulfur mustard analog. Int. Immunopharmacol 80. 10.1016/j.intimp.2019.105879. [DOI] [PubMed] [Google Scholar]
- 72.Feng Y, Xu Q, Yang Y, et al. 2019. The therapeutic effects of bone marrow-derived mesenchymal stromal cells in the acute lung injury induced by sulfur mustard. Stem. Cell Res. Ther 10: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cruz FF & Rocco PRM. 2019. Cell therapy for acute respiratory distress syndrome patients: the START study. J. Thorac. Dis 11: S1329–S1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haines DD & Fox SC. 2014. Acute and long-term impact of chemical weapons: lessons from the Iran–Iraq War. Forensic Sci. Rev 26: 97–114. [PubMed] [Google Scholar]
- 75.Pauluhn J, Carson A, Costa DL, et al. 2007. Workshop summary: phosgene-induced pulmonary toxicity revisited: appraisal of early and late markers of pulmonary injury from animal models with emphasis on human significance. Inhal. Toxicol 19: 789–810. [DOI] [PubMed] [Google Scholar]
- 76.Diller WF 1985. Pathogenesis of phosgene poisoning. Toxicol. Ind. Health 1: 7–15. [DOI] [PubMed] [Google Scholar]
- 77.Lim SC, Yang JY, Jang AS, et al. 1996. Acute lung injury after phosgene inhalation. Korean J. Intern. Med 11: 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hardison LS, Wright E & Pizon AF. 2014. Phosgene exposure: a case of accidental industrial exposure. J. Med. Toxicol 10: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang C, Yang C, Xiaogang Y, et al. 2019. Management of phosgene-induced acute lung injury (ALI) by personalized protective PEEP-ECMO: what can we learn from animal bioassays? J. Integrat. Cardiol Open Access 1–9. [Google Scholar]
- 80.Vaish AK, Consul S, Agrawal A, et al. 2013. Accidental phosgene gas exposure: a review with background study of 10 cases. J. Emerg. Trauma Shock 6: 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aggarwal S, Jilling T, Doran S, et al. 2019. Phosgene inhalation causes hemolysis and acute lung injury. Toxicol. Lett 312: 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sciuto AM, Stotts RR & Hurt HH. 1996. Efficacy of ibuprofen and pentoxifylline in the treatment of phosgene-induced acute lung injury. J. Appl. Toxicol 16: 381–384. [DOI] [PubMed] [Google Scholar]
- 83.Ji L, Liu R, Zhang XD, et al. 2010. N-acetylcysteine attenuates phosgene-induced acute lung injury via upregulation of Nrf2 expression. Inhal. Toxicol 22: 535–542. [DOI] [PubMed] [Google Scholar]
- 84.Filipczak PT, Senft AP, Seagrave J, et al. 2015. NOS-2 inhibition in phosgene-induced acute lung injury. Toxicol. Sci 146: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laskin DL, Sunil VR, Fakhrzadeh L, et al. 2010. Macrophages, reactive nitrogen species, and lung injury. Ann. N.Y. Acad. Sci 1203: 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li W-L, Hai C-X & Pauluhn J. 2011. Inhaled nitric oxide aggravates phosgene model of acute lung injury. Inhal. Toxicol 23: 842–852. [DOI] [PubMed] [Google Scholar]
- 87.Zhang J, Shao Y, He D, et al. 2016. Evidence that bone marrow-derived mesenchymal stem cells reduce epithelial permeability following phosgene-induced acute lung injury via activation of wnt3a protein-induced canonical wnt/β-catenin signaling. Inhal. Toxicol 28: 572–579. [DOI] [PubMed] [Google Scholar]
- 88.Zhang L, Zhang F, He D, et al. 2017. Melatonin attenuates phosgene-induced acute lung injury via the upregulation Wnt/β-catenin pathway. Int. J. Clin. Exp. Pathol. 10:11281–11287. [PMC free article] [PubMed] [Google Scholar]
- 89.Shao Y, Zhou F, He D, et al. 2019. Overexpression of CXCR7 promotes mesenchymal stem cells to repair phosgene-induced acute lung injury in rats. Biomed. Pharmacother 109: 1233–1239. [DOI] [PubMed] [Google Scholar]
- 90.Xu N, Shao Y, Ye K, et al. 2019. Mesenchymal stem cell-derived exosomes attenuate phosgene-induced acute lung injury in rats. Inhal. Toxicol 31: 52–60. [DOI] [PubMed] [Google Scholar]
- 91.Simoes DCM, Vassilakopoulos T, Toumpanakis D, et al. 2008. Angiopoietin-1 protects against airway inflammation and hyperreactivity in asthma. Am. J. Respir. Crit. Care Med 177: 1314–1321. [DOI] [PubMed] [Google Scholar]
- 92.He D-K, Chen J-F, Shao Y-R, et al. 2018. Adenovirus-delivered angiopoietin-1 ameliorates phosgene-induced acute lung injury via inhibition of NLRP3 inflammasome activation. Inhal. Toxicol 30: 187–194. [DOI] [PubMed] [Google Scholar]
- 93.He D-K, Shao Y-R, Zhang L, et al. 2014. Adenovirus-delivered angiopoietin-1 suppresses NF-κB and p38 MAPK and attenuates inflammatory responses in phosgene-induced acute lung injury. Inhal. Toxicol 26: 185–192. [DOI] [PubMed] [Google Scholar]
- 94.Shao Y, Shen J, Zhou F, et al. 2018. Mesenchymal stem cells overexpressing Ang1 attenuates phosgene-induced acute lung injury in rats. Inhal. Toxicol 30: 313–320. [DOI] [PubMed] [Google Scholar]
- 95.Carlisle M, Lam A, Svendsen ER, et al. 2016. Chlorine-induced cardiopulmonary injury. Ann. N.Y. Acad. Sci 1374: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Svendsen ER 2018. Chlorine countermeasures: supplemental oxygen equals supplemental lung injury? Am. J. Respir. Cell Mol. Biol 58: 10–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoyle GW, Chen J, Schlueter CF, et al. 2016. Development and assessment of countermeasure formulations for treatment of lung injury induced by chlorine inhalation. Toxicol. Appl. Pharmacol 298: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Evans RB 2005. Chlorine: state of the art. Lung 183: 151–167. [DOI] [PubMed] [Google Scholar]
- 99.White CW & Martin JG. 2010. Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proc. Am. Thorac. Soc 7: 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Withers RMJ & Lees FP. 1985. The assessment of major hazards: the lethal toxicity of chlorine. J. Hazard. Mater 12: 283–302. [Google Scholar]
- 101.Das R & Blanc PD. 1993. Chlorine gas exposure and the lung: a review. Toxicol. Ind. Health 9: 439–455. [DOI] [PubMed] [Google Scholar]
- 102.Van Sickle D, Wenck MA, Belflower A, et al. 2009. Acute health effects after exposure to chlorine gas released after a train derailment. Am. J. Emerg. Med 27: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Batchinsky AI, Martini DK, Jordan BS, et al. 2006. Acute respiratory distress syndrome secondary to inhalation of chlorine gas in sheep. J. Trauma 60: 944–956; discussion 956–957. [DOI] [PubMed] [Google Scholar]
- 104.Musah S, Schlueter CF, Humphrey DM, et al. 2017. Acute lung injury and persistent small airway disease in a rabbit model of chlorine inhalation. Toxicol. Appl. Pharmacol 315: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mo Y, Chen J, Humphrey DM, et al. 2015. Abnormal epithelial structure and chronic lung inflammation after repair of chlorine-induced airway injury. Am. J. Physiol. Lung Cell Mol. Physiol 308: L168–L178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zarogiannis SG, Wagener BM, Basappa S, et al. 2014. Postexposure aerosolized heparin reduces lung injury in chlorine-exposed mice. Am. J. Physiol. Lung Cell Mol. Physiol 307: L347–L354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Massa CB, Scott P, Abramova E, et al. 2014. Acute chlorine gas exposure produces transient inflammation and a progressive alteration in surfactant composition with accompanying mechanical dysfunction. Toxicol. Appl. Pharmacol 278: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aggarwal S, Lazrak A, Ahmad I, et al. 2020. Heme impairs alveolar epithelial sodium channels post toxic gas inhalation. Physiology 10.1101/2020.01.22.909879. [DOI] [Google Scholar]
- 109.Menaouar A, Anglade D, Baussand P, et al. 1997. Chlorine gas induced acute lung injury in isolated rabbit lung. Eur. Respir. J 10: 1100–1107. [DOI] [PubMed] [Google Scholar]
- 110.Elfsmark L, Ågren L, Akfur C, et al. 2018. 8-isoprostane is an early biomarker for oxidative stress in chlorine-induced acute lung injury. Toxicol. Lett 282: 1–7. [DOI] [PubMed] [Google Scholar]
- 111.Martin JG, Campbell HR, Iijima H, et al. 2003. Chlorine-induced injury to the airways in mice. Am. J. Respir. Crit. Care Med 168: 568–574. [DOI] [PubMed] [Google Scholar]
- 112.Leustik M, Doran S, Bracher A, et al. 2008. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am. J. Physiol. Lung Cell Mol. Physiol 295: L733–L743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zarogiannis SG, Jurkuvenaite A, Fernandez S, et al. 2011. Ascorbate and deferoxamine administration after chlorine exposure decrease mortality and lung injury in mice. Am. J. Respir. Cel Mol. Biol 45: 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Samal AA, Honavar J, Brandon A, et al. 2012. Administration of nitrite after chlorine gas exposure prevents lung injury: effect of administration modality. Free Radic. Biol. Med 53: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jurkuvenaite A, Benavides GA, Komarova S, et al. 2015. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic. Biol. Med 85: 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aggarwal S, Ahmad I, Lam A, et al. 2018. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight 3: e120694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hamanaka K, Jian M-Y, Weber DS, et al. 2007. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am. J. Physiol. Lung Cell Mol. Physiol 293: L923–L932. [DOI] [PubMed] [Google Scholar]
- 118.Balakrishna S, Song W, Achanta S, et al. 2014. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol 307: L158–L172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hoyle GW 2010. Mitigation of chlorine lung injury by increasing cyclic AMP levels. Proc. Am. Thorac. Soc 7:284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chang W, Chen J, Schlueter CF, et al. 2012. Inhibition of chlorine-induced lung injury by the type 4 phosphodiesterase inhibitor rolipram. Toxicol. Appl. Pharmacol 263: 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Song W, Wei S, Liu G, et al. 2011. Postexposure administration of a {beta}2-agonist decreases chlorine-induced airway hyperreactivity in mice. Am. J. Respir. Cell Mol Biol 45: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen J, Mo Y, Schlueter CF, et al. 2013. Inhibition of chlorine-induced pulmonary inflammation and edema by mometasone and budesonide. Toxicol. Appl. Pharmacol 272: 408–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wigenstam E, Koch B, Bucht A, et al. 2015. N-acetyl cysteine improves the effects of corticosteroids in a mouse model of chlorine-induced acute lung injury. Toxicology 328: 40–47. [DOI] [PubMed] [Google Scholar]
- 124.Audi J, Belson M, Patel M, et al. 2005. Ricin poisoning: a comprehensive review. JAMA 294: 2342–2351. [DOI] [PubMed] [Google Scholar]
- 125.Gal Y, Mazor O, Falach R, et al. 2017. Treatments for pulmonary ricin intoxication: current aspects and future prospects. Toxins (Basel) 9. 10.3390/toxins9100311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sapoznikov A, Gal Y, Falach R, et al. 2019. Early disruption of the alveolar–capillary barrier in a ricin-induced ARDS mouse model: neutrophil-dependent and -independent impairment of junction proteins. Am. J. Physiol. Lung Cell Mol. Physiol 316: L255–L268. [DOI] [PubMed] [Google Scholar]
- 127.Katalan S, Falach R, Rosner A, et al. 2017. A novel swine model of ricin-induced acute respiratory distress syndrome. Dis. Model. Mech 10: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moshiri M, Hamid F & Etemad L. 2016. Ricin toxicity: clinical and molecular aspects. Rep. Biochem. Mol. Biol 4: 60–65. [PMC free article] [PubMed] [Google Scholar]
- 129.Wilhelmsen CL & Pitt ML. 1996. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol 33: 296–302. [DOI] [PubMed] [Google Scholar]
- 130.Pratt TS, Pincus SH, Hale ML, et al. 2007. Oropharyngeal aspiration of ricin as a lung challenge model for evaluation of the therapeutic index of antibodies against ricin A—chain for post-exposure treatment. Exp. Lung Res 33: 459–481. [DOI] [PubMed] [Google Scholar]
- 131.Poli MA, Rivera VR, Pitt ML, et al. 1996. Aerosolized specific antibody protects mice from lung injury associated with aerosolized ricin exposure. Toxicon 34: 1037–1044. [DOI] [PubMed] [Google Scholar]
- 132.Cavalli G & Dinarello CA. 2018. Anakinra therapy for non-cancer inflammatory diseases. Front. Pharmacol 9: 1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lindauer ML, Wong J, Iwakura Y, et al. 2009. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J. Immunol 183: 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gal Y, Mazor O, Alcalay R, et al. 2014. Anti-body/doxycycline combined therapy for pulmonary ricinosis: attenuation of inflammation improves survival of ricin-intoxicated mice. Toxicol. Rep 1: 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gal Y, Sapoznikov A, Falach R, et al. 2016. Potent antiedematous and protective effects of ciprofloxacin in pulmonary ricinosis. Antimicrob. Agents Chemother 60: 7153–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Smallshaw JE, Richardson JA & Vitetta ES. 2007. RiVax, a recombinant ricin subunit vaccine, protects mice against ricin delivered by gavage or aerosol. Vaccine 25: 7459–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Griffiths GD, Phillips GJ & Bailey SC. 1999. Comparison of the quality of protection elicited by toxoid and peptide liposomal vaccine formulations against ricin as assessed by markers of inflammation. Vaccine 17: 2562–2568. [DOI] [PubMed] [Google Scholar]
- 138.Roy CJ, Brey RN, Mantis NJ, et al. 2015. Thermostable ricin vaccine protects rhesus macaques against aerosolized ricin: epitope-specific neutralizing antibodies correlate with protection. Proc. Natl. Acad. Sci. U.S.A 112: 3782–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]